
Abstract
Chagas disease and leishmaniasis are neglected tropical disorders caused by the protozoans Trypanosoma cruzi and Leishmania spp. Carbonic anhydrases (CAs, EC 4.2.1.1) from these protozoans (α-TcCA and β-LdcCA) have been validated as promising targets for chemotherapic interventions. Many anti-protozoan agents, such as nitroimidazoles, nifurtimox, and benznidazole possess a nitro aromatic group in their structure which is crucial for their activity. As a continuation of our previous work on N-nitrosulfonamides as anti-protozoan agents, we investigated benzenesulfonamides bearing a nitro aromatic moiety against TcCA and LdcCA, observing selective inhibitions over human off-target CAs. Selected derivatives were assessed in vitro in different developmental stages of T. cruzi and Leishmania spp. A lack of significant growth inhibition has been found, which has been connected to the low permeability of this class of derivatives through cell membranes. Further strategies necessarily need to be designed for targeting Chagas disease and leishmaniasis with nitro-containing CA inhibitors.
1. Introduction
Chagas disease (American trypanosomiasis) and leishmaniasis are potentially life-threatening illnesses that have been included in the list of neglected tropical diseases (NTDs) by the World Health Organization (WHO). These infections belong to the vector-borne diseases affecting 20 million people and killing more than 50,000 every year and are caused by parasites of the kinetoplastida family (Trypanosoma cruzi and Leishmania sp.)Citation1. Kissing bugs of the Triatoma and Rhodnius genera naturally transmit T. cruzi that is primarily diffused in Latin America. Chagas disease progresses by damaging organs in the cardiac, digestive, or nervous systemsCitation1. The bite of infected phlebotomines instead is the main cause of Leishmania transmission and potentially generates skin or visceral fatal damages. Leishmaniasis is the first-in-class NTD in terms of mortality and morbidityCitation1.
To date, a limited arsenal of anti-protozoan agents is available for the treatment of these NTDs. These drugs are marked by high toxicity and limited efficacy, and resistance phenomena are constantly increasing worldwideCitation2–4. The poor interest shown by the pharmaceutical industry in searching new effective drugs for NTDs treatment is related to high costs and expected low financial return. On the contrary, it should be considered a priority to find new approaches in the treatment of these parasitosisCitation2,Citation5. Large-scale analysis on the completely known genome sequence of both protozoans have recently provided the identification of new enzymatic targetsCitation6,Citation7.
The enzymes carbonic anhydrases (CAs, EC 4.2.1.1) identified in these protozoans, TcCA in T. cruzi and LdcCA in L. donovani (a parasite from the Leishmania complex, causing visceral leishmaniasis) have recently been recognised as suitable targets to fight these infectionsCitation6,Citation8,Citation9. CAs are natural catalysts that speed up the rate of CO2 conversion to bicarbonate and proton. This reaction was shown to be basic in the growth and virulence of pathogenic microorganismsCitation9. TcCA and LdcCA were both cloned and characterised in 2013Citation10–12. Many inhibitors of these isoforms have been identified, which represent potential anti-protozoan agents acting by a new mechanism of action which is probably devoid of cross-resistance to the existing drugs.
TcCA is an α-class enzyme that contains the three highly conserved histidines (His94, His96, and His119) coordinating to zinc ion in the enzyme active site, and glutamic acid (Glu 106) as the gate-keeping residueCitation10. Measurement of the catalytic activity of TcCA in CO2 hydration showed a kcat of 1.21 × 106 s–1, Km of 8.1 × 10−3 M and kcat/Km of 1.49 × 108 M–1 s–1 Citation10. TcCA is inhibited in the nanomolar range by many CA inhibitory chemotypes such as aromatic/heterocyclic sulfonamidesCitation10,Citation13,Citation14, sulfamatesCitation10, thiolsCitation10, anionsCitation15, dithiocarbamatesCitation15, hydroxamatesCitation16, benzoxaborolesCitation17, and N-nitrosulfonamidesCitation18. Thiols, hydroxamates, and N-nitrosulfonamides show in vitro anti-trypanosomal activity, deterring multiple phases in the life cycle of the pathogenCitation10,Citation16,Citation18.
LdcCA is a β-class CA whose catalytic activity evaluation reported a kcat of 9.35 × 105 s–1, Km of 15.8 × 10−3 M, and kcat/Km of 5.9 × 107 M–1 s–1 Citation12. LdcCA was shown to be efficiently inhibited by sulfonamides, heterocyclic thiols, and N-nitrosulfonamides with nanomolar inhibition constantsCitation12,Citation18. Some compounds of the two latter classes showed in vitro anti-leishmanial activity in preliminary assays, causing the reduction of the parasites growth and their deathCitation12,Citation18.
N-Nitrosulfonamides have been designed by us based on the presence of the nitro group in the structure of many anti-protozoan agents, such as the nitroimidazoles, this moiety being pivotal for the drug mechanism of actionCitation18,Citation19. For instance, nifurtimox and benznidazole (Bzn) have been the first effective drugs for treating acute-phase human Chagas infection, with the first being no longer available on the market because of undesirable side effectsCitation6. Considering that sulfonamides are the most effective CAIs known to dateCitation20, we first attached the nitro group on the sulfonamide itself, providing the N-nitro derivatives as a new chemotype exhibiting a selective inhibition of protozoan CAs over human ubiquitous isoformsCitation18. As second design strategy, we report herein, consists in the incorporation of the nitro group on the benzene scaffold bearing the sulfonamide, driven by the aromatic character shown by the nitro moieties present in many anti-protozoan agents, mentioned above. A set of 3-nitrobenzenesulfonamide bearing a variety of substituents on the main scaffold has thus been reported. This set has been recently evaluated also for the inhibition of the human tumour-associated CA IX and XII over the ubiquitous CA I and II and for hypoxia-enhanced anti-proliferative activity on tumour cell linesCitation21. In fact, nitroaromatic groups are subjected to bioreduction processes in hypoxic tissues, which can be exploited to selectively generate cytotoxins against tumour cellsCitation21. Here, the set of nitro-benzenesulfonamides has been screened for the inhibition of TcCA and LdcCA and the most effective derivatives were studied in vitro against different species of Leishmania and T. cruzi.
2. Materials and methods
2.1. Chemistry
The synthesis of 3-nitro-4-hydroxy-sulfonamides 4–24 was reported earlier by our groupCitation21.
2.2. Carbonic anhydrase inhibition
An Applied Photophysics stopped-flow instrument has been used for assaying the CA-catalysed CO2 hydration activityCitation22. Phenol red (at a concentration of 0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 20 mM Hepes (pH 7.5) as buffer, and 20 mM Na2SO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalysed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor, at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The uncatalysed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (0.1 mM) were prepared in distilled-deionised water and dilutions up to 0.01 nM were done thereafter with the assay buffer. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature prior to assay, in order to allow for the formation of the E–I complex. The inhibition constants were obtained by nonlinear least-squares methods using PRISM 3 and the Cheng–Prusoff equation, as reported earlier, and represent the mean from at least three different determinationsCitation23–26. All CA isoforms were recombinant ones obtained in-house as reported earlierCitation27,Citation28.
2.3. Biological assays
2.3.1. Cell cultures
2.3.1.1. Trypansoma cruzi and Leishmania parasites cultures
Epimastigote forms of the T. cruzi clone Dm28cCitation29 and T. cruzi YCitation30 strain were obtained from the Laboratory of Cellular Ultrastructure, FIOCRUZ. L. infantum MHOM/BR/1974/PP75 and L. amazonensis IFLA/BR/1967/PH8 were donated by the Leishmania Type Culture Collection (LTCC) of Oswaldo Cruz Institute/Fiocruz (Rio de Janeiro, Brazil). The parasites were maintained by weekly subcultures in PBHIL medium supplemented with 10% foetal bovine serum (FBS) at 28 °CCitation16.
2.3.1.2. RAW 264.7 macrophage cell line cultures
RAW 264.7 murine macrophages were obtained from the National Institute of Metrology, Quality and Technology (Instituto Nacional de Metrologia, Qualidade e Tecnologia, INMETRO, Rio de Janeiro, Brazil) and maintained in DMEM medium supplemented with 10% FBS at 37 °C in a 5% controlled CO2 atmosphere. Cell maintenance was performed every 48–72 h, time necessary for cells to achieve confluent monolayers.
2.3.2. Inhibitory activity on epimastigotes of Trypanosoma cruzi and promastigotes of Leishmania
The evaluation of anti-parasites activity was performed in 96 well plates where the synthetic compounds were serially diluted in the PHBIL medium supplemented with 10% FBS in concentrations ranging from 2 to 400 µM. Then, parasites (1.8 × 106) were added to each well and the plates incubated for 48 h at 28 °C. The experiment controls were: negative control (culture medium without parasite) and positive culture (culture medium with parasite). Benznidazole and amphotericin B (Amp) were used as reference drugs of T. cruzi and Leishmania, respectively. The minimum inhibitory concentration (MIC) for epimastigotes (T. cruzi DM28c and Y) and promastigotes (L. amazonensis and L. infantum) was performed using resazurin (125 µM) as an indicator of cellular metabolic function. MIC was determined as the lowest concentration of the inhibitor capable of inhibiting in vitro growth of the parasites by spectrophotometric analysis at 490 and 595Citation31. The concentration of drug which reduces parasites number by 50% (IC50) was determined by regression analysis using Microsoft Excel 2013.
2.3.3. Cytotoxicity assay in macrophages
Cytotoxicity was performed using tetrazolium dye (MTT) colorimetric assay. RAW 264.7 macrophages cells were harvested after confluent monolayer achievementCitation32. The cells were washed twice with PBS and a cellular suspension of 106 cells/ml was prepared in fresh DMEM culture medium. Aliquots of 100 µl of the cellular suspension were placed into polystyrene 96-well plates, and then incubated at 37 °C in a 5% CO2 atmosphere for 6 h in order to allow macrophage adherence. After this period, the adherent cells were subjected to treatment with several concentrations of the drugs (2–256 µM), and then incubated for additional 48 h. Finally, 20 µl of MTT solution (5 mg/ml) were added to each well and the plates incubated for 4 h. Macrophage viability was determined after formazan crystals solubilisation with DMSO followed by the absorbance measurement at 570 nm using a SpectraMax M5 spectrophotometer (Molecular Devices, Sunnyvale, CA).
2.3.4. Determination of selectivity index
The selectivity index (SI) of tested drugs was calculated as a ratio of RAW 264.7 macrophages CC50 to parasites IC50. Benznidazole (Sigma-Aldrich, Milan, Italy) and Amp were used as reference drugs.
3. Results and discussion
3.1. Chemistry
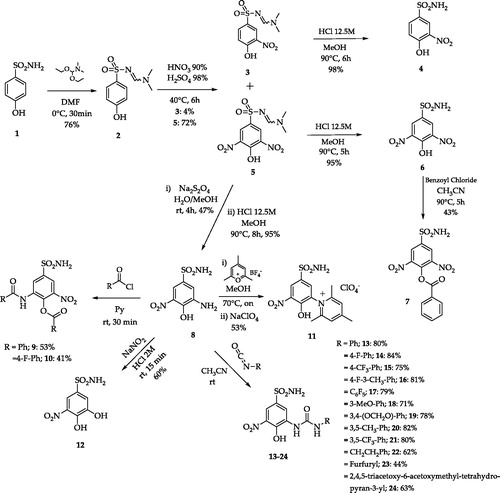
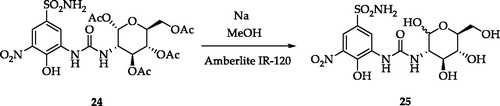
A set of variably substituted 3-nitrobenzenesulfonamides was prepared starting from 4-hydrobenzenesulfonamide 1Citation21. The sulfonamide moiety was protected (compound 2) to avoid decomposition to sulfonic acid that occurs in the nitrating conditions. Both mono- and di-nitro derivatives were obtained in different yields and deprotected in acidic media (compounds 4 and 6). 3,5-Dinitro compound 6 was benzoylated to afford 7. A key intermediate, 3-amino-4-hydroxy-5-nitro-benzenesulfonamide 8 was achieved by reduction of a unique nitro group of 5 with Na2S2O4 and sequential sulfonamide deprotection in acidic media. Intermediate 8 was subjected to several functionalisation reactions. Acylation reactions produced the di-benzoyl compounds 9 and 10. The pyridinium salt 11 was prepared by the reaction of 8 with the proper pyrylium salt. The light-sensitive derivative 12 was obtained by diazonium salt formation and N2 release in aqueous NaNO2Citation21. A set of ureas (13–24) was prepared by the reaction of 8 with commercially available isocyanates, in addition to the freshly prepared one obtained from 1,3,4,6-tetra-O-acetyl-glucosamineCitation21. Compound 24 was de-acetylated with sodium methoxide to give the glycoside 25 (Schemes 1 and 2).
Scheme 1. Synthetic routes to 3-nitrobenzenesulfonamides 3–24Citation21.
Scheme 2. Synthesis of derivative 25.
3.2. Carbonic anhydrase inhibition
The TcCA and LdcCA inhibitory profiles of compounds 4–25 were evaluated by applying a stopped flow carbon dioxide hydrase assayCitation22 in comparison to AAZ as standard CAI and compared to those against the human off-target CA I and II. The following SAR can be built from the inhibition data shown in .
Table 1. Inhibition data of TcCA, LdCA and human CA I and II with sulfonamides reported here and the standard inhibitor acetazolamide (AAZ) by a stopped flow CO2 hydrase assay.
TcCA was effectively inhibited by most 3-nitrobenzenesulfonamides investigated here. Inhibition constants (KIs) span in medium nanomolar to low micromolar range between 0.08 and 10.7 µM. The derivative showing the lowest steric hindrance, namely 4, acts as the most potent TcCA inhibitor with a KI of 80 nM. The incorporation of a nitro, amino or hydroxy moiety in position 5 of compound 4 as in 6, 8, and 11 lowered the inhibition efficacy to 160, 240, and 110 nM, respectively. Lowering of inhibition potency was observed by benzoylation of 6 as in 7 (KI of 2.5 µM) or dybenzoylation of 8 as in 9 and 10 (KIs of 3.5 and 4.8 µM). Hence, enhancement of steric hindrance at position 4 has a deleterious effect on the compounds binding to TcCA. Incorporation of a positively charged moiety, such as pyridinium at position 5 in compound 11, caused an evident drop of inhibitory efficacy (KI of 10.7 µM). Within the set of ureas, the unsubstituted phenyl derivative 13 and the pentafluorinated one 17 showed the most effective inhibition, with KIs of 0.32 and 0.28 µM. All other substitutions on the ureido-aromatic ring negatively affect the inhibition to 0.35–1.35 µM. Insertion of aliphatic linkers between the urea and the outer aromatic portion also has a negative outcome on the KIs of 22 and 23, which increase to 0.74 and 0.4 µM. The glycosidic derivatives 24 and 25 are micromolar TcCA inhibitors with KIs of 2.47 and 2.14 µM.
LdcCA inhibition profiles show analogies with those against TcCA. Again, the simplest derivatives 4, 6, 8, and 12 act as the best LdcCA inhibitors with KIs of 0.21, 0.34, 0.46, and 0.39 µM, respectively. Benzoylation of the hydroxy moiety at position 4 markedly reduced the LdcCA inhibitory properties of 7, 9, and 10 (KIs of 4.68, 3.87, and 8.49 µM) as well as incorporation of the charged pyridinium portion as in 11 (KI of 6.57 µM). Ureido derivatives 13–25 inhibited LdcCA in a rather flat range spanning from 0.86 to 3.65 µM.
As a general trend, most compounds were more effective against TcCA than CA I, with an SI from 2 to >150, with the exception of 9 and 10 (). On the other hand, only few derivatives (8, 12, 13, 18, 19, 22, and 23) inhibited TcCA more efficiently than CA II, with SI spanning between 2.5 and 6. LdcCA was found to be better inhibited than hCA I by most compound, though the SIs were lower than those TcCA/CA I, and spanned in the range of 2–50. Most compounds inhibited CA II better than LdcCA. All compounds inhibited the screened isoforms worse than the standard AAZ, but the latter did not show selectivity for the target TcCA and LdcCA compared to the ubiquitous hCAs ().
Table 2. Selectivity index (SI) for target protozoan CAs over hCA I and II.
3.3. Anti-protozoan activity
3.3.1. Trypanosoma cruzi strain DM28c and Y
Ten selected derivatives bearing different substituents at the 3-nitrobenzenesulfonamide scaffold (4, 6, 8, 10, 11, 17, 18, 19, 21, and 24) were screened for their inhibition activity different species of Leishmania and Trypanosoma cruzi.
The MIC and IC50 values against T. cruzi epimastigote forms of these compounds are shown in . The experiments showed that no compounds significantly affect the growth of the pathogen below 256 µM. The reference drug Bzn showed IC50 values against Dm28c clone and Y strains of 16.56 ± 1.51 and 6.54 ± 1.82 µM, respectively. The assessment of the toxicity of the selected 3-nitrobenzensulfonamides for Raw 267.4 macrophages cells showed that most derivatives were less toxic than Bzn (CC50 of 115.14 ± 9.48 µM) with CC50 above 172.65 ± 10.44 µM. Compounds 4 and 6 showed instead comparable toxicity with Bnz with CC50 values of 97.65 ± 11.13 and 100.21 ± 17.27 µM.
Table 3. Minimum inhibitory concentration (MIC), IC50 values derived from growth inhibition assays of T. cruzi Dm 28c and Y, determination of cytotoxicity (CC50), selectivity index (SI50) of compounds 4, 6, 8, 10, 11, 17, 18, 19, 21, and 24.
3.3.2. L. amazonensis and L. infantum
The MIC and IC50 values of the selected compounds against two Leishmania species are shown in . The experiments on L. amazonensis and L. infantum strains did not show MIC values below 400 µM. Despite the compounds showed micromolar inhibition of LdcCA, their efficacy turned out to be insignificant when translated in vitro against the pathogen cell cultures. The reference drug Amp exhibited IC50 values against the two strains of 1.65 ± 0.28 and 1.77 ± 0.35 µM, respectively. The tested sulfonamides showed anyhow remarkably minor toxicity for Raw 267.4 macrophages cells compared to Amp, that has a CC50 of 1 µM against both strains.
Table 4. Minimum inhibitory concentration (MIC), IC50 values derived from growth inhibition assays of L. amazonensis and L. infantum, determination of cytotoxicity (CC50) and selectivity index (SI50) of compounds 4, 6, 8, 10, 11, 17, 18, 19, 21, and 24.
Unfortunately, the tested 3-nitrobenzenesulfonamides turned out to be ineffective in vitro against strains of T. cruzi and Leishmania. The lack of activity is not a totally new issue in the field of sulfonamide CAIs against pathogens. For instance, some sulfonamide derivatives demonstrated remarkable in vitro efficacy in inhibiting the β-CA from the yeast Malassezia globosa, arousing anyhow complications in vivo because of permeability problems through biological membranesCitation33.
In the context of T. cruzi and Leishmania, some previously tested sulfonamides showed an absence of anti-protozoan efficacy, which has been related to the lack of permeability through the biological membranes of the pathogenCitation34–36. Hence, a formulation of such sulfonamides in nano-emulsions (NEs) of clove oil was attempted to enhance their bioavailability and penetrability through membranesCitation34,Citation35. The drugs–NEs formulations potently inhibited the growth of T. cruzi and Leishmania in vitro, with a huge increase of efficacy over the sulfonamide CAI alone. NEs turned out as a novel vehicle for the delivery of such hydrophilic drugs.
Indeed, it should be noted that 3-nitro-4-hydroxybenzenesulfonamides reported here are even more hydrophilic, which can cause difficulties for the compounds to cross the protozoa cell membrane and inhibit the cytoplasmatic CAs or exert further actions due to the nitro group. Hence, formulation to enhance the compounds bioavailability, such as NEs, is being prepared to evaluate the real anti-protozoan efficacy of these set of nitroaromatic CAIs.
4. Conclusions
We proposed here nitroaromatic sulfonamides for the treatment of Chagas disease and leishmaniasis based on CA inhibition. As a continuation of a previous work of us on N-nitrosulfonamides as anti-protozoan agents, we studied here benzenesulfonamides (4–24) bearing a nitro moiety on the aromatic scaffold against TcCA from T. cruzi, responsible of Chagas disease, and LdcCA from Leishmania spp. The compounds reported valuable micromolar inhibition of these two enzymes, in some cases even selective for the target CAs over the human ubiquitous CA I and II. Unfortunately, a selected set of such derivatives tested in vitro against multiple strains of T. cruzi and Leishmania did not produce growth inhibition of the parasites. The lack of anti-protozoan efficacy of sulfonamide type derivatives had been already reported by us and justified by low permeability of this class of derivatives through the cell membranes. The use of carriers such as nanoemulsions allowed to overcome this issue. The application of this approach has been being carried out for 3-nitrosulfonamides 4–24 to elucidate whether the combination of CA inhibition and further anti-protozoan actions related to the nitro group could be a winning anti-infective strategy.
Disclosure statement
No potential conflict of interest was reported by the authors.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
Related Research Data
References
- WHO. World Health Organization. Available from: http://www.who.int/chagas/en/; http://www.who.int/leishmaniasis/en/.
- Mackey TK, Liang BA, Cuomo R, et al. Emerging and reemerging neglected tropical diseases: a review of key characteristics, risk factors, and the policy and innovation environment. Clin Microbiol Rev 2014;27:949–79.
- Barrett MP, Croft SL. Management of trypanosomiasis and leishmaniasis. Br Med Bull 2012;104:175–96.
- Guedes PM, Silva GK, Gutierrez FR, et al. Current status of Chagas disease chemotherapy. Expert Rev Anti Infect Ther 2011;9:609–20.
- WHO. World Health Organization. Sustaining the drive to overcome the global impact of neglected tropical diseases. Second WHO report on neglected tropical diseases; 2013. Available from: http://www.who.int/neglected_diseases/9789241564540/en/.
- Vermelho AB, Capaci GR, Rodrigues IA, et al. Carbonic anhydrases from Trypanosoma and Leishmania as anti-protozoan drug targets. Bioorg Med Chem 2017;25:1543–55.
- Ortiz C, Moraca F, Medeiros A, et al. Binding mode and selectivity of steroids towards glucose-6-phosphate dehydrogenase from the pathogen Trypanosoma cruzi. Molecules 2016;21:368.
- Supuran CT. Inhibition of carbonic anhydrase from Trypanosoma cruzi for the management of Chagas disease: an underexplored therapeutic opportunity. Future Med Chem 2016;8:311–24.
- (a) Capasso C, Supuran CT. An overview of the alpha-, beta-and gamma-carbonic anhydrases from Bacteria: can bacterial carbonic anhydrases shed new light on evolution of bacteria? J Enzyme Inhib Med Chem 2015;30:325–32. (b) Del Prete S, Vullo D, Fisher GM, et al. Discovery of a new family of carbonic anhydrases in the malaria pathogen Plasmodium falciparum – the η-carbonic anhydrases. Bioorg Med Chem Lett 2014;24:4389–96. (c) Supuran CT. Structure-based drug discovery of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2012;27:759–72. (d) Ozensoy Guler O, Capasso C, Supuran CT. A magnificent enzyme superfamily: carbonic anhydrases, their purification and characterization. J Enzyme Inhib Med Chem 2016;31:689–94. (e) Capasso C, Supuran CT. Bacterial, fungal and protozoan carbonic anhydrases as drug targets. Expert Opin Ther Targets 2015;19:1689–704.
- Pan P, Vermelho AB, Capaci GR, et al. Cloning, characterization, and sulfonamide and thiol inhibition studies of an a-carbonic anhydrase from Trypanosoma cruzi, the causative agent of Chagas disease. J Med Chem 2013;56:1761–71.
- De Menezes DR, Calvet CM, Rodrigues GC, et al. Hydroxamic acid derivatives: a promising scaffold for rational compound optimization in Chagas disease. J Enzyme Inhib Med Chem 2016;31:964–73.
- Syrjanen L, Vermelho AB, Rodrigues IA, et al. Cloning, characterization, and inhibition studies of a b-carbonic anhydrase from Leishmania donovani chagasi, the protozoan parasite responsible for leishmaniasis. J Med Chem 2013;56:7372–81.
- Guzel-Akdemir O, Akdemir A, Pan P, et al. A class of sulfonamides with strong inhibitory action against the a-carbonic anhydrase from Trypanosoma cruzi. J Med Chem 2013;56:5773–81.
- Alafeefy AM, Ceruso M, Al-Jaber NA, et al. A new class of quinazoline-sulfonamides acting as efficient inhibitors against the a-carbonic anhydrase from Trypanosoma cruzi. J Enzyme Inhib Med Chem 2015;30:581–5.
- Pan P, Vermelho AB, Scozzafava A, et al. Anion inhibition studies of the a-carbonic anhydrase from the protozoan pathogen Trypanosoma cruzi, the causative agent of Chagas disease. Bioorg Med Chem 2013;21:4472–6.
- Rodrigues GC, Feijo DF, Bozza MT, et al. Design, synthesis, and evaluation of hydroxamic acid derivatives as promising agents for the management of Chagas disease. J Med Chem 2014;57:298–308.
- Nocentini A, Cadoni R, Dumy P, et al. Carbonic anhydrases from Trypanosoma cruzi and Leishmania donovani chagasi are inhibited by benzoxaboroles. J Enzyme Inhib Med Chem 2018;33:286–9.
- Bonardi A, Vermelho AB, da Silva Cardoso V, et al. N-nitrosulfonamides as carbonic anhydrase inhibitors: a promising chemotype for targeting Chagas disease and leishmaniasis. ACS Med Chem Lett 2019;10:413–8.
- Nocentini A, Vullo D, Bartolucci G, et al. N-Nitrosulfonamides: a new chemotype for carbonic anhydrase inhibition. Bioorg Med Chem 2016;24:3612–7.
- (a) Supuran CT. Structure and function of carbonic anhydrases. Biochem J 2016;473:2023–32. (b) Supuran CT. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin Drug Discov 2017;12:61–88. (c) Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81. (d) Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77. (e) Supuran CT, Vullo D, Manole G, et al. Designing of novel carbonic anhydrase inhibitors and activators. Curr Med Chem Cardiovasc Hematol Agents 2004;2:49–68. (f) Alterio V, Di Fiore A, D’Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68.
- Nocentini A, Trallori E, Singh S, et al. 4-Hydroxy-3-nitro-5-ureido-benzenesulfonamides selectively target the tumor-associated carbonic anhydrase isoforms IX and XII showing hypoxia-enhanced antiproliferative profiles. J Med Chem 2018;61:10860–74.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. J Biol Chem 1971;246:2561–73.
- (a) Nocentini A, Cadoni R, Del Prete S, et al. Benzoxaboroles as efficient inhibitors of the β-carbonic anhydrases from pathogenic fungi: activity and modeling study. ACS Med Chem Lett 2017;8:1194–8. (b) Köhler K, Hillebrecht A, Schulze Wischeler J, et al. Saccharin inhibits carbonic anhydrases: possible explanation for its unpleasant metallic aftertaste. Angew Chem Int Ed Engl 2007;46:7697–9. (c) Supuran CT. Carbonic anhydrase activators. Future Med Chem 2018;10:561–73. (d) Clare BW, Supuran CT. Carbonic anhydrase activators. 3: structure–activity correlations for a series of isozyme II activators. J Pharm Sci 1994;83:768–73. (e) De Simone G, Supuran CT. (In)organic anions as carbonic anhydrase inhibitors. J Inorg Biochem 2012;111:117–29.
- (a) Vullo D, Del Prete S, Nocentini A, et al. Dithiocarbamates effectively inhibit the β-carbonic anhydrase from the dandruff-producing fungus Malassezia globosa. Bioorg Med Chem 2017;25:1260–5. (b) Borras J, Scozzafava A, Menabuoni L, et al. Synthesis of water-soluble, topically effective intraocular pressure lowering aromatic/heterocyclic sulfonamide containing 8-quinoline-sulfonyl moieties: is the tail more important than the ring? Bioorg Med Chem 1999;7:2397–406. (c) Supuran CT. Carbon-versus sulphur-based zinc binding groups for carbonic anhydrase inhibitors? J Enzyme Inhib Med Chem 2018;33:485–95. (d) Pastorekova S, Casini A, Scozzafava A, et al. Carbonic anhydrase inhibitors: the first selective, membrane-impermeant inhibitors targeting the tumor-associated isozyme IX. Bioorg Med Chem Lett 2004;14:869–73.
- (a) Supuran CT. Carbonic anhydrases: from biomedical applications of the inhibitors and activators to biotechnological use for CO2 capture. J Enzyme Inhib Med Chem 2013;28:229–30. (b) Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31:345–60. (c) Ibrahim HS, Allam HA, Mahmoud WR, et al. Dual-tail arylsulfone-based benzenesulfonamides differently match the hydrophobic and hydrophilic halves of human carbonic anhydrases active sites: selective inhibitors for the tumor-associated hCA IX isoform. Eur J Med Chem 2018;152:1–9. (d) Supuran CT. Applications of carbonic anhydrases inhibitors in renal and central nervous system diseases. Expert Opin Ther Pat 2018;28:713–21. (e) Supuran CT. Carbonic anhydrase inhibitors and their potential in a range of therapeutic areas. Expert Opin Ther Pat 2018;28:709–12.
- (a) Entezari Heravi Y, Bua S, Nocentini A, et al. Inhibition of Malassezia globosa carbonic anhydrase with phenols. Bioorg Med Chem 2017;25:2577–82. (b) Tars K, Vullo D, Kazaks A, et al. Sulfocoumarins (1,2-benzoxathiine 2,2-dioxides): a class of potent and isoform-selective inhibitors of tumor-associated carbonic anhydrases. J Med Chem 2013;56:293–300. (c) Pustenko A, Stepanovs D, Žalubovskis R, et al. 3H-1,2-benzoxathiepine 2,2-dioxides: a new class of isoform-selective carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2017;32:767–75. (d) Briganti F, Pierattelli R, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. Part 37. Novel classes of carbonic anhydrase inhibitors and their interaction with the native and cobalt-substituted enzyme: kinetic and spectroscopic investigations. Eur J Med Chem 1996;31:1001–10. (e) Carta F, Scozzafava A, Supuran CT. Sulfonamides: a patent review (2008–2012). Expert Opin Ther Pat 2012;22:747–58.
- (a) Nocentini A, Gratteri P, Supuran CT. Phosphorus versus sulfur: discovery of benzenephosphonamidates as versatile sulfonamide-mimic chemotypes acting as carbonic anhydrase inhibitors. Chem Eur J 2019;25:1188–92. (b) Supuran CT. Carbonic anhydrase inhibitors in the treatment and prophylaxis of obesity. Expert Opin Ther Pat 2003;13:1545–50. (c) Winum JY, Temperini C, El Cheikh K, et al. Carbonic anhydrase inhibitors: clash with Ala65 as a means for designing inhibitors with low affinity for the ubiquitous isozyme II, exemplified by the crystal structure of the topiramate sulfamide analogue. J Med Chem 2006;49:7024–31.
- Nocentini A, Ceruso M, Bua S, et al. Discovery of β-adrenergic receptors blocker-carbonic anhydrase inhibitor hybrids for multitargeted antiglaucoma therapy. J Med Chem 2018;61:5380–94.
- Aymerich S, Goldenberg S. The karyotype of Trypanosoma cruzi Dm 28c: comparison with other T. cruzi strains and trypanosomatids. Exp Parasitol 1989;69:107–15.
- Alvarenga NJ, Bronfen E. Metaciclogênese do Trypanosoma cruzi como parâmetro de interação do parasita com o triatomíneo vetor. Rev Soc Bras Med Trop 1997;30:247–50.
- Rólon M, Vega C, Escario JA, et al. Development of resazurin microtiter assay for drug sensibility testing of Trypanosoma cruzi epimastigotes. J Parasitol Res 2006;99:103–7.
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983;983:55–63.
- Nocentini A, Bua S, Del Prete S, et al. Natural polyphenols selectively inhibit β-carbonic anhydrase from the dandruff-producing fungus malasseziaglobosa: activity and modeling studies. ChemMedChem 2018;13:816–23.
- Vermelho AB, da Silva Cardoso V, Ricci Junior E, et al. Nanoemulsions of sulfonamide carbonic anhydrase inhibitors strongly inhibit the growth of Trypanosoma cruzi. J Enzyme Inhib Med Chem 2018;33:139–46.
- da Silva Cardoso V, Vermelho AB, Ricci Junior E, et al. Antileishmanial activity of sulphonamide nanoemulsions targeting the β-carbonic anhydrase from Leishmania species. J Enzyme Inhib Med Chem 2018;33:850–7.
- Rogato A, Del Prete S, Nocentini A, et al. Phaeodactylum tricornutum as a model organism for testing the membrane penetrability of sulphonamide carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2019;34:510–8.