
Abstract
To develop multifunctional aldose reductase (AKR1B1) inhibitors for anti-diabetic complications, a novel series of 2-phenoxypyrido[3,2-b]pyrazin-3(4H)-one derivatives were designed and synthesised. Most of the derivatives were found to be potent and selective against AKR1B1, and 2-(7-chloro-2-(3,5-dihydroxyphenoxy)-3-oxopyrido[3,2-b]pyrazin-4(3H)-yl) acetic acid (4k) was the most active with an IC50 value of 0.023 µM. Moreover, it was encouraging to find that some derivatives showed strong antioxidant activity, and among them, the phenolic 3,5-dihydroxyl compound 4l with 7-bromo in the core structure was proved to be the most potent, even comparable to that of the well-known antioxidant Trolox. Thus the results suggested success in the construction of potent and selective AKR1B1 inhibitors with antioxidant activity.
Introduction
Diabetes mellitus (DM) is a metabolic disorder resulting from defects in insulin secretion, insulin action, or bothCitation1. People suffering from DM are vulnerable to chronic diabetic complications, including neuropathy, retinopathy, nephropathy, and cataracts, which are the major menace to diabetic patientsCitation2,Citation3. Increasing clinical research indicated that the abnormal polyol pathway flux of blood glucose is obviously related to pathogenesis of diabetes complicationsCitation4. Normally, the blood glucose is predominantly converted to glucose-6-phosphate by hexokinase and then enters the glycolytic pathway. However, at high glucose concentration, specifically in diabetics, the combining capacity of aldose reductase (AKR1B1) to glucose is motivated and about one-third of the total glucose is metabolised through the polyol pathway in tissues such as lens, kidney, retina, and peripheral nervesCitation5–7. In polyol pathway (), AKR1B1 is the first enzyme and catalyses the reduction of glucose by NADPH to sorbitol, which can, in turn, be oxidised by the enzyme sorbitol dehydrogenase with concomitant reduction of NAD+. In the tissues implicated in these pathologies, the increased polyol pathway flux would directly cause the accumulation of sorbitol, which is hard to penetrate through cellular membranes, resulting in osmotic imbalance, cell swelling, and membrane permeability changes. All these AKR1B1-mediated biochemical alterations contribute to the pathogenesis of diabetic complicationsCitation8–10. Therefore, design and synthesis aldose reductase inhibitors (ARIs) to inhibit the activity of AKR1B1 and further regulate the polyol pathway of glucose are likely to be a promising therapy to prevent the development of diabetic complications.
Figure 1. Polyol pathway and pathogenesis of diabetic complications.
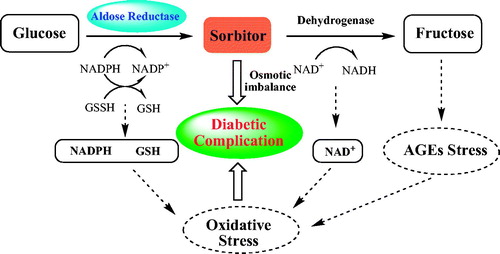
Numerous variously ARIs () have been developed and some of them are endowed with excellent inhibitory activityCitation11–15. However, most of the ARIs have failed clinically due to inadequate efficacies or pharmacokinetic drawbacks. Although the underlying mechanism of the low efficacy is unclear at present, it is speculated that only inhibiting the accumulation of sorbitol is not enough to prevent and treat pathological changes in all the tissues. As the reduction of glucose consumes NADPH, which is required for regenerating reduced glutathione (GSH), this could induce intracellular oxidative stress (). In addition, the abnormal decrease of NAD+ gives rise to changes in cellular redox potentials and the activity of enzymes such as nitric oxide synthase (NOS) and GSH reductase would further exacerbate intracellular oxidative stressCitation16–18. The pathogenesis of diabetic complications and hyperglycaemia-induced oxidative stress always promote to each other. Thus, it will be particularly important to develop ARIs having antioxidant activity, which keeps the enzyme in its reduced form and decreases the damage due to oxidative stress, and this could enhance the overall efficacy of ARIs targeted at diabetic complications.
Figure 2. Structures of ARIs.
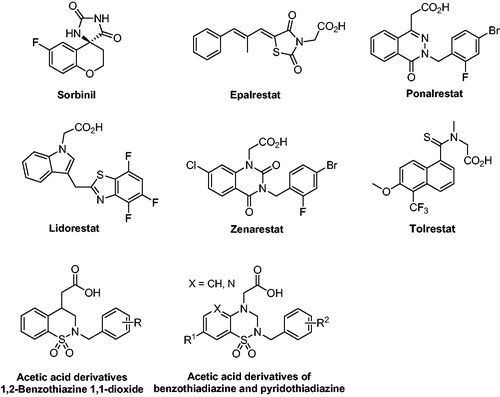
Based on our previous work of structurally different ARIsCitation19–22, we have engaged in preparing a novel series of 2-phenoxypyrido[3,2-b]pyrazin-3(4H)-one derivatives as the new multifunctional ARIs with combined AKR1B1 inhibition and antioxidant activity. This study will focus on the further optimisation of the C2 side chain and the introduction of halogen substituent to the C7 position of the core structure.
Chemistry
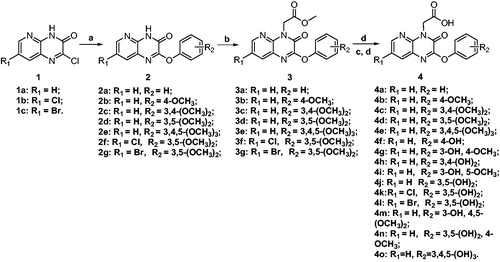
The designed compounds with an acetic acid substituent at N4 position and a variety of phenoxyl substituents at C2 position of the pyrido[2,3-b]pyrazin-3(4H)-one scaffold were obtained by the syntheses starting from compounds 1a–c, which was prepared according to our previous reported methodologyCitation20,Citation21. As shown in Scheme 1, compounds 1a–c were firstly reacted with different phenolate to obtain compounds 2a–g. Then the phenol methyl ethers 2a–g were alkylated with methyl bromoacetate at the N4 position to form 3a–g as key intermediates. Direct hydrolysis of 3a–e with lithium hydroxide yielded compounds 4a–e, and demethylation of partial or all the methoxyl with AlCl3 followed by hydrolysis of 3b–g gave the target compounds 4f–o. The detailed experimental data was depicted in the supplemental material.
Scheme 1. a: PhOH, DMF, K2CO3, 75 °C; b: BrCH2COOCH3, K2CO3, CH3CN, 55 °C; c: AlCl3, CH2Cl2, 45 °C; d: LiOH, H2O, THF, r.t., then 0.1 N HCl.
Results and discussion
All new synthetic compounds 4a–o were tested for their potential inhibitory activity of AKR1B1 isolated from rat lenses. Besides, the inhibitory activity of aldehyde reductase (AKR1A1) isolated from rat kidneys was also investigated to evaluate the selectivity for AKR1B1. The enzyme AKR1A1 is closely related to AKR1B1, and plays an important role in physiological detoxification. The validity of the inhibition results was assessed with respect to epalrestat as a positive ARI. Additionally, the antioxidant activity was assessed by using the model reaction with the stable free radical of 2,2-diphenyl-1-picrylhydrazyl (DPPH) according to the modified method, which was first employed by BloisCitation23.
Inhibition of enzymes
As shown in , most of the 2-phenoxypyrido[3,2-b]pyrazin-3(4H)-one derivatives showed significant AKR1B1 inhibition and selectivity. Of all the compounds, 2–(7-chloro-2–(3,5-dihydroxyphenoxy)-3-oxopyrido[3,2-b]pyrazin-4(3H)-yl)acetic acid (4k) was the most active having an IC50 value of 0.023 µM and was more potent than epalrestat. In contrast, compound 4a, which has no structural modification on the core structure and C2 phenoxyl side chain, was the lowest effective with an IC50 value of 8.596 µM. It is encouraging to find that introduction of phenolic hydroxyl or methoxyl to the C2 phenoxyl ring of 4a could enhance the inhibitory activity. Compounds 4b–e, 4g, 4i, 4m, and 4n containing one or more methoxyl on the C2-phenoxyl side chain displayed relatively low AKR1B1 inhibition with IC50 values ranging from 2.357 to 7.569 µM. Further demethylation of all methoxyl group leading to compounds 4f, 4h, 4j, and 4o showed an obvious enhancement in AKR1B1 inhibition with IC50 values in the range between 0.087 and 0.859 µM. The phenolic hydroxyl in C2 phenoxyl ring had an effect on AKR1B1 inhibition with the rank order of 3,5-(OH)2>4-OH > 3,4-(OH)2>3,4,5-(OH)3>3,5-(OH)2, 4-OCH3>3-OH, 4-OCH3>3-OH, 5-OCH3>3-OH, 4,5-(OCH3)2. Furthermore, the halogen substituent (Cl or Br) was introduced at the C7 position of compound 4j giving 4k and 4l, which largely increased the inhibitory activity. Analysis of the structure–activity relationship (SAR) indicated that introduction of both phenolic hydroxyl to the C2 phenoxyl ring and halogen substituent at the C7 position of the core enhanced the AKR1B1 inhibitory activity. In addition, all compounds were also tested for their inhibition ability against AKR1A1, and showed low activity with IC50 values more than 15 µM, demonstrating good selectivity for AKR1B1.
Table 1. Enzyme inhibition activity of 2–(3-oxo-2-phenoxypyrido[3,2-b]pyrazin-4(3H)- yl)acetic acid derivatives.
The antioxidant activity
The antioxidant activity of all synthesised compounds was determined with the DPPH model reaction and 6-hydroxy-2,5,7,8-chroman-2-carboxylic acid (Trolox) was employed as a positive control (). Most of the derivatives containing phenolic hydroxyl showed good DPPH radical scavenging activity ranging from 21.5 to 80.6% at the concentration of 50 µM. Of all tested compounds, 4l with 3,5-dihydroxyl on the C2 phenoxyl ring and 7-bromo on the core structure showed the best scavenging activity, that is, 95.3, 80.6, and 46.8% at concentrations of 100, 50, and 10 µM, respectively, which had an almost similar activity compared with Trolox at high concentrations indicating the potency of antioxidant (). SAR study of compounds 4b vs. 4f, 4c vs. 4h, 4d vs. 4j, and 4e vs. 4o indicated that the substitution of methoxyl with phenolic hydroxyl observably enhanced the radical scavenging activity. Regarding the position of phenolic hydroxyl on the C2 phenoxyl side chain, the 3,5-dihydroxyl substituent was the most effective in activity enhancement when comparing all the phenolic hydroxyl derivatives. However, comparison of compounds 4j, 4k, and 4l revealed that the C7-halogen substituent had little impact on the radical scavenging activity.
Figure 3. Comparison on DPPH radical scavenging activity.
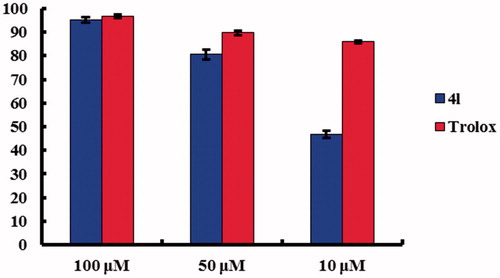
Table 2. Antioxidant activity of 2–(3-oxo-2-phenoxypyrido[3,2-b]pyrazin-4(3H) -yl)acetic acid derivatives.
Molecular docking
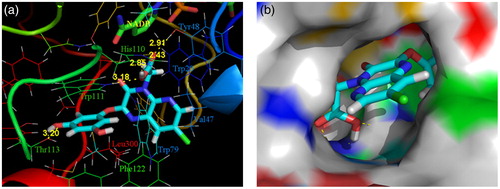
Compound 4k endowed with excellent activities both in the AKR1B1 inhibition and antioxidant reaction was docked with the conformation of the human AKR1B1/NADP+/lidorestat complex (PDB code: 1Z3N), to understand the mechanistic details and the above-described SARs. As shown in , compound 4k fitted well to the active site of AKR1B1. The carboxylate group was inserted deeply in the anion binding site by forming tight hydrogen-bonding interactions with Tyr48 (2.91 Å), Trp111 (3.18 Å), and His110 (2.85 and 2.43 Å) and engaging in a stabilizing electrostatic interaction with the positively charged nicotinamide moiety of the NADP cofactor (N–O = 4.12 Å). Besides, the 3-hydroxyl oxygen atom of the C2 phenoxyl side chain formed an additional hydrogen bond with the side chain of Thr113 (3.20 Å), confirming the importance of phenolic hydroxyl on the activity enhancement of AKR1B1 inhibition. Moreover, the 3,5-dihydroxyphenoxyl ring of the C2 side chain was well placed into the specificity pocket and paralleled to the indole ring of Trp111 forming a stable stacking interaction. Meanwhile, the pyrido[2,3-b]pyrazine-3(4H)-one core structure matched very well the hydrophobic pocket and the 7-chloro substituent pointed towards the opening of the active cleft.
Figure 4. Docking of 4k into the active site of AKR1B1. (a) The protein structure is shown in ribbon and tube representation with selected residues labeled and shown in line representation, ligand, and NADP are shown as stick models. The docked pose of 4k is shown in cyan (C), red (O), blue (N), and green (Cl). Hydrogen bonds are shown as yellow dashed lines. (b) Protein residues are in surface representation.
Conclusion
In conclusion, a series of novel ARI candidates (4a–o) based on pyrido[2,3-b]pyrazin-3(4H)-one core were synthesised and biologically evaluated for AKR1B1 inhibition and selectivity, as well as anti-oxidative properties through DPPH radical scavenging test. All compounds exhibited excellent AKR1B1 inhibitory activity in the IC50 range of 0.023 to 8.569 µM. Therein, compounds 4j, 4k, and 4l containing phenolic 3,5-dihydroxyl on the C2 phenoxyl side chain were not only sufficient to inhibit AKR1B1 but also effective for DPPH radical scavenging, which indicated success in the development of potent ARIs with antioxidant activity. Compound 4k was the most potent against AKR1B1 and much less active against AKR1A1 suggesting distinguished selectivity, while compound 4l showed the best DPPH scavenging activity even comparable with Trolox at high concentrations. The SAR studies indicated that the combination of phenolic 3,5-dihydroxyl in the C2-phenoxy group and C7-halogen (Cl or Br) in the core structure largely increased the AKR1B1 inhibitory activity, and the 3,5-dihydroxyl substituent contributed greatly to the activity enhancement of anti-oxidation when comparing all the phenolic hydroxyl derivatives, which provided a beneficial strategy for the discovery of new potent ARIs with antioxidant activity.
Supplemental Material
Download MS Word (123 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Related Research Data
References
- Williamson JR, Chang K, Frangos M, et al. Hyperglycemic pseudohypoxia and diabetic complications. Diabetes 1993;42:801–13.
- American Diabetes Association. Prevention or delay of type 2 diabetes: standards of medical care in diabetes-2018. Diabetes Care 2018;41:51.
- Davidson EP, Coppey LJ, Shevalye H, et al. Vascular and neural complications in type 2 diabetic rats: improvement by sacubitril/valsartan greater than valsartan alone. Diabetes 2018;67:1616–26.
- Brownlee M. Biochemistry and molecular biology of diabetic complications. Nature 2001;414:813–20.
- Boulton AJ. The pathogenesis of diabetic foot problems: an overview. Diabetes 1996;13:58–61.
- Nakayama M, Nakamura J, Hamada Y, et al. Aldose reductase inhibition ameliorates pupillary light reflex and f-wave latency in patients with mild diabetic neuropathy. Diabetes Care 2001;24:1093–8.
- Alexiou P, Pegklidou K, Chatzopoulou M, et al. Aldose reductase enzyme and its implication to major health problems of the 21(st) century. Curr Med Chem 2009;16:734–52.
- Alim Z, Kilinç N, Şengül B, Beydemir Ş. Inhibition behaviours of some phenolic acids on rat kidney aldose reductase enzyme: an in vitro study. J Enzym Inhib Med Chem 2017;32:277–84.
- Han Z, Hao X, Gao Z, et al. Multifunctional aldose reductase inhibitors based on 2h-benzothiazine 1,1-dioxide. RSC Adv 2016;6:12761–9.
- Balestri F, Quattrini L, Coviello V, et al. Acid derivatives of pyrazolo[1,5-a]pyrimidine: the first class of aldose reductase differential inhibitors. Cell Chem Biol 2018;25:1414–8.
- Da Settimo F, Primofiore G, La Motta C, et al. Naphtho[1,2-d]isothiazole acetic acid derivatives as a novel class of selective aldose reductase inhibitors. J Med Chem 2005;48:6897–907.
- Sartini S, Cosconati S, Marinelli L, et al. Benzofuroxane derivatives as multi-effective agents for the treatment of cardiovascular diabetic complications. Synthesis, functional evaluation, and molecular modeling studies. J Med Chem 2012;55:10523.
- Maccari R, Ottana R. Targeting aldose reductase for the treatment of diabetes complications and inflammatory diseases: new insights and future directions. J Med Chem 2015;58:2047–67.
- Nicolaou I, Demopoulos VJ. Substituted pyrrol-1-ylacetic acids that combine aldose reductase enzyme inhibitory activity and ability to prevent the nonenzymatic irreversible modification of proteins from monosaccharides. J Med Chem 2003;46:417–26.
- Chatzopoulou M, Mamadou E, Juskova M, et al. Structure-activity relations on [1-(3,5-difluoro-4-hydroxyphenyl)-1H -pyrrol-3-yl]phenylmethanone. The effect of methoxy substitution on aldose reductase inhibitory activity and selectivity. Bioorgan Med Chem 2011;19:1426–33.
- Maccari R, Vitale RM, Ottana R, et al. Structure–activity relationships and molecular modelling of new 5-arylidene-4-thiazolidinone derivatives as aldose reductase inhibitors and potential anti-inflammatory agents. Eur J Med Chem 2014;81:1–14.
- Polyxeni A, Demopoulos VJ. A diverse series of substituted benzenesulfonamides as aldose reductase inhibitors with antioxidant activity: design, synthesis, and in vitro activity. J Med Chem 2010;53:7756–66.
- La Motta C, Sartini S, Salerno S, et al. Acetic acid aldose reductase inhibitors bearing a five-membered heterocyclic core with potent topical activity in a visual impairment rat model. J Med Chem 2008;51:3182–93.
- Hao X, Qin X, Hussain S, et al. Chiral resolution, determination of absolute configuration, and biological evaluation of (1,2-benzothiazin-4-yl)acetic acid enantiomers as aldose reductase inhibitors. J Enzym Inhib Med Chem 2015;30:846–51.
- Qin X, Hao X, Han H, et al. Design and synthesis of potent and multifunctional aldose reductase inhibitors based on quinoxalinones. J Med Chem 2015;58:1254–67.
- Han Z, Hao X, Ma B, Zhu C. A series of pyrido[2,3-b] pyrazin-3(4 h) -one derivatives as aldose reductase inhibitors with antioxidant activity. Eur J Med Chem 2016;121:308–17.
- Zhu S, Zhang S, Hao X, et al. Pyridothiadiazine derivatives as aldose reductase inhibitors having antioxidant activity. J Enzym Inhib Med Chem 2016;31:126–30.
- Silva JP, Areias FM, Proença FM, Coutinho OP. Oxidative stress protection by newly synthesized nitrogen compounds with pharmacological potential. Life Sci 2006;78:1256–67.