
Abstract
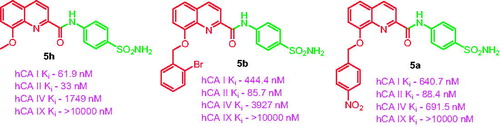
A series of novel 8-substituted-N-(4-sulfamoylphenyl)quinoline-2-carboxamides was synthesised by the reaction of 8-hydroxy-N-(4-sulfamoylphenyl) quinoline-2-carboxamide with alkyl and benzyl halides. The compounds were assayed for carbonic anhydrase (CA) inhibitory activity against four hCA isoforms, hCA I, hCA II, hCA IV, and hCA IX. Barring hCA IX, all the isoforms were inhibited from low to high nanomolar range. hCA I was inhibited in the range of 61.9–8126 nM, with compound 5h having an inhibition constant of KI = 61.9 nM. hCA II was inhibited in the range of 33.0–8759 nM, with compound 5h having an inhibition constant of 33.0 nM and compounds 5a and 5b having inhibition constants of 88.4 and 85.7 nM, respectively. hCA IV was inhibited in the range of 657.2–6757 nM. Hence, compound 5h, possessing low nanomolar hCA I and II inhibition, can be selected as a lead for the design of novel CA I and II inhibitors.
Graphical Abstract
1. Introduction
Carbonic anhydrases (CAs, EC 4.2.1.1) are a group of convergently evolved, ubiquitous metalloenzymes, which play a pivotal role in the maintenance of pH homoeostasis across all living phylaCitation1–4. They catalyse the reversible interconversion of carbon dioxide to bicarbonate and protons under physiological conditions via a ping pong mechanism, which is normally very slow under non-catalytic conditions (reaction 1):
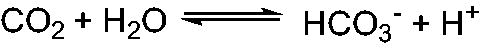 (1)
(1)
By efficiently catalysing the above reaction, CAs play an important role in many metabolic processes like facilitating the transport of CO2 between the metabolising tissues and lungs, lipogenesis, gluconeogenesis, and ureagenesisCitation1–4.
Up until now, eight genetically distinct families of CAs are known to be present across all phyla. These seven families are α-, β-, γ-, δ-, ζ-, η-, θ-, and the recently reported ι-classCitation5. The metal ion at the active site, which plays a highly important role in the catalytic activity of the enzyme, is variable among the different families, and can be Zn(II), Cd(II), Fe(II), and even Mn(II) in the ι-CAsCitation1–5. The α-CAs are predominantly found in vertebrates, protozoa, algae, bacteria, and the cytoplasm of green plants. The β-CAs are bacteria, algae, fungi, some archaea, and the chloroplasts of monocots and dicots. The γ-CAs are found in archaea and bacteria. The δ-, ζ-, -, θ-, and ι-CAs are present in marine diatoms. The η-CAs are present in various Plasmodium speciesCitation5.
α-CAs are widely distributed in many organisms and their number of isoforms is rather high, with 15 such CAs being present in humans and other primates and 16 in other mammals. In humans, the sub-cellular localisation of the isoforms is as follows: CA I, CA II, CA III, CA VII, CA VIII, CA X, CA XI, and CA XIII are cytosolic, CA IV, CA IX, CA XII, CAX IV, and CA XV are membrane-bound, CA VA and VB are mitochondrial, whereas CA VI is secreted in milk and salivaCitation1–4. Different isoforms are implicated in different diseases; hence, selective inhibition of a particular isoform may lead to the rectification of that particular disease in which it plays a major roleCitation1–4.
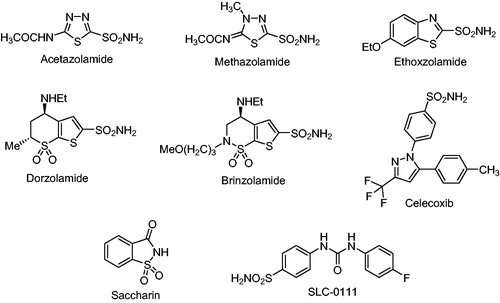
Sulfonamides and their isosteres (sulfamates and sulfamides) have been known for many years for their effective inhibition of many CA isoformsCitation5–8. Their mode of inhibition is by binding to the metal ion present in the active site, in a deprotonated form, as sulfonamidate anion. About 20 compounds incorporating the sulfonamide moiety are in clinical use for many years, with one of the compounds, SLC-0111, developed by one of our groups, being in phase II clinical trials. Some of the sulfonamide molecules in clinical use since many years are shown in Citation6–11.
Figure 1. Structures of some clinically used sulfonamides.
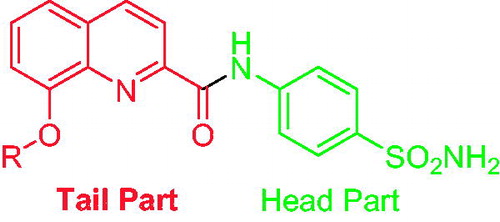
Quinoline is an aromatic, heterocyclic, nitrogen containing compound which is having a benzene ring fused with a pyridine ring at two adjacent carbon atomsCitation12. The quinoline nucleus exhibit diverse biological activities such as anticancer, antimalarial, antitubercular, antibacterial, antiprotozoal, antiproliferative, anti-inflammatory, antihypertensive, and anti-HIV activityCitation13. One of the potent quinoline derivatives is 8-hydroxy quinoline. It is obtained from plants as well as by synthesis. It is basically a small, planar and lipophilic molecule having an array of biological activities and also good metal chelating propertiesCitation14. Supuran and coworkers have previously investigated the quinoline scaffold, wherein they found it to exhibit potent activities on various CA isoformsCitation15–17. Hence, in order to further probe the efficacy of the quinoline scaffold for CA inhibition, the “tail approach” () was adopted and novel 8-substituted-N-(4-sulfamoylphenyl) quinoline-2-carboxamides were synthesised and assayed for their CA inhibitory activity against four CA isoforms, namely, CA I, II, IV, and IX. The drug acetazolamide (AAZ) was used as a drug standard.
Figure 2. The “Tail approach” method for the design of novel CA inhibitors in this work.
2. Materials and methods; chemistry part
2.1. General
All the chemicals and solvents were procured and utilised as such from the suppliers. Wherever necessary, anhydrous solvents were used. Thin layer chromatography (TLC) analysis was done by utilising Merck silica gel 60 F254 aluminium plates. Stuart digital melting point apparatus (SMP 30) was used in determining the melting points of the compounds, which are uncorrected. 1H and 13C NMR spectra were recorded using Bruker Avance 500 MHz and 125 MHz respectively using DMSO-d6 as the solvent. Chemical shift values are recorded in ppm using TMS as the internal standard. HRMS were determined by Agilent QTOF mass spectrometer 6540 series instrument and were performed using ESI techniques at 70 eV.
2.1.1. General procedure for the preparation of 8-hydroxy-N-(4-sulfamoylphenyl)quinoline-2-carboxamide
To a stirred solution of intermediate 3 (1 g, 5 mmol 1 eq.) in dry DMF (10 ml) HATU was added (3 g, 7.9 mmol, 1.5 eq.) at 0 °C. The resultant solution was stirred for one hour at 0 °C. Thereafter, sulfanilamide (1 g, 5.5 mmol, 1.1 eq.) and DIPEA (2 g, 15 mmol, 3 eq.) were added to the reaction mixture and the resultant solution was allowed to stir for overnight at room temperature. The completion of the reaction was monitored by TLC. On completion of the reaction mixture as evidenced by the TLC, it was dumped into crushed ice. The precipitated solid was collected by filtration and it was subjected to column chromatography using silica gel 60–120 mesh as the stationary phase and EtOAc:hexane 6:4 as mobile phase to afford intermediate 4 as a beige solid. Yield (60%).
2.1.2. General procedure for the preparation of 8-substituted-N-(4-sulfamoylphenyl)quinoline-2-carboxamidederivatives (5a–h)
To a stirred solution of intermediate 4 (80 mg, 0.2 mmol, 1 eq.) in acetone (5 ml) K2CO3 was added (22 mg, 0.4 mmol, 2 eq.) and the resultant solution was allowed to stir for 15 min. Thereafter, alkyl or benzyl halide (1.5 eq.) was added to the reaction mixture and it was allowed to stir for overnight at room temperature. The completion of the reaction was monitored by TLC. The reaction solvent was distilled off under vacuum and the crude residue was subjected to column chromatography using silica gel 60–120 mesh as the stationary phase and EtOAc:hexane 4:6 as mobile phase to afford the final compounds 5a–h.
2.1.3. 8-((4-Nitrobenzyl)oxy)-N-(4-sulfamoylphenyl)quinoline-2-carboxamide (5a)
Yellow solid, yield: 55%; mp: 257–259 °C; 1H NMR (500 MHz, DMSO) δ 10.78 (s, 1H), 8.62 (d, J = 8.5 Hz, 1H), 8.31 (d, J = 8.5 Hz, 2H), 8.26 (d, J = 8.5 Hz, 1H), 8.01 (d, J = 8.6 Hz, 2H), 7.97 (d, J = 8.4 Hz, 2H), 7.90 (d, J = 8.6 Hz, 2H), 7.68 (d, J = 7.0 Hz, 2H), 7.42 (d, J = 6.0 Hz, 1H), 7.35 (s, 2H), 5.62 (s, 2H). 13C NMR (125 MHz, DMSO) δ 163.46, 154.24, 148.83, 147.53, 145.62, 141.35, 139.69, 138.78, 138.45, 130.82, 129.49, 128.39, 127.34, 124.04, 120.81, 119.86, 119.69, 112.24, 69.58. HRMS (ESI): m/z calculated for C23H18N4O6S 479.1025, found 479.1030 [M + H]+.
2.1.4. 8-((2-Bromobenzyl)oxy)-N-(4-sulfamoylphenyl)quinoline-2-carboxamide (5b)
Yellow solid, yield: 40%; mp: 270–272 °C 1H NMR (500 MHz, DMSO) δ 10.76 (s, 1H), 8.62 (d, J = 8.5 Hz, 1H), 8.25 (d, J = 8.5 Hz, 1H), 7.98 (d, J = 8.7 Hz, 2H), 7.92–7.85 (m, 2H), 7.68 (d, J = 6.4 Hz, 2H), 7.65 (s, 3H), 7.41 (dd, J = 6.2, 2.6 Hz, 1H), 7.33 (s, 2H), 5.45 (s, 2H). 13C NMR (125 MHz, DMSO) δ 162.43, 153.40, 147.68, 140.27, 138.61, 137.71, 137.46, 136.14, 130.76, 129.73, 128.78, 128.47, 126.30, 120.25, 119.52, 118.71, 118.53, 111.26, 68.88. HRMS (ESI): m/z calculated for C23H18BrN3O4S 512.0280, found 514.0265 [M + 2]+.
2.1.5. 8-(Benzyloxy)-N-(4-sulfamoylphenyl)quinoline-2-carboxamide (5c)
Yellow solid, yield: 60%; mp: 247–249 °C1H NMR (500 MHz, DMSO) δ 10.79 (s, 1H), 8.60 (t, J = 8.4 Hz, 1H), 8.24 (d, J = 8.4 Hz, 1H), 7.97 (d, J = 8.6 Hz, 2H), 7.91 (d, J = 8.6 Hz, 2H), 7.70 (d, J = 7.4 Hz, 2H), 7.67 (d, J = 4.2 Hz, 2H), 7.47 (t, J = 7.4 Hz, 2H), 7.44–7.41 (m, 1H), 7.40 (d, J = 7.4 Hz, 1H), 7.35 (s, 2H), 5.46 (s, 2H). 13C NMR (125 MHz, DMSO) δ 163.40, 154.65, 148.55, 141.33, 139.67, 138.75, 138.53, 137.72, 129.58, 128.91, 128.27, 127.64, 127.41, 120.70, 120.39, 119.61, 119.48, 112.14, 70.64, 48.96. HRMS (ESI): m/z calculated for C23H19N3O4S434.1175, found 434.1175 [M + H]+.
2.1.6. 8-((3,5-Dimethylbenzyl)oxy)-N-(4-sulfamoylphenyl)quinoline-2-carboxamide (5d)
Yellow solid, yield: 50%; mp: 290–292 °C 1H NMR (500 MHz, DMSO) δ 10.79 (s, 1H), 8.61 (d, J = 8.5 Hz, 1H), 8.24 (d, J = 8.4 Hz, 1H), 7.97 (d, J = 8.6 Hz, 2H), 7.87 (d, J = 8.7 Hz, 2H), 7.69–7.66 (m, 2H), 7.42 (dd, J = 5.7, 2.7 Hz, 1H), 7.35 (s, 2H), 7.30 (s, 2H), 7.01 (s, 1H), 5.35 (s, 2H), 2.32 (s, 6H), 2.09 (s, 3H).13C NMR (125 MHz, DMSO) δ 163.45, 154.72, 148.58, 141.36, 139.66, 138.77, 138.47, 137.96, 137.48, 130.79, 129.65, 127.27, 125.24, 120.36, 119.67, 119.54, 112.01, 70.75, 21.43. HRMS (ESI): m/z calculated for C25H23N3O4S462.1488, found 462.1492 [M + H]+.
2.1.7. 8-((3-Chlorobenzyl)oxy)-N-(4-sulfamoylphenyl)quinoline-2-carboxamide (5e)
Yellow solid, yield: 60%; mp: 279–281 °C 1H NMR (500 MHz, DMSO) δ 10.85 (s, 1H), 8.66 (d, J = 8.2 Hz, 1H), 8.29 (d, J = 8.2 Hz, 1H), 8.06 (d, J = 8.0 Hz, 2H), 7.93 (d, J = 8.9 Hz, 3H), 7.73 (s, 2H), 7.66 (d, J = 6.7 Hz, 1H), 7.58–7.45 (m, 3H), 7.39 (s, 2H), 5.50 (s, 2H). 13C NMR (125 MHz, DMSO) δ 163.36, 154.41, 148.62, 141.31, 140.30, 139.66, 138.77, 138.42, 133.76, 130.79, 129.56, 128.18, 127.31, 127.21, 126.09, 120.59, 119.68, 119.55, 112.01, 69.69.
2.1.8. 8-((2,5-Difluorobenzyl)oxy)-N-(4-sulfamoylphenyl)quinoline-2-carboxamide (5f)
Yellow solid, yield: %;65 mp: 260–262 °C 1H NMR (500 MHz, DMSO) δ 10.84 (s, 1H), 8.63 (d, J = 8.5 Hz, 1H), 8.25 (d, J = 8.5 Hz, 1H), 8.02 (d, J = 8.7 Hz, 2H), 7.87 (d, J = 8.6 Hz, 4H), 7.71 (d, J = 6.7 Hz, 2H), 7.51 (dd, J = 6.5, 2.2 Hz, 1H), 7.35–7.31 (m, 3H), 5.49 (s, 2H). 13C NMR (125 MHz, DMSO) δ 163.38, 159.82, 157.91, 157.19, 155.27, 154.19, 148.75, 141.38, 139.66, 138.81, 138.49, 130.77, 129.57, 127.29, 120.98, 119.59, 117.45, 117.19, 116.62, 116.16, 112.48, 64.56. HRMS (ESI): m/z calculated for C23H17F2N3O4S 470.0986, found 470.0994 [M + H]+.
2.1.9. 8-(Prop-2-yn-1-yloxy)-N-(4-sulfamoylphenyl)quinoline-2-carboxamide (5g)
Yellow solid, yield: 30%; mp: 240–242 °C 1H NMR (500 MHz, DMSO) δ 10.76 (s, 1H), 8.62 (d, J = 8.5 Hz, 1H), 8.24 (d, J = 8.5 Hz, 1H), 8.07 (d, J = 8.7 Hz, 2H), 7.89 (t, J = 8.7 Hz, 2H), 7.74–7.70 (m, 2H), 7.49–7.43 (m, 1H), 7.33 (s, 2H), 5.17 (d, J = 2.1 Hz, 2H), 3.67 (t, J = 2.1 Hz, 1H). 13C NMR (125 MHz, DMSO) δ 163.79, 153.50, 149.21, 141.47, 139.69, 138.72, 138.51, 130.78, 129.25, 127.22, 121.08, 120.26, 119.81, 112.64, 79.58, 79.34, 57.15. HRMS (ESI): m/z calculated for C19H15N3O4S 382.0862, found 404.0684 [M + Na]+.
2.1.10. 8-Methoxy-N-(4-sulfamoylphenyl)quinoline-2-carboxamide (5h)
Yellow solid, yield: 50%; mp: 261–263 °C; 1H NMR (500 MHz, DMSO) δ 10.77 (s, 1H), 8.59 (d, J = 8.5 Hz, 1H), 8.23 (d, J = 8.4 Hz, 1H), 8.10 (dd, J = 18.5, 8.6 Hz, 2H), 7.85 (dd, J = 24.8, 8.6 Hz, 2H), 7.72–7.63 (m, 2H), 7.40–7.31 (m, 3H), 4.08 (s, 3H). 13C NMR (125 MHz, DMSO) δ 163.82, 155.77, 148.91, 141.50, 139.65, 138.55, 138.25, 130.67, 129.59, 128.29, 127.20, 120.49, 120.27, 119.92, 119.76, 109.97, 56.44. HRMS (ESI): m/z calculated for C17H15N3O4S 358.0862, found 358.0865 [M + H]+. Spectral data are provided in the Supplemental data, available online on the journal website.
2.2. CA inhibition assay
An SX.18V-R Applied Photophysics (Oxford, UK) stopped flow instrument has been used to assay the catalytic/inhibition of various CA isozymesCitation18. Phenol Red (at a concentration of 0.2 mM) has been used as an indicator, working at an absorbance maximum of 557 nm, with 10 mM Hepes (pH 7.4) as a buffer, 0.1 M Na2SO4 or NaClO4 (for maintaining constant the ionic strength; these anions are not inhibitory in the used concentration), following the CA-catalysed CO2 hydration reaction for a period of 5–10 s. Saturated CO2 solutions in water at 25 °C were used as substrate. Stock solutions of inhibitors were prepared at a concentration of 10 mM (in DMSO–water 1:1, v/v) and dilutions up to 0.01 nM done with the assay buffer mentioned above. At least seven different inhibitor concentrations have been used for measuring the inhibition constant. Inhibitor and enzyme solutions were pre-incubated together for 10 min at room temperature prior to assay, in order to allow for the formation of the E–I complex. Triplicate experiments were done for each inhibitor concentration, and the values reported throughout the paper is the mean of such results. The inhibition constants were obtained by non-linear least squares methods using the Cheng–Prusoff equation, as reported earlier, and represent the mean from at least three different determinations. All CA isozymes used here were recombinant proteins obtained as reported earlier by our groupCitation19,Citation20.
3. Results and discussion
3.1. Chemistry
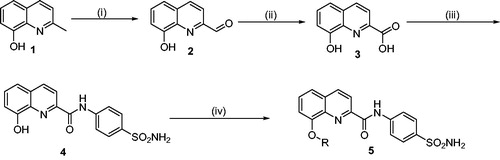
A series of structurally diverse 8-substituted quinoline-2-carboxamides were synthesised according to general synthetic route as illustrated in Scheme 1. In brief, 8-hydroxy quinaldine (1) was treated with selenium dioxide to yield 8-hydroxyquinoline-2-carbaldehyde (2) which was further treated with H2O2 in the presence of formic acid to yield 8-hydroxyquinoline-2-carboxylic acidCitation21 (3). This intermediate was further subjected to acid-amine coupling with sulfanilamide to give the amide product (4). This was further subjected to O-alkylation using various aliphatic and benzylic halides to afford the final products (5a–h). All the final products were confirmed using various analytical and spectral techniques.
Scheme 1. General synthetic route for the synthesis of 8-substituted quinoline-linked sulfonamide derivatives (5a–h). Reagents and conditions: (i) SeO2, 1,4-dioxane, 110 °C, 12 h, (ii) H2O2, Formic acid, 0 °C, 12 h, (iii) Sulfanilamide, HATU, DIPEA, DMF, 0 °C-rt, 12–15 h, and (iv) R-X, K2CO3, Acetone, r.t., 12–15 h.
3.2. CA inhibition studies
The newly synthesised 8-substituted quinoline-linked sulfonamide derivatives were evaluated for their inhibitory activity against the cytosolic isoforms hCA I and II, the membrane-bound isoform hCA IV and the transmembrane isoform hCA IX. Acetazolamide was used as a standard drug. The following structure–activity relationship (SAR) may be inferred from the inhibition data shown in :
Table 1. CA inhibition data with the synthesised compounds 5a–5h and acetazolamide as standard drug, by a stopped flow CO2 hydrase assay.
The ubiquitous cytosolic isoform, hCA I, which is localised in the erythrocytes, gastrointestinal tract and eye, was inhibited from low to high nanomolar range, with the inhibition constants ranging from 61.9 to 8126 nM. The best inhibition constant was shown by compound 5h, possessing a methyl substitution on the 8-OH group. It elicited an inhibition of more than four times compared to the standard AAZ, with a KI of 61.9 nM.
The cytosolic isoform, hCA II, which is also one of the physiologically relevant isoforms, was inhibited in a diverse manner by the compounds 5a–h, with the inhibition constants ranging from 33.0 to 8759 nM. The most potent inhibitor was again found to be compound 5h, with an inhibition constant of 33.0 nM. Compounds 5a and 5b, possessing a 4-nitro benzyl and 2-bromo benzyl substitution at 8-OH position, also showed low nanomolar inhibitory potencies with KI values of 88.4 and 85.7 nM, respectively.
The membrane-bound isoform, hCA IV, was inhibited with a moderate to weak inhibition profile by the compounds 5a–h. The inhibition constants ranged from 657.2 to 6757 nM, as compared to the standard, AAZ, which showed a much stronger inhibition at 74 nM.
The tumour-associated isoform, hCA IX, was not at all inhibited by the compounds 5a–h (KI > 10,000 nM).
4. Conclusions
A series of novel 8-substituted quinoline-linked sulfonamide derivatives (5a–h) were synthesised and assayed for inhibitory activity against a series of CA isoforms, namely, hCA I and II which are cytosolic, hCA IV which is membrane-bound and hCA IX which is tumour-associated isoform. Except for hCA IX, the synthesised compounds exhibited a variable degree of inhibition profiles for the other isoforms, ranging from low nanomolar to high nanomolar. Among all the compounds, compound 5h exhibited low nanomolar inhibitory profiles for hCA I, with KI 61.9 nM (wherein it proved to be four times more potent than the standard AAZ) and hCA II, with KI of 33.0 nM. Compounds 5a and 5b showed inhibition constants of 88.4 and 85.7 nM respectively for hCA II. Apart from that, all the compounds showed moderate to high nanomolar inhibition for hCA I, II, and IV. Hence, compound 5h can be developed further as a lead compound to design more effective hCA I and hCA II inhibitors.
Supplemental Material
Download PDF (1.1 MB)Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- (a) Alterio V, Di Fiore A, D’Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68. (b) Guler OO, Capasso C, Supuran CT. A magnificent enzyme superfamily: carbonic anhydrases, their purification and characterization. J Enzyme Inhib Med Chem 2015;31:689694. (c) Supuran CT. Structure and function of carbonic anhydrases. Biochem J 2016;473:2023–32. (d) Supuran CT. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin Drug Discov 2017;12:61–88. (e) Supuran CT. Carbonic anhydrase activators. Future Med Chem 2018;10:561–73.
- (a) Supuran CT, Capasso C. The η-class carbonic anhydrases as drug targets for antimalarial agents. Expert Opin Ther Targets 2015;19:551–63. (b) Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81. (c) Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77. (d) Supuran CT, Vullo D, Manole G, et al. Designing of novel carbonic anhydrase inhibitors and activators. Curr Med Chem Cardiovasc Hematol Agents 2004;2:49–68.
- (a) Supuran CT. Carbonic anhydrase inhibitors and activators for novel therapeutic applications. Future Med Chem 2011;3:1165–80. (b) Capasso C, Supuran CT. An overview of the alpha-, beta- and gamma-carbonic anhydrases from bacteria: can bacterial carbonic anhydrases shed new light on evolution of bacteria? J Enzyme Inhib Med Chem 2015;30:325–32. (c) Del Prete S, Vullo D, Fisher GM, et al. Discovery of a new family of carbonic anhydrases in the malaria pathogen Plasmodium falciparum – the η-carbonic anhydrases. Bioorg Med Chem Lett 2014;24:4389–96. (d) Supuran CT. Structure-based drug discovery of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2012;27:759–72. (e) Clare BW, Supuran CT. Carbonic anhydrase activators. 3: structure–activity correlations for a series of isozyme II activators. J Pharm Sci 1994;83:768–73.
- (a) Supuran CT. Carbonic anhydrase inhibitors and their potential in a range of therapeutic areas. Expert Opin Ther Pat 2018;28:709–12. (b) Supuran CT. Applications of carbonic anhydrases inhibitors in renal and central nervous system diseases. Expert Opin Ther Pat 2018;28:713–21. (c) Winum JY, Temperini C, El Cheikh K, et al. Carbonic anhydrase inhibitors: clash with Ala65 as a means for designing inhibitors with low affinity for the ubiquitous isozyme II, exemplified by the crystal structure of the topiramate sulfamide analogue. J Med Chem 2006;49:7024–31. (d) Supuran CT. Carbonic anhydrase inhibitors in the treatment and prophylaxis of obesity. Expert Opin Ther Pat 2003;13:1545–50.
- Jensen EL, Clement R, Kosta A, et al. A new widespread subclass of carbonic anhydrase in marine phytoplankton. ISME J 2019. DOI:10.1038/s41396-019-0426-8
- (a) Supuran CT. Carbon-versus sulphur-based zinc binding groups for carbonic anhydrase inhibitors? J Enzyme Inhib Med Chem 2018;33:485–95. (b) Pastorekova S, Casini A, Scozzafava A, et al. Carbonic anhydrase inhibitors: the first selective, membrane-impermeant inhibitors targeting the tumor-associated isozyme IX. Bioorg Med Chem Lett 2004;14:869–73. (c) Melis C, Meleddu R, Angeli A, et al. Isatin: a privileged scaffold for the design of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2017;32:68–73. (d) Gul HI, Mete E, Eren SE, et al. Designing, synthesis and bioactivities of 4-[3-(4-hydroxyphenyl)-5-aryl-4,5-dihydro-pyrazol-1-yl]benzenesulfonamides. J Enzyme Inhib Med Chem 2017;32:169–75. (e) Gul HI, Mete E, Taslimi P, et al. Synthesis, carbonic anhydrase I and II inhibition studies of the 1,3,5-trisubstituted-pyrazolines. J Enzyme Inhib Med Chem 2017;32:189–92.
- (a) Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31:345–60. (b) da Silva Cardoso V, Vermelho AB, Ricci Junior E, et al. Antileishmanial activity of sulphonamide nanoemulsions targeting the β-carbonic anhydrase from Leishmania species. J Enzyme Inhib Med Chem 2018;33:850–7. (c) Akocak S, Lolak N, Vullo D, et al. Synthesis and biological evaluation of histamine Schiff bases as carbonic anhydrase I, II, IV, VII, and IX activators. J Enzyme Inhib Med Chem 2017;32:1305–12. (d) Supuran CT. Acetazolamide for the treatment of idiopathic intracranial hypertension. Expert Rev Neurother 2015;15:851–6.
- (a) Nocentini A, Cadoni R, Dumy P, et al. Carbonic anhydrases from Trypanosoma cruzi and Leishmania donovani chagasi are inhibited by benzoxaboroles. J Enzyme Inhib Med Chem 2018;33:286–9. (b) Kumar R, Sharma V, Bua S, et al. Synthesis and biological evaluation of benzenesulphonamide-bearing 1,4,5-trisubstituted-1,2,3-triazoles possessing human carbonic anhydrase I, II, IV, and IX inhibitory activity. J Enzyme Inhib Med Chem 2017;32:1187–94. (c) Bozdag M, Bua S, Osman SM, et al. Carbonic anhydrase I, II, IV and IX inhibition with a series of 7-amino-3,4-dihydroquinolin-2(1H)-one derivatives. J Enzyme Inhib Med Chem 2017;32:885–92. (d) Bertol E, Vaiano F, Mari F, et al. Advances in new psychoactive substances identification: the U.R.I.To.N. Consortium. J Enzyme Inhib Med Chem 2017;32:841–9. (e) Supuran CT. Drug interaction considerations in the therapeutic use of carbonic anhydrase inhibitors. Expert Opin Drug Metab Toxicol 2016;12:423–31.
- (a) Abbate F, Supuran CT, Scozzafava A, et al. Nonaromatic sulfonamide group as an ideal anchor for potent human carbonic anhydrase inhibitors: role of hydrogen-bonding networks in ligand binding and drug design. J Med Chem 2002;45:3583–7. (b) Briganti F, Pierattelli R, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. Part 37. Novel classes of carbonic anhydrase inhibitors and their interaction with the native and cobalt-substituted enzyme: kinetic and spectroscopic investigations. Eur J Med Chem 1996;31:1001–10. (c) Carta F, Scozzafava A, Supuran CT, Sulfonamides R. Sulfonamides: a patent review (2008–2012). Expert Opin Ther Pat 2012;22:747–58. (d) Scozzafava A, Carta F, Supuran CT. Secondary and tertiary sulfonamides: a patent review (2008–2012). Expert Opin Ther Pat 2013;23:203–13.
- (a) Lou Y, McDonald PC, Oloumi A, et al. Targeting tumor hypoxia: suppression of breast tumor growth and metastasis by novel carbonic anhydrase IX inhibitors. Cancer Res 2011;71:3364–76. (b) Pastorekova S, Parkkila S, Pastorek J, Supuran CT. Carbonic anhydrases: current state of the art, therapeutic applications and future prospects. J Enzyme Inhib Med Chem. 2004;19:199–229. (c) Supuran CT. Carbonic anhydrase inhibition and the management of hypoxic tumors. Metabolites 2017;7:E48. (d) Supuran CT. Carbonic anhydrases and metabolism. Metabolites 2018;8:25. (e) Iessi E, Logozzi M, Mizzoni D, et al. Rethinking the combination of proton exchanger inhibitors in cancer therapy. Metabolites 2017;8:2. (f) Kuchuk O, Tuccitto A, Citterio D, et al. pH regulators to target the tumor immune microenvironment in human hepatocellular carcinoma. Oncoimmunology 2018;7:e1445452.
- Eldehna WM, Abo-Ashour MF, Berrino E, et al. Novel synthesized SLC-0111 thiazole and thiadiazole analogues: determination of their carbonic anhydrase inhibitory activity and molecular modeling studies. Bioorg Chem 2019;87:794–802.
- Keri RS, Patil SA. Quinoline: a promising antitubercular target. Biomed Pharmacother 2014;68:1161–75.
- Nainwal LM, Tasneem S, Akhtar W, et al. Green recipes to quinoline: a review. Eur J Med Chem 2019;164:121–70.
- Prachayasittikul V, Prachayasittikul S, Rachirawat S, Prachayasittikul V. 8-Hydroxyquinolines: a review of their metal chelating properties and medicinal applications. Drug Des Devel Ther 2013;4:1157–78.
- Borras J, Scozzafava A, Menabuoni L, et al. Synthesis of water-soluble, topically effective intraocular pressure lowering aromatic/heterocyclic sulfonamide containing 8-quinoline-sulfonyl moieties: is the tail more important than the ring? Bioorg Med Chem 1999;7:2397–406.
- Agrawal VK, Bano S, Supuran CT, Khadikar PV. QSAR study on carbonic anhydrase inhibitors: aromatic/heterocyclic sulfonamides containing 8-quinoline-sulfonyl moieties, with topical activity as antiglaucoma agents. Eur J Med Chem 2004;39:593–600.
- Buemi MR, De Luca L, Ferro S, et al. Carbonic anhydrase inhibitors: design, synthesis and structural characterization of new heteroaryl-N-carbonylbenzenesulfonamides targeting druggable human carbonic anhydrase isoforms. Eur J Med Chem 2015;102:223–32.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flowkinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73.
- (a) Krasavin M, Shetnev A, Baykov S, et al. Pyridazinone-substituted benzenesulfonamides display potent inhibition of membrane-bound human carbonic anhydrase IX and promising anti-proliferative effects against cancer cell lines. Eur J Med Chem 2019;168:301–14. (b) Dilworth JR, Pascu SI, Waghorn PA, et al. Synthesis of sulfonamide conjugates of Cu(II), Ga(III), In(III), Re(V) and Zn(II) complexes: carbonic anhydrase inhibition studies and cellular imaging investigations. Dalton Trans 2015;44:4859–73. (c) Sneddon D, Niemans R, Bauwens M, et al. Synthesis and in vivo biological evaluation of (68)Ga-labeled carbonic anhydrase IX targeting small molecules for positron emission tomography. J Med Chem 2016;59:6431–43. (d) Ramya PVS, Angapelly S, Angeli A, et al. Discovery of curcumin inspired sulfonamide derivatives as a new class of carbonic anhydrase isoforms I, II, IX, and XII inhibitors. J Enzyme Inhib Med Chem 2017;32:1274–81.
- (a) Thacker PS, Alvala M, Arifuddin M, et al. Design, synthesis and biological evaluation of coumarin-3-carboxamides as selective carbonic anhydrase IX and XII inhibitors. Bioorg Chem 2019;86:386–92. (b) Supuran CT. Carbonic anhydrase inhibition and the management of neuropathic pain. Expert Rev Neurother 2016;16:961–8. (c) Mboge MY, Mahon BP, Lamas N, et al. Structure activity study of carbonic anhydrase IX: selective inhibition with ureido-substituted benzenesulfonamides. Eur J Med Chem 2017;132:184–91. (d) Stanica L, Gheorghiu M, Stan M, et al. Quantitative assessment of specific carbonic anhydrase inhibitors effect on hypoxic cells using electrical impedance assays. J Enzyme Inhib Med Chem 2017;32:1079–90. (e) Abdoli M, Angeli A, Bozdag M, et al. Synthesis and carbonic anhydrase I, II, VII, and IX inhibition studies with a series of benzo[d]thiazole-5- and 6-sulfonamides. J Enzyme Inhib Med Chem 2017;32:1071–8.
- Dodd RH, Hyaric ML. The oxidation of aromatic aldehydes to carboxylic acids using hydrogen peroxide in formic acid. Synthesis 1993;3:295–7.