
Abstract
A library of 4-[(3-methyl-4-aryl-2,3-dihydro-1,3-thiazol-2-ylidene)amino]benzene-1-sulphonamides (EMAC8002a–m) was designed and synthesised to evaluate the effect of substituents in the positions 3 and 4 of the dihydrothiazole ring on the inhibitory potency and selectivity toward human carbonic anhydrase isoforms I, II, IX, and XII. Most of the new compounds preferentially inhibit the isoforms II and XII. Both electronic and steric features on the aryl substituent in the position 4 of the dihydrothiazole ring concur to determine the overall biological activity of these new derivatives.
Introduction
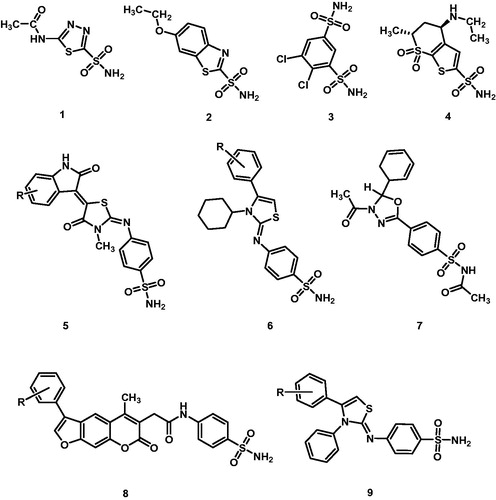
1,3-thiazole and their hydrogenated analogues are important molecular subunits in diverse classes of biologically active molecules and thus can be found in several drugs approved for clinical use. Not surprisingly, this moiety has been extensively studied and both natural and synthetic thiazole derivatives are in therapeutic use or have shown potential therapeutic application toward several pathologies and targets such as bacteriaCitation1,Citation2, tumoursCitation3–6, HIV-1 protease and reverse transcriptaseCitation7–12, fungiCitation13–15, neurodegeneration and related pathologiesCitation12,Citation16,Citation17, and protozoal infectionsCitation18. Recently, we reported on benzenesulphonamide dihydrothiazole derivatives as inhibitors of human carbonic anhydrase (hCA) isozymes I, II, IX, and XII19. This enzyme family catalyses the reversible hydration of carbon dioxide to bicarbonate and protonsCitation19 and, therefore, plays an essential role in CO2-related metabolism and in its transportation across biological membranesCitation20,Citation21. Due to their simple but essential role, hCAs have been recognised as main actors in a number of physiological processes and pathologiesCitation22–30. Not surprisingly, hCA inhibitors have been intensively studied and several are in clinical use for diverse pathologiesCitation31–37. Although different mechanisms of inhibition of hCA have been reported (e.g. coumarins, phenols, primary amines, COOMe derivatives)Citation38–42, benzenesulphonamides and their isosters are the most represented molecular class of inhibitorsCitation32,Citation43–48. These inhibitors belong to the so called zinc binders. They bind the zinc cofactor as conjugated bases and therefore, the acidity of the sulphonamide group influences their potency. Thus, by conjugating the benzenesulphonamide group to an electron withdrawing heterocyclic ring, the activity could be favourably influenced. Moreover, the introduction of further substituents in the heterocyclic core may influence the isozyme selectivity. On these bases, and according to our previous observationCitation31,Citation47–50, we have synthesised and evaluated for the inhibition activity toward hCA I, II, IX, and XII isoforms a series of the 4-[(3-methyl-4-aryl-2,3-dihydro-1,3-thiazol-2-ylidene)amino]benzenesulphonamides ().
Figure 1. Carbonic anhydrase inhibitors in clinical use and previously reported EMAC derivatives: (1) acetazolamide (2) ethoxzolamide, (3) dichlorphenamide, (4) dorzolamide, (5) EMAC10020Citation47, (6) EMAC8001Citation31, (7) EMAC8000Citation48, (8) EMAC10153Citation50, (9) EMAC10111Citation49.
Methods
Materials and apparatus
Starting materials and reagents were obtained from commercial suppliers and were used without purification. All melting points were determined on a Stuart SMP11 melting points apparatus and are uncorrected. Electron ionisation mass spectra were obtained by a Fisons QMD 1000 mass spectrometer (Danvers, MA) (70 eV, 200 mA, ion source temperature 200 °C). Samples were directly introduced into the ion source. Melting points, yield of reactions, and analytical data of derivatives EMAC8002a–l are reported in .
Table 1. Chemical, analytical, and physical data of derivatives EMAC8002 a–m.
1H-NMR () were registered on a Bruker AMX (300 MHz) (chemical shifts in δ values) or on a Unity Inova 500NB high-resolution spectrometer (Agilent Technologies, CA) (500 MHz) All samples were measured in DMSO. Chemical shifts are reported referenced to the solvent in which they were measured. Coupling constants J are expressed in hertz (Hz). Elemental analyses were obtained on a Perkin–Elmer 240 B microanalyser. Analytical data of the synthesised compounds are in agreement within ± 0.4% of the theoretical values. TLC chromatography was performed using silica gel plates (Merck F 254), spots were visualised by UV light.
Table 2. 1H NMR data of derivatives EMAC8002a–m.
General procedure for the synthesis of compound EMAC8002a–m
Synthesis of 1-methyl-3–(4-sulfamoylethyl)thiourea
To an ethanolic solution of 4-aminobenzenesulphonamide (1 eq), methyl isothiocyanate (2 eq) was added dropwise. The mixture was heated under reflux until the completion of the reaction (10 h). The progress of the reaction was monitored by TLC (ethyl acetate/n-hexane 2/1). Then the reaction was cooled overnight in the fridge. A precipitate was formed which was collected by filtration under vacuum and crystallised from ethanol to afford the desired product.
Synthesis of 4-[(3-methyl-4-aryl-2,3-dihydro-1,3-thiazol-2-ylidene)amino]benzenesulphonamide
A mixture of 1-methyl-3–(4-sulfamoylphenyl)thiourea (1 eq) and α-halogenoketone (1 eq) was reacted in ethanol solution. Different reaction conditions have been employed. Thus, while in the presence of α-bromoketones the reaction temperature was kept between 30 and 50 °C, refluxing conditions were used when α-chloroketones were reacted. The mixture was reacted until completion (TLC, ethyl acetate/n-hexane 2/1). By cooling to room temperature, a precipitate was formed. The crude product was filtered and crystallised from the appropriate solvent. Analytical and spectral data of compounds EMAC8002a–m are reported in .
Molecular modelling
The new ligand EMAC8002i was built by means of Maestro GUICitation51 in E configuration. Then a conformational search analysis was performed using MCMM method allowing 5000 iterations in implicit solventCitation52.
Docking experiments were performed by means of Glide Quantum-Mechanical Polarised DockingCitation53,Citation54. The crystallographic model with the best resolution was considered (pdb code 5MSA, 1.2 Å). The protein was prepared with Preparation Wizard protocol. The Grid box was centred on the co-crystallised ligand and all parameters were set up as default.
The best pose complex was then minimised to consider the induced fit phenomena and used to analyse the ligand-binding mode. 10,000 steps of the Polak-Ribier conjugate gradient (PRCG) minimisation method were conducted on the top ranked theoretical complex using OPLS_2005 force fieldCitation55.
The optimisation process was performed up to the derivative convergence criterion equal to 0.05 kcal/(mol*Å)−1.
Biological activity
Carbonic anhydrase inhibition assay
The purification of cytosolic CA isoenzymes (CA I and CA II) was previously described with a simple one-step method by a Sepharose-4B-L tyrosine-sulphanilamide affinity chromatographyCitation56.
The protein quantity in the column effluents was determined spectrophotometrically at 280 nm. Sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) was applied with a Bio-Rad Mini Gel system Mini-PROTEINVR system (Hercules, CA), Bio-Rad Laboratories, Inc., China after purification of both CA isoenzymes. Briefly, it was performed in acrylamide for the running (10%) and the stacking gel (3%) contained SDS (0.1%), respectively. Activities of CA isoenzymes were determined according to a method by Verporte et al.Citation57. The increase in absorbance of the reaction medium was spectrophotometrically recorded at 348 nm. Also, the quantity of protein was determined at 595 nm according to the Bradford methodCitation58. Bovine serum albumin was used as standard protein. The IC50 values were obtained from activity (%) versus compounds plotsCitation59. For calculation of KI values, three different concentrations were used. The Lineweaver–Burk curves were drawn and calculations were realisedCitation59. The biological data are reported in .
Table 3. Inhibition data towards hCA I, II, IX, and XII of compounds EMAC8002 a–m.
Results and discussion
As a continuation of our ongoing research in the field of carbonic anhydrase and anticancer agentsCitation31,Citation47–49,Citation60, we have synthesised a new series of 4-[(3-methyl-4-aryl-2,3-dihydro-1,3-thiazol-2-ylidene)amino]benzenesulphonamides, namely compounds EMAC8002a–m, to evaluate their activity and selectivity toward CA isozymes and to gain information on the structure–activity relationships of these derivatives. All the synthesised derivatives are characterised by the presence of a benzenesulphonamide moiety as zinc binder group, conjugated with the position 2 of a dihydrothiazole heterocyclic core. A methyl substituent is always present on the heterocyclic nitrogen atom, while a differently substituted aromatic ring occupies the position 4.
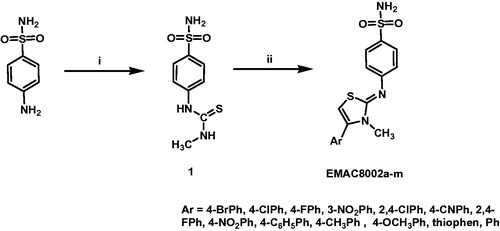
The synthetic procedure to obtain compounds EMAC8002a–m is depicted in Scheme 1. Briefly, it consists of two steps: the synthesis of the 3‐methyl‐1‐(4‐sulfamoylphenyl)thiourea (1) by reaction of the 4-aminobenzensufonamide with methyl isocyanate. The second step is the formation of the 4-aryl dihydrothiazole nucleus. It was accomplished by reacting 1 with the appropriate α-halogenoketone in ethanol solution. EMAC8002a–m were characterised by means of analytical and spectroscopic methods ( and ) and then submitted to enzymatic evaluation toward hCA I, II, IX, and XII.
Scheme 1. Synthetic pathway to compounds EMAC8002a–m. Reagents and conditions: (i) ethanol, methylisothiocyanate; (ii) ethanol, α-halogenoarylketone.
The results are summarised in . Accordingly with our previous observations with similar derivatives, none of the EMAC8002 compounds was active toward hCA I isozyme. On the contrary, when isozymes II, IX, and XII are investigated, some consideration regarding the structure–activity relationships could be done. When compounds EMAC8002 are tested on hCA II, the introduction of a halogen atom, in position 4 of the phenyl moiety, in position 4 of the dihydrothiazole ring, appeared beneficial for the activity. However, the larger is the atomic radius of the halogen, the lower the activity. So far, the Ki values are in the following order: 4-F (EMAC8002c) 5.3 nM <4-Cl (EMAC8002b) 13.1 nM <4-Br (EMAC8002a) 45.8 nM. A similar behaviour was observed in the case of hCAXII: 4-F (EMAC8002c) 5.3 nM <4-Cl (EMAC8002b) 13.1 nM <4-Br (EMAC8002a) 45.8 nM. On the contrary, when the same compounds were evaluated on hCA IX, a totally reversed trend was observed. In fact, the larger is the halogen atom the higher the activity. Accordingly, the Ki values are 4-F (EMAC8002c) 2482 nM > 4-Cl (EMAC8002b) 1213 nM > 4-Br (EMAC8002a) 58.1 nM. The introduction of a second halogen atom in the position 2 of the phenyl ring is beneficial only for the activity toward the hCA XII isoform. Thus, in the case of compounds EMA8002e (2,4-Cl) and EMAC8002g (2,4-F) the Ki values are 10.5 and 9.0 nM, respectively. Accordingly, when the 4-Cl derivative (EMAC8002b) is compared with the 2,4-diCl one (EMAC8002e), a 9-fold gain in potency toward the hCA XII isozyme was observed. Similarly, a 4-fold increase in potency toward hCA XII was observed when 4-F (EMAC8002c) and 2,4-diF (EMAC8002g) are compared. On the contrary, the inverse was observed when hCA II isozyme is considered. A decrease in the activity of 7 folds was observed when 2,4-diF (EMAC8002g) was compared with 4-F (EMAC8002c). Analogously a decrease of the inhibition potency of 30 folds was measured when 2,4-diCl (EMAC8002e) was compared with 4-Cl (EMAC8002b). On these bases, we can summarise that by introducing specific halogen atoms in specific positions of the 4-phenyl ring, it is possible to modulate the activity toward different hCA isozymes. The introduction of a nitro group is tolerated both in the 3 and 4 position of the phenyl ring when hCA II is considered. On the contrary, when the activity on hCA XII is measured, the introduction of the nitro group is only tolerated in the 4 position. The introduction of electron-donating groups such as methyl or methoxy, as in compounds EMAC8002j and EMAC8002k, respectively, led to most active compounds when hCA II, IX, and XII are considered. Unfortunately, none of the two substitutions led to selective compounds. The presence of a nitrile in the position 4 as in the case of compound EMAC8002f, is generally detrimental for the activity, but for hCAII, where this substitution is tolerated. By introducing a biphenyl group in the position 4 of the dihydrothiazole, the most selective compound toward hCA XII (EMAC8002i) was obtained, with a selective index hCA II/hCA XII higher than 52. When compared with 4-phenyldihydrothiazole (EMAC8002m), the isosteric introduction of a thiophene-2-yl moiety in the position 4 of the dihydrothiazole (EMAC8002l) was beneficial only for the activity toward hCA I, although with high values of Ki.
These results, together with our previous findings, indicate that the 4-[(3-methyl-4-aryl-2,3-dihydro-1,3-thiazol-2-ylidene)amino]benzenesulphonamide scaffold could be rationally and efficiently decorated in order to achieve potent and selective hCA inhibitors.
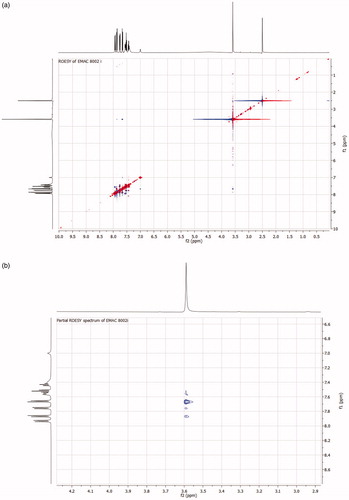
The possible formation of both E and Z diastereoisomers along the C=N double bond was investigated by 2D NMR experiments. To this end, the ROESY spectrum of the most interesting compound of the series EMAC8002i was recorded (). This derivative showed good selectivity toward hCA XII and inhibitory activity toward this enzyme in the low nM range. ROESY cross-peak from the methyl group at δH 3.59 (3H, s) to the aromatic protons H-2 and H-3 at δH 7.56 (2H, d, J = 8 Hz) permitted to assign the configuration around the double bond as E. In fact, examination of a molecular model confirms that, in the case of (Z) configuration, H-2 and H-3 aromatic protons would be too far to the methyl group and correlation should not be observed. Analogous experiments were performed along the full series of compounds and, as expected, the (E) configuration was assigned to all derivatives.
Figure 2. (a) ROESY spectrum of compound EMAC8002i; (b) Partial ROESY spectrum of compound EMAC8002i.
In order to predict the binding mode of compound EMAC8002i, a molecular docking experiment was performed. The most selective was well docked into the catalytic site of CA XII with binding energies of −10.194 kcal/mol. The complex has been energy minimised and the putative binding mode is depicted in .
Figure 3. Three-dimensional representation of the putative binding mode as obtained by docking experiments of: (a) EMAC8002-i and (b) relative 2D representation of the complex stabilising interactions with the residues of the binding site.
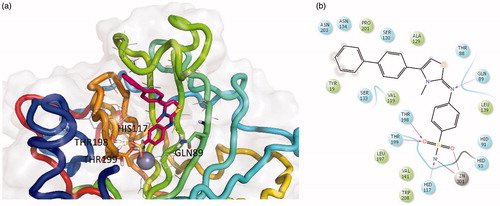
The ligand well fit into the binding pocket. The benzenesulphonamide moiety tightly interacts with the deep cavity and the Zn (II) ion, being stabilised by the metal chelation and an array of hydrogen bonds with the residues around the ion: Thr198, Thr199, and His117. Furthermore, the nitrogen atom of the aminobenzenesulphonamide moiety interacts with Gln89. Moreover, the substituent in the position 4 of the thiazolino portion interacts with the external part of the cavity. The analysis of the putative binding mode highlighted the presence of extra space in correspondence of the N methyl substituent which will be exploited to increase the ligand complementarity and probably its activity and selectivity.
Conclusions
We have designed and synthesised a series of 4-[(3-methyl-4-aryl-2,3-dihydro-1,3-thiazol-2-ylidene)amino]benzenesulphonamides and evaluate their activity on hCA I, II, IX, and XII isozymes. While these derivatives were weak inhibitors of the hCA I isoform, interestingly, the nature and substitution pattern of the aromatic group in the position 4 of the dihydrothiazole core was relevant for the activity and the selectivity between the hCA II and XII isoforms. Nevertheless, we observed that the introduction of a 4-methylphenyl or a 4-methoxyphenyl moiety in the position 4 of the dihydrothiazole ring is beneficial for the activity toward hCA II, IX, and XII isozymes. These data prompted us to further investigate on these scaffolds in order to optimise both the activity and the isozyme selectivity.
Acknowledgements
The authors wish to acknowledge the “Ufficio Valorizzazione dei Risultati della Ricerca” of Sardegna Ricerche Technological Park, Pula (CA) – Italy. The authors also thank the COST action CA15135 (Multitarget Paradigm for Innovative Ligand Identification in the Drug Discovery Process MuTaLig) for support.
Disclosure statement
No potential conflict of interest was reported by the authors.
Related Research Data
References
- Lott WA, Bergeim FH. 2-(p-Aminobenzenesulfonamido)-thiazole: a new chemotherapeutic agent. J Am Chem Soc 1939;61:3593–4.
- Ball AP, Geddes AM, Davey PG, et al. Clavulanic acid and amoxycillin: a clinical, bacteriological, and pharmacological study. Lancet 1980;1:620–3.
- Oslob JR, Allen DA, Baskaran S, et al. Discovery of a potent and selective aurora kinase inhibitor. Bioorg Med Chem Lett 2008;18:4880–4.
- Claussen CA, Long EC. Nucleic acid recognition by metal complexes of bleomycin. Chem Rev 1999;99:2797–816.
- Tricot G, Jayaram HN, Weber G, et al. Tiazofurin: biological effects and clinical uses. Int J Cell Clon 1990;8:161–70.
- Hara M, Asano K, Kawamoto I, et al. Leinamycin, a new antitumor antibiotic from streptomyces: producing organism, fermentation and isolation. J Antibiot 1989;42:1768–74.
- Kempf DJ, Marsh KC, Denissen JF, et al. ABT-538 is a potent inhibitor of human immunodeficiency virus protease and has high oral bioavailability in humans. Proc Natl Acad Sci USA 1995;92:2484–8.
- Meleddu R, Distinto S, Corona A, et al. Isatin thiazoline hybrids as dual inhibitors of HIV-1 reverse transcriptase. J Enzyme Inhib Med Chem 2017;32:130–6.
- Meleddu R, Distinto S, Corona A, et al. (3Z)-3-(2-[4-(aryl)-1,3-thiazol-2-yl]hydrazin-1-ylidene)-2,3-dihydro-1H-indol-2-one derivatives as dual inhibitors of HIV-1 reverse transcriptase. Eur J Med Chem 2015;93:452–60.
- Corona A, Meleddu R, Esposito F, et al. Ribonuclease H/DNA polymerase HIV-1 reverse transcriptase dual inhibitor: mechanistic studies on the allosteric mode of action of isatin-based compound RMNC6. PLoS One 2016;11:e0147225.
- Distinto S, Maccioni E, Meleddu R, et al. Molecular aspects of the RT/drug interactions. perspective of dual inhibitors. Curr Pharm Design 2013;19:1850–9.
- Distinto S, Esposito F, Kirchmair J, et al. Identification of HIV-1 reverse transcriptase dual inhibitors by a combined shape-, 2D-fingerprint- and pharmacophore-based virtual screening approach. Eur J Med Chem 2012;50:216–29.
- Meleddu R, Distinto S, Corona A, et al. Exploring the thiazole scaffold for the identification of new agents for the treatment of fluconazole resistant Candida. J Enzyme Inhib Med Chem 2016;31:1672–7.
- Ojika M, Suzuki Y, Tsukamoto A, et al. Cystothiazoles A and B, new bithiazole-type antibiotics from the myxobacterium Cystobacter fuscus. J Antibiot 1998;51:275–81.
- Marquez BL, Watts KS, Yokochi A, et al. Structure and absolute stereochemistry of hectochlorin, a potent stimulator of actin assembly. J Nat Prod 2002;65:866–71.
- Costa FH, Rosso AL, Maultasch H, et al. Depression in Parkinson's disease: diagnosis and treatment. Arquivos de Neuro-Psiquiatria 2012;70:617–20.
- Chimenti F, Maccioni E, Secci D, et al. Synthesis, stereochemical identification, and selective inhibitory activity against human monoamine oxidase-B of 2-methylcyclohexylidene-(4-arylthiazol-2-yl)hydrazones. J Med Chem 2008;51:4874–80.
- Willcox RR. Treatment of vaginal trichomoniasis with aminitrozole and trichomycin given orally. Gynaecologia 1960;149:122–7.
- Domsic JF, Avvaru BS, Kim CU, et al. Entrapment of carbon dioxide in the active site of carbonic anhydrase II. J Biol Chem 2008;283:30766–71.
- Boron WF. Evaluating the role of carbonic anhydrases in the transport of HCO3−-related species. Biochimica et Biophysica Acta 2010;1804:410–21.
- Geers C, Gros G. Carbon dioxide transport and carbonic anhydrase in blood and muscle. Physiol Rev 2000;80:681–715.
- Horie K, Kawakami K, Fujita Y, et al. Exosomes expressing carbonic anhydrase 9 promote angiogenesis. Biochem Biophys Res Commun 2017;492:356–61.
- Supuran CT. Structure and function of carbonic anhydrases. Biochem J 2016;473:2023–32.
- McIntyre A, Hulikova A, Ledaki I, et al. Disrupting hypoxia-induced bicarbonate transport acidifies tumor cells and suppresses tumor growth. Cancer Res 2016;76:3744–55.
- Jiang J, Zhao JH, Wang XL, et al. Correlation between carbonic anhydrase IX (CA-9), XII (CA-12) and hypoxia inducible factor-2α (HIF-2α) in breast cancer. Neoplasma 2015;62:456–63.
- Imtaiyaz Hassan M, Shajee B, Waheed A, et al. Structure, function and applications of carbonic anhydrase isozymes. Bioorg Med Chem 2013;21:1570–82.
- Supuran CT. Inhibition of carbonic anhydrase IX as a novel anticancer mechanism. World J Clin Oncol 2012;3:98–103.
- Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77.
- Swietach P, Vaughan-Jones RD, Harris AL. Regulation of tumor pH and the role of carbonic anhydrase 9. Cancer Metast Rev 2007;26:299–310.
- Zhou Y, Mokhtari RB, Pan J, et al. Carbonic anhydrase II mediates malignant behavior of pulmonary neuroendocrine tumors. Am J Respir Cell Mol Biol 2015;52:183–92.
- Meleddu R, Maccioni E, Distinto S, et al. New 4-[(3-cyclohexyl-4-aryl-2,3-dihydro-1,3-thiazol-2-ylidene)amino]benzene-1-sulphonamides, synthesis and inhibitory activity toward carbonic anhydrase I, II, IX, XII. Bioorg Med Chem Lett 2015;25:3281–4.
- Eldehna WM, Fares M, Ceruso M, et al. Amido/ureidosubstituted benzenesulphonamides-isatin conjugates as low nanomolar/subnanomolar inhibitors of the tumor-associated carbonic anhydrase isoform XII. Eur J Med Chem 2016;110:259–66.
- Ceruso M, Bragagni M, AlOthman Z, et al. New series of sulphonamides containing amino acid moiety act as effective and selective inhibitors of tumor-associated carbonic anhydrase XII. J Enzyme Inhib Med Chem 2015;30:430–4.
- Supuran CT. Structure-based drug discovery of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2012;27:759–72.
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81.
- Supuran CT. Carbonic anhydrase inhibitors in the treatment and prophylaxis of obesity. Expert Opin Ther Pat 2003;13:1545–50.
- Picard F, Deshaies Y, Lalonde J, et al. Topiramate reduces energy and fat gains in lean (Fa/?) and obese (fa/fa) Zucker rats. Obes. Res 2000;8:656–63.
- Maresca A, Temperini C, Pochet L, et al. Deciphering the mechanism of carbonic anhydrase inhibition with coumarins and thiocoumarins. J Med Chem 2010;53:335–44.
- Maresca A, Temperini C, Vu H, et al. Non-zinc mediated inhibition of carbonic anhydrases: coumarins are a new class of suicide inhibitors. J Am Chem Soc 2009;131:3057–62.
- Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31:345–60.
- De Simone G, Alterio V, Supuran CT. Exploiting the hydrophobic and hydrophilic binding sites for designing carbonic anhydrase inhibitors. Expert Opin Drug Discov 2013;8:793–810.
- Alterio V, Di Fiore A, D'Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68.
- Pala N, Micheletto L, Sechi M, et al. Carbonic anhydrase inhibition with benzenesulphonamides and tetrafluorobenzenesulphonamides obtained via click chemistry. ACS Med Chem Lett 2014;5:927–30.
- Suthar SK, Bansal S, Lohan S, et al. Design and synthesis of novel 4-(4-oxo-2-arylthiazolidin-3-yl)benzenesulphonamides as selective inhibitors of carbonic anhydrase IX over I and II with potential anticancer activity. Eur J Med Chem 2013;66:372–9.
- Guzel-Akdemir O, Akdemir A, Karali N, et al. Discovery of novel isatin-based sulphonamides with potent and selective inhibition of the tumor-associated carbonic anhydrase isoforms IX and XII. Org Biomol Chem 2015;13:6493–9.
- Ibrahim HS, Abou-Seri SM, Tanc M, et al. Isatin-pyrazole benzenesulphonamide hybrids potently inhibit tumor-associated carbonic anhydrase isoforms IX and XII. Eur J Med Chem 2015;103:583–93.
- Melis C, Meleddu R, Angeli A, et al. Isatin: a privileged scaffold for the design of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2017;32:68–73.
- Bianco G, Meleddu R, Distinto S, et al. N-Acylbenzenesulphonamide dihydro-1,3,4-oxadiazole hybrids: seeking selectivity toward carbonic anhydrase isoforms. ACS Med Chem Lett 2017;8:792–6.
- Meleddu R, Distinto S, Cottiglia F, et al. Tuning the dual inhibition of carbonic anhydrase and cyclooxygenase by dihydrothiazole benzensulphonamides. ACS Med Chem Lett 2018;9:1045.
- Melis C, Distinto S, Bianco G, et al. Targeting tumor associated carbonic anhydrases IX and XII: highly isozyme selective coumarin and psoralen inhibitors. ACS Med Chem Lett 2018;9:725–9.
- Schrödinger LLC, New York, NY. 2018.
- Mohamadi F, Richards NG, Guida WC, et al. MacroModel—an integrated software system for modeling organic and bioorganic molecules using molecular mechanics. J Comput Chem 1990;11:440–67.
- Cho AE, Guallar V, Berne BJ, et al. Importance of accurate charges in molecular docking: quantum mechanical/molecular mechanical (QM/MM) approach. J Comput Chem 2005;26:915–31.
- Chung JY, Hah JM, Cho AE. Correlation between performance of QM/MM docking and simple classification of binding sites. J Chem Inform Model 2009;49:2382–7.
- Jorgensen WL. OPLS force fields. In Schleyer PvR, ed. Encyclopedia of Computational Chemistry. New York: Wiley; 1998:1986–9.
- Akbaba Y, Akıncıoğlu A, Göçer H, et al. Carbonic anhydrase inhibitory properties of novel sulphonamide derivatives of aminoindanes and aminotetralins. J Enzyme Inhib Med Chem 2014;29:35–42.
- Verpoorte JA, Mehta S, Edsall JT. Esterase activities of human carbonic anhydrases B and C. J Biol Chem 1967;242:4221–9.
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248–54.
- Senturk M, Gulcin I, Beydemir S, et al. In vitro inhibition of human carbonic anhydrase I and II isozymes with natural phenolic compounds. Chem Biol Drug Des 2011;77:494–9.
- Meleddu R, Petrikaite V, Distinto S, et al. Investigating the anticancer activity of isatin/dihydropyrazole hybrids. ACS Med Chem Lett 2018;10:571–6.