
Abstract
A series of sulfonamide benzoquinazolinones 5–18 was synthesized and evaluated for cytotoxic activity against MDA-MB-231 cell line. The compounds showed IC50 ranging from 0.26 to 161.49 µM. The promising compounds were evaluated for their inhibitory profile against epidermal growth factor (EGFR) and HER2 enzymes. Compound 10 showed more potent activity on both EGFR and HER2 than erlotinib (IC50 3.90 and 5.40 µM versus 6.21 and 9.42 µM). The pro-apoptotic activity of 10 was evaluated against caspase-3, Bax, B-cell lymphoma protein 2 (Bcl-2) expression levels, and cell cycle analysis. Compound 10 increased the level of caspase-3 by 10 folds, Bax level by 9 folds, decreased the level of the Bcl-2 by 0.14 and arrested the cell cycle in the G2/M phase. The radio-sensitizing activity of 10 was measured using a single dose of 8 Gy gamma radiation (IC50 decreased from 0.31 to 0.22 µM). Molecular docking was performed on EGFR and HER2 receptors.
Introduction
The major challenge in cancer therapy is the induction of apoptosis through anticancer agentsCitation1–3. Apoptosis is a crucial process in maintaining normal tissue homeostasis in the human body, mediated by signal transduction pathways. The two major apoptotic pathways are extrinsic and intrinsic. The extrinsic pathway is induced by the trans-membrane death receptors, while the intrinsic is through mitochondrial stress caused by DNA damage and heat shockCitation4. Activated caspases are the executioners of apoptosisCitation5. So, more effective therapeutic strategies for better understanding of signaling pathways and molecular targets should be further provided.
Breast cancer is the world’s second leading cause of cancer-related deathCitation6. The overexpression of the HER2 enzyme in breast cancer is correlated with poor prognosis and drug resistanceCitation7. HER2 belongs to the epidermal growth factor family (EGFR), also called ErbB. It is a member of receptor tyrosine kinases (TKs) involved in signaling pathways controlling angiogenesis, cell differentiation, and proliferationCitation8. The EGFR consists of a subfamily of EGFR (HER1), HER2, HER3, and HER4, that are only expressed at low levels in normal human tissuesCitation9. Although most patients with EGFR mutant cancers respond to therapies, the patients develop resistance after an average of one year on treatmentCitation10. The resistance to HER2 targeted therapies is associated with the overexpression of EGFR family enzymesCitation11. It is obvious that HER family is interdependent and shows functional redundancy. The blockage of one HER receptor can be compensated by another HER family memberCitation9,Citation12. The cross-linking and compensatory activities of the HER family members can provide a strong rationale for co-targeting of both EGFR and HER2 enzymes.
Molecular hybridization is a simple and effective tool to combine covalently two drug pharmacophores into a single moleculeCitation13. Lately, it has been observed that benzo[g]quinazoline and sulfonamides demonstrated profound growth inhibitory activity against different cancer cells and TK enzymesCitation14,Citation15. The quinazoline is a privileged scaffold that constitutes an important class of heterocyclic compounds owing to its varies pharmacological propertiesCitation16,Citation17. Afatinib, lapatinib, gefitinib, and erlotinib () are the representative drugs in this class in clinical use for targeted anticancer therapiesCitation18–21. The use of them has paved the way to develop new quinazoline-based molecules acting as EGFR inhibitors. Also, it is well-known that sulfonamides are strongly related to anticancer activityCitation22,Citation23. They have several targets, most of which are directly connected to oncogenesisCitation24. They proved to exhibit good activity through many mechanisms as carbonic anhydraseCitation24, matrix metalloprotienaseCitation25, NADPH reductaseCitation26, histone deacetylaseCitation27, and PI3K inhibitionCitation28.
Figure 1. Examples of dual EGFR/HER2 inhibitors.
In this context, we desire to exploit newer leads with tuneable anticancer activity and low toxicityCitation14,Citation29. A series of substituted benzo[g]quinazolinone benzenesulfonamide hybrids were designed, synthesized, and evaluated as dual EGFR/HER2 inhibitors. The apoptotic activity of the most potent compound was evaluated through the activation of the proteolytic caspase-3, Bax and B-cell lymphoma protein 2 (Bcl2) expression levels, cell cycle analysis, and radio-sensitizing activity. Molecular docking was carried out inside the binding site of EGFR and HER2 receptors in order to confirm their possible mechanism of action.
Materials and methods
Melting points were uncorrected and measured on a Gallen Kamp melting point apparatus (Sanyo Gallen Kamp, UK). Precoated silica gel plates (Kieselgel 0.25 mm, 60 F254, Merck, Germany) were used for TLC with a developing solvent system of chloroform/methanol (7:3) and detected by the UV lamp. IR spectra were recorded using FT-IR spectrophotometer (Perkin Elmer, USA). NMR spectra were scanned on an NMR spectrophotometer (Bruker AXS Inc., Switzerland) operating at 500 MHz for 1H and 125.76 MHz for 13C. Chemical shifts are expressed in δ-values (ppm) relative to TMS as an internal standard, using DMSO-d6 as a solvent. Mass spectra were recorded on ISQ LT Thermo Scientific GCMS model (Massachusetts, USA). Elemental analyses were performed on a model 2400 CHNSO analyser (Perkin Elmer, USA). All the values were within ±0.4% of the theoretical values. All reagents were obtained from Sigma-Aldrich of AR grade.
Chemistry
2-[(4-Oxo-3-(4-sulfamoylphenyl)-3,4-dihydrobenzo[g]quinazolin-2-yl)thio]-N-substituted acetamide derivatives (5–18)
General procedure
A mixture of 4 (0.383 g, 0.001 mol) and 2-chloro-N-substituted acetamide derivatives (0.001 mol) in dry acetone (30 ml) and anhydrous K2CO3 (0.138 g, 0.001 mol) was stirred at room temperature for 10 h. The mixture was filtered and the product formed was crystallized from ethanol to give 5–18.
N-(5-Methylisoxazol-3-yl)-2-[(4-oxo-3–(4-sulfamoylphenyl)-3,4-dihydrobenzo[g]quinazolin-2-yl)thio]acetamide (5): Yield, 68%; m.p. 292.4 °C. IR: 3403, 3305, 3190 (NH2, NH), 3095 (arom.), 2980, 2922 (aliph.), 1693, 1665 (2CO), 1631 (CN), 1340, 1161 (SO2). 1HNMR: 2.10 (s, 3H, CH3), 4.21 (s, 2H, S-CH2), 7.02 (s, 1H, CH isoxazole), 7.61–8.20 (m, 10H, Ar-H), 8.81 (s, 2H, SO2NH2), 9.50 (s, 1H, NH). 13CNMR: 18.5, 30.2, 92.7, 119.3, 119.9 (2), 124.1, 126.8 (2), 126.9, 127.7 (2), 128.0, 128.4, 130.6, 131.8, 133.7, 135.8, 145.9, 149.1, 161.2, 162.5, 169.7, 170.2. MS m/z (%): 521 (M+), 383 (100). Anal. Calcd. for C24H19N5O5S2 (521.08): C, 55.27; H, 3.67; N, 13.43. Found: C, 55.49; H, 3.98; N, 13.76.
2-[(4-Oxo-3-(4-sulfamoylphenyl)-3,4-dihydrobenzo[g]quinazolin-2-yl)thio]-N-(thiazol-2-yl)acetamide (6): Yield, 73%; m.p. 304.0 °C. IR: 3410, 3381, 3111 (NH2, NH), 3100 (arom.), 2970, 2881 (aliph.), 1741, 1693 (2CO), 1601 (CN), 1365, 1163 (SO2). 1HNMR: 4.20 (s, 2H, S-CH2), 7.01–8.20 (m, 12H, Ar-H), 8.82–8.88 (m, 3H, SO2NH2+NH). 13CNMR: 27.3, 113.3, 119.4, 123.3 (2), 124.4 (2), 126.6, 128.1, 128.7 (2), 129.4, 129.9, 131.0, 136.8, 137.9, 139.1 (2), 142.8, 155.4, 161.2, 167.1, 168.2. MS m/z (%): 523 (M+) (0.72), 156 (100). Anal. Calcd. for C23H17N5O4S3 (523.61): C, 52.76; H, 3.27; N, 13.38. Found: C, 52.98; H, 3.48; N, 13.74.
N-(6-Ethoxybenzo[d]thiazol-2-yl)-2-[(4-oxo-3-(4-sulfamoylphenyl)-3,4-dihydrobenzo[g]quinazolin-2-yl)thio]acetamide (7): Yield, 78%; m.p. 255.9 °C. IR: 3336, 3210, 3169 (NH2, NH), 3059 (arom.), 2978, 2931 (aliph.), 1680, 1678 (2CO), 1602 (CN), 1355, 1161 (SO2). 1HNMR: 1.32 (t, 3H, J = 10 Hz, CH3 ethoxy), 3.90 (s, 2H, S-CH2), 4.12 (q, 2H, J = 10.5 Hz, CH2 ethoxy), 6.99–8.10 (m, 13H, Ar-H), 8.82–8.86 (m, 3H, SO2NH2+NH). 13CNMR: 15.2, 27.3, 63.9, 105.6, 114.1, 119.3, 120.0, 123.4 (2), 126.5, 127.5 (2), 128.1, 128.6 (2), 129.2, 129.8, 130.9, 131.1, 134.0, 136.9, 139.5, 143.0, 144.4, 154.4, 156.5, 161.4, 170.3, 172.9. MS m/z (%): 617 (M+), 383 (100). Anal. Calcd. for C29H23N5O5S3 (617.09): C, 56.39; H, 3.75; N, 11.34. Found: C, 56.68; H, 4.09; N, 11.71.
N-(6-Nitrobenzo[d]thiazol-2-yl)-2-[(4-oxo-3-(4-sulfamoylphenyl)-3,4-dihydrobenzo[g]quinazolin-2-yl)thio]acetamide (8): Yield, 70%; m.p. 278.3 °C. IR: 3363, 3274, 3220 (NH2, NH), 3071 (arom.), 2929, 2840 (aliph.), 1710, 1695 (2CO), 1597 (CN), 1566, 1336 (NO2), 1336, 1165 (SO2). 1HNMR: 4.30 (s, 2H, S-CH2), 7.51–8.20 (m, 13H, Ar-H), 8.71 (s, 2H, SO2NH2), 8.90 (s, 1H, NH). 13CNMR: 31.1, 119.1, 119.3, 121.8 (2), 122.4 (2), 126.0, 127.4 (2), 128.8, 129.5 (2), 129.8 (2), 131.1 (2), 139.1 (3), 143.0 (2), 157.6 (2), 161.0, 169.2 (2). MS m/z (%): 618 (M+) (4.78), 124 (100). Anal. Calcd. for C27H18N6O6S3 (618.04): C, 52.42; H, 2.93; N, 13.58. Found: C, 52.78; H, 3.21; N, 13.82.
2-[(4-Oxo-3-(4-sulfamoylphenyl)-3,4-dihydrobenzo[g]quinazolin-2-yl)thio]-N-(5-(trifluoromethyl)-1,3,4-thiadiazol-2-yl)acetamide (9): Yield, 81%; m.p. 257.0 °C. IR: 3444, 3284, 3246 (NH2, NH), 3091 (arom.), 2910, 2835 (aliph.), 1715, 1695 (2CO), 1600 (CN), 1400, 1174 (SO2). 1HNMR: 4.20 (s, 2H, S-CH2), 7.63–8.10 (m, 10H, Ar-H), 8.81 (s, 2H, SO2NH2), 11.83 (s, 1H, NH). 13CNMR: 26.9, 119.4 (2), 123.5 (2), 126.5, 127.4 (2), 128.1, 128.6, 129.2 (2), 129.8 (2), 131.1, 136.9, 139.4, 145.7, 156.2 (2), 161.4 (2), 172.4. MS m/z (%): 592 (M+) (2.11), 350 (100). Anal. Calcd. for C23H15F3N6O4S3 (592.03): C, 46.62; H, 2.55; N, 14.18. Found: C, 46.30; H, 2.21; N, 13.93.
N-(3,4-Dimethylphenyl)-2-[(4-oxo-3-(4-sulfamoylphenyl)-3,4-dihydrobenzo[g]quinazolin-2-yl)thio]acetamide (10): Yield, 77%; m.p. 232.8 °C. IR: 3416, 3289, 3143 (NH2, NH), 3063 (arom.), 2948, 2842 (aliph.), 1718, 1691 (2CO), 1631 (CN), 1390, 1160 (SO2). 1HNMR: 2.15 (s, 3H, CH3), 2.18 (s, 3H, CH3), 4.12 (s, 2H, S-CH2), 7.03–8.21 (m, 13H, Ar-H), 8.80 (s, 2H, SO2NH2), 10.31 (s, 1H, NH). 13CNMR: 19.2, 20.0, 27.9, 117.2, 119.4, 120.9, 123.4 (2), 126.6, 127.4 (2), 128.1, 128.8, 129.4 (2), 129.9 (2), 130.0, 131.0, 131.7, 136.8 (2), 136.9, 137.1, 145.8, 155.4, 161.3, 165.6. MS m/z (%): 544 (M+) (1.24), 310 (100). Anal. Calcd. for C28H24N4O4S2 (544.12): C, 61.75; H, 4.44; N, 10.29. Found: C, 62.04; H, 4.69; N, 10.56.
N-(2,5-Dimethylphenyl)-2-[(4-oxo-3-(4-sulfamoylphenyl)-3,4-dihydrobenzo[g]quinazolin-2-yl)thio]acetamide (11): Yield, 78%; m.p. 279.3 °C. IR: 3388, 3269, 3212 (NH2, NH), 3051 (arom.), 2982, 2844 (aliph.), 1693, 1655 (2CO), 1600 (CN), 1328, 1157 (SO2). 1HNMR: 2.02 (s, 3H, CH3), 2.21 (s, 3H, CH3), 4.20 (s, 2H, S-CH2), 7.18–8.34 (m, 13H, Ar-H), 8.86 (s, 2H, SO2NH2), 11.16 (s, 1H, NH). 13CNMR: 19.3, 22.6, 30.2, 110.7, 119.2, 119.9 (2), 122.7, 124.6, 125.2 (2), 127.0, 127.4, 128.6, 128.9 (2), 129.0 (2), 129.9, 130.9, 133.8, 134.6, 135.9, 136.5, 145.2, 155.8, 161.4, 169.0. MS m/z (%): 544 (M+) (2.88), 340 (100). Anal. Calcd. for C28H24N4O4S2 (544.12): C, 61.75; H, 4.44; N, 10.29. Found: C, 61.62; H, 4.11; N, 10.07.
N-(2,6-Dimethylphenyl)-2-[(4-oxo-3-(4-sulfamoylphenyl)-3,4-dihydrobenzo[g]quinazolin-2-yl)thio]acetamide (12): Yield, 89%; m.p. 300.5 °C. IR: 3361, 3269, 3132 (NH2, NH), 3049 (arom.), 2972, 2871 (aliph.), 1699, 1653 (2CO), 1600 (CN), 1355, 1155 (SO2). 1HNMR: 1.78 (s, 6H, 2CH3), 4.22 (s, 2H, S-CH2), 7.54–8.32 (m, 13H, Ar-H), 8.81–8.85 (m, 3H, SO2NH2+NH). 13CNMR: 15.0 (2), 31.1, 119.4, 123.3 (2), 126.6 (2), 127.4 (4), 128.1, 128.8 (2), 129.4, 129.8, 131.0 (2), 136.9 (2), 139.1 (2), 142.9, 145.4, 155.0, 161.3, 166.2. MS m/z (%): 544 (M+) (1.80), 340 (100). Anal. Calcd. for C28H24N4O4S2 (544.12): C, 61.75; H, 4.44; N, 10.29. Found: C, 61.42; H, 4.18; N, 9.99.
N-(2-Methyl-4-nitrophenyl)-2-[(4-oxo-3-(4-sulfamoylphenyl)-3,4-dihydrobenzo[g]quinazolin-2-yl)thio]acetamide (13): Yield, 85%; m.p. 293.8 °C. IR: 3441, 3358, 3240 (NH2, NH), 3057 (arom.), 2978, 2916 (aliph.), 1697, 1664 (2CO), 1627 (CN), 1539, 1340 (NO2), 1357, 1161 (SO2). 1HNMR: 2.04 (s, 3H, CH3), 4.30 (s, 2H, S-CH2), 7.53–8.25 (m, 13H, Ar-H), 8.81 (s, 2H, SO2NH2), 10.03 (s, 1H, NH). 13CNMR: 18.3, 27.4, 105.2, 119.4, 121.2, 123.4 (2), 123.7, 123.9, 126.7 (2), 127.4, 128.2, 128.8 (2), 129.4, 129.9, 131.8, 136.8, 134.7, 139.3, 142.8, 143.8, 145.9, 155.3, 161.3, 167.0. MS m/z (%): 575 (M+) (8.50), 79 (100). Anal. Calcd. for C27H21N5O6S2 (575.09): C, 56.34; H, 3.68; N, 12.17. Found: C, 56.72; H, 3.77; N, 12.50.
N-(2,4-Dioxo-1,2,3,4-tetrahydropyrimidin-5-yl)-2-[(4-oxo-3-(4-sulfamoylphenyl)-3,4-dihydrobenzo[g]quinazolin-2-yl)thio]acetamide (14): Yield, 76%; m.p. 280.0 °C. IR: 3409, 3261, 3217 (NH2, NH), 3100 (arom.), 2972, 2841 (aliph.), 1741, 1701, 1681, 1653 (4CO), 1582 (CN), 1396, 1159 (SO2). 1HNMR: 4.13 (s, 2H, S-CH2), 5.20 (s, 1H, CH uracil), 7.51–8.22 (m, 10H, Ar-H), 8.75 (s, 2H, SO2NH2), 9.42 (s, 2H, 2NH), 10.81 (s, 1H, CONHCO). 13CNMR: 28.2, 78.2, 119.3, 123.9 (2), 126.6, 127.4 (2), 128.2, 128.5 (2), 129.3, 129.8, 131.0, 136.8 (2), 139.0, 142.7, 146.1, 155.2, 161.4 (2), 165.4 (2). MS m/z (%): 550 (M+) (4.50), 79 (100). Anal. Calcd. for C24H18N6O6S2 (550.07): C, 52.36; H, 3.30; N, 15.26. Found: C, 52.72; H, 3.67; N, 15.50.
N-(1,3-Dimethyl-2,6-dioxo-1,2,3,6-tetrahydropyrimidin-4-yl)-2-[(4-oxo-3-(4-sulfamoylphenyl)-3,4-dihydrobenzo[g]quinazolin-2-yl)thio]acetamide (15): Yield, 83%; m.p. 307.7 °C. IR: 3410, 3334, 3171 (NH2, NH), 3086 (arom.), 2963, 2831 (aliph.), 1708, 1691, 1678, 1645 (4CO), 1618 (CN), 1398, 1155 (SO2). 1HNMR: 3.41 (s, 6H, 2CH3), 4.13 (s, 2H, S-CH2), 6.58 (s, 1H, CH uracil), 7.50–8.22 (m, 10H, Ar-H), 8.81 (s, 2H, SO2NH2), 11.30 (s, 1H, NH). 13CNMR: 26.4, 28.6, 31.2, 73.8, 119.3, 123.4 (2), 126.7, 127.4 (2), 128.1, 128.8 (2), 129.4, 129.9, 131.0, 136.8, 139.1 (2), 142.7, 145.8, 155.1, 158.5, 161.3, 166.6, 170.1. MS m/z (%): 578 (M+) (3.42), 89 (100). Anal. Calcd. for C26H22N6O6S2 (578.10): C, 53.97; H, 3.83; N, 14.52. Found: C, 53.68; H, 3.59; N, 14.31.
2-[(4-Oxo-3-(4-sulfamoylphenyl)-3,4-dihydrobenzo[g]quinazolin-2-yl)thio]-N-(pyrazin-2-yl)acetamide (16): Yield, 81%; m.p. 205.7 °C. IR: 3429, 3325, 3246 (NH2, NH), 3060 (arom.), 2959, 2825 (aliph.), 1695, 1681 (2CO), 1629 (CN), 1338, 1157 (SO2). 1HNMR: 4.21 (s, 2H, S-CH2), 7.53–8.42 (m, 13H, Ar-H), 8.87 (s, 2H, SO2NH2), 9.24 (s, 1H, NH). 13CNMR: 28.9, 119.4, 123.4 (2), 126.6, 127.4 (2), 128.0, 128.8 (2), 129.4, 129.9 (2), 131.0, 136.7, 136.8 (2), 139.1, 142.7, 145.9, 149.2, 155.2, 161.3, 167.5. MS m/z (%): 518 (M+) (1.09), 129 (100). Anal. Calcd. for C24H18N6O4S2 (518.08): C, 55.59; H, 3.50; N, 16.21. Found: C, 55.28; H, 3.19; N, 16.03.
N-(Naphthalene-1-yl)-2-[(4-oxo-3-(4-sulfamoylphenyl)-3,4-dihydrobenzo[g]quinazolin-2-yl)thio]acetamide (17): Yield, 78%; m.p. 241.6 °C. IR: 3412, 3296, 3166 (NH2, NH), 3059 (arom.), 2981, 2860 (aliph.), 1741, 1658 (2CO), 1627 (CN), 1348, 1161 (SO2). 1HNMR: 4.33 (s, 2H, S-CH2), 7.45–8.24 (m, 17H, Ar-H), 8.86 (s, 2H, SO2NH2), 10.34 (s, 1H, NH). 13CNMR: 31.3, 108.2, 119.0, 121.2, 121.4, 122.4 (2), 123.4, 126.0 (2), 126.2, 126.5, 126.7 (2), 127.4, 128.1, 128.4 (2), 128.6, 129.5, 129.9, 131.1, 136.9 (2), 141.0 (2), 143.8, 157.9, 161.4, 166.9. MS m/z (%): 566 (M+) (7.12), 114 (100). Anal. Calcd. for C30H22N4O4S2 (566.11): C, 63.59; H, 3.91; N, 9.89. Found: C, 63.78; H, 4.02; N, 10.12.
N-(9,10-Dioxo-9,10-dihydroanthracen-2-yl)-2-[(4-oxo-3-(4-sulfamoylphenyl)-3,4-dihydrobenzo[g]quinazolin-2-yl)thio]acetamide (18): Yield, 85%; m.p. 256.7 °C. IR: 3442, 3279, 3134 (NH2, NH), 3061 (arom.), 2976, 2833 (aliph.), br. 1693, 1670 (4CO), 1629 (CN), 1332, 1161 (SO2). 1HNMR: 4.15 (s, 2H, S-CH2), 7.23–8.16 (m, 17H, Ar-H), 8.87 (s, 2H, SO2NH2), 10.43 (s, 1H, NH). 13CNMR: 31.1, 114.2, 119.6, 119.9 (2), 123.0, 125.6 (2), 125.7, 126.8 (2), 126.9, 127.1, 127.5, 128.8 (2), 128.9, 129.7, 130.4 (2), 131.0, 131.9 (2), 132.9, 133.3, 133.7, 136.0, 143.0, 144.1, 160.6, 160.9, 167.4, 187.1 (2). MS m/z (%): 646 (M+) (5.29), 128 (100). Anal. Calcd. for C34H22N4O6S2 (646.10): C, 63.15; H, 3.43; N, 8.66. Found: C, 63.41; H, 3.70; N, 9.02.
Biological evaluation
MTT assay
MDA-MB-231 breast cancer cells and 184A1 normal breast cells of American Type Culture Collection were obtained from VACSERA, Egypt. Cells were cultured using Dulbecco’s Modified Eagle’s Medium (Invitrogen/Life Technologies) supplemented with 10% FBS (Hyclone), 10 µg/mL of insulin, and 1% penicillin–streptomycin. Cells were seeded in 96-well plate with cells density 1.2–1.8 × 10,000 cells/well, in a volume of 100 µL complete growth medium + 100 µL of the tested compound per well and the plate was incubated for 24 h before the MTT assay. The cell layer was rinsed with 0.25% (w/v) trypsin, 0.53 mM EDTA solution, incubated for 2 h, then the absorbance was measured at a wavelength of 570 nmCitation30. IC50 was calculated according to the equation of Boltzmann sigmoidal concentration-response curve using Graph Pad Prism 5.
In vitro enzymatic activity assay
EGFR and HER2 kinase kits were purchased from Invitrogen. EGFR (PV3872), 0.200 mg/mL and HER2 (PV3366), 0.192 mg/mL were used. ATP solution and a kinase/peptide mixture were prepared. The plate was incubated for 1 h at room temperature. About 5 mL of the developing solution was added to each well. The plate was incubated for 1 h and then read by ELISA Reader (PerkinElmer, USA). Every experiment was repeated three times. Data represented as means ± SE from three independent experiments. Curve fitting was performed using Graph Pad Prism 5.
The effect on caspase-3
The Quantikine-Human active caspase-3 immunoassay (R&D Systems Inc., USA) is used to measure the active caspase-3 level, by adding 100 µL of the standard diluent to the zero standard wells. Cover and incubate for 2 h at room temperature. Add 100 μL of caspase-3 (active) detection antibody solution into each well except the chromogen blank. Incubate for 1 h then add 100 µL anti-rabbit IgG HRP working solution to each well and incubate for 30 min. The absorbance of each well was measured at 450 nm.
The effect on BAX and Bcl-2 levels
Cells were grown in RPMI 1640 containing 10% foetal bovine serum at 37 °C, stimulated with the compounds to be tested for Bax, and lysed with cell extraction buffer. This lysate was diluted in the standard diluent buffer over the range of the assay and measured for human active Bax and Bcl2 content according to the reported methodCitation31.
Analysis of the cell cycle distribution
To determine the effect of compound 10 and erlotinib on the cell cycle distribution of MDA-MB-231 cell line; cell cycle analysis was performed using the CycleTEST™ PLUS DNA Reagent Kit (Becton Dickinson Immunocytometry Systems, San Jose, CA, USA). Control cells with known DNA content (peripheral blood mononuclear cells) were used as a reference point for determining the DNA index for the test samples. The cells were stained with propidium iodide stain following the procedure provided by the kit then incubated at room temperature for 5 min in the dark and run on the DNA cytometer. Cell cycle distribution was calculated using CELLQUEST software (Becton Dickinson Immunocytometry Systems, San Jose, CA, USA).
Radiosensitizing activity
Irradiation was performed at the National Center for Radiation Research and Technology (NCRRT), Egyptian Atomic Energy Authority (EAEA), using gamma cell-40 (137Cs) source. Compound 10 was selected to be re-evaluated for the in vitro antiproliferative activity in combination with γ-irradiation using MTT assay. Cells were incubated with compound 10 in molar concentrations of 0.01, 0.1, 1.0, and 10 µM. After 2 h, cells were subjected to a single dose of 8 Gy of γ-radiation at a dose rate of 0.758 rad/s for 17.73 min, and then the anti-proliferative activity was measured 48 h after irradiation. The IC50 of the tested compounds was calculated after irradiation.
Molecular docking
Molecular modeling was performed using the Molecular Operating Environment (MOE, 10.2008) software. The protein data bank files (PDB: 1M17 and 3RCD) were selected for this purpose. Water molecules were ignored and hydrogen atoms were added. The co-crystallized ligands in both receptors were re-docked into the active site for method standardization. The structure of compound 10 was drawn on ChemDraw and copied as smiles to MOE. Energy minimizations were performed for compound 10 using MMFF94X force field and the partial charges were calculated. Docking of 10 inside the active site of the enzyme to generate one hundred conformations. Top-scored conformation was captured by 2D and 3D images.
Results and discussion
Chemistry
The synthesis of the target compounds 5–18 was described in Scheme 1. The starting compound 4-(2-mercapto-4-oxobenzo[g]quinazolin-3(4H)-yl) benzenesulfonamide 4Citation29was prepared from the reaction of 3-amino-2-naphthoic acid 3 with 4-isothiocyanatobenzenesulfonamide 2Citation22. The reaction of 4 with 2-chloro-N-substituted acetamide derivatives in dry acetone containing an equimolar amount of anhydrous K2CO3 yielded the appropriate N-(substituted)-2-[(4-oxo-3-(4-sulfamoylphenyl)-3,4-dihydrobenzo[g]quinazolin-2-yl)thio]acetamides 5–18 (Scheme 1). IR spectra of 5–18 revealed NH, CH aliphatic, and CO bands at their specified regions. 1H-NMR spectra of 5–18 revealed two singlets at 3.90–4.33 ppm attributed to the CH2 and 8.81–11.83 ppm attributed to the NH protons and the disappearance of SH singlet at 2.01 ppm of 4. 13C-NMR spectra of 5–18 exhibited two downfield signals attributed to the C–S and CO carbons. The 1HNMR spectrum of 5 revealed two singlets at 2.10 and 7.02 ppm corresponding to the CH3 and CH isoxazole. 13C-NMR of 5 showed an up-field signal at 18.5 ppm due to CH3. 1HNMR of 7 showed triplet at 1.32 ppm and quartet at 4.12 ppm due to the ethoxy group. 13C-NMR of 7 showed two up-field signals at 15.2 and 63.9 ppm due to the ethoxy carbons. IR of 8 revealed the NO2 peaks at 1566 and 1336 cm−1. 13C-NMR of 9 showed a signal at 119.4 ppm for the CF3 carbon. 1HNMR of 10–12 revealed singlets at the range of 1.78–2.21 ppm due to the 2CH3 and 13C-NMR showed two signals in the range of 15.0–22.6 ppm. IR of 13 showed the NO2 peaks at 1539 and 1340 cm−1. 1HNMR of 13 revealed a singlet at 2.04 ppm for the CH3. IR of 14 and 15 showed 4 CO peaks in their specified regions. 1HNMR of 14 revealed two singlets at 5.20 and 10.81 ppm corresponding to the CH uracil and CONHCO, respectively. 1HNMR of 15 revealed two singlets at 3.41 and 6.58 ppm due to 2CH3 and CH uracil, respectively. 13C-NMR of 15 showed two signals at 28.6 and 31.2 ppm for the 2CH3. 13C-NMR of 18 revealed a signal at 187.1 due to the 2CO of anthraquinone.
Scheme 1. Synthesis of the benzo[g]quinazolinone derivatives 4–18.
![Scheme 1. Synthesis of the benzo[g]quinazolinone derivatives 4–18.](/cms/asset/75efb37e-3849-40ee-b3ec-aeed85696faa/ienz_a_1609469_sch0001_b.jpg)
Biological evaluation
In vitro cytotoxic activity against MDA-MB-231 cell line
The in vitro cytotoxicity of the targeted compounds 5–18 was measured using MTT assay against human breast cancer cell line (MDA-MB-231), and erlotinib was used as the reference drug. indicates that compounds 5–18 showed IC50 values in the range of 0.26–161.49 µM, in comparison to erlotinib (IC50= 0.48 µM). Compounds 10, 11, 13, 14, 16, and 18 were more active than the reference drug, with IC50 values in the range of 0.26–0.40 µM. The 9,10-dioxo-9,10-dihydroanthracene derivative 18 was the most active followed by the 2,5-dimethyl phenyl 11, the 3,4-dimethyl phenyl 10, the pyrazinyl 16, the 2,4-dioxopyrimidinyl 14, and the 2-methyl-4-nitrophenyl derivative 13. The EGFR inhibitory profile of the synthesized compounds 5–18 was measured and reported in . The results showed that the tested compounds exhibited inhibitory activity towards EGFR, ranging from 72.90% to 8.71%. The most cytotoxic compounds showed the highest inhibitory profile. Compound 14 showed the highest percentage inhibition followed by 11, 10, erlotinib, 13, and 18 (percentage inhibition ranging from 72.90% to 67.26%).
Table 1. The cytotoxic activity and percentage inhibition of compounds 5–18 on EGFR against MDA-MB-231 breast cancer cell line.
EGFR and HER2 inhibition
The IC50 values of the compounds showing the highest percentage inhibition towards EGFR were determined. Compounds 10, 11, 13, 14, and 18 were screened on both EGFR and HER2 enzymes in reference to erlotinib. From the results in , we can conclude that all the tested compounds together with erlotinib have better inhibitory activity and lower IC50 on EGFR than HER-2 enzyme except for compound 18 (IC50 ranges from 2.55 to 10.20 µM towards EGFR versus 3.20–31.31 µM towards HER2). The 3,4-dimethyl phenyl derivative 10 was more potent than erlotinib on both EGFR and HER2 (IC50 3.90 and 5.40 µM versus 6.21 and 9.42 µM, respectively). Compound 11 was the most potent towards EGFR (IC50 2.55 µM), while compound 18 was the most potent towards HER2 (IC50 3.20 µM).
Table 2. IC50 of compounds 10, 11, 13, 14, and 18 against EGFR and HER2 enzymes.
Activation of caspase-3
Caspase-3 is a member of the cysteine-aspartic acid protease family that plays a crucial role in apoptosisCitation32. It is an inactive proenzyme converted to the active form through caspases 8, 9, and 10Citation33. Caspase-3 is activated in the apoptotic cell by both extrinsic (death ligand) and intrinsic (mitochondrial) pathwaysCitation34 by cleaving multiple proteins in the cells leading to cell deathCitation35. The effect of compound 10 on caspase-3 was evaluated in reference to erlotinib. Compound 10 showed an increase in the level of the active caspase 3 by 10 folds compared to the control cells. While erlotinib increases the level of caspase 3 by 9 folds ().
Table 3. The effect of compound 10 on the level of caspase-3.
Effects on Bcl-2 family proteins
The Bcl-2 family plays a central role in tumour progression or inhibition of mitochondrial intrinsic apoptotic pathwayCitation36. The pro-apoptotic Bax is essential for cell apoptosis. However, the anti-apoptotic Bcl-2 overexpression enhances cell survival by suppressing apoptosisCitation37. Thus, the balance between these two different proteins determines the cell fateCitation38,Citation39. Increments in the Bax/Bcl2 ratio trigger a cascade of caspases that leads to the activation of caspase 3; the apoptosis executionerCitation40. In this study, MDA-MB-231 breast cells were treated with compound 10 and their effect on the expression levels of Bcl2, and Bax were illustrated in .
Table 4. The effect of compound 10 on Bax/Bcl2 expression levels.
Compound 10 and erlotinib boosted the level of the pro-apoptotic protein Bax by 9 and 11 folds, respectively, compared to the control cells. On the other hand, they markedly reduced the levels of the anti-apoptotic proteins Bcl2 by 0.14 and 0.07 folds, respectively. The results showed that both compound 10 and erlotinib markedly boosted the Bax level and down-regulated Bcl2 level proving their pro-apoptotic effect.
Cell cycle analysis
Cell cycle progression is responsible for normal cell growth and proliferation. DNA damage can lead to either DNA repair or cell death through apoptosis. The condition of the cells is assessed at certain checkpoints that act as control mechanisms to ensure the proper cell division. Cell cycle checkpoints are the G1 (restriction), the S (metaphase), and the G2/MCitation41. The role of anticancer agents is to stop the cell division at these checkpoints. Treatment with the anticancer agents can determine at which phase apoptosis occurs in the cell cycle. In this study, MDA-MB-231 cells were treated with compound 10 at its IC50. The results in indicate that compound 10 arrested the cell cycle at the G2/M phase when compared to the untreated control (17.52% and 6.44%, respectively; ). While erlotinib arrested the cell cycle at the G2/M phase by 24.81% (). Also, the cell population in G1 and S phases decreases after treatment (49.36% and 18.28% versus 69.55% and 23.04%, respectively) in case of compound 10 compared to control. While in the case of erlotinib, the cell population in G1 and S phases markedly decreases after treatment to (41.55% and 16.31%, respectively). These results reveal that in MDA-MB-231 cells, cell cycle arrest occurs in the G2/M phase in the case of compound 10 and erlotinib.
Figure 2. The effect of inhibitors on the phases of the cell cycle (A) compound 10, (B) erlotinib, and (C) control MDA-MB-231 cells.
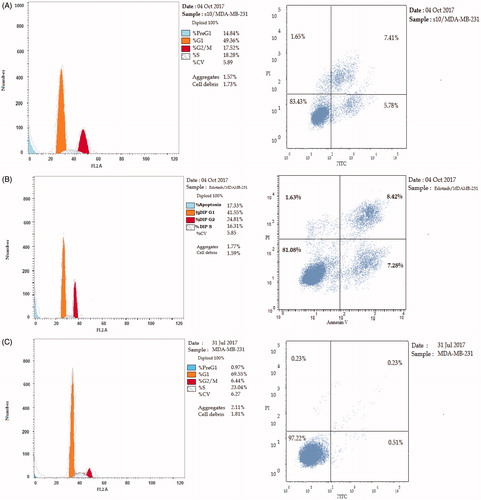
Table 5. The effect of compound 10 and erlotinib on the phases of cell cycle.
Cytotoxicity against normal breast cells
The cytotoxicity of compound 10 compared to erlotinib was measured against 184A1 normal breast cells using MTT assay in order to determine the relative safety of compound 10 on normal tissues. Compound 10 and erlotinib showed mild cytotoxic effect with an IC50 of 84.5 and 101.9 µM, respectively ().
Table 6. The cytotoxicity of compound 10 and erlotinib on 184A1 normal breast cells
Radiosensitizing evaluation
Most cancer patients receive radiation therapy during the course of treatment. Gamma rays are high energy radiation used in therapy to shrink tumours and kill malignant cells by damaging their DNA either directly or indirectly through free radicals formation. The major drawback of radiation therapy is that they cannot differentiate between normal and cancerous cells. So, the use of radiotherapy and selective chemotherapy are required in order to eliminate normal tissue damageCitation42.
The cytotoxicity of compound 10 was measured on MDA-MB-231 cell line before and after being subjected to a single dose of 8 Gy γ-irradiation. The ability of compound 10 to enhance the cell-killing effect of γ-irradiation was examined. The results showed that compound 10 is able to sensitize the cancer cells to the lethal effects of gamma radiation ().
Table 7. IC50 of compound 10 on MDA-MB-231 cells before and after being subjected to a single dose of 8 Gy γ-radiation.
Molecular docking
Molecular docking was performed using MOE 10.2008 inside the active site of EGFR (PDB ID: 1M17)Citation43 and HER2 receptors (PDB ID: 3RCD)Citation44. In order to rationalize the biological results and to gain insight into the SAR of the target compounds, an attempt to interpret the observed enzymatic activities of the tested compounds on the basis of the ligand-protein interactions was done. The enzymatic activity of EGFR and HER2 inhibitors depends on the ability of the compound to properly dock into the binding site and to establish interactions with the key amino acids. Accordingly, the active compound in this study should attain the same binding mode observed for the ligand.
Docking on EGFR
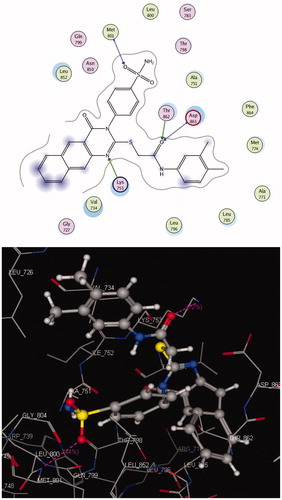
The EGFR catalytic domain consists of an N-terminal lobe, which consists mainly of one α-helix and C-helix. The C-terminal lobe is mainly α-helical, and a short strand termed the hinge region connects the two lobesCitation45. The N-(3-ethynylphenyl)-6,7-bis(2-methoxyethoxy)quinazolin-4-amine (erlotinib) is the co-crystallized ligand inside the EGFR receptorCitation46. Erlotinib was located well in the ATP pocket and interacts with Met 769 by a hydrogen bond of 2.70 A° length, and hydrophobic interactions with Leu 694, Leu 820, Lys 721, and Thr 766 (hinge region; ). Compound 10 was docked in the active site of the enzyme and bound in the same manner as the ligand. Compound 10 binds with energy score (S = −9.88 Kcal/mol) and interact with the active site through Met 769 by a hydrogen bond of 0.85 A°, Cys 773 with the CO of quinazolinone and Phe 699 with the phenyl ring of the acetamide through a π–π interaction (, ).
Figure 3. 2 D and 3 D ligand interactions of erlotinib inside the active site of 1M17.
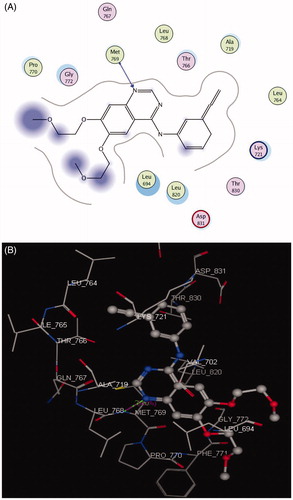
Figure 4. 2 D and 3 D interaction maps of compound 10 inside the active site of 1M17.
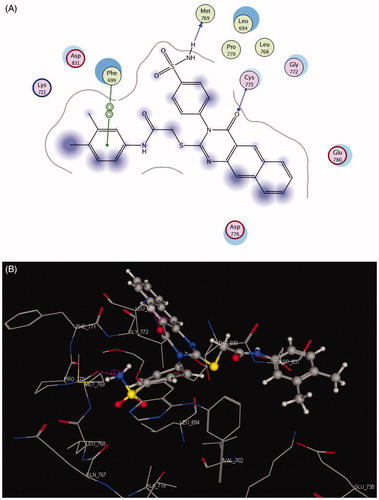
Table 8. Docking results of compound 10 inside 1M17 and 3RCD active sites.
Docking on HER2
The crystal structure of HER2 complexed with TAK-285 (PDB ID: 3RCD) showed that Ala 751, Leu 800, Met 801, Leu 852, and Asp 863 are the key amino acids. The X-ray co-crystallized structure of TAK-285 with HER2 demonstrated that it binds to the ATP pocket through an H-bond with Met 801 and to the hinge region by a series of hydrophobic interactions with Leu 852, Leu 726, Phe 1004, Thr 798, Thr 862, and Leu 785Citation47 (). Compound 10 pursued the similar binding pattern in HER2 with Met 801 by the SO2 of the sulfonamide group, Thr 862 and Asp 863 by the CO of the acetamide and Lys 753 with the N-1 of quinazolinone (, ).
Figure 5. 2 D and 3 D interaction maps of TAK-285 inside the active site of 3RCD.
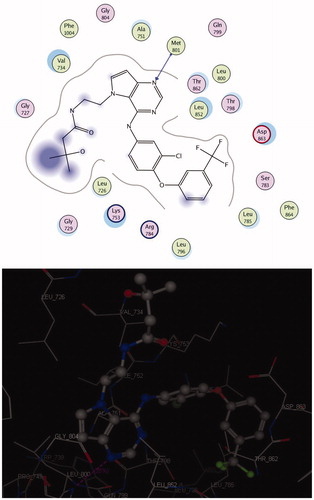
Figure 6. 2 D and 3 D interaction maps of 10 inside the active site of 3RCD.
Conclusion
An array of new 3,4-dihydrobenzo[g]quinazolinone derivatives containing sulfonamide moiety was designed, synthesized, and evaluated for their cytotoxic effect on MDA-MB-231 breast cancer cell line. The tested compounds showed IC50 values ranging from 0.26 to 161.49 µM on MDA-MB-231. The new compounds were tested for their inhibitory profile against EGFR and HER2 enzymes. The 3,4-dimethyl phenyl derivative 10 was more potent than erlotinib on both EGFR and HER2 (IC50 3.90 and 5.40 µM versus 6.21 and 9.42 µM, respectively). The 2,5-dimethyl phenyl derivative 11 was the most potent towards EGFR, while the anthraquinone derivative 18 was the most potent towards HER2. Compound 10 was evaluated as an apoptosis inducer through the activation of the proteolytic caspase-3, Bax and Bcl-2 expression levels, and cell cycle analysis. It was found that compound 10 increases the level of caspase-3 by 10 folds, Bax level by 9 folds, decreases the level of Bcl-2 by 0.14 folds and arrested the cell cycle in the G2/M phase. The radiosensitizing activity of 10 was measured on MDA-MB-231 cell line after being irradiated by a single dose of 8 Gy. IC50 decreased from 0.31 to 0.22 µM after being irradiated. Docking of 10 inside the active site of EGFR and HER2 receptors revealed that it binds in the same manner as that of the co-crystallized ligand.
Acknowledgments
A. M. Soliman and M. M. Ghorab appreciate the staff members of gamma irradiation unit at the National Center for Radiation Research and Technology (NCRRT) for carrying the irradiation process. A. S. Alqahtani is thankful to the Research center, College of Pharmacy, and Deanship of Scientific Research at King Saud University, Riyadh, Saudi Arabia.
Disclosure statement
No potential conflict of interest was reported by the authors.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
References
- Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 1972;26:239–57.
- Fischer U, Janssen K, Schulze-Osthoff K. Cutting-edge apoptosis-based therapeutics. Biodrugs 2007;21:273–97.
- Chen Z, Liang X, Zhang H, et al. A new class of naphthalimide-based antitumor agents that inhibit topoisomerase II and induce lysosomal membrane permeabilization and apoptosis. J Med Chem 2010;53:2589–600.
- Tian Z, Xie S, Du Y, et al. Synthesis, cytotoxicity and apoptosis of naphthalimide polyamine conjugates as antitumor agents. Eur J Med Chem 2009;44:393–9.
- Bredesen DE. Apoptosis: overview and signal transduction pathways. J Neurotrauma 2000;17:801–10.
- DeSantis C, Ma J, Bryan L, Jemal A. Breast cancer statistics, 2013. CA Cancer J Clin 2014;64:52–62.
- Slamon DJ, Clark GM, Wong SG, et al. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987;235:177–82.
- Clauditz TS, Gontarewicz A, Lebok P, et al. Epidermal growth factor receptor (EGFR) in salivary gland carcinomas: potentials as therapeutic target. Oral Oncol 2012;48:991–6.
- Arteaga CL, Engelman JA. ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell 2014;25:282–303.
- Ohashi K, Sequist LV, Arcila ME, et al. Characteristics of lung cancers harboring NRAS mutations. Clin Cancer Res 2013;19:1–8.
- Narayan M, Wilken JA, Harris LN, et al. Trastuzumab-induced HER reprogramming in “resistant” breast carcinoma cells. Cancer Res 2009;69:2191–4.
- Olayioye MA, Neve RM, Lane HA, Hynes NE. The ErbB signaling network: receptor heterodimerization in development and cancer. Embo J 2000;19:3159–67.
- Nagarsenkar A, Prajapti SK, Guggilapu SD, et al. Investigation of triazole-linked indole and oxindole glycoconjugates as potential anticancer agents: novel Akt/PKB signaling pathway inhibitors. Med Chem Comm 2016;7:646–53.
- Alsaid MS, Al-Mishari AA, Soliman AM, et al. Discovery of benzo [g] quinazolin benzenesulfonamide derivatives as dual EGFR/HER2 inhibitors. Eur J Med Chem 2017;141:84–91.
- Ghorab MM, Alsaid MS, Soliman AM, Al-Mishari AA. Benzo[g]quinazolin-based scaffold derivatives as dual EGFR/HER2 inhibitors . J Enzyme Inhib Med Chem 2018;33:67–73.
- Elkamhawy A, Farag AK, Viswanath ANI, et al. Targeting EGFR/HER2 tyrosine kinases with a new potent series of 6-substituted 4-anilinoquinazoline hybrids: design, synthesis, kinase assay, cell-based assay, and molecular docking. Bioorg Med Chem Lett 2015;25:5147–54.
- Zhang C, Li W. Regioselective synthesis of 6-aryl-benzo [h][1, 2, 4]-triazolo [5, 1-b] quinazoline-7, 8-diones as potent antitumoral agents. Bioorg Med Chem Lett 2013;23:5002–5.
- Schuler M, Yang JH, Park K, et al. Afatinib beyond progression in patients with non-small-cell lung cancer following chemotherapy, erlotinib/gefitinib and afatinib: phase III randomized LUX-Lung 5 trial. Ann Oncol 2016;27:417–23.
- Lee CK, Brown C, Gralla RJ, et al. Impact of EGFR inhibitor in non-small cell lung cancer on progression-free and overall survival: a meta-analysis. J Natl Cancer Inst 2013;105:595–605.
- Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA 2004;101:13306–11.
- Yin S, Tang C, Wang B, et al. Design, synthesis and biological evaluation of novel EGFR/HER2 dual inhibitors bearing a oxazolo [4, 5-g] quinazolin-2 (1H)-one scaffold. Eur J Med Chem 2016;120:26–36.
- Ghorab MM, El Ella DAA, Heiba HI, Soliman AM. Synthesis of certain new thiazole derivatives bearing a sulfonamide moiety with expected anticancer and radiosensitizing activities. J Mater Sci Eng A 2011;1:684–91.
- El Ella DAA, Ghorab MM, Heiba HI, Soliman AM. Synthesis of some new thiazolopyrane and thiazolopyranopyrimidine derivatives bearing a sulfonamide moiety for evaluation as anticancer and radiosensitizing agents. Med Chem Res 2012;21:2395–407.
- Supuran CT, Scozzafava A. Carbonic anhydrase inhibitors. Curr Med Chem Immunol Endocr Metab Agents 2001;1:61–97.
- Casini A, Scozzafava A, Supuran CT. Sulfonamide derivatives with protease inhibitory action as anticancer, anti-inflammatory and antiviral agents. Expert Opin Ther Pat 2002;12:1307–27.
- Villar R, Encio I, Migliaccio M, et al. activity of lipophilic sulfonamide derivatives of the benzo [b] thiophene 1, 1-dioxide. Bioorg Med Chem 2004;12:963–8.
- Payne JE, Bonnefous C, Hassig CA, et al. Identification of KD5170: a novel mercaptoketone-based histone deacetylase inhibitor. Bioorg Med Chem Lett 2008;18:6093–6.
- Wurz RP, Liu L, Yang K, et al. Synthesis and structure–activity relationships of dual PI3K/mTOR inhibitors based on a 4-amino-6-methyl-1, 3, 5-triazine sulfonamide scaffold. Bioorg Med Chem Lett 2012;22:5714–20.
- Ghorab MM, Alsaid MS, Soliman AM, Ragab FA. VEGFR-2 inhibitors and apoptosis inducers: synthesis and molecular design of new benzo [g] quinazolin bearing benzenesulfonamide moiety. J Enzyme Inhib Med Chem 2017;32:893–907.
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983;65:55–63.
- Emily HYC, Wei MC, Weiler S, et al. BCL-2, BCL-X L sequester BH3 domain-only molecules preventing BAX-and BAK-mediated mitochondrial apoptosis. Mol Cell 2001;8:705–11.
- Alnemri ES, Livingston DJ, Nicholson DW, et al. Human ICE/CED-3 protease nomenclature. Cell 1996;87:171.
- Harrington HA, Ho KL, Ghosh S, Tung K. Construction and analysis of a modular model of caspase activation in apoptosis. Theor Biol Med Model 2008;5:26–41.
- Ghavami S, Hashemi M, Ande SR, et al. Apoptosis and cancer: mutations within caspase genes. J Med Genet 2009;46:497–510.
- Rudel T. Caspase inhibitors in prevention of apoptosis. Herz 1999;24:236–41.
- Tsujimoto Y. Cell death regulation by the Bcl-2 protein family in the mitochondria. J Cell Physiol 2003;195:158–67.
- Hasnan J, Yusoff M, Damitri T, et al. Relationship between apoptotic markers (Bax and Bcl-2) and biochemical markers in type 2 diabetes mellitus. Singapore Med J 2010;51:50–6.
- Callagy GM, Webber MJ, Pharoah PD, Caldas C. Meta-analysis confirms BCL2 is an independent prognostic marker in breast cancer. BMC Cancer 2008;8:153–8.
- McDonnell TJ, Korsmeyer SJ. Progression from lymphoid hyperplasia to high-grade malignant lymphoma in mice transgenic for the t(14; 18)). Nature 1991;349:254–6.
- Zhu H, Zhang J, Xue N, et al. Novel combretastatin A-4 derivative XN0502 induces cell cycle arrest and apoptosis in A549 cells. Invest New Drugs 2010;28:493–501.
- MacLachlan TK, Sang N, Giordano A. Cyclins, cyclin-dependent kinases and cdk inhibitors: implications in cell cycle control and cancer. Crit Rev Eukaryot Gene Expr 1995;5:127–56.
- DeVita VT, Lawrence TS, Rosenberg SA. Cancer: principles & practice of oncology: primer of the molecular biology of cancer. Philadelphia, PA: Lippincott Williams & Wilkins; 2012.
- Stamos J, Sliwkowski MX, Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J Biol Chem 2002;277:46265–72.
- Ishikawa T, Seto M, Banno H, et al. Design and synthesis of novel human epidermal growth factor receptor 2 (HER2)/epidermal growth factor receptor (EGFR) dual inhibitors bearing a pyrrolo [3, 2-d] pyrimidine scaffold. J Med Chem 2011;54:8030–50.
- Lin J, Shen W, Xue J, et al. Novel oxazolo[4,5-g]quinazolin-2(1H)-ones: dual inhibitors of EGFR and Src protein tyrosine kinases. Eur J Med Chem 2012;55:39–48.
- Halmos B, Pennell NA, Fu P, et al. Randomized phase II trial of erlotinib beyond progression in advanced erlotinib-responsive non-small cell lung cancer. Oncologist 2015;20:1298–303.
- Yousuf Z, Iman K, Iftikhar N, Mirza MU. Structure-based virtual screening and molecular docking for the identification of potential multi-targeted inhibitors against breast cancer. Breast Cancer (Dove Med Press) 2017;9:447–59.