
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
A series of histamine bis-Schiff bases and bis-spinaceamine derivatives were synthesised and investigated as activators of four human (h) carbonic anhydrase (CA, EC 4.2.1.1) isoforms, the cytosolic hCA I, II and VII, and the membrane-associated hCA IV. All isoforms were effectively activated by the new derivatives, with activation constants in the range of 4.73–10.2 µM for hCA I, 6.15–42.1 µM for hCA II, 2.37–32.7 µM for hCA IV and 32 nM–18.7 µM for hCA VII, respectively. The nature of the spacer between the two histamine/spinaceamine units of these molecules was the main contributor to the diverse activating efficacy, with a very different fine tuning for the diverse isoforms. As CA activators recently emerged as interesting agents for enhancing cognition, in the management of CA deficiencies, or for therapy memory and artificial tissues engineering, our compounds may be considered as candidates for such applications.
1. Introduction
In previous research from our groupsCitation1,Citation2, we reported two novel classes of activators of the enzyme carbonic anhydrase (CA, EC 4.2.1.1): the histamine Schiff basesCitation1 and the spinaceamine derivativesCitation2. As all CA activators (CAAs), these new classes of enzyme modulators also participate in the catalytic cycle of the enzymeCitation3–6. Indeed, CAs are metalloenzymes, usually using Zn(II) ions within their active site for performing the efficient hydration of CO2 to bicarbonate and protons. A water molecule coordinated to the zinc ion becomes highly nucleophilic, and as hydroxide ion attacks the CO2 molecule bound within the active site of the enzyme, with formation of bicarbonate coordinated to the zinc (EquationEquation (1)(1)
(1) )Citation7–10. Another incoming water molecule subsequently displaces the bound bicarbonate, liberating it in solution, and leading to the formation of the acidic species of the enzyme, with water as zinc ligand EquationEquation (1)
(1)
(1) . In order to obtain the nucleophilic species of the enzyme, with the hydroxide coordinated to the zinc EquationEquation (2)
(2)
(2) , a proton transfer reaction must occur, which is the rate determining step of the entire catalytic cycleCitation3,Citation7–10.
(1)
(1)
(2)
(2)
It has been demonstrated that the activators A in EquationEquation (3)(3)
(3) intervene in this step, providing an alternative pathway for the release of protons and formation of the zinc hydroxide species of the enzymeCitation3–6.
(3)
(3)
The activator molecule participates to the rate-determining step of the catalytic cycle, that is, the proton shuttling between the zinc-coordinated water molecule and the environment, with the formation of the zinc hydroxide species of the enzymeCitation3–6,Citation11. In many CA isoforms, it has been shown that residue His64 placed in the middle of the active site cavity is involved in this phenomenon, acting as a natural proton shuttle residue during the catalytic cycleCitation11. Confirmation that CAAs have a similar role to His64, that is, shuttling of the protons from the active site to the environment and facilitation of the formation of the nucleophilic enzyme species, came from many X-ray crystallographic studies of isoforms CA I and II complexed with amine and amino acid activatorsCitation3–6. Histamine, the first CAA investigated by means of X-ray crystallographyCitation3, was observed to bind at the entrance of the active site cavity, distant from the zinc ion, and participating in a network of hydrogen bonds involving several water molecules, which, as for His64, favour the release of the proton from the water molecule coordinated to the zinc, to the reaction mediumCitation3. X-ray crystal of other CAAs reinforced the above findings: all activators bind in the same active site region, at the entrance of the cavity, from where they can enhance the formation of the zinc hydroxide species of the enzyme, by favouring the proton shuttling between the cavity and the reaction mediumCitation3–6. Furthermore, recently, it has also been shown the CAAs may have pharmacological applications for enhancing cognition, in the management of CA deficiencies, for therapy memory and for obtaining artificial tissuesCitation12. Thus, there is a strong interest in designing CAAs belonging to various chemical classes, in order to detect compounds with high efficacy and eventually isoform-selective action, considering that in humans at least 15 CA isoforms were described so farCitation7. Here, we report some new CAAs obtained by considering our previous findings, that is, histamine Schiff bases and spinaceamine derivatives, which posses efficient CA activating propertiesCitation1,Citation2.
2. Materials and methods
2.1. Chemistry
All chemicals and anhydrous solvents were purchased from Sigma-Aldrich, Merck, Alfa Aesar and TCI and used without further purification. Melting points (mp) were determined with SMP30 melting point apparatus in open capillaries and are uncorrected. FT-IR spectra were recorded by using Perkin Elmer Spectrum 100 FT-IR spectrometer. Nuclear Magnetic Resonance (1H-NMR and 13 C-NMR) spectra of compounds were recorded using a Bruker Advance III 300 MHz spectrometer in DMSO-d6 and TMS as an internal standard operating at 300 MHz for 1H-NMR and 75 MHz for 13 C-NMR. Thin layer chromatography (TLC) was carried out on Merck silica gel 60 F254 plates.
2.1.1. General procedure for the synthesis of bis-histamine schiff bases H1, H2, H3 and H4
Potassium hydroxide (10 mmol) was added to a stirred suspension of histamine dihydrochloride (5 mmol) in dry EtOH (10–15 ml) at room temperature. After stirring for 2 h, the precipitate salt (KCl) was filtered off and the filtrate was treated with a solution of bis-aldehydes (2.5 mmol) in dry EtOH (20–25 ml). The homogeneous mixture was stirred overnight at room temperature. The completion of the reaction was monitored by TLC and FT-IR. The excess solvent was evaporated and the oily residue was crystallized with ethyl acetate and diethylether to obtain the corresponding bis-histamine Schiff base derivatives. The desired final products H1, H2, H3, and H4 were dried under vacuum and fully characterised by FT-IR, 1H-NMR, 13 C-NMR, elemental analysis and melting points.
1,4-Phenylenebis(methanylylidene))bis(2-(1H-imidazol-4-yl)ethanamine) (H1): Yield: 65%; Colour: cream powder, mp: 165–1680 C; FT-IR (cm−1): 3085, 3020, 2841, 2631, 1636 (-C = N-), 1453, 1292, 821; 1H-NMR (DMSO-d6, 300 MHz, δ ppm): 8.68 (s, 2H, -N=CH-), 8.08 (d, 2H, J = 2.4, H-2 Im), 7.98 (d, 4H, J = 7.5, Ar-H), 7.55 (s, 2H, H-5 Im), 3.75 (t, 4H, J = 5.5, -CH2CH2-Im), 2.95 (t, 4H, J = 5.5, -CH2CH2-Im): 13 C-NMR (DMSO-d6, 75 MHz, δ ppm): 167.5 (-N = CH-), 135.4, 133.2, 131.7, 130.4, 116.3, 55.5, 29.8;
1,3-Phenylenebis(methanylylidene))bis(2-(1H-imidazol-4-yl)ethanamine) (H2): Yield: 48%; Colour: cream powder, mp: 210-2120 C; FT-IR (cm−1): 3120, 3024, 2922, 2851, 1615 (-C = N-), 1437, 1290, 822; 1H-NMR (DMSO-d6, 300 MHz, δ ppm): 8.70 (s, 2H, -N=CH-), 8.25 (s, 1H, Ar-H), 8.12 (d, 2H, J = 2.8, H-2 Im), 8.00 (d, 2H, J = 7.2, Ar-H), 7.62 (s, 2H, H-5 Im), 7.58 (m, 1H, Ar-H), 3.78 (t, 4H, J = 5.8, -CH2CH2-Im), 2.98 (t, 4H, J = 5.8, -CH2CH2-Im): 13 C-NMR (DMSO-d6, 75 MHz, δ ppm): 167.2 (-N = CH-), 138.6, 136.2, 133.6, 131.2, 130.8, 128.6, 116.6, 55.8, 29.9;
1,2-Phenylenebis(methanylylidene))bis(2-(1H-imidazol-4-yl)ethanamine)(H3): Yield: 35%; Colour: cream powder, mp: 222–2250 C; FT-IR (cm−1): 3117, 2926, 2849, 1648 (-C = N-), 1434, 1223, 828; 1H-NMR (DMSO-d6, 300 MHz, δ ppm): 8.71 (s, 2H, -N=CH-), 8.15 (d, 2H, J = 6.9, Ar-H), 8.10 (d, 2H, J = 2.8, H-2 Im), 7.60 (s, 2H, H-5 Im), 7.48 (m, 2H, Ar-H), 3.76 (t, 4H, J = 5.5, -CH2CH2-Im), 2.97 (t, 4H, J = 5.5, -CH2CH2-Im): 13 C-NMR (DMSO-d6, 75 MHz, δ ppm): 166.8 (-N = CH-), 138.4, 135.8, 132.9, 131.7, 130.4, 128.3, 116.9, 55.9, 29.7;
Furan-2,5-diylbis(methanylylidene))bis(2-(1H-imidazol-4-yl)ethanamine) (H4): Yield: 32%; Colour: brown powder, mp: 114-1170 C; FT-IR (cm−1): 3107, 3016, 2926, 2853, 1621 (-C = N-), 1433, 1224, 816; 1H-NMR (DMSO-d6, 300 MHz, δ ppm): 8.88 (s, 2H, -N=CH-), 7.99 (d, 2H, J = 2.2, H-2 Im), 7.74 (s, 2H, H-5 Im), 6.85 (d, 2H, J = 2.8, furan), 3.52 (t, 4H, J = 5.8, -CH2CH2-Im), 2.88 (t, 4H, J = 5.8, -CH2CH2-Im): 13 C-NMR (DMSO-d6, 75 MHz, δ ppm): 166.2 (-N = CH-), 151.4, 144.9, 138.3, 134.7, 119.5, 113.2, 56.3, 31.5;
2.1.2. General procedure for the synthesis of bis-Spinaceamine substituted compounds SPH1, SPH2, and SPH4
To a solution of 5 mmol of histamine dihydrochloride in 10 ml of water were added solutions of 15 mmol of sodium hydroxide (NaOH) and 2.5 mmol of appropriate bis-aldehyde derivatives in 15 ml of ethanol. The reaction mixture was heated overnight at 80 °C. The completion of the reaction was monitored by TLC and FT-IR. After that, the mixture was allowed to cool to room temperature and the formed precipitate was filtered off. The crude powders were recrystallized from hot water and dried under vacuum at 40 °C to afford SPH1, SPH2, and SPH4 which were fully characterized by FT-IR, 1H-NMR, 13 C-NMR, and melting points.
1,4-Bis(4,5,6,7-tetrahydro-3H-imidazo[4,5-c]pyridin-4-yl)benzene (SPH1): Yield: 56%; Colour: cream powder, mp: 95-980 C; FT-IR (cm−1): 3122, 3024, 2956, 2971, 2926, 1606, 1455, 963, 817; 1H-NMR (CD3OD, 300 MHz, δ ppm): 7.78 (s, 2H, H-2 Im), 7.46 (d, 4H, Ar-H), 5.10 (s, 2H, -CH-), 3.35 (m, 4H,-CH2CH2-Im), 2.92 (t, 4H, J = 5.2, -CH2CH2-Im): 13 C-NMR (CD3OD, 75 MHz, δ ppm): 141.7, 135.9, 134.2, 129.8, 128.1, 127.3, 113.9, 64.2, 39.5, 30.0;
1,3-Bis(4,5,6,7-tetrahydro-3H-imidazo[4,5-c]pyridin-4-yl)benzene (SPH2): Yield: 45%; Colour: white powder, mp: 190-1920 C; FT-IR (cm−1): 3117, 3036, 2922, 2852, 1611, 1448, 947, 817; 1H-NMR (CD3OD, 300 MHz, δ ppm): 7.81 (s, 2H, H-2 Im), 7.52 (m, 1H, Ar-H), 7.22 (d, 2H, Ar-H), 7.05 (s, 1H, Ar-H), 5.08 (s, 2H, -CH-), 3.33 (m, 4H,-CH2CH2-Im), 2.93 (t, 4H, J = 5.5, -CH2CH2-Im): 13 C-NMR (CD3OD, 75 MHz, δ ppm): 141.2, 136.5, 134.1, 129.4, 127.9, 127.7, 113.5, 64.4, 39.3, 29.7;
2,5-Bis(4,5,6,7-tetrahydro-3H-imidazo[4,5-c]pyridin-4-yl)furan (SPH4): Yield: 48%; Colour: orange mp: 185-1880 C; FT-IR (cm−1): 3012, 2919, 2849, 1608, 1379, 1322, 1135, 1011, 813; 1H-NMR (CD3OD, 300 MHz, δ ppm): 7.92 (s, 2H, H-2 Im), 6.65 (d, 2H, J = 5.5, furyl), 5.25 (s, 2H, -CH-), 3.36 (m, 4H,-CH2CH2-Im), 2.97 (t, 4H, J = 5.8, -CH2CH2-Im): 13 C-NMR (CD3OD, 75 MHz, δ ppm): 151.9, 143.4, 139.8, 134.7, 118.6, 114.2, 106.8, 63.2, 39.4, 29.2.
2.2. CA activation
An Sx.18Mv-R Applied Photophysics (Oxford, UK) stopped-flow instrument has been used to assay the catalytic activity of various CA isozymes for CO2 hydration reactionCitation13. Phenol red (at a concentration of 0.2 mM) was used as indicator, working at the absorbance maximum of 557 nm, with 10 mM Hepes (pH 7.5) as buffer, 0.1 M NaClO4 (for maintaining constant ionic strength), following the CA-catalysed CO2 hydration reaction for a period of 10 s at 25 °C. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each activator at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The uncatalysed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of activators (at 0.1 mM) were prepared in distilled-deionised water and dilutions up to 1 nM were made thereafter with the assay buffer. Enzyme and activator solutions were pre-incubated together for 15 min prior to assay, in order to allow for the formation of the enzyme–activator complexes. The activation constant (KA), defined similarly with the inhibition constant KI, can be obtained by considering the classical Michaelis–Menten equation (EquationEquation (4)(4)
(4) , which has been fitted by non-linear least squares by using PRISM 3:
(4)
(4)
where [A]f is the free concentration of activator.
Working at substrate concentrations considerably lower than KM ([S] ≪KM), and considering that [A]f can be represented in the form of the total concentration of the enzyme ([E]t)and activator ([A]t), the obtained competitive steady-state equation for determining the activation constant is given by EquationEquation (5)(5)
(5) :
(5)
(5)
where v0 represents the initial velocity of the enzyme-catalyzed reaction in the absence of activatorCitation14–18. All CAs were recombinant proteins obtained as reported earlierCitation18.
3. Results and discussion
3.1. Chemistry
The rationale for designing CAAs presented in this work is based on our previous data which showed efficient CA VII activating effects for derivatised histamine Schiff base compounds and spinaceamine derivativesCitation1,Citation2. Therefore, in this work, a number of structurally diverse bis-histamine Schiff bases and bis-Spinaceamine substituted compounds(ring-closure product of histamine Schiff bases) were synthesised according to general synthetic routes as illustrated in Scheme 1. In order to generate chemical diversity, different bis-aldehydes were chosen, possessing aromatic and heterocyclic moieties, and they were reacted with histamine leading to the bis-histamine Schiff bases and bis-Spinaceamine substituted compounds SH1, H2, H3, H4, SPH1, SPH2, and SPH4 (Scheme 1). All the synthesised compounds were fully characterised by using several analytical and spectral data (see experimental part for details).
Scheme 1. General synthetic route for the synthesis of the bis-histamine Schiff bases and bis-spinaceamine substituted compounds (incorporating the fused imidazole ring system).
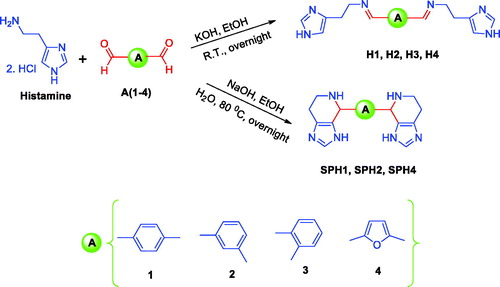
In the current work, the synthesis of a series of bis-histamine Schiff bases and bis-spinaceamine substituted compounds was carried out with some modifications of the literature proceduresCitation2. Briefly, histamine dihydrochloride was coupled with substituted aromatic and heterocyclic bis-aldehydes, leading to the formation of of bis-histamine Schiff bases and ring-closure products of histamine. The structures of bis-histamine Schiff bases and bis-spinaceamine substituted compounds were confirmed by using several analytical and spectral data (FT-IR, 1H-NMR, 13 C-NMR, and melting points) as described in the experimental part.
3.2. CA activation
Considering the fact that the new heterocyclic derivatives H1-H4 and SPH1, 2 and4 reported here incorporate in their molecules two functionalities with a pKa appropriate for acting as proton shuttles in the CA catalytic cycleCitation3,Citation12, we have investigated them as CAAs against the following four CA isoforms with important physiological functions: the three cytosolic enzymes (h = human), hCA I, II and VIICitation19, and the membrane-associated hCA IVCitation20. They are involved in various pathologies, both in the CNS, kidneys, eyes and other organs in which they are highly abundantCitation21–23.
The following structure–activity relationship (SAR) can be evidenced from data of :
Table 1. CA activation data with bis-histamines H1-H4 and bis-spinaceamines SPH(1, 2 and4) and histamine(HST) as a standard activator, by a stopped-flow CO2 hydrase assayCitation13.
Among the four investigated isoforms, hCA VII was the most sensitive to these activators (similar to the lead compounds used for obtaining these derivativesCitation1,Citation2), followed by hCA IV and I, whereas hCA II was the least sensitive to the activating effects of these compounds. However, these new derivatives reported here – H1-H4 and SPH(1, 2 and4) – were much more effective as hCA II activators compared to histamine (HST), a standard activatorCitation3 ().
The slow cytosolic isoform hCA I was activated efficiently by H1-H4 and SPH(1, 2 and4), with KAs ranging between 4.73–18.4 µM. Similar activities were observed for the bis-histamine Schiff bases and the bis-spinaceamine derivatives, with the main factor influencing activity being the spacer between the two imidazole moieties. Indeed, for this isoform, the p-phenylene spacer present in H1 and the m-phenylene one, present in H2 and SPH2, led to the most effective activators ():
The fast cytosolic enzyme hCA II was also effectively activated by the new derivatives, with KAs ranging between 6.15 and42.1 µM (compared to a KA of 125 µM for histamine). The rationale of our drug design was in fact to introduce two proton shuttling moieties, of the histamine/spinaceamine type, in order to enhance the affinity for the enzyme and to facilitate the rate-determining step of the catalytic cycle. Although for hCA I this is not obvious, for hCA II the activating effects of the bis-derivatives investigated here are indeed much higher compared to the mono-derivatives incorporating just one proton shuttling moiety, as histamine. In fact, the best bis-activator of hCA II, compound SPH2, is 20.3-times a more effective activator compared to histamine ().
(iv)The membrane-anchored hCA IV was activated by the new derivatives with activation constants ranging between 2.37 and 32.7 µM. Many of the new activators (e.g., H1, H2, H4 and SPH4) were more effective than histamine (KA of 4.03 µM) whereas the remaining ones were slightly less effective. Again the spacer between the two imidazole(-like) units was the main factor responsible of these effects, with the 2,5-furylene one leading to effective hCA IV activators (H4 and SPH4).
The most activatable isoform was the brain-associated hCA VII, for which the new activators reported here showed KAs ranging between 32 nM and 18.7 µM. One histamine bis-Schiff base (H4) and all three bis-spinaceamines SPH1, 2 and 4, were nanomolar hCA VII activators, with affinities of 32-85 nM (). Thus, for these last derivatives, the nature of the spacer had less influence on activity, as all of them show a behavior of potent activators, whereas for the histamine derivatives only the furyl-containing compound (H4) was an effective activator, with the phenylene ones H1-H3 being several orders of magnitude less effective.
4. Conclusions
We report here a small series of histamine bis-Schiff bases and bis-spinaceamine derivatives, which were synthesized by original procedures and investigated as activators of four hCA isoforms involved in a variety of diseases, the cytosolic hCA I, II and VII, and the membrane-associated hCA IV. All these isoforms were effectively activated by the new derivatives, with activation constants in the range of 4.73–10.2 µM for hCA I, 6.15–42.1 µM for hCA II, 2.37–32.7 µM for hCA IV and 32 nM -18.7 µM for hCA VII, respectively. The nature of the spacer between the two histamine/spinaceamine units of these molecules was the main contributor to the diverse activating efficacy, with a very different fine tuning for the diverse isoforms. As CA activators recently emerged as interesting agents for enhancing cognition, in the management of CA deficiencies, or for therapy memory and artificial tissues engineering, our compounds may be considered as candidates for such applications.
Disclosure statement
The authors declare no conflict of interest.
Additional information
Funding
Related Research Data
References
- Akocak S, Lolak N, Vullo D, et al. Synthesis and biological evaluation of histamine Schiff bases as carbonic anhydrase I, II, IV, VII, and IX activators. J Enzyme Inhib Med Chem 2017;32:1305–12.
- Akocak S, Lolak N, Bua S, et al. α-Carbonic anhydrases are strongly activated by spinaceamine derivatives. Bioorg Med Chem 2019;27:800–4.
- a) Supuran CT. Carbonic anhydrase activators. Future Med Chem 2018;10:561–73. b) Temperini C, Scozzafava A, Supuran CT. Carbonic anhydrase activation and the drug design. Curr Pharm Des 2008;14:708–15. c) Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81.
- a) Clare BW, Supuran CT. Carbonic anhydrase activators. 3: Structure‐activity correlations for a series of isozyme II activators. J Pharm Sci 1994;83:768–73. b) Ozensoy Guler O, Capasso C, Supuran CT. A magnificent enzyme superfamily: carbonic anhydrases, their purification and characterization. J Enzyme Inhib Med Chem 2016;31:689–94. c) Briganti F, Mangani S, Orioli P, et al. Carbonic anhydrase activators: X-ray crystallographic and spectroscopic investigations for the interaction of isozymes I and II with histamine. Biochemistry 1997;36:10384–92.
- a) Temperini C, Scozzafava A, Vullo D, Supuran CT. Carbonic anhydrase activators. Activation of isozymes I, II, IV, VA, VII, and XIV with l- and d-histidine and crystallographic analysis of their adducts with isoform II: engineering proton-transfer processes within the active site of an enzyme. Chemistry 2006;12:7057–66. b) Temperini C, Scozzafava A, Vullo D, Supuran CT. Carbonic anhydrase activators. Activation of isoforms I, II, IV, VA, VII, and XIV with L- and D-phenylalanine and crystallographic analysis of their adducts with isozyme II: stereospecific recognition within the active site of an enzyme and its consequences for the drug design. J Med Chem 2006;49:3019–27. c) Temperini C, Innocenti A, Scozzafava A, Supuran CT. Carbonic anhydrase activators: kinetic and X-ray crystallographic study for the interaction of D- and L-tryptophan with the mammalian isoforms I-XIV. Bioorg Med Chem 2008;16:8373–8. d) Temperini C, Innocenti A, Scozzafava A, et al. Carbonic anhydrase activators: L-Adrenaline plugs the active site entrance of isozyme II, activating better isoforms I, IV, VA, VII, and XIV. Bioorg Med Chem Lett 2007;17:628–35.
- a) Bhatt A, Mondal UK, Supuran CT, et al. Crystal structure of carbonic anhydrase II in complex with an activating ligand: implications in neuronal function. Mol Neurobiol 2018;55:7431–7. b) Temperini C, Scozzafava A, Supuran CT. Carbonic anhydrase activators: the first X-ray crystallographic study of an adduct of isoform I. Bioorg Med Chem Lett 2006;16:5152–6. c) Temperini C, Scozzafava A, Puccetti L, Supuran CT. Carbonic anhydrase activators: X-ray crystal structure of the adduct of human isozyme II with L-histidine as a platform for the design of stronger activators. Bioorg Med Chem Lett 2005;15:5136–41. d) Draghici B, Vullo D, Akocak S, et al. Ethylene bis-imidazoles are highly potent and selective activators for isozymes VA and VII of carbonic anhydrase, with a potential nootropic effect. Chem. Commun 2014;50:5980–3.
- a) Supuran CT. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin Drug Discov 2017;12:61–88. b) Supuran CT. Structure and function of carbonic anhydrases. Biochem J 2016;473:2023–32. c) Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77. d) Supuran CT, Vullo D, Manole G, et al. Designing of novel carbonic anhydrase inhibitors and activators. Curr Med Chem Cardiovasc Hematol Agents 2004;2:49–68.
- a) Alterio V, Di Fiore A, D'Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68. b) Supuran CT. Carbonic anhydrase inhibitors and their potential in a range of therapeutic areas. Expert Opin Ther Pat 2018;28:709–12. c) Briganti F, Pierattelli R, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. Part 37. Novel classes of carbonic anhydrase inhibitors and their interaction with the native and cobalt-substituted enzyme: kinetic and spectroscopic investigations. Eur J Med Chem 1996;31:1001–10. d) Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31:345–60.
- a) Supuran CT. Carbonic anhydrase inhibitors in the treatment and prophylaxis of obesity. Expert Opin Ther Pat 2003;13:1545–50. b) Winum JY, Temperini C, El Cheikh K, et al. Carbonic anhydrase inhibitors: clash with Ala65 as a means for designing inhibitors with low affinity for the ubiquitous isozyme II, exemplified by the crystal structure of the topiramate sulfamide analogue. J Med Chem 2006;49:7024–31. c) Gul HI, Mete E, Eren SE, et al. Designing, synthesis and bioactivities of 4-[3-(4-hydroxyphenyl)-5-aryl-4,5-dihydro-pyrazol-1-yl]benzenesulfonamides. J Enzyme Inhib Med Chem 2017;32:169–75. d) Alper Türkoğlu E, Şentürk M, Supuran CT, Ekinci D. Carbonic anhydrase inhibitory properties of some uracil derivatives. J Enzyme Inhib Med Chem 2017;32:74–7.
- a) Supuran CT. Applications of carbonic anhydrases inhibitors in renal and central nervous system diseases. Expert Opin Ther Pat 2018;28:713–21. b) Supuran CT. Carbonic anhydrase inhibitors and their potential in a range of therapeutic areas. Expert Opin Ther Pat 2018;28:709–12. c) Supuran CT. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin Drug Discov 2017;12:61–88.
- Tu CK, Silverman DN, Forsman C, et al. Role of histidine 64 in the catalytic mechanism of human carbonic anhydrase II studied with a site-specific mutant. Biochemistry 1989;28:7913–8.
- a) Canto de Souza L, Provensi G, Vullo D, et al. Carbonic anhydrase activation enhances object recognition memory in mice through phosphorylation of the extracellular signal-regulated kinase in the cortex and the hippocampus. Neuropharmacology 2017;118:148–56. b) Wang X, Schröder HC, Schlossmacher U, et al. Modulation of the initial mineralization process of SaOS-2 cells by carbonic anhydrase activators and polyphosphate. Calcif Tissue Int 2014;94:495–509. c) Sanku RKK, John JS, Ilies MA, Walker EA. Potential learning and memory disruptors and enhancers in a simple, 1-day operant task in mice. Behavioural Pharmacol 2018;29:482–92.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73.
- a) Angeli A, Del Prete S, Alasmary FAS, et al. The first activation studies of the η-carbonic anhydrase from the malaria parasite Plasmodium falciparum with amines and amino acids. Bioorg Chem 2018;80:94–8. b) Angeli A, Kuuslahti M, Parkkila S, Supuran CT. Activation studies with amines and amino acids of the α-carbonic anhydrase from the pathogenic protozoan Trypanosoma cruzi. Bioorg Med Chem 2018;26:4187–90. c) Stefanucci A, Angeli A, Dimmito MP, et AL. Activation of β- and γ-carbonic anhydrases from pathogenic bacteria with tripeptides. J Enzyme Inhib Med Chem 2018;33:945–50.
- a) Angeli A, Alasmary FAS, Del Prete S, et al. The first activation study of a δ-carbonic anhydrase: TweCAδ from the diatom Thalassiosiraweissflogii is effectively activated by amines and amino acids. J Enzyme Inhib Med Chem 2018;33:680–5. b) Tanini D, Capperucci A, Supuran CT, Angeli A. Sulfur, selenium and tellurium containing amines act as effective carbonic anhydrase activators. Bioorg Chem 2019;87:516–22. c) Rami M, Winum JY, Supuran CT, et al. (Hetero)aryl substituted thiazol-2,4-yl scaffold as human carbonic anhydrase I, II, VII and XIV activators. J Enzyme Inhib Med Chem 2019;34:224–9.
- a) Bua S, Haapanen S, Kuuslahti M, et al. Activation studies of the β-carbonic anhydrase from the pathogenic protozoan Entamoeba histolytica with amino acids and amines. Metabolites 2019;9:26. b) Angeli A, Buonanno M, Donald WA, et al. The zinc - but not cadmium - containing ζ-carbonic from the diatom Thalassiosira weissflogii is potently activated by amines and amino acids. Bioorg Chem 2018;80:261–5.
- a) Angeli A, Del Prete S, Osman SM, et al. Activation studies with amines and amino acids of the β-carbonic anhydrase encoded by the Rv3273 gene from the pathogenic bacterium Mycobacterium tuberculosis. J Enzyme Inhib Med Chem 2018;33:364–9. b) Angeli A, Del Prete S, Donald WA, et al. The γ-carbonic anhydrase from the pathogenic bacterium Vibrio cholerae is potently activated by amines and amino acids. Bioorg Chem 2018;77:1–5. c) Angeli A, Chiaramonte N, Manetti D, et al. Investigation of piperazines as human carbonic anhydrase I, II, IV and VII activators. J Enzyme Inhib Med Chem 2018;33:303–8. d) Angeli A, Del Prete S, Osman SM, et al. Activation studies of the α- and β-carbonic anhydrases from the pathogenic bacterium Vibrio cholerae with amines and amino acids. J Enzyme Inhib Med Chem 2018;33:227–33.
- a) Vullo D, Del Prete S, Osman SM, et al. Comparison of the amine/amino acid activation profiles of the β- and γ-carbonic anhydrases from the pathogenic bacterium Burkholderia pseudomallei. J Enzyme Inhib Med Chem 2018;33:25–30. b) Le Duc Y, Licsandru E, Vullo D, et al. Carbonic anhydrases activation with 3-amino-1H-1,2,4-triazole-1-carboxamides: Discovery of subnanomolar isoform II activators. Bioorg Med Chem 2017;25:1681–6. c) Licsandru E, Tanc M, Kocsis I, et al. A class of carbonic anhydrase I - selective activators. J Enzyme Inhib Med Chem 2017;32:37–46.
- a) Buonanno M, Di Fiore A, Langella E, et al. The Crystal Structure of a hCA VII Variant Provides Insights into the Molecular Determinants Responsible for Its Catalytic Behavior. Int J Mol Sci 2018;19:1571. b) Monti DM, De Simone G, Langella E, et al. Insights into the role of reactive sulfhydryl groups of Carbonic Anhydrase III and VII during oxidative damage. J Enzyme Inhib Med Chem 2017;32:5–12.
- a) Kumar R, Sharma V, Bua S, et al. Synthesis and biological evaluation of benzenesulphonamide-bearing 1,4,5-trisubstituted-1,2,3-triazoles possessing human carbonic anhydrase I, II, IV, and IX inhibitory activity. J Enzyme Inhib Med Chem 2017;32:1187–94. b) Bozdag M, Bua S, Osman SM, et al. Carbonic anhydrase I, II, IV and IX inhibition with a series of 7-amino-3,4-dihydroquinolin-2(1H)-one derivatives. J Enzyme Inhib Med Chem 2017;32:885–92. c) Bertol E, Vaiano F, Mari F, et al. Advances in new psychoactive substances identification: the U.R.I.To.N. Consortium. J Enzyme Inhib Med Chem 2017;32:841–9.
- a) Angeli A, Vaiano F, Mari F, et al. Psychoactive substances belonging to the amphetamine class potently activate brain carbonic anhydrase isoforms VA, VB, VII, and XII. J Enzyme Inhib Med Chem 2017;32:1253–9. b) El-Gazzar MG, Nafie NH, Nocentini A, et al. Carbonic anhydrase inhibition with a series of novel benzenesulfonamide-triazole conjugates. J Enzyme Inhib Med Chem 2018;33:1565–74. c) Krasavin M, Korsakov M, Ronzhina O, et al. Primary mono- and bis-sulfonamides obtained via regiospecific sulfochlorination of N-arylpyrazoles: inhibition profile against a panel of human carbonic anhydrases. J Enzyme Inhib Med Chem 2017;32:920–34.
- a) Pustenko A, Stepanovs D, Žalubovskis R, et al. 3H-1,2-benzoxathiepine 2,2-dioxides: a new class of isoform-selective carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2017;32:767–75. b) Entezari Heravi Y, Sereshti H, Saboury AA, et al. 3D QSAR studies, pharmacophore modeling, and virtual screening of diarylpyrazole-benzenesulfonamide derivatives as a template to obtain new inhibitors, using human carbonic anhydrase II as a model protein. J Enzyme Inhib Med Chem 2017;32:688–700. c) D'Ascenzio M, Guglielmi P, Carradori S, et al. Open saccharin-based secondary sulfonamides as potent and selective inhibitors of cancer-related carbonic anhydrase IX and XII isoforms. J Enzyme Inhib Med Chem 2017;32:51–9.
- a) Melis C, Meleddu R, Angeli A, et al. Isatin: a privileged scaffold for the design of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2017;32:68–73. b) Gul HI, Mete E, Eren SE, et al. Designing, synthesis and bioactivities of 4-[3-(4-hydroxyphenyl)-5-aryl-4,5-dihydro-pyrazol-1-yl]benzenesulfonamides. J Enzyme Inhib Med Chem 2017;32:169–75. c) Gul HI, Mete E, Taslimi P, et al. Synthesis, carbonic anhydrase I and II inhibition studies of the 1,3,5-trisubstituted-pyrazolines. J Enzyme Inhib Med Chem 2017;32:189–92.