
Abstract
Carbonic anhydrases (CAs, EC 4.2.1.1) are crucial metalloenzymes that are involved in diverse bioprocesses. We report the synthesis and biological evaluation of novel series of benzenesulfonamides incorporating un/substituted ethyl quinoline-3-carboxylate moieties. The newly synthesised compounds were in vitro evaluated as inhibitors of the cytosolic human (h) isoforms hCA I and II. Both isoforms hCA I and II were inhibited by the quinolines reported here in variable degrees: hCA I was inhibited with KIs in the range of 0.966–9.091 μM, whereas hCA II in the range of 0.083–3.594 μM. The primary 7-chloro-6-flouro substituted sulphfonamide derivative 6e (KI = 0.083 μM) proved to be the most active quinoline in inhibiting hCA II, whereas, its secondary sulfonamide analog failed to inhibit the hCA II up to 10 μM, confirming the crucial role of the primary sulphfonamide group, as a zinc-binding group for CA inhibitory activity.
Introduction
Carbonic anhydrases (CA) (CAs, EC 4.2.1.1) are zinc-containing metalloenzymes that are present in most organisms all over the tree of lifeCitation1,Citation2. These metalloenzymes efficiently catalyse the rapid interconversion of carbon dioxide and water to bicarbonate and protons. In humans, this fundamental reaction encompasses three simple chemical entities, CO2, HCO3−, and H+, essential in a host of physiological and pathological processes, such as calcification, bone resorption, electrolyte secretion, pH and CO2 homeostasis, tumorigenicity, and several biosynthetic reactionsCitation3–5. Eight distinct genetic enzymatic families were identified; the α-, β-, γ-, δ-, ζ-. η-, θ- and ι-CAsCitation3–5. To date, 15 human (h) isoforms of CA have been identified, which have all belong to the α-class and have different patterns of tissue distribution and cellular localisation as the following; cytosolic (I, II, III, VII, and XIII), membrane-bound (IV, IX, XII, and XIV), secreted (VI) and mitochondrial (VA and VB) formsCitation3–5. CA I and II are present at high concentrations comparing to other CA isoforms in the erythrocytes cytosol and several other tissues.
Several important pathological consequences result from the dysfunction of hCA II activity, thus this isoform is an established drug target for a multitude of diseases, such as oedemaCitation6, epilepsyCitation7, acute mountain sicknessCitation8, and glaucoma, where excessive aqueous humour is secreted within the eye, with the subsequent increase in the intraocular pressure (IOP)Citation9–11. CA inhibitors (CAIs) are able to diminish IOP by decreasing the rate of bicarbonate formation and thus secretion of the aqueous humour. For more than 60 years, carbonic anhydrase inhibitors are in clinical use for the treatment of glaucoma, such as the topically acting dorzolamide and brinzolamide drugs, and the systemic acetazolamide and methazolamide drugsCitation9 ().
Figure 1. Structures of some approved CAIs antiglaucoma drugs, and the target quinolines 6a–f and 11.
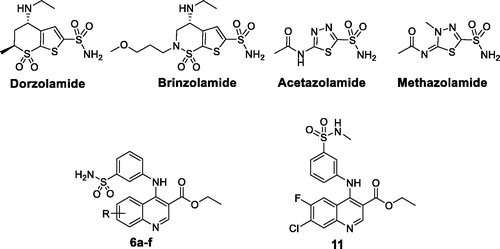
Pertaining to its prevalence in diverse natural products, such as alkaloids, and in different pharmacologically active substances, quinoline stands out as a promising privileged scaffold that is endowed with a wide spectrum of biological activities. Just to name a few, antimalarialCitation12, antileishmanialCitation13, anti-tubercularCitation14, antidepressantCitation15, anticancerCitation16,Citation17 and antiglaucomaCitation18 actions were reported for quinoline derivatives. Accordingly, medicinal chemists embarked on exploring various quinoline-based molecules comprehending their potential to develop promising and efficient bioactive compoundsCitation19,Citation20. These efforts led to FDA approval for several quinoline-based drugs such as the anticancer agent lenvatinib, the anti-asthmathic drug montelukast, the antiviral Clioquinol, and the anaesthetic Dibucain.
In the present study, we report a new series of primary benzenesulfonamides incorporating un/substituted ethyl quinoline-3-carboxylate (6a–6f, ) as well as the secondary benzenesulfonamide analogue (11, ), with the prime goal of developing effective quinoline-based antiglaucoma candidates targeting the cystolic isoform hCA II. These quinoline-based benzenesulfonamides were evaluated in vitro for their inhibitory activity towards the physiologically relevant hCA isoforms I and II, using stopped-flow CO2 hydrase assay.
Materials and methods
Chemistry
All reaction and manipulations were performed in nitrogen atmosphere using standard Schlenk techniques. All reaction solvents and reagents were purchased from commercial suppliers and used without further purification. Microwave-assisted synthesis was carried out in a Biotage Initiator + apparatus operating in single mode, the microwave cavity producing controlled irradiation at 2.45 GHz (Biotage AB, Uppsala, Sweden). The reactions were run in sealed vessels. These experiments were performed by employing magnetic stirring and a fixed hold time using variable power to reach (during 1 − 2 min) and then maintain the desired temperature in the vessel for the programed time period. The temperature was monitored by an IR sensor focused on a point on the reactor vial glass. The IR sensor was calibrated to internal solution reaction temperature by the manufacturer. The NMR spectra were obtained on Bruker Avance 400 (400 MHz 1H and 101 MHz 13 C NMR). 1H NMR spectra were referenced to tetramethylsilane (δ = 0.00 ppm) as an internal standard and were reported as follows: chemical shift, multiplicity (b = broad, s = singlet, d = doublet, t = triplet, dd = doublet of doublet, m = multiplet). Column chromatography was performed on Merck Silica Gel 60 (230–400 mesh) and eluting solvents for all of these chromatographic methods were noted as appropriated-mixed solvent with given volume-to-volume ratios. TLC was carried out using glass sheets pre-coated with silica gel 60 F254 purchased by Merk. High-resolution spectra were performed on Waters ACQUITY UPLC BEH C18 1.7 μ–Q-TOF SYNAPT G2-Si High Definition Mass Spectrometry. Compounds 3a-f, 4a-fCitation21–22 and 10Citation23 were previously prepared.
General procedure for preparation of compounds 4a–f
A solution of compounds 3a–f (1.0 mmol) in POCl3 (6 ml) was refluxed for 1 h. The mixture was evaporated in vacuo and the residue was extracted with methylene chloride, crushed ice and aqueous NH3. The organic layer was dried over Na2SO4 and concentrated. The residue was purified by column chromatography (SiO2, ethyl acetate (EA): n-Hex 10: 1) to get key intermediates 4a-fCitation21,Citation22.
General procedures for preparation of the target quinolines 6a–f and 11
To a MW vial, were successively added the appropriate ethyl 4-chloroquinoline-3-carboxylate derivative 4a-f (0.21 mmol), 3-aminobenzenesulfonamide 5 (0.036 gm, 0.21 mmol) or 3-amino-N-methylbenzenesulfonamide 10 (0.040 gm, 0.21 mmol), and ethanol (12 ml) at room temperature. The MW vial was sealed and heated under MW conditions for 30 min at 150 °C. The mixture was evaporated in vacuo and the residue was extracted with EA and NaHCO3 (aq). The organic layer was dried over Na2SO4 and concentrated. The residue was purified by column chromatography (SiO2, EA: n-Hex), in a gradient elution with 1:5 (EA: n-hex) ratio, to furnish quinolines 6a–f and 11, respectively.
Ethyl 4-((3-sulphamoylphenyl)amino)quinoline-3-carboxylate (6a)
White solid, yield: 49%, mp: 183.6 − 185.0 °C; 1H NMR (DMSO-d6, 400 MHz) δ ppm: 1.16 (t, J = 6.8 Hz, 3H, CH2 CH3), 4.05 (q, J = 6.8 Hz, 2H, CH2CH3), 7.16–7.18 (m, 1H, H-2 of benzenesulfonamide), 7.34 (s, 2H, SO2NH2), 7.45–7.56 (m, 4H, H-4,5,6 of benzenesulfonamide and H-6 quinoline), 7.80–7.84 (m, 1H, H-7 quinoline), 8.01 (d, J = 8.0 Hz, 1H, H-5 quinoline), 8.10 (d, J = 8.4 Hz, 1H, H-8 quinoline), 9.01 (s, 1H, H-2 quinoline), 9.75 (s, 1H, NH); 13 C NMR (DMSO-d6, 101 MHz) δ ppm: 14.31 (CH3), 61.48, 111.07 (quinoline C-3), 116.42 (benzenesulfonamide C-2), 119.82 (benzenesulfonamide C-4), 121.40 (quinoline C-10), 121.92 (benzenesulfonamide C-6), 124.88 (quinoline C-5), 126.55 (quinoline C-6), 130.05 (quinoline C-8), 130.22 (benzenesulfonamide C-5), 131.95 (quinoline C7), 144.44 (benzenesulfonamide C-3), 145.60 (quinoline C-4), 148.12 (benzenesulfonamide C-1), 150.36 (quinoline C-2), 151.38 (quinoline C-9), 166.81 (C = O); HRMS (ESI) for C18H18N3O4S: calcd 372.1018, found: 372.1017 [M + H]+.
Ethyl 6-methyl-4-((3-sulphamoylphenyl)amino)quinoline-3-carboxylate (6 b)
Yellow solid, yield: 97%, mp: 223.0 − 224.5 °C; 1H NMR (DMSO-d6, 400 MHz) δ ppm: 1.09 (t, J = 5.6 Hz, 3H, CH2 CH3), 2.40 (s, 3H, CH3), 3.89 (q, J = 5.6 Hz, 2H, CH2CH3), 7.25 (s, 1H, H-2 of benzenesulfonamide), 7.39 (s, 2H, SO2NH2), 7.47–7.55 (m, 3H, H-4,5,6 of benzenesulfonamide), 7.73–7.74 (m, 1H, H-5 quinoline), 7.96 (s, 1H, quinoline H-7), 8.26 (s, 1H, H-8 quinoline), 8.88 (s, 1H, H-2 quinoline), 10.25 (s, 1H, NH); 13 C NMR (DMSO-d6, 101 MHz) δ ppm: 14.22 (CH2 CH3), 21.73 (CH3), 61.57 (CH2), 111.34 (quinoline C-3), 116.98 (benzenesulfonamide C-2), 120.58 (quinoline C-10), 121.17 (benzenesulfonamide C-4), 122.55 (benzenesulfonamide C-6), 123.97 (quinoline C-6), 127.10 (quinoline C-5), 130.19 (benzenesulfonamide C-5), 134.72 (quinoline C-8), 136.89 (quinoline C-7), 143.66 (benzenesulfonamide C-3), 145.60 (benzenesulfonamide C-1), 144.74 (quinoline C-2), 148.89 (quinoline C-9), 166.02 (C = O); HRMS (ESI) for C19H20N3O4S: calcd 386.1175, found: 386.1170 [M + H]+.
Ethyl 6-methoxy-4-((3-sulphamoylphenyl)amino)quinoline-3-carboxylate (6c)
White solid, yield: 61%, mp: 214.9 − 216.3 °C; 1H NMR (DMSO-d6, 400 MHz) δ ppm: 1.13 (t, J = 6.8 Hz, 3H, CH2 CH3), 3.73 (s, 3H, OCH3), 3.99 (q, J = 6.8 Hz, 2H, CH2CH3), 7.14–7.16 (m, 1H, H-2 of benzenesulfonamide), 7.33 (s, 2H, SO2NH2), 7.42–7.48 (m, 4H, H-4,5,6 of benzenesulfonamide and H-7 quinoline), 7.96 (s, 1H, H-5 quinoline), 7.91–7.93 (m, 1H, H-8 quinoline), 8.84 (s, 1H, quinoline H-2), 9.95 (s, 1H, NH); 13 C NMR (DMSO-d6, 101 MHz) δ ppm: 14.31 (CH2 CH3), 21.73 (CH3), 55.92 (OCH3) 61.40 (CH2), 103.51 (quinoline C-4), 111.54 (quinoline C-3), 116.36 (benzenesulfonamide C-2), 119.51 (benzenesulfonamide C-4), 121.78 (quinoline C-10), 122.35 (benzenesulfonamide C-6), 123.78 (quinoline C-7), 130.17 (benzenesulfonamide C-5), 131.56 (quinoline C-8), 144.36 (benzenesulfonamide C-3), 145.55 (quinoline C-9), 146.16 (benzenesulfonamide C-1), 146.59 (quinoline C-2), 148.90 (quinoline C-4), 157.51 (quinoline C-6), 166.90 (C = O); HRMS (ESI) for C19H20N3O5S: calcd 402.1124, found: 402.1126 [M + H]+.
Ethyl 6-bromo-4-((3-sulphamoylphenyl)amino)quinoline-3-carboxylate (6d)
Yellow solid, yield: 71%, mp: 235.6 − 237.2 °C; 1H NMR (DMSO-d6, 400 MHz) δ ppm: 1.07 (t, J = 5.6 Hz, 3H, CH2 CH3), 3.88 (q, J = 5.6 Hz, 2H, CH2CH3), 7.17 (s, 1H, H-2 of benzenesulfonamide), 7.36 (s, 2H, SO2NH2), 7.45–7.51 (m, 3H, H-4,5,6 of benzenesulfonamide), 7.94 (s, 2H, H-7,8 quinoline), 8.53 (s, 1H, H-5 quinoline), 8.92 (s, 1H, H-2 quinoline), 9.71 (s, 1H, NH); 13 C NMR (DMSO-d6, 101 MHz) δ ppm: 14.22 (CH3), 61.49 (CH2), 111.61 (quinoline C-3), 116.40 (benzenesulfonamide C-2), 119.81 (quinoline C-10), 119.95, (benzenesulfonamide C-4), 121.47 (benzenesulfonamide C-6), 123.08 (quinoline C-6), 126.60 (quinoline C-5), 130.21 (benzenesulfonamide C-5), 132.13 (quinoline C-8), 134.76 (quinoline C-7), 144.01 (benzenesulfonamide C-3), 145.68 (benzenesulfonamide C-1), 146.37 (quinoline C-2), 148.91 (quinoline C-9), 152.06 (quinoline C-4), 166.33 (C = O); HRMS (ESI) for C18H17BrN3O4S: calcd 450.0123, found: 450.0127 [M + H]+.
Ethyl 7-chloro-6-fluoro-4-((3-sulphamoylphenyl)amino)quinoline-3-carboxylate (6e)
Yellow solid, yield: 55%, mp: 190.0 − 191.0 °C; 1H NMR (DMSO-d6, 400 MHz) δ ppm: 1.08 (t, J = 7.2 Hz, 3H, CH2CH3), 3.91 (q, J = 7.2 Hz, 2H, CH2CH3), 7.18–7.20 (m, 1H, H-2 of benzenesulfonamide), 7.45–7.49 (m, 3H, H-4,5,6 of benzenesulfonamide), 7.36 (s, 2H, SO2NH2), 8.18 (d, J = 11.2 Hz, H-8 quinoline), 8.25 (d, J = 7.6 Hz, 1H, H-5 quinoline), 8.91 (s, 1H, H-2 quinoline), 9.68 (s, 1H, NH); 13 C NMR (DMSO-d6, 101 MHz) δ ppm: 14.22 (CH3), 61.58 (CH2), 110.16, 110.40 (quinoline C-5), 111.47 (quinoline C-3), 116.52 (benzenesulfonamide C-2), 120.18 (quinoline C-10), 121.24 (benzenesulfonamide C-4), 121.85 (quinoline C-10), 125.21 (benzenesulfonamide C-6), 124.41 (benzenesulfonamide C-5), 130.29 (quinoline C-7), 131.58 (quinoline C-8), 143.76 (benzenesulfonamide C-3), 145.69 (benzenesulfonamide C-1), 147.05, 147.52 (quinoline C-6), 152.27 (quinoline C-9), 153.76 (quinoline C-2), 156.21 (quinoline C-4), 166.26 (C = O); HRMS (ESI) for C18H16ClFN3O4S: calcd 424.0534, found: 424.0525 [M + H]+.
Ethyl 5,7-dichloro-4-((3-sulphamoylphenyl)amino)quinoline-3-carboxylate (6f)
Yellow solid, yield: 98%, mp: 228.7 − 230.3 °C; 1H NMR (DMSO-d6, 400 MHz) δ ppm: 1.21 (t, J = 7.2 Hz, 3H, CH2CH3), 4.17 (q, J = 7.2 Hz, 2H, CH2CH3), 6.97–6.99 (m, 1H, H-2 of benzenesulfonamide), 7.28–7.37 (m, 3H, H-4,5,6 of benzenesulfonamide), 7.72 (d, J = 6.0 Hz, 1H, H-6 quinoline), 8.08 (s, 1H, H-8 quinoline), 9.09 (s, 1H, H-2 quinoline), 9.83 (s, 1H, NH); 13 C NMR (DMSO-d6, 101 MHz) δ ppm: 14.22 (CH3) 62.08 (CH2), 114.54 (quinoline C-3), 117.87 (benzenesulfonamide C-2), 118.79 (quinoline C-10), 120.52 (benzenesulfonamide C-4), 127.44 (benzenesulfonamide C-6), 129.58 (quinoline C-6), 130.43 (quinoline C-5, 8), 131.42 (benzenesulfonamide C-5), 136.15 (quinoline C-7), 136.62 (benzenesulfonamide C-1), 145.22 (quinoline C-4), 148.09 (quinoline C-9), 152.40 (quinoline C-2), 166.39 (C = O); HRMS (ESI) for C18H16Cl2N3O4S: calcd 440.0239, found: 440.0237 [M + H]+.
Ethyl 7-chloro-6-fluoro-4-((3-(N-methylsulphamoyl)phenyl)amino)quinoline-3-carboxylate (11)
Yellow solid; yield: 40%, 1H NMR (DMSO-d6, 400 MHz) δ ppm: 1.07 (t, J = 6.8 Hz, 3H, CH2CH3), 2.40 (s, 3H, NHCH3), 3.90 (q, J = 6.8 Hz, 2H, CH2CH3), 7.27 (d, J = 7.6 Hz, 1H, H-2 of benzenesulfonamide), 7.41 (s, 1H, NHCH3), 7.49–7.53 (m, 3H, H-4,5,6 of benzenesulfonamide), 8.19 (d, J = 11.6 Hz, 1H, H-5 quinoline), 8.26 (d, J = 7.2 Hz, 1H, H-8 quinoline), 8.91 (s, 1H, H-2 quinoline), 9.79 (s, 1H, NH); 13 C NMR (DMSO-d6, 101 MHz) δ ppm: 14.19 (CH2CH3), 29 (NHCH3), 61.52 (CH2), 110.18 (quinoline C-5), 110.42 (quinoline C-5), 111.56 (quinoline C-3), 117.44 (benzenesulfonamide C-2), 121.17 (benzenesulfonamide C-4), 121.25 (quinoline C-10), 121.33 (quinoline C-10), 122.63 (benzenesulfonamide C-6), 125.22 (quinoline C-7), 125.43 (quinoline C-7), 130.61 (benzenesulfonamide C-5), 131.58 (quinoline C-8), 140.89 (quinoline C-8), 144.09 (benzenesulfonamide C-3), 146.97 (benzenesulfonamide C-1), 147.02 (quinoline C-9), 147.54 (quinoline C-2), 152.26 (quinoline C-4), 153.76 (quinoline C6), 156.21 (quinoline C-6), 166.24 (C = O); HRMS (ESI) for C19H18ClFN3O4S: calcd 438.0691, found: 438.0693 [M + H]+.
CA inhibitory assay
An Applied Photophysics stopped-flow instrument has been used for assaying the CA catalyzed CO2 hydration activityCitation24. Phenol red (at a concentration of 0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 20 mM Hepes (pH 7.5) as buffer, and 20 mM Na2SO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalyzed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The uncatalysed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (0.1 mM) were prepared in distilled-deionised water and dilutions up to 0.01 nM were done thereafter with the assay buffer. Inhibitor and enzyme solutions were pre-incubated together for 15 min at room temperature prior to assay, in order to allow for the formation of the E-I complex. The inhibition constants were obtained by non-linear least-squares methods using PRISM 3 and the Cheng-Prusoff equation, as reported earlierCitation25–29 and represent the mean from at least three different determinations.
Results and discussion
Chemistry
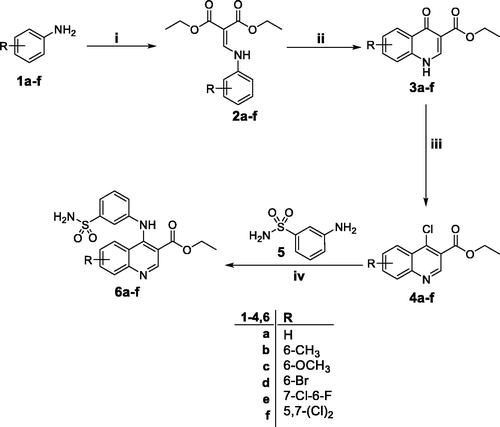
The methods adopted for synthesis of the target quinolines 6a–f and 11 are depicted in Schemes 1 and 2. Firstly, anilines 1a–f were heated with diethyl ethoxymethylenemalonate in refluxing ethanol to furnish diethyl 2-((phenylamino)methylene)malonate derivatives 2a–f which thermally cyclised to the corresponding ethyl 4-oxo-1,4-dihydroquinoline-3-carboxylates 3a–f via heating in diphenyl ether. Next, chlorination of quinolinones 3a–f was carried out under anhydrous condition through heating with excess of phosphorus oxychloride to afford the key intermediates ethyl 4-chloroquinoline-3-carboxylates 4a–f. The target primary 3-(quinolin-4-ylamino)benzenesulfonamides 6a–f were obtained through a MW assisted nucleophilic substitution reaction of 3-aminobenzenesulfonamide 5 with the appropriate key intermediate 4a–f in ethyl alcohol (Scheme 1).
Scheme 1. Synthesis of target quinolines 6a–f; Reagents and conditions: (i) DEEMM/Ethanol/reflux 1 h; (ii) Diphenyl ether/250 °C/45 min; (iii) POCl3/reflux 1 h; (iv) Absolute ethyl alcohol/reflux 4 h.
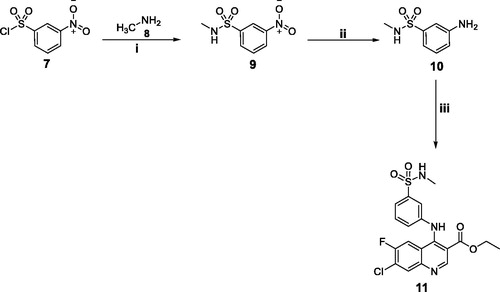
In Scheme 2, 3-amino-N-methylbenzenesulfonamide 10 was prepared as reported earlierCitation13 through a nucleophilic substitution for 3-nitrobenzenesulphonyl chloride 7 with methylamine, followed by a catalytic hydrogenation to the nitro function. The later reacted with the key intermediate 4e in refluxing ethanol to afford the target secondary benzenesulfonamide 11 (Scheme 2).
Scheme 2. Synthesis of target quinoline 11; Reagents and conditions: (i) Hunig's Base/THF/stirring at r.t./1 h; (ii) H2/10% Pd/C/MeOH/r.t.; (iii) Compound 4e/Absolute ethyl alcohol/reflux 4 h.
The structures of the newly prepared quinolines 6a–f and 11 were confirmed and elucidated by NMR spectroscopy and high resolution mass spectroscopy, which were in full agreement with the postulated structures (Supplementary material).
1H NMR spectra of quinolines 6a-f showed new characteristic signals at δ 7.33– 7.37 ppm, and 9.68–10.25 ppm corresponding to NH2 and NH groups, respectively, that distinguished the target quinolines 6a-f from the key intermediates chloroquinolines 4a-f. Also, the 1H NMR of 7-chloro-6-fluoro-4–(3-methanesulphonylaminophenyamino)-quinoline-3-carboxylic acid ethyl ester (11) displayed three significant signals at δ 2.99, 9.63 and 9.79 ppm assigned to -NHCH3, -SO2NH- and -NH- protons, respectively.
Biological evaluation
Carbonic anhydrase inhibition
The newly prepared 3-(quinolin-4-ylamino)benzenesulfonamides 6a–f and 11 were evaluated for their ability to inhibit the physiologically relevant hCA cytosolic isoforms, hCA I and II, by a stopped-flow CO2 hydrase assayCitation24. The inhibition data of the prepared quinolines and the sulfonamide acetazolamide AAZ (as a standard inhibitor) against the two examined isoforms are summarised in . The following structure-activity relationship (SAR) could be noted regarding the inhibition data reported in :
Table 1. Inhibition data of human CA isoforms hCA I and II for quinolines 6a–f and 11, determined by stopped-flow CO2 hydrase assay, using acetazolamide (AAZ) as a standard drug.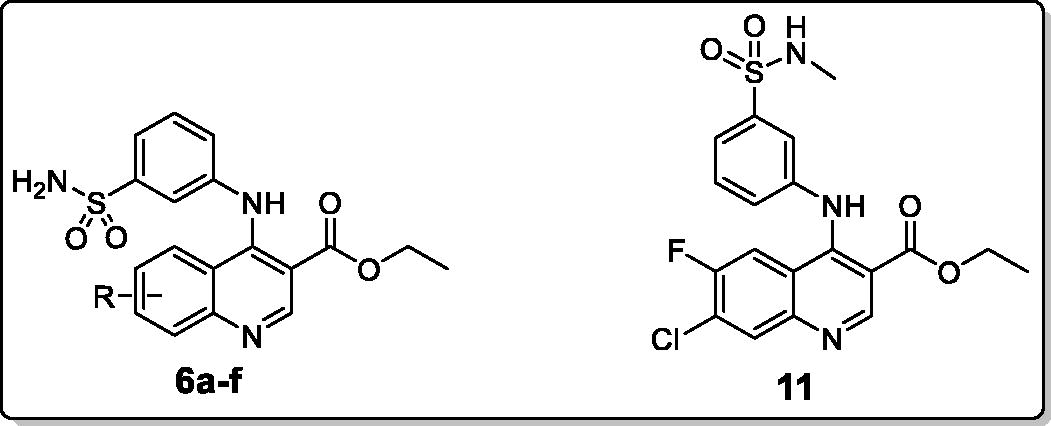
The secondary sulfonamide reported here (11) failed to inhibit the tested hCA isoforms (hCA I and hCA II) up to 10 μM, which confirmed the crucial role of the primary sulfonamide as a zinc-anchoring group, with the additional two hydrogen bonds with Thr199 and Thr200 residues within the enzyme active site.
The data presented in ascribed to the prepared primary sulfonamides (6a-6e) weak potency in inhibiting the ubiquitous cytosolic isoform hCA I with inhibition constants (KIs) in the micromolar range, in detail, between 4.233 and 9.091 μM, except for the 6-methoxy substituted analog 6c which arose as a submicromolar hCA I inhibitor with a KI equals 0.966 μM, which represents 3.8-fold decreased efficacy to the reference drug AAZ (KI equals 0.250 μM towards hCA I). On contrary, the 5,7-dichloro substituted primary sulfonamide 6f failed to inhibit the hCA I up to 10 μM.
Noteworthy, the SAR outcomes highlighted that grafting the strong electron-donating 6-metoxy group (compound 6c; KI = 0.966 μM) resulted in 4.4-fold efficacy enhancement in comparison to the unsubstituted analogue 6a (KI = 4.233 μM). Regarding the impact of substitution of the quinoline moiety within the primary sulfonamides series 6a-6f; the inhibitory activities were decreased in the order of 6-OCH3 >6-CH3 >7-Cl-6-F > 6-Br >5,7-(Cl)2.
The second ubiquitous cytosolic isoform examined here was hCA II. It was apparent from the displayed results () that the tested primary sulfonamides (6a-6e) effectively interfere with hCA II catalytic activities in submicromolar/micromolar concentration range (KI values of 0.083 – 3.594 μM), whereas, no significant inhibition towards hCA II was revealed for quinoline 6f (KI >10 μM). Nevertheless, among the tested quinolines, 7-chloro-6-flouro substituted compound 6e (KI = 0.083 μM) proved to be the most active quinoline in inhibiting hCA II. Moreover, grafting a 6-methoxy group within the quinoline scaffold (compound 6c; KI = 0.083 μM) was advantageous for the inhibitory activity toward hCA II, similarly to the SAR for hCA I inhibition. Regarding the substitution effect for the quinoline moiety; the inhibitory activities towards hCA II were decreased in the order of 7-Cl-6-F > 6-OCH3 > 6-CH3 > 6-Br > 5,7-(Cl)2.
Conclusion
In summary, we successfully synthesised new benzenesulfonamides, bearing un/substituted ethyl quinoline-3-carboxylate scaffold (6a-f and 11), which were evaluated for their inhibition of hCA I and hCA II. Both the examined isoforms were inhibited by the quinolines reported here in variable degrees; hCA I was inhibited with KIs in the range of 0.966–9.091 μM, whereas hCA II in the range of 0.083–3.594 μM. Among the tested compounds, the primary 7-chloro-6-flouro substituted sulfonamide derivative 6e (KI = 0.083 μM) proved to be the most active quinoline in inhibiting hCA II, whereas, its secondary sulfonamide analogue 11 failed to inhibit the hCA II up to 10 μM, confirming the crucial role of the primary sulphonamido group, as a ZBG, for CA inhibitory activity.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- (a) Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81., (b) Alterio V, Di Fiore A, D’Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68., (c) Supuran CT. Applications of carbonic anhydrases inhibitors in renal and central nervous system diseases. Expert Opin Ther Pat 2018;28:713–21., (d) Supuran CT. Carbonic anhydrase inhibitors and their potential in a range of therapeutic areas. Expert Opin Ther Pat 2018;28:709–12., (e) Supuran CT. Carbonic anhydrase inhibitors as emerging agents for the treatment and imaging of hypoxic tumors. Expert Opin Investig Drugs 2018;27:963–70., (f) Nocentini A, Supuran CT. Carbonic anhydrase inhibitors as antitumor/antimetastatic agents: a patent review (2008-2018). Expert Opin Ther Pat 2018;28:729–40.
- (a) Supuran CT. Structure-based drug discovery of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2012;27:759–72., (b) De Simone G, Supuran CT. (In)organic anions as carbonic anhydrase inhibitors. J Inorg Biochem 2012;111:117–29., (c) Supuran CT. Carbonic anhydrase inhibition and the management of hypoxic tumors. Metabolites 2017;7:E48, (d) Supuran CT. Carbonic anhydrases and metabolism. Metabolites 2018;8:E25., (e) Supuran CT. Carbon- versus sulphur-based zinc binding groups for carbonic anhydrase inhibitors? J Enzyme Inhib Med Chem 2018;33:485–95.
- (a) Supuran CT. Carbonic anhydrase inhibitors. Bioorg Med Chem Lett 2010;20:3467–74., (b) Briganti F, Pierattelli R, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. Part 37. Novel classes of carbonic anhydrase inhibitors and their interaction with the native and cobalt-substituted enzyme: kinetic and spectroscopic investigations. Eur J Med Chem 1996;31:1001–10., (c) Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31:345–60., (d) Supuran CT. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin Drug Discov 2017;12:61–88., (e) Supuran CT. Structure and function of carbonic anhydrases. Biochem J 2016;473:2023–32., (f) Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77., (g) Supuran CT, Vullo D, Manole G, et al. Designing of novel carbonic anhydrase inhibitors and activators. Curr Med Chem Cardiovasc Hematol Agents 2004;2:49–68.
- (a) Pastorekova S, Parkkila S, Pastorek J, Supuran CT. Carbonic anhydrases: current state of the art, therapeutic applications and future prospects. J Enzyme Inhib Med Chem 2004;19:199–229., (b) Supuran CT, Capasso C. The eta-class carbonic anhydrases as drug targets for antimalarial agents. Expert Opin Ther Targets 2015;19:551–63., (c) Capasso C, Supuran CT. Bacterial, fungal and protozoan carbonic anhydrases as drug targets. Expert Opin Ther Targets 2015;19:1689–704., (d) Supuran CT, Capasso C. Biomedical applications of prokaryotic carbonic anhydrases. Expert Opin Ther Pat 2018;28:745–54., (e) Supuran CT, Capasso C. An overview of the bacterial carbonic anhydrases. Metabolites 2017;7:56.
- (a) Supuran CT. Carbonic anhydrase inhibitors and activators for novel therapeutic applications. Future Med Chem 2011;3:1165–80., (b) Akocak S, Lolak N, Bua S, et al. Discovery of novel 1,3-diaryltriazene sulfonamides as carbonic anhydrase I, II, VII, and IX inhibitors. J Enzyme Inhib Med Chem 2018;33:1575–80., (c) Winum JY, Temperini C, El Cheikh K, et al. Carbonic anhydrase inhibitors: clash with Ala65 as a means for designing inhibitors with low affinity for the ubiquitous isozyme II, exemplified by the crystal structure of the topiramate sulfamide analogue. J Med Chem 2006;49:7024–31., (d) Supuran CT. Carbonic anhydrase inhibitors in the treatment and prophylaxis of obesity. Expert Opin Ther Pat 2003;13:1545–50.
- (a) Carta F, Supuran CT. Diuretics with carbonic anhydrase inhibitory action: a patent and literature review (2005–2013). Expert Opin Ther Pat 2013;23:681–91., (b) da Silva Cardoso V, Vermelho AB, Ricci Junior E, et al. Antileishmanial activity of sulfonamide nanoemulsions targeting the β-carbonic anhydrase from Leishmania species. J Enzyme Inhib Med Chem 2018;33:850–7., (c) Akocak S, Lolak N, Vullo D, et al. Synthesis and biological evaluation of histamine Schiff bases as carbonic anhydrase I, II, IV, VII, and IX activators. J Enzyme Inhib Med Chem 2017;32:1305–12., (d) Supuran CT. Acetazolamide for the treatment of idiopathic intracranial hypertension. Expert Rev Neurother 2015;15:851–6.
- Aggarwal M, Kondeti B, McKenna R. Anticonvulsant/antiepileptic carbonic anhydrase inhibitors: a patent review. Expert Opin Ther Pat 2013;23:717–24.
- Kayser B, Dumont L, Lysakowski C, et al. Reappraisal of acetazolamide for the prevention of acute mountain sickness: a systematic review and meta-analysis. High Alt Med Biol 2012;13:82–92.
- (a) Masini E, Carta F, Scozzafava A, Supuran CT. Antiglaucoma carbonic anhydrase inhibitors: a patent review. Expert Opin Ther Pat 2013;23:705–16., (b) Borras J, Scozzafava A, Menabuoni L, et al. Synthesis of water-soluble, topically effective intraocular pressure lowering aromatic/heterocyclic sulfonamide containing 8-quinoline-sulfonyl moieties: is the tail more important than the ring? Bioorg Med Chem 1999;7:2397–406., (c) Stanica L, Gheorghiu M, Stan M, et al. Quantitative assessment of specific carbonic anhydrase inhibitors effect on hypoxic cells using electrical impedance assays. J Enzyme Inhib Med Chem 2017;32:1079–90., (d) Abdoli M, Angeli A, Bozdag M, et al. Synthesis and carbonic anhydrase I, II, VII, and IX inhibition studies with a series of benzo[d]thiazole-5- and 6-sulfonamides. J Enzyme Inhib Med Chem 2017;32:1071–8.
- (a) Fabrizi F, Mincione F, Somma T, et al. A new approach to antiglaucoma drugs: carbonic anhydrase inhibitors with or without NO donating moieties. Mechanism of action and preliminary pharmacology. J Enzyme Inhib Med Chem 2012;27:138–47., (b) Supuran CT. Carbonic anhydrase activators. Future Med Chem 2018;10:561–73.
- Chiaramonte N, Bua S, Ferraroni M, et al. 2-Benzylpiperazine: a new scaffold for potent human carbonic anhydrase inhibitors. Synthesis, enzyme inhibition, enantioselectivity, computational and crystallographic studies and in vivo activity for a new class of intraocular pressure lowering agents. Eur J Med Chem 2018;151:363–75.
- Egan TJ. Quinoline antimalarials. Expert Opin Ther Pat 2001;11:185–209.
- Sangshetti JN, Khan FA, Kulkarni AA, et al. Antileishmanial drug discovery: comprehensive review of the last 10 years. Rsc Advanc 2015;5:32376–415.
- Keri RS, Patil SA. Quinoline: a promising antitubercular target. Biomed Pharmacother 2014;68:1161–75.
- Zajdel P, Marciniec K, Maślankiewicz A, et al. Antidepressant and antipsychotic activity of new quinoline-and isoquinoline-sulfonamide analogs of aripiprazole targeting serotonin 5-HT1A/5-HT2A/5-HT7 and dopamine D2/D3 receptors. Eur J Med Chem 2013;60:42–50.
- R Solomon V, Lee H. Quinoline as a privileged scaffold in cancer drug discovery. Curr Med Chem 2011;18:1488–508.
- (a) Afzal O, Kumar S, Haider MR, et al. A review on anticancer potential of bioactive heterocycle quinolone. Eur J Med Chem 2015;97:871–910., (b)Lenoci A, Tomassi S, Conte M, et al. Quinoline A ‐based p300 histone acetyltransferase inhibitors with pro‐apoptotic activity in human leukemia U937 cells. ChemMedChem 2014;9:542–8.
- Old DW, Preparation of alkyl or hydroxyalkyl-1- naphthamide or quinoline derivatives useful in treatment of glaucoma and ocular hypertension patent US Pat. 20150175586A1. 2015.
- Nainwal LM, Tasneem S, Akhtar W, et al. Green recipes to quinoline: a review. Eur J Med Chem 2019;164:121–70.
- Chu XM, Wang C, Liu W, et al. Quinoline and quinolone dimers and their biological activities: an overview. Eur J Med Chem 2019;161:101–17.
- Rivilli MJL, Turina AV, Bignante EA, et al. Synthesis and pharmacological evaluation of pyrazolo [4, 3-c] quinolinones as high affinity GABAA-R ligands and potential anxiolytics. Bioorg Med Chem 2018;26:3967–74.
- Medapi B, Suryadevara P, Renuka J, et al. 4-Aminoquinoline derivatives as novel Mycobacterium tuberculosis GyrB inhibitors: structural optimization, synthesis and biological evaluation. Eur J Med Chem 2015;103:1–16.
- Lawrence HR, Kazi A, Luo Y, et al. Synthesis and biological evaluation of naphthoquinone analogs as a novel class of proteasome inhibitors. Bioorg Med Chem 2010;18:5576–92.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase I. Stopflow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73.
- (a) Abo-Ashour MF, Eldehna WM, Nocentini A, et al. Novel hydrazido benzenesulfonamides-isatin conjugates: synthesis, carbonic anhydrase inhibitory activity and molecular modeling studies. Eur J Med Chem 2018;157:28–36., (b) Krall N, Pretto F, Decurtins W, et al. A small‐molecule drug conjugate for the treatment of carbonic anhydrase IX expressing tumors. Angew Chem Int Ed Engl 2014;53:4231–5., (c) Pacchiano F, Aggarwal M, Avvaru BS, et al. Selective hydrophobic pocket binding observed within the carbonic anhydrase II active site accommodate different 4-substituted-ureido-benzenesulfonamides and correlate to inhibitor potency. Chem Commun (Camb) 2010;46:8371–3., (d) Köhler K, Hillebrecht A, Schulze Wischeler J, et al. Saccharin inhibits carbonic anhydrases: possible explanation for its unpleasant metallic aftertaste. Angew Chem Int Ed Engl 2007;46:7697–9.
- (a) Eldehna WM, Abo-Ashour MF, Berrino E, et al. SLC-0111 enaminone analogs, 3/4-(3-aryl-3-oxopropenyl) aminobenzenesulfonamides, as novel selective subnanomolar inhibitors of the tumor-associated carbonic anhydrase isoform IX. Bioorg Chem 2019;83:549–58., (b) Alterio V, Esposito D, Monti SM, et al. Crystal structure of the human carbonic anhydrase II adduct with 1-(4-sulfamoylphenyl-ethyl)-2,4,6-triphenylpyridinium perchlorate, a membrane-impermeant, isoform selective inhibitor. J Enzyme Inhib Med Chem 2018;33:151–7., (c) Innocenti A, Vullo D, Scozzafava A, et al. Carbonic anhydrase inhibitors. Inhibition of mammalian isoforms I–XIV with a series of substituted phenols including paracetamol and salicylic acid. Bioorg Med Chem 2008;16:7424–8., (d) Nocentini A, Bonardi A, Gratteri P, et al. Steroids interfere with human carbonic anhydrase activity by using alternative binding mechanisms. J Enzyme Inhib Med Chem 2018;33:1453–9., (e) Tars K, Vullo D, Kazaks A, et al. Sulfocoumarins (1,2-benzoxathiine-2,2-dioxides): a class of potent and isoform-selective inhibitors of tumor-associated carbonic anhydrases. J Med Chem 2013;56:293–300.
- (a) Eldehna WM, Abo-Ashour MF, Nocentini A, et al. Enhancement of the tail hydrophobic interactions within the carbonic anhydrase IX active site via structural extension: design and synthesis of novel N-substituted isatins-SLC-0111 hybrids as carbonic anhydrase inhibitors and antitumor agents. Eur J Med Chem 2019;162:147–60., (b) Gul HI, Mete E, Taslimi P, et al. Synthesis, carbonic anhydrase I and II inhibition studies of the 1,3,5-trisubstituted-pyrazolines. J Enzyme Inhib Med Chem 2017;32:189–92.
- (a) Eldehna WM, Nocentini A, Al-Rashood ST, et al. Tumor-associated carbonic anhydrase isoform IX and XII inhibitory properties of certain isatin-bearing sulfonamides endowed with in vitro anticancer activity towards colon cancer. Bioorg Med Chem 2018;81:425–32., (b) Pustenko A, Stepanovs D, Žalubovskis R, et al. 3H-1,2-benzoxathiepine 2,2-dioxides: a new class of isoform-selective carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2017;32:767–75.
- (a) Fares M, Eladwy RA, Nocentini A, et al. Synthesis of bulky-tailed sulfonamides incorporating pyrido[2,3-d][1,2,4]triazolo[4,3-a]pyrimidin-1(5H)-yl) moieties and evaluation of their carbonic anhydrases I, II, IV and IX inhibitory effects. Bioorg Med Chem 2017;25:2210–7., (b) Güzel-Akdemir Ö, Angeli A, Demir K, et al. Novel thiazolidinone-containing compounds, without the well-known sulfonamide zinc-binding group acting as human carbonic anhydrase IX inhibitors. J Enzyme Inhib Med Chem 2018;33:1299–308., (c) Ramya PVS, Angapelly S, Angeli A, et al. Discovery of curcumin inspired sulfonamide derivatives as a new class of carbonic anhydrase isoforms I, II, IX, and XII inhibitors. J Enzyme Inhib Med Chem 2017;32:1274–81., (d) Entezari Heravi Y, Sereshti H, Saboury AA, et al. 3D QSAR studies, pharmacophore modeling, and virtual screening of diarylpyrazole-benzenesulfonamide derivatives as a template to obtain new inhibitors, using human carbonic anhydrase II as a model protein. J Enzyme Inhib Med Chem 2017;32:688–700., (e) D'Ascenzio M, Guglielmi P, Carradori S, et al. Open saccharin-based secondary sulfonamides as potent and selective inhibitors of cancer-related carbonic anhydrase IX and XII isoforms. J Enzyme Inhib Med Chem 2017;32:51–9., (f) Krasavin M, Korsakov M, Ronzhina O, et al. Primary mono- and bis-sulfonamides obtained via regiospecific sulfochlorination of N-arylpyrazoles: inhibition profile against a panel of human carbonic anhydrases. J Enzyme Inhib Med Chem 2017;32:920–34.