
Abstract
Ethoxzolamide (EZA), acetazolamide, and methazolamide are clinically used sulphonamide drugs designed to treat non-bacteria-related illnesses (e.g. glaucoma), but they also show antimicrobial activity against the gastric pathogen Helicobacter pylori. EZA showed the highest activity, and was effective against clinical isolates resistant to metronidazole, clarithromycin, and/or amoxicillin, suggesting that EZA kills H. pylori via mechanisms different from that of these antibiotics. The frequency of single-step spontaneous resistance acquisition by H. pylori was less than 5 × 10−9, showing that resistance to EZA does not develop easily. Resistance was associated with mutations in three genes, including the one that encodes undecaprenyl pyrophosphate synthase, a known target of sulphonamides. The data indicate that EZA impacts multiple targets in killing H. pylori. Our findings suggest that developing the approved anti-glaucoma drug EZA into a more effective anti-H. pylori agent may offer a faster and cost-effective route towards new antimicrobials with a novel mechanism of action.
Introduction
Helicobacter pylori persistently colonises the epithelium of the stomach in approximately half of the world’s populationCitation1. Colonisation can lead to the development of gastric and duodenal ulcers, mucosa-associated B-cell lymphoma, and gastric adenocarcinomaCitation2,Citation3. When left untreated, up to 3% of H. pylori infections progress to gastric cancerCitation4. Treatment of H. pylori infection involves complete eradication of the organism from the host. The efficacy of existing drug regimes has significantly declined over the yearsCitation5. In 2017, clarithromycin-resistant H. pylori was ranked as a high priority pathogen for antibiotic research development by the World Health Organisation (WHO)Citation6, highlighting the pressing need for novel anti-H. pylori therapies.
New treatment strategies may target adaptation mechanisms of H. pylori to the acidic pH of the stomach. H. pylori is a neutralophile, but it is capable of maintaining its cytoplasmic pH at near-neutral levels during short-term exposure to pH as low as 1.4Citation7. This is achieved via the combined action of H. pylori urease and two carbonic anhydrasesCitation8–10. Urease converts urea to NH3 and CO2, which have acid neutralising and buffering properties. CO2 generated as a result of urease activity is hydrated in the periplasm and cytoplasm by α- and β-carbonic anhydrases (HpαCA and HpβCA), respectively, resulting in the production of protons (H+) and bicarbonate (HCO3−). The protons react with NH3 to form NH4+ ions. The resultant NH3/NH4+ and CO2/HCO3− acid-base couples buffer the cytoplasm and periplasm at pH close to neutralCitation10. Recent detection of HpαCA in the outer membrane vesicles produced by H. pyloriCitation11 suggested that this enzyme could have an additional, as yet unknown, role in initiating or regulating pathogenesis in the host.
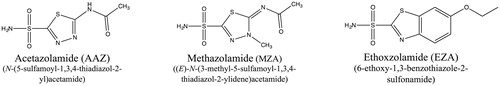
HpαCA and HpβCA are strongly inhibited by primary sulphonamides RSO2NH2, including acetazolamide (AAZ), ethoxzolamide (EZA), and methazolamide (MZA) () that have been originally developed as inhibitors of human CAs and used clinically as diuretics, and antiglaucoma or antiulcer drugs known under the names Diamox (AAZ), Cardrase (EZA), and Neptazane (MZA)Citation12,Citation13. Analysis of the crystal structures of HpαCA in complex with either AAZ or MZACitation14 revealed that these sulphonamides act as active-site inhibitors that mimic the transition state of the reaction catalysed by the enzyme. Furthermore, the crystal structures of HpαCA in complex with a series of AAZ-related sulphonamides, including EZA, revealed that the mode of sulphonamide binding to HpαCA correlates well with their inhibitory activitiesCitation15. Cumulatively, these data have raised a question of whether the HpαCA inhibition by sulphonamides would result in killing H. pylori.
Figure 1. The chemical structures of acetazolamide, methazolamide and ethoxzolamide.
Indeed, MZA has been shown to suppress growth of H. pylori strains SS1 and 11637 in vitroCitation13. Furthermore, the results of treatment of gastroduodenal ulcers with EZACitation16 and AAZCitation17,Citation18 suggested that these drugs inhibit H. pylori growth in vivo. For example, administration of EZA for 3 weeks at 5–10 mg/kg body weight/day resulted in ulcer healing in 98% of the patientsCitation16. This could be, in part, attributed to inhibition of human CA activity in the parietal cells of the stomach which resulted in a reduced basal secretion of gastric acid (antacid action). However, it has been recognised that the EZA and AAZ treatment also likely eradicated the H. pylori infection which caused ulcer disease in the first place, because, two years after treatment, the ulcer recurrence rate in patients treated with EZA (11%)Citation16 or AAZ (6%)Citation17 was significantly lower than that with classical antacid drugs (34–79%) and close to that achieved by the triple H. pylori eradication therapyCitation19.
Given the growing resistance of H. pylori to clinically used antibiotics, these findings have highlighted the potential of sulphonamide inhibitors of HpαCA as lead compounds for developing novel anti-infective agents. Moreover, since EZA, AAZ, and MZA have been used since the 1950s as drugs to treat various human conditions, their pharmacokinetic properties are well understood, and repurposing these drugs as antimicrobials to treat multi-resistant bacterial infections would be cost-effective. However, HpαCA has not, as yet, been validated as a drug target. In this study, we have examined the potency of the HpαCA inhibitors MZA, AAZ and EZA against several H. pylori laboratory and clinical strains, and isolated and characterised a mutant resistant to the most potent compound, EZA, which has led to the identification of the genetic determinants that confer resistance and to the understanding of the aspects of the mechanism of anti-H. pylori activity of EZA.
Materials and methods
Bacterial strains and culture conditions
H. pylori laboratory strains P12Citation20, 26695Citation21, SS1Citation22, and J99Citation23, and clinical isolates from gastric biopsies (CH425, CH426, and CH427) were used in the study. H. pylori strains were grown on horse blood agar (HBA) prepared using Columbia blood agar base (Oxoid) supplemented with 5% (v/v) defibrinated horse blood and an antibiotic cocktail comprising 10 µg/mL vancomycin, 5 µg/mL cefsulodin, 2.5 U/mL polymyxin B, 5 µg/mL trimethroprim, and 8 µg/mL amphotericin B. Plates were incubated at 37 °C for 48–72 h under microaerophilic conditions generated using the CampyGen (Oxoid) system. Liquid cultures were grown at 37 °C with shaking at 120 rpm in Brucella broth (Becton Dickinson) containing 10 µg/mL vancomycin and 10% (v/v) foetal bovine serum (FBS), in microaerophilic conditions. Antibiotic solutions were prepared according to Clinical and Laboratory Standards Institute (CLSI) guidelinesCitation24. AAZ, MZA, and EZA were dissolved in dimethyl sulfoxide (DMSO). AAZ, MZA, EZA, antibiotics, and DMSO were purchased from Sigma-Aldrich.
Determining minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC)
MICs and MBCs of sulphonamide compounds were determined as previously describedCitation25. Liquid cultures of H. pylori were grown to an optical density of 0.4–0.6 at 600 nm (OD600). Cells were pelleted, washed, resuspended in antibiotic-free medium to an OD600 of 0.05, and aliquoted in 1 ml volumes supplemented with various concentrations of sulphonamides (0–15 mM AAZ, 0–5 mM MZA, or 0–1 mM EZA), or 1% (v/v) DMSO as a control. The cultures were incubated for 24 h, and initial and final colony forming units (CFU) were quantified by plating out serial dilutions and counting the colonies. Cell survival after 24 h was calculated as follows: CFU survival (%)=(CFU24h/CFU0h)*100. H. pylori P12 had the highest sensitivity to all three compounds and thus was chosen for further studies.
MICs of antibiotics commonly used to treat H. pylori infections (metronidazole, clarithromycin, amoxicillin, and tetracycline) against the clinical strains were measured using E-test (bioMérieux)Citation26.
Bactericidal kinetics of AAZ, MZA, and EZA
A liquid culture of H. pylori P12 at OD600 of 0.05 was prepared as above, and supplemented with AAZ, MZA, or EZA at concentrations corresponding to their respective 1 × MBC or 2 × MBC. Cultures were grown for 48 h, and sampled at 0, 6, 12, 18, 24, 36, and 48 h for CFU quantification.
To compare activities of AAZ, MZA, and EZA against H. pylori P12 under neutral (pH 6.8) and acidic (pH 4.5) conditions, time-kill curves were generated using 2 × MBC of each inhibitor in Brucella broth/10% FBS at pH 6.8 or in the same medium adjusted to pH 4.5 using 0.2 M phosphate-citrate buffer. Liquid cultures were grown to an OD600 of 0.4–0.6. The cells were then pelleted, washed, and resuspended in liquid medium supplemented with 2 × MBC of AAZ, MZA, or EZA at either pH 6.8 or pH 4.5, to an OD600 of 0.05. The cultures were grown as described above, and sampled at 0, 3, 6, 12, 18, 24, and 36 h to quantify CFU.
Isolation and characterisation of an H. pylori P12 mutant resistant to EZA
An H. pylori P12 mutant clone resistant to EZA (hereafter referred to as MutE) was obtained by iterative selection for progressive resistanceCitation27,Citation28 to 0.05 mM (0.25 × MIC), 0.1 mM, and then 2 mM EZA. The frequency of pre-existing spontaneous mutations allowing growth at 2 mM EZA was estimated by single-step selection. Plates containing the inhibitor were inoculated with 2 × 106, 2 × 107, or 2 × 108 CFU. Plates with 10 µg/mL rifampicin, a compound with a known frequency of spontaneous resistant mutants in H. pylori, were used as controlsCitation29. The mutation frequency was calculated as the average CFU generated on the inhibitor-supplemented plate, divided by the CFU in the inoculum.
To assess the stability of the resistant phenotype, 5 colonies of MutE were picked from plates containing the highest inhibitor concentration, and passaged 5 times on inhibitor-free plates. The resulting isolates showed no significant change in MIC or MBC values relative to the starting ones.
To construct growth curves, liquid cultures of H. pylori P12 WT and MutE at starting OD600 of 0.05 were grown for 48 h in the absence of EZA. Samples were taken at 0, 3, 6, 9, 12, 18, 24, 30, 36, and 48 h, and CFU enumerated. Values were analysed in GraphPad Prism version 7.02 using two-way ANOVA with Dunnett’s multiple comparison test.
MICs of commercial antibiotics against the EZA-resistant mutant and its WT parent
MICs of the clinically used antibiotics against H. pylori P12 WT and MutE were determined by the agar dilution methodCitation24. Ten µL of the starter cultures at OD600 0.05 were plated onto HBA containing various concentrations of antibiotics (0.0075–0.96 µg/mL amoxicillin and clarithromycin, 0.5–16 µg/mL metronidazole, and 0.06–4 µg/mL tetracycline). CFU were determined as described above.
Confirmation of resistance-conferring mutations by transformation
In order to separate the mutations responsible for the resistant phenotype from all other spontaneous mutations in MutE, its genomic DNA was isolated and transformed into the WT P12 strain. WT genomic DNA and buffer were used as negative controls. Four independent transformant colonies displaying resistance to EZA (hereafter referred to as MutETF1, MutETF2, MutETF3, and MutETF4) were selected on Columbia blood agar plates containing 2 mM EZA.
Genome sequencing and analysis
Genomic DNA was extracted using GenElute Bacterial Genomic DNA kit (Sigma-Aldrich). Further sample preparation steps and genome sequencing were performed at the Micromon High-Throughput Sequencing Facility (Monash University). Sequencing libraries were constructed using the Illumina NexteraXT (Illumina) and quantified using the Qubit DNA HS kit (Invitrogen). Sequencing was performed on the Illumina MiSeq platform with a paired end configuration and average read length of 150 bp.
Sequence analysis was performed using CLC genomics Workbench v. 7.0.3 (Qiagen). Reads were aligned to the reference genome of H. pylori strain P12 (NCBI accession number NC_011498.1). Differences between the WT parental strain and MutE, identified using the Probabilistic Variant Detection and the Quality Based Variant Detection analysis tools in CLC genomics Workbench, were confirmed by Sanger sequencing at Micromon (see below).
Sanger sequencing
The genes of interest were PCR-amplified from genomic DNA, purified using the Wizard SV gel and PCR clean-up kit and sequenced using the conventional Sanger method. Sequences were aligned with BioEdit v 7.2.5 (http://www.mbio.ncsu.edu/bioedit/bioedit.html).
Results and discussion
Antimicrobial activity of EZA, AAZ, and MZA against H. pylori
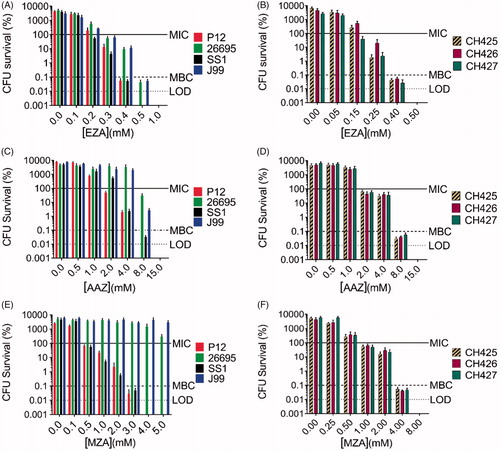
We assessed the antimicrobial activities of sulphonamide drugs AAZ, MZA, and EZA against the H. pylori laboratory strains P12, 26695, SS1, and J99, and against the clinical isolates resistant to metronidazole, clarithromycin, and/or amoxicillin (CH425, CH426, and CH427, ). Growth inhibition assays showed that most tested strains were sensitive to high-micromolar or low-millimolar concentrations of sulphonamides (, . EZA had the highest anti-H. pylori activity of all tested compounds and inhibited growth of all strains. The lowest MIC/MBC values for EZA were observed with the strains P12 and SS1 (MIC = 0.2 mM, MBC = 0.4 mM for both strains). MIC values for AAZ were approximately one order of magnitude higher than those of EZA in all tested strains (, ). Strains 26695 and J99 were the least sensitive to all CA inhibitors used, and neither showed measurable sensitivity to MZA in the tested concentrations range. Importantly, although the three clinical strains have different resistance profiles to the commercial first-line antibiotics (amoxicillin, clarithromycin, metronidazole, and tetracycline), their sensitivity to each sulphonamide was equivalent. Since H. pylori strain P12 showed the greatest sensitivity to all compounds tested, it was selected for the subsequent mechanistic studies.
Table 1. Sensitivity profiles of H. pylori clinical isolate strains CH425, CH426, and CH427 to clinically used antibiotics.
Table 2. MIC and MBC values of three sulphonamide drugs against four laboratory strains and three clinical isolates.
Figure 2. The antimicrobial effects of carbonic anhydrase inhibitors EZA, AAZ, and MZA on various H. pylori strains. Sensitivity of the laboratory strains P12, 26695, SS1, and J99 to (A) EZA, (C) AAZ, and (E) MZA and sensitivity of the clinical strains CH425, CH426, CH427 to (B) EZA, (D) AAZ, and (F) MZA are represented as percentage survival after 24 h (CFU survival (%)=(CFU24h/CFU0h)*100). The CFU survival levels corresponding to MIC (100.1% survival), MBC (0.1% survival) and limit of detection (LOD) are indicated by horizontal lines. Error bars represent the standard error of the mean for three independent biological replicates.
H. pylori growth inhibition by EZA, AAZ and MZA is time-, concentration- and pH-dependent
To determine the duration of inhibitor treatment required to kill H. pylori at neutral pH (under conditions optimal for H. pylori growth), time-dependent killing kinetics were assessed for each sulphonamide at concentrations corresponding to their respective 1 × MBC and 2 × MBC (. For EZA, 18-h incubation with 2 × MBC (1 mM) of the compound was sufficient to kill 99.9% of cells, while a 36-h exposure was required when 1 × MBC was used. Bactericidal kinetics for MZA were similar, except that a 24-h exposure was required to kill 99.9% of cells at 2 × MBC of the compound (6 mM). In comparison to EZA and MZA, the bactericidal action of AAZ was slower: the time required for 1 × MBC of the inhibitor to kill 99.9% of cells was 48 h (36 h for 2 × MBC). This analysis has also demonstrated that anti-H. pylori activity of all three sulphonamides is concentration-dependent.
Figure 3. Analysis of the time and dose dependency of the antimicrobial action of AAZ, MZA, and EZA on H. pylori P12. (A) Bactericidal kinetics for 1 × MBC and 2 × MBC of the respective sulphonamide, measured at neutral pH (B) Bactericidal kinetics for 2 × MBC of the respective sulphonamide under neutral (pH 6.8) and acidic (pH 4.5) conditions. The horizontal dashed line represents the limit of detection (100 cells) and the horizontal solid line corresponds to 99.9% cell death. Error bars represent the standard error of the mean for three independent biological replicates.
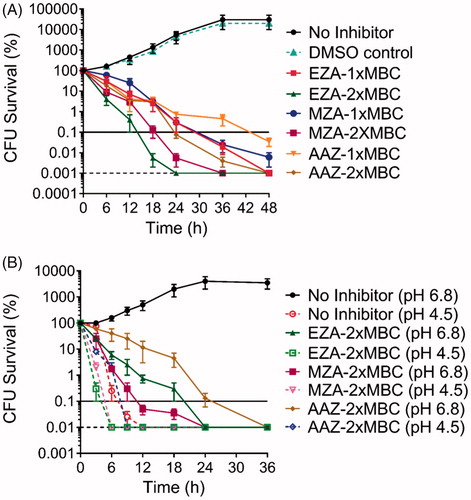
To determine the effect of low pH on the bactericidal activity of the sulphonamide inhibitors, time-dependent killing curves were also generated using the medium buffered at pH 4.5 (approximating conditions to which H. pylori is exposed during the initial colonisationCitation30) (. Firstly, the results confirm that in vitro, H. pylori would not withstand low pH conditions for long, as even in the absence of inhibitors no bacteria survived after 12 h at pH 4.5. Secondly, 2 × MBC of EZA, AAZ, or MZA accelerated the elimination of H. pylori at pH 4.5, which occurred after 6 h for EZA, and 9 h for MZA and AAZ (. Thus, sulphonamides exerted detectable antimicrobial activity under both neutral and low pH conditions. However, the reduced bacterial viability at low pH precluded direct quantitative comparisons.
The observation that the sulphonamide compounds display bactericidal activity at both neutral and acidic pH has not been expected, as the inhibitors of H. pylori α- and β-carbonic anhydrases were thought to affect the cell viability only at acidic pH, when the functions of these enzymes are known to be essential. As neutral pH approximates the conditions under which H. pylori persists in the mucous layer adjacent to the gastric epithelium, we have addressed the mechanism of bacterial killing at neutral pH by isolating and characterising a spontaneous mutant resistant to the most potent compound in the series, EZA.
Isolation and characterisation of H. pylori P12 mutant with decreased susceptibility to EZA
Selection by serial passages of H. pylori P12 in the presence of sub-lethal concentrations of EZA enabled isolation of a mutant significantly more resistant to this compound than the parental wild type. The EZA-resistant strain MutE had an MIC >2 mM (10 × WT MIC). The resistance phenotype was stable during growth in the absence of EZA for at least 15 days.
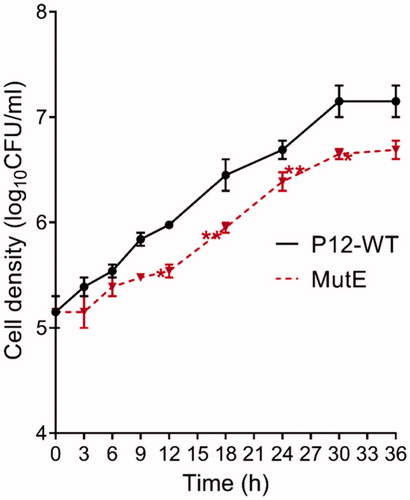
Estimation of the frequency of spontaneous resistant mutants using a single selection step with 2 mM EZA yielded a value of <5 × 10−9, which is significantly lower than the previously reported frequencies of spontaneous mutations leading to H. pylori resistance to rifampicin (10−6)Citation29, metronidazole, or tetracycline (10−5–10−6)Citation31. This observation prompted us to determine whether the mutation(s) associated with resistance to EZA incurred a fitness cost. Indeed, MutE showed reduced growth in comparison to the parental strain (); the doubling time of P12 WT was 5 h, whereas the doubling time of MutE was 6 h.
Figure 4. Growth curves for H. pylori P12 WT and MutE measured over 36 h. Error bars represent the standard error of the mean for three independent biological replicates. Significant differences compared to wild type P12 are indicated; *p < .05, **p < .01. All other differences are not significant.
Identification of genetic determinants linked to EZA resistance
As carbonic anhydrases were considered the likely targets for the anti-H. pylori activity of sulfonamidesCitation12–15, the genes for α- and β-carbonic anhydrases were sequenced in the parental strain and in MutE. No mutations were found, eliminating the possibility that amino acid changes in these enzymes caused the resistance phenotype.
To investigate the genetic basis for the phenotypically stable resistance to sulphonamides, we therefore determined and compared the full genome sequences of P12 WT and MutE. All observed genomic differences were single nucleotide polymorphisms in 12 genes (listed in Supplementary Table 1). To separate the mutations that caused EZA resistance from unlinked random mutations, we performed natural transformation of the mutant chromosomal DNA back into a WT background, and selected four resultant EZA-resistant recombinants (MutETF1–MutETF4, ) for Sanger sequencing of the candidate genes. MutETF1, MutETF2, MutETF3, and MutETF4 retained mutations in four, four, four and five genes, respectively. Only three mutations were common to all four EZA-resistant transformants. One mutation (Glu173Lys) was in the gene HPP12_RS06100 that encodes undecaprenyl pyrophosphate synthase UppS, an enzyme essential for cell wall biosynthesis. The second mutation (Cys29Arg) was in the regulatory gene HPP12_RS07625 encoding transcription termination factor NusA. The third mutation (a frameshift) was in the gene HPP12_RS01490 encoding an inner membrane protein of unknown function (). This result suggests that acquisition of resistance by H. pylori to EZA at neutral pH was associated with mutations in these three genes, and was likely the result of a combination of different mechanisms involving modifications of cellular proteins and systems other than HpαCA and HpβCA.
Table 3. Nucleotide changes in H. pylori P12 EZA-resistant mutants generated by transformation of WT P12 with MutE chromosomal DNA.
Our study allows the proposal of putative resistance mechanisms and discussion of the implications for the mode of antimicrobial action of EZA. One possible resistance mechanism is alteration of a putative sulphonamide target. We note that sulphonamides (albeit other than EZA) have been shown to inhibit H. pylori undecaprenyl pyrophosphate synthase (UppS) with micromolar IC50Citation32. In our experiments, selection for resistance to EZA resulted in a mutant with the Glu173Lys substitution in UppS. The respective residue in E. coli UppS (Glu198) is proximal to the catalytic Mg2+ ion in the active siteCitation33. We, therefore, postulate that EZA binds in the H. pylori UppS active site through coordination of Mg2+, in competition with the natural substrate, thus acting as a competitive inhibitor. The Glu173Lys mutation likely removes favourable interactions or introduces a steric clash with the inhibitor, conferring resistance.
Changes in cell physiology may also contribute to EZA resistance. MutE contains a mutation in a gene regulating transcription (a single amino-acid substitution Cys29Arg in the transcription termination factor NusA), which likely affects the global regulation of metabolic enzymes, aiding resistance. The third resistance-linked mutation, found in the gene HPP12_RS01490 encoding an inner membrane protein that shares no sequence similarity with any protein of a known function, may affect an entry pathway of EZA into H. pylori.
There is no cross-resistance between EZA and clinically used anti-H. pylori antibiotics
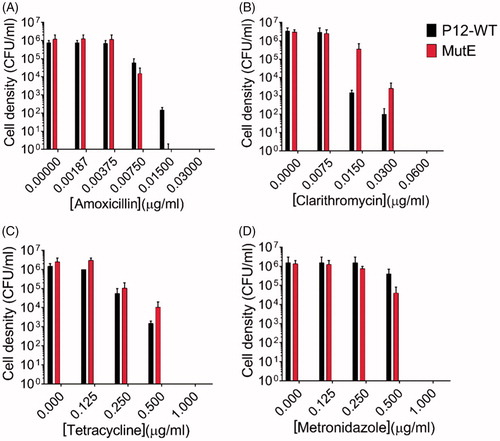
To gain further insight into whether there were common mechanisms between the EZA-resistant mutant generated in this study and the known antibiotic resistances in H. pylori, cross-resistance to commercial antibiotics was investigated. As described in an earlier section, clinical strains resistant to amoxicillin, clarithromycin, and/or metronidazole were sensitive to EZA. Evaluation of the bactericidal activity of these compounds against MutE showed that the reverse is also true: MIC and MBC values of the first line antibiotics for the WT strain were not significantly different from those for MutE (), indicating that there are no shared resistance mechanisms. This result suggests that EZA kills H. pylori via mechanisms that are different from the mode of action of current first-line antibiotics (amoxicillin, clarithromycin, metronidazole, and tetracycline) used to treat H. pylori infections.
Figure 5. Sensitivity of H. pylori P12 WT and its sulphonamide-resistant mutants MutE to clinically used antibiotics amoxicillin, clarithromycin, tetracycline, and metronidazole. Error bars represent the standard error of the mean for two independent biological replicates. (A) The MIC values of both the WT strain and MutE for amoxicillin were 0.0075 µg/ml, with the MBC value for MutE (0.015 µg/ml) being 2-fold lower than for the WT strain. (B) The P12 WT strain and MutE showed very close sensitivities to clarithromycin (MIC = 0.015 µg/ml, MBC = 0.06 µg/ml). (C) The sensitivity assay for tetracycline yielded the MIC and MBC values of 0.25 µg/ml and 1 µg/ml, respectively for both the WT strain and MutE. (D) The P12 WT strain and MutE showed the same sensitivity pattern for metronidazole (MIC = 0.5 µg/ml, MBC = 1 µg/ml).
Conclusion
The sulphonamide drugs AAZ, MZA, and EZA displayed bactericidal activity against both laboratory strains and clinical antibiotic-resistant isolates of H. pylori, with EZA showing the greatest activity. Importantly, the mechanism of action of EZA is different from that of conventional antibiotics used to treat H. pylori infections, and is not restricted to impacting carbonic anhydrase function. Our findings suggest that EZA impacts multiple targets within H. pylori, and that alterations in several functions may be required for resistance to emerge. Indeed, the frequency of spontaneous resistant mutants was found to be <5 × 10−9, indicating that resistance does not develop easily. The low frequency of spontaneous resistant mutants was also in agreement with our observation that resistance to EZA incurred a significant fitness cost. EZA is an inexpensive and relatively safe drug that was used clinically since 1950s to treat various non-bacteria-related illnesses (e.g. glaucomaCitation34); its side effects are generally tolerableCitation35, and its pharmacokinetic properties are well understood. Taken together, our findings suggest that developing EZA into a more effective anti-H. pylori agent may offer a faster and cost-effective route towards new antimicrobials with a novel mechanism of action, and that investigations of the in vivo antimicrobial action of this compound are warranted. A number of potential limitations to such approach need to be considered and addressed in follow-up studies. Activity of EZA against H. pylori is relatively weak in comparison to existing antibiotics, and a systematic structure-activity relationship study is needed to determine if it can serve as a starting point for a drug discovery programme. In addition, it was beyond the scope of this work to examine potential antimicrobial activity of EZA against other bacteria, which leaves open the question of specificity towards H. pylori.
Supplemental Material
Download PDF (20.5 KB)Acknowledgements
We thank Melanie L. Hutton (Monash University) for her contribution to the design of experiments. J.G.-B. was supported by a Larkins Fellowship from Monash University.
Disclosure statement
No potential conflict of interest was reported by the authors.
Related Research Data
References
- Hooi JKY, Lai WY, Ng WK, et al. Global prevalence of Helicobacter pylori infection: systematic review and meta-analysis. Gastroenterology 2017;153:420–9.
- Take S, Mizuno M, Ishiki K, et al. Seventeen-year effects of eradicating Helicobacter pylori on the prevention of gastric cancer in patients with peptic ulcer; a prospective cohort study. J Gastroenterol 2015;50:638–44.
- Graham DY. Helicobacter pylori update: gastric cancer, reliable therapy, and possible benefits. Gastroenterology 2015;148:719–31.
- Uemura N, Okamoto S, Yamamoto S, et al. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med 2001;345:784–9.
- Shiota S, Reddy R, Alsarraj A, et al. Antibiotic resistance of Helicobacter pylori among male United States veterans. Clin Gastroenterol Hepatol 2015;13:1616–24.
- Savoldi A, Carrara E, Graham DY, et al. Prevalence of antibiotic resistance in Helicobacter pylori: a systematic review and meta-analysis in World Health Organization regions. Gastroenterology 2018;155:1372–82.
- Marcus EA, Scott DR. Gastric colonization by H. pylori. In: Kim N, ed. Helicobacter pylori. Singapore: Springer; 2016:23–34.
- Eaton KA, Krakowka S. Effect of gastric pH on urease-dependent colonization of gnotobiotic piglets by Helicobacter pylori. Infect Immun 1994;62:3604–7.
- Eaton KA, Brooks CL, Morgan DR, Krakowka S. Essential role of urease in pathogenesis of gastritis induced by Helicobacter pylori in gnotobiotic piglets. Infect Immun 1991;59:2470–5.
- Sachs G, Weeks DL, Wen Y, et al. Acid acclimation by Helicobacter pylori. Physiology 2005;20:429–38.
- Ronci M, Del Prete S, Puca V, et al. Identification and characterization of the α-CA in the outer membrane vesicles produced by Helicobacter pylori. J Enzyme Inhib Med Chem 2019;34:189–95.
- Nishimori I, Minakuchi T, Morimoto K, et al. Carbonic anhydrase inhibitors: DNA cloning and inhibition studies of the alpha-carbonic anhydrase from Helicobacter pylori, a new target for developing sulfonamide and sulfamate gastric drugs. J Med Chem 2006;49:2117–26.
- Nishimori I, Onishi S, Takeuchi H, Supuran CT. The alpha and beta classes carbonic anhydrases from Helicobacter pylori as novel drug targets. Curr Pharm Des 2008;14:622–30.
- Modak JK, Modakh JK, Liu YC, et al. Structural basis for the inhibition of Helicobacter pylori α-carbonic anhydrase by sulfonamides. PLoS One 2015;10:e0127149.
- Modak JK, Liu YC, Supuran CT, Roujeinikova A. Structure-activity relationship for sulfonamide inhibition of Helicobacter pylori α-carbonic anhydrase. J Med Chem 2016;59:11098–109.
- Puscas I, Buzas G. Treatment of duodenal ulcers with ethoxzolamide, an inhibitor of gastric mucosa carbonic anhydrase. Int J Clin Pharmacol Ther Toxicol 1986;24:97–9.
- Vălean S, Vlaicu R, Ionescu I. Treatment of gastric ulcer with carbonic anhydrase inhibitors. Ann N Y Acad Sci 1984;429:597–600.
- Puscas I. Treatment of gastroduodenal ulcers with carbonic anhydrase inhibitors. Ann N Y Acad Sci 1984;429:587–91.
- Graham D, Lew GM, Klein D, et al. Effect of treatment of Helicobacter pylori infection on the long-term recurrence of gastric or duodenal ulcer. A randomized, controlled study. Ann Intern Med 1992;116:705–8.
- Haas R, Meyer TF, van Putten JP. Aflagellated mutants of Helicobacter pylori generated by genetic transformation of naturally competent strains using transposon shuttle mutagenesis. Mol Microbiol 1993;8:753–60.
- Akopyants NS, Eaton KA, Berg DE. Adaptive mutation and cocolonization during Helicobacter pylori infection of gnotobiotic piglets. Infect Immun 1995;63:116–21.
- Lee A, O'Rourke J, De Ungria MC, et al. A standardized mouse model of Helicobacter pylori infection: introducing the Sydney strain. Gastroenterology 1997;112:1386–97.
- Alm RA, Ling LS, Moir DT, et al. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature 1999;397:176–80.
- Clinical and Laboratory Standards Institute. 2015. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically. Approved standard, 10th ed, supplement M07-A10. Wayne, PA: Clinical and Laboratory Standards Institute.
- Makobongo MO, Kovachi T, Gancz H, et al. In vitro antibacterial activity of acyl-lysyl oligomers against Helicobacter pylori. Antimicrob Agents Chemother 2009;53:4231–9.
- Clinical and Laboratory Standards Institute. 2016. Methods for antimicrobial dilution and disk susceptibility testing of infrequently isolated or fastidious bacteria. 3rd ed. CLSI guideline M45. Wayne, PA: Clinical and Laboratory Standards Institute.
- Haas CE, Nix DE, Schentag JJ. In vitro selection of resistant Helicobacter pylori. Antimicrob Agents Chemother 1990;34:1637–41.
- Wang G, Wilson TJ, Jiang Q, Taylor DE. Spontaneous mutations that confer antibiotic resistance in Helicobacter pylori. Antimicrob Agents Chemother 2001;45:727–33.
- Kulick S, Moccia C, Kraft C, Suerbaum S. The Helicobacter pylori mutY homologue HP0142 is an antimutator gene that prevents specific C to A transversions. Arch Microbiol 2008;189:263–70.
- Quigley EM, Turnberg LA. pH of the microclimate lining human gastric and duodenal mucosa in vivo. Studies in control subjects and in duodenal ulcer patients. Gastroenterology 1987;92:1876–84.
- Petschow BW, Batema RP, Ford LL. Susceptibility of Helicobacter pylori to bactericidal properties of medium-chain monoglycerides and free fatty acids. Antimicrob Agents Chemother 1996;40:302–6.
- Nishizawa T, Suzuki H. Mechanisms of Helicobacter pylori antibiotic resistance and molecular testing. Front Mol Biosci 2014;1:19.
- Kuo CJ, Guo RT, Lu IL, et al. Structure-based inhibitors exhibit differential activities against Helicobacter pylori and Escherichia coli undecaprenyl pyrophosphate synthases. J Biomed Biotechnol 2008;2008:841312.
- Maren TH, Brechue WF, Bar-Ilan A. Relations among IOP reduction, ocular disposition and pharmacology of the carbonic anhydrase inhibitor ethoxzolamide. Exp Eye Res 1992;55:73–9.
- Swenson ER. Safety of carbonic anhydrase inhibitors. Expert Opin Drug Saf 2014;13:459–72.