
Abstract
The 3-hydroxy-3-methylglutaryl-CoA reductase, a key enzyme of the mevalonate pathway for the synthesis of cholesterol in mammals (ergosterol in fungi), is inhibited by statins, a class of cholesterol lowering drugs. Indeed, statins are in a wide medical use, yet statins treatment could induce side effects as hepatotoxicity and myopathy in patients. We used Saccharomyces cerevisiae as a model to investigate the effects of statins on mitochondria. We demonstrate that statins are active in S.cerevisiae by lowering the ergosterol content in cells and interfering with the attachment of mitochondrial DNA to the inner mitochondrial membrane. Experiments on murine myoblasts confirmed these results in mammals. We propose that the instability of mitochondrial DNA is an early indirect target of statins.
1. Introduction
Cholesterol is a vital component of cell membranes, essential for the synthesis of steroid hormones, bile acids, and vitamin D, but high cholesterol levels are associated with an elevated risk of cardiovascular diseases. Cholesterol (ergosterol in yeast) is the end-product of the mevalonate pathwayCitation1. The enzyme 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase is the rate-limiting step in the synthesis of cholesterol and it catalyses the conversion of HMG-CoA to mevalonate; the mevalonate pathway and the HMG-CoA enzyme, as well as its regulation are conserved from yeast to humansCitation2,Citation3. HMG-CoA is the target of statinsCitation4, the most prescribed drug class for lowering elevated LDL-cholesterol, generally effective and well tolerated; however, patients can experience muscle adverse effects such as myopathyCitation5,Citation6. Genetic factorsCitation7 and drug-drug interactionsCitation8 also contribute as risk factors for the development of muscle side effects in patientsCitation9,Citation10.
Mitochondrial dysfunction has been described as responsible for several statins side effects and serious adverse reactions; in particular, various mitochondrial phenotypes were described such as the decrease of respiratory chain function, alteration of calcium homeostasis, ROS production, alteration of mitochondrial volume, however, the primary effect of statins remains to be elucidatedCitation11,Citation12. The activity of statins was investigated previously in the yeast Candida glabrataCitation13 and in Saccharomyces cerevisiaeCitation14; the yeast S. cerevisiae is an excellent unicellular model to gain insights into human diseasesCitation15 in particular, those resulting from impaired mitochondrial functionCitation16–18, because S. cerevisiae is able, utilising fermentable carbon sources to survive without mitochondrial respiration. We previously demonstrated that ketoconazole, which blocks the ergosterol biosynthetic pathway downstream the HMG-CoA reductase, reduced the ergosterol content in yeast cells and induced the loss of mitochondrial DNA; this phenomenon is coupled with a re-localisation of Erg27 enzyme from Lipid Droplets (LDs) to Endoplasmic Reticulum (ER)Citation19. Here, we expand the analysis of mitochondrial function and the stability of mitochondrial DNA using statins in S. cerevisiae: we showed that these molecules cause a reduction of ergosterol, inducing loss of mitochondrial DNA and resulting in a reduction of mitochondrial respiration/function coupled with the Erg27 re-localisation in ER. In murine myoblasts, upon statins treatment, we also observed a reduction of mitochondrial DNA copy number associated with an impaired differentiation capacity.
2. Materials and methods
2.1. Yeast strains and growth conditions
The strains used in this study: the S. cerevisiae W303 (MAT a, his3–11, ade2–1, leu2–3, −112, ura3–1, trp1–1, can1–100), W303-ERG27-GFP, MValC25T (MATa, ade2–1, ura3–52, leu2, kar1–1, syn); the K. lactis PM6-7A (MATa uraA1-1 adeT-600a), PM6-7A/Δpda1 (Klpda1::Tn5BLE). The plasmid pVT100U-mitoGFP was used to visualise mitochondria.
Yeast culture media: YPD (1% bacto peptone, 1% yeast extract and 2% glucose), was used as rich medium. YPG (1% bacto peptone, 1% yeast extract and 3% glycerol), was the medium used to verify the respiratory competence of yeast colonies. All media were supplemented with 2.3% bacto agar (Difco) for solid media.
YPD medium supplemented with 0.5 mg/ml of ergosterol. Ergosterol and Tween 80 were dissolved in pure ethanol to final concentration of 10 mg/ml for ergosterol and 42% for Tween80, and steamed at 100 °C for 10 min, always protected from light, before being added to the medium. Yeast cultures were grown and analysed in exponential phase at 28 °C, unless otherwise specified.
2.2. C2C12 cell culture
C2C12 myoblasts were grown at 5% CO2 and 37 ̊C in Dulbecco’s modified Eagle’s medium (DMEM; Gibco) supplemented with 1% penicillin/streptomycin (Gibco) and 10% foetal bovine serum (Gibco). To induce differentiation, cells were cultured with Dulbecco’s modified Eagle’s medium and 2% horse serum (Gibco) for 48 h, when more than 90% of the cells had fused into myotubes. C2C12 were treated for 48 with 25uM Atorvastatin in growth medium and then induced to differentiate for further 48 h either in the presence or absence (ctrl) of the compound.
2.3. Rho° production
Strains (W303) devoid of mitochondrial DNA were produced as follows: cells were grown at the density of 1 × 106cells/ml on YPD medium, phosphate buffer pH 6.5 was added in1 ml of culture, at the final concentration of 0.05 M, and ethidium bromide at final concentration of 50 μg/ml. The cultures were incubated at 28 °C for 24 h. Cells were washed twice, plated on YPD and incubated at 28 °C. The absence of mitochondrial DNA was assessed by DAPI staining and absence of growth on glycerol containing medium as a carbon source.
2.4. S. cerevisiae cell culture
The cells were pre-activated in YPD and YPD rich medium supplemented with different statin up to the stationary phase. Subsequently, the cultures were all diluted to a concentration of 104 cells/ml with the respective media and incubated at 28 °C. The growth trend was monitored with the measurement of the optical density OD600 and/or with the cell count with the Bürker chamber.
2.5. Petites assay
Cells were grown in YPD until the stationary phase. At the concentration of 1 × 107cells/ml and 1 × 108cells/ml, 100 cells were spotted in YPD plates and incubated at 28 °C. After two days, colonies were replicated on YPG plates. The lack of rho° cell growth was assessed on glycerol as the carbon source and the absence of mtDNA by 4′,6-diamidino-2-phenylindole (DAPI; Sigma) staining.
2.6. Microscopy
Cells were observed with a Zeiss Axio Imager Z1 Fluorescence Microscope with AxioVision 4.8 Digital Image Processing System, the objective lens used was 63× Oil. Filter sets: 38HE (GFP), 43HE (DsRed). Filters for GFP (470/40 nm excitation and 525/50 nm emission) and DAPI (365-nm excitation and 445/450-nm emission), were used Metamorph software (Universal Imaging, West Chester, PA) was used to deconvolute Z-series and treat the imagesCitation20.
2.7. Cells treatment
Pravastatin and simvastatin were hydrolysed in ethanolic NaOH [15% (v/v) ethanol and 0.25% (w/v) NaOH] at 60 °C for 1 h. Atorvastatin and rosuvastatin were dissolved in H2Odd supplemented with 1% DMSO and 0.9% NaCl. Q10 was hydrolysed in 100% acetone.
Cells were grown to a density of 1 × 107cells/ml in YPD rich medium supplemented with: pravastatin 150 µg/ml, atorvastatin 100 µg/ml, rosuvastatin 50 µg/ml and Q10 10, 50 µg/ml. The cells treated were used for several assays.
2.8. Mass spectrometric analyses
Dry cell pellets were deep frozen by immersion in liquid nitrogen and quickly ground to a powder using an IKA A11 basic laboratory mill (IKA, Staufen, Germany). 300 mg of cell pellet were transferred to glass vials with 10 ml Folch solvent (chloroform/methanol 2:1, v/v) containing 0.01% butylated hydroxytoluene followed by the addition of10μl of internal standard (19-hydroxy-cholesterol, 2 mg/ml in methanol) as internal standards (IS). The homogenisation and extraction (60 min) were carried out at room temperature (RT). The extracts were evaporated under a stream of N2then the dry residue was dissolved in1 ml hydrolysis solution containing 5% KOH in (MeOH/H2O, 95:5) and subjected to saponification (1 h, 80 °C). The hydrolysed solution was extracted with 0.5 ml distilled H2O and 4 ml diethyl ether. The ether phase was dried under a stream of N2at RT. The samples were then reconstituted with 25 μl ethylacetate and vortex mixed. The fractions to be analysed were converted to their trimethyl-silylated derivatives by heating with 40 μl BSTFA containing 1% TMCS at 70 °C for 45 min. The sample was dried under a stream of N2. The residue was dissolved in200 ml hexane and the clear hexane phase was transferred into a glass vial for GC–MS injection. GC–MS analyses were performed with an Agilent 6850 A gas chromatograph coupled to a 5973 N quadrupole mass selective detector (Agilent Technologies, Palo Alto, CA, USA).
Chromatographic separations were carried out with an Agilent HP5msfused-silica capillary column (30 m × 0.25 mm i.d.) coated with 5%-phenyl-95%-dimethylpolysiloxane (film thickness 0.25 μm) as stationary phase. Injection mode: splitless at a temperature of 280 °C. Column temperature programme: 120 °C (1 min) then to 320 °C at a rate of 5 °C/min and held for 5 min. The carrier gas was helium at a constant flow of 1.0 ml/min. The spectra were obtained in the electron impact mode at 70 eV ionisation energy; ion source 280 °C; ion source vacuum10-5 Torr. MS analysis was performed simultaneously in TIC (mass range scan from m/z50 to 800 at a rate of 0.42 scans s-1) and SIM mode. For GC-SIM-MS analysis, ion m/z 363 was selected for ergosterol and ion m/z 353 was selected for the internal standard.
2.9. O2 consumption measurement
Respiration studies were performed using a Clark oxygen electrode (Hansatech Instruments) as described in De Luca et al. (2009)Citation21. Cells (1 × 108) were collected, washed with 1 ml sterile water, suspended in 1 ml sodium phosphate buffer (10 mM pH 7.4 containing 4 g l−1 glucose) and loaded in the reaction vessel of the previously calibrated oxygen electrodCitation22.
2.10. Immunofluorescence
For the immunofluorescence on C2C12 cells were fixed with 4% paraformaldehyde in PBS for 20 min at RT, and permeabilized with 0.5% Triton X-100 in PBS. After 1 h incubation with blocking solution (4% BSA in PBS), primary antibody for MyHC (DSHB, clone MF20; 1:50 in blocking solution) was incubated overnight (O/N) at 4 °C. A goat anti-mouse secondary antibody coupled to Alexa Fluor 488 (Thermo Fisher; A21202; 1:250 in blocking solution) was then incubate for 1 h. DNA was counterstained with DAPI (Sigma; D9542). Coverslips were mounted in ProLongTM Diamond Antifade Mountant (Invitrogen; p36970). ImageJ was used to quantify the percentage of nuclei inside the myotubes compared to control (Differentiation index). Images were taken with Nikon eclipse TE300 microscope. Pictures showed in the figures are representative of all examined fields.
2.11. Quantitative analysis of mitochondrial DNA
Total DNA was isolated using phenol/chloroform-extraction. mtDNA was quantified by q-PCR using PowerUpSYBR Green PCR Master mix (Applied Biosystems; 4367659) and analysed on a StepOne Plus Real-Time PCR System (Applied Biosystems). To evaluate the amount of mtDNA present per nuclear genome, we quantified the relative copy number differences through the ΔΔCt methodCitation23, comparing mtDNA and nuclear DNA using the following primers sequences: mtDNA CYTOCHROME C (mt-CO1) FW: 5′-TCATCCCTTGACATCGTGCT-3’ RV: 5′-GTCTGAGTAGCGTCGTGGTA-3’; nuclear DNA ACTIN BETA (ACTB) FW: 5′-CCCTGAGTGTTTCTTGTGGC-3′ RV: 5′-GTCTCCGGAGTCCATCACAA-3′.
3. Results
3.1. Statins induced a petite phenotype in S. cerevisiae
As a potent HMG-CoA inhibitor, statins reduce cellular ergosterol levels also in S. cerevisiae. Indeed, the effect of lovastatin on yeast was previously investigated in the S. cerevisiae wild type 2180–1 ACitation14,Citation24 and BY4741 (derivative of S288C) model laboratory strainsCitation25. Similarly, the effect of the additional statins, including rosuvastatin, atorvastatin, simvastatin and fluvastatin was tested in the BY4742 genotype, expressing the human orthologue of HMG-CoACitation26.
Herein, we used S. cerevisiae W303 yeast cells to investigate the effects of human HMG-CoA reductase inhibitors; W303 is an excellent model to study sterols because the level of sterols is higher than in S288C and derivative strainsCitation27,Citation28.
To assess the effect of statins on mitochondria, we selected pravastatin, rosuvastatin, and atorvastatin for our studies, at non-toxic concentrations (Supplementary Figure 1) previously used to study the effect of statins on yeastCitation26. The treatment of wild type W303 cells with statins on agar YPD, led to the production of high percentage of small colonies. These colonies are referred to as petites, a phenotype which indicates a mitochondrial dysfunction, hence we assessed the effects of statins on yeast mitochondrial function using a petite assay (Supplementary Figure 2). The petite phenotype refers to the size of a S. cerevisiae colony with rearrangements (rho−) or the lack of mitochondrial DNA (rho°). We monitored petite production plating the cultures at 24, 48, 72 h of growth. At the concentration used, the statins induced a high rate of petite phenotype, especially atorvastatin and rosuvastatin (), demonstrating that the statins induced a defect in mitochondrial function, similar to ketoconazole.
Figure 1. Statins induced a high percentage of petites production in yeast cells. (a) Percentage of petites production of wild type W303 and W303 rho° grown in YPD medium and wild type W303 grown in YPD medium with pravastatin 150 µg/ml, atorvastatin 100 µg/ml and rosuvastatin 50 µg/ml. (b) The petite phenotype is suppressed by ergosterol. The same statins treatments as in (a) didn’t show a petite phenotype production in cells grown in a medium supplemented with ergosterol (0,5 mg/ml, YPDE). Cultures grown 24, 48 and 72 hours were plated on YPD plates and petites were counted after a replica plating on glycerol containing medium. Data are the average of 3 independent experiments and standard deviation is indicated. The asterisks indicate significant modulations (p < 0.05) according to Student’s T-test.
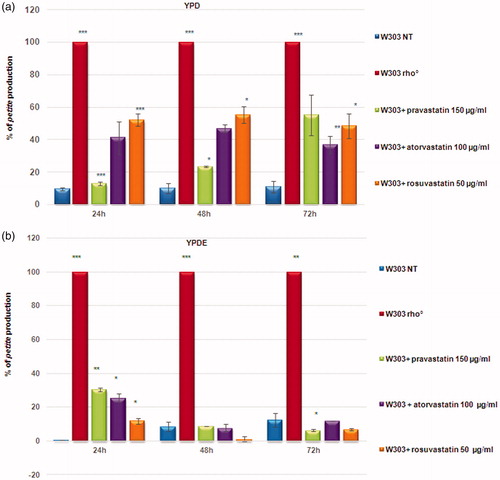
We previously show that in yeast, the presence of ergosterol (0.5 mg/ml) in growth medium suppressed the petite phenotype induced by ketoconazoleCitation19. We verified if the generation of petite colonies in presence of statins could be counteracted by adding ergosterol, at a concentration used to suppress the ketoconazole effects. Indeed, the percentage of petite was very low in presence of ergosterol, in similar to the wild type strain with functional mitochondria ().
Because treated cells showed different size, we verified cellular vitality by counting cells number to construct growth curves, which is not consider with optical density measure (Supplementary Figure 3(A)). Of note, treatments of W303 with statins resulted in intermediate growth curves, located between the non-treated W303 strain and the strain devoid of mitochondrial DNA (W303 rho°), while the presence of ergosterol in the medium restores a wild type growth in statin treated cells and in W303 rho° (Supplementary Figure 3(B)). These results suggest that statins treatment induced mitochondrial dysfunction in S. cerevisiae by lowering ergosterol content. To verify this hypothesis, we measured the content of ergosterol in W303 wild type cells treated with statins, using, as a control, W303 rho° strain, which has a low ergosterol contentCitation17. Ergosterol content was measured in yeast cells treated with statins by gas chromatography-mass spectrometry (GC-MS) (Figure S4). The results revealed that ergosterol content was significantly lower in statin-treated cells, compared with the non-treated wild type strain. These results are in agreement with a previous study in Candida glabrata, demonstrates that simvastatin reduces ergosterol quantities in cellsCitation17.
3.2. The petite colonies induced by statin treatment contain rho° cells, devoid of mitochondrial DNA
To establish if statins act directly on the stability of mitochondrial DNA, because of low ergosterol content, we visualised mitochondrial DNA from petite colonies by DAPI staining. Results, reported in , showed that wild type cells contained mitochondrial DNA, W303 rho° cells had only nuclear DNA and wild type cells treated with statins showed a high percentage of cells devoid of mitochondrial DNA. The quantitative analysis of rho° cells generated by statins is reported in : histograms report the percentage of cells without mitochondrial DNA. The same experiment was performed in parallel with cultures grown in YPDE medium.
Figure 2. Statins treatments induced the loss of mitochondrial DNA and the ergosterol suppressed this phenotype. (a) DAPI staining of cells presented in Fig. 1; wild type W303, W303 rho° and wild type W303 treated with pravastatin 150 µg/ml, atorvastatin 100 µg/ml and rosuvastatin 50 µg/ml grown in YPD and YPDE media (YPD supplemented with 0,5mg/ml ergosterol). Bar: 2 µm. The magnification is the same in each picture. (b) The histogram shows the percentage of cells without mitochondrial DNA. An average of 300 cells were counted.
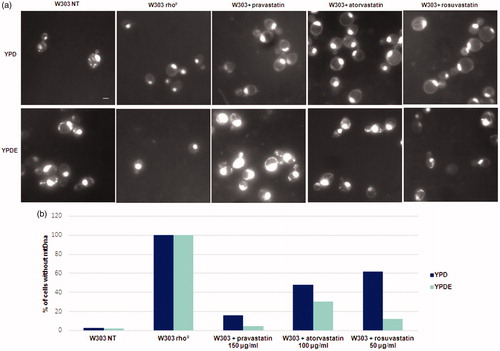
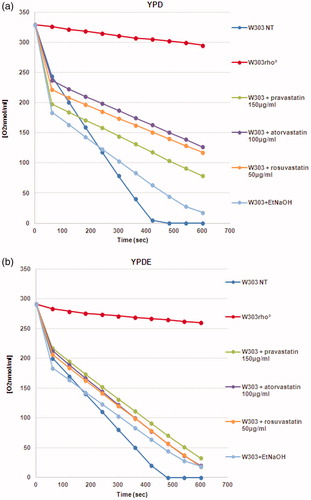
Results demonstrated that statins induced the loss of mitochondrial DNA and that the presence of ergosterol suppressed this phenotype; we also verified that the colonies with mitochondrial DNA were able to grow on glycerol medium (not shown). This result indicate that loss of mitochondrial DNA was correlated to low concentration of ergosterol in the mitochondrial membranes and that the exogenous addition of ergosterol was sufficient to stabilise the mitochondrial DNA, counteracting the statins effect. We then measured the oxygen consumption of cells treated with statins; reports that the respiration rate of wild type strain treated with statins is lower if compared with the non-treated wild type cells; when the same cultures were grown in YPD medium supplemented with ergosterol, the respiration rate was higher, indicating that the presence of ergosterol compensate for the effect of statins (). This result suggests an impairment of the respiratory chain function when cells were treated with statins (see Discussion).
Figure 3. The oxygen consumption rate of yeast cells treated with statins is low compared to the wild type strain. (a) Oxygen consumption of W303, W303 rho° and W303 cells treated with statins in YPD medium showed low oxygen consumption in cells treated with statins; (b) The respiration rate is higher when the same cultures were grown in YPD medium supplemented with ergosterol. The oxygen consumption rate is expressed as O2 nmol/ml, and the respiration rates was normalized for cell number.
3.3. Statins treatment resulted in mitochondrial morphology defects
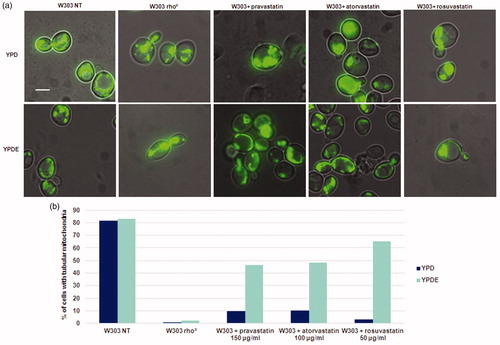
In yeast, cells lacking mitochondrial DNA or a conditional mutant (ERG27) of the ergosterol pathway show defects in mitochondrial morphologyCitation19 and a down regulation of the ergosterol genes results in mitochondrial membrane alterationCitation29,Citation30. We therefore analysed mitochondrial morphology after statins treatment (). Accordingly, wild type strain transformed with a vector expressing the mitoGFP for green fluorescent labelling of mitochondria in living cells, and mitochondrial morphology was assessed upon treatment with statins at exponential phase, in glucose-containing medium with or without ergosterol. By fluorescence microscopy, we observed a relevant percentage of aggregated vs tubular mitochondria in wild type strain treated with statins. This phenotype is reminiscent of mutants lacking mitochondrial DNA, or of W303 rho° strainCitation19. The addition of ergosterol partially suppressed the aggregated mitochondrial morphology phenotype, partially restoring a tubular mitochondrial structure (). By staining cells with DAPI, we were able to verify the presence of mitochondrial DNA in the strains expressing mitoGFP, treated with statins, and supplemented with ergosterol (Supplementary Figure 5).
Figure 4. Statins treatments in S. cerevisiae results in an aberrant mitochondrial morphology phenotype, similar to that of rho° cells and this phenotype is suppressed by ergosterol. (a) W303 was treated with pravastatin 150 µg/ml, atorvastatin 100µg/ml and rosuvastatin 50 µg/ml grown in YPD and YPDE media in exponential growth phase; mitochondria were visualized with mitoGFP. (b) The histogram shows the percentage of cells with tubular mitochondria. As a control, W303 rho° strain was used. Bar: 2 µm. The magnification is the same in each picture.
3.4. Statins induced a cellular re-localisation of Erg27, a sensor of mitochondrial dysfunction
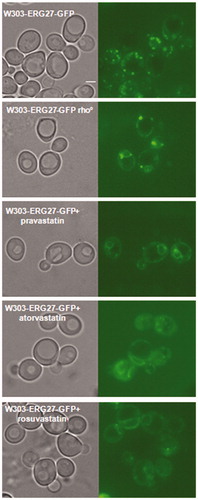
Erg27 is an essential enzyme of the ergosterol biosynthetic pathway that plays as a sensor of mitochondrial function: in wild type cells the enzyme is mainly found in Lipid Droplets (LDs), while in cells with a block of respiration or in rho° cells, it moves to the nuclear and cortical Endoplasmic Reticulum (ER)Citation19. To assess a possible mitochondrial defect, we determined the cellular localisation of Erg27 following statins treatment. Wild type W303 and W303 rho° strains, harbouring the ERG27-GFP fusion integrated in the genome, were treated with statins and analysed by fluorescence microscopy. Results reported in showed that in wild type cells, Erg27-GFP decorated lipid droplets, while in cells treated with statins, Erg27-GFP re-localised in nuclear and cortical ER, as in W303 rho° cells. The re-localisation of Erg27-GFP demonstrated that statins, similarly to a block of respiration, induced the movement of Erg27 from lipid droplets to the endoplasmic reticulum.
Figure 5. Statins treatments induced the re-localization of Erg27, a sensor of mitochondrial state, from lipid droplets to nuclear and cortical ER. The Erg27 localization in wild type cells is predominantly in lipid droplets, while in rho° cells the enzyme is mainly in nuclear and cortical endoplasmic reticulum. Wild type W303 with the ERG27-GFP fusion integrated in the genome was visualized at fluorescence microscopy after statins treatments, pravastatin 150µg/ml, atorvastatin 100µg/ml and rosuvastatin 50µg/ml in exponential growth phase in YPD medium. Bar: 2 µm. The magnification is the same in each picture.
3.5. Effect of statins on the respiratory obligate yeast Kluyveromyces lactis
The yeast Kluyveromyces lactis has a metabolism similar to that of human cells and the regulation between fermentative and respiratory metabolism depends on oxygen availability; moreover, in K. lactis, as in human cells, cells cannot survive without mitochondrial DNACitation31. We analysed the effect of statins on K. lactis growth, in order to enlighten the effects of statins on respiration in a yeast that could mimic the human cell behaviour. The K. lactis Δpda1, a mutant strain lacking the pyruvate dehydrogenase E1α subunitCitation19, was used to mimic the growth behaviour of a S. cerevisiae rho° strain. Statins treatments resulted in a slight delay in growth, intermediate between the untreated wild type and the Δpda1 mutant strain with a defective respiration (Supplementary Figure 6). This experiment demonstrated that statins are effective also in a yeast unable to lose spontaneously mitochondrial DNA, as for human cells.
3.6. Effect of statins in a yeast model of human MELAS mitochondrial disease
Statins are not recommended for patients affected by mitochondrial diseasesCitation32 and patients with Mitochondrial Encephalomyopathy, Lactic Acidosis and Stroke-like episodes (MELAS) experienced myotoxicityCitation33,Citation34. A yeast model for MELAS mitochondrial disease exists: the strain C25T containing the C25T mutation in the mitochondrial tRNALeu (UUR), is the yeast counterpart of the human mitochondrial C3256T mutation in the tRNALeu (UUR) gene, identified in MELAS patientsCitation35. The C25T mitochondrial mutation induces a reduced growth on a respiratory carbon source and shows mitochondrial morphology defectsCitation36. In order to verify if the effect of statins treatment could be additive on the yeast MELAS phenotypes, we followed the growth of C25T cells with statins (Supplementary Figure 7). The treatment with statins causes a mild effect on growth in exponential phase, intermediate between rho° cells and the non-treated mutant. Of note, pravastatin seems to exert a greater effect, inducing a lag phase in growth. This experiment showed that statins exacerbated the mitochondrial dysfunction of a yeast strain with impaired mitochondrial function.
3.7. In vitro effects of statins on C2C12 murine cells
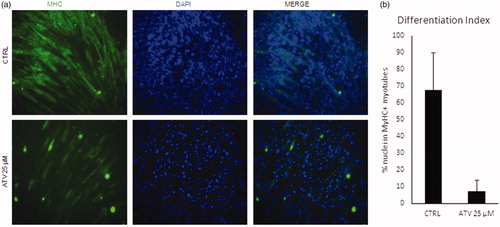
In light of the evidence that statins assumption is often associated with muscle-related side effects, such as fatigue, muscle pain and muscle weakness, we sought to verify the effect of statins on a well-established in vitro model of myogenesis, the C2C12 murine myoblasts cell lineCitation37. To this end, we assessed the differentiation capacity of C2C12 upon atorvastatin (25 µM) treatment, as compared to untreated cells (Ctrl) by immunofluorescence for Myosin heavy chain, a marker of terminal myogenic differentiation. As shown in , atorvastatin dramatically reduced C2C12 myogenic differentiation (), as quantified evaluating differentiation index (). To verify if the observed differentiation defects were also accompanied with mitochondrial dysfunction, we measured the mitochondrial DNA content by qRT-PCR (see Methods). The threshold of amplification between mtDNA (mt-CO1 gene) and nuclear DNA (using ACTB as reference) was measured; the average of ratio of treated/non-treated was 0.96 ± 0.015 (mean ± standard deviation) (mean of n = 4 replicates), indicated that atorvastatin induced a dose-dependent reduction of mitochondrial DNA, in agreement with data obtained in yeast cells.
Figure 6. Statins block C2C12 differentiation and induce loss of mtDNA. (a) C2C12 were treated for 48 with 25uM atorvastatin (ATV) in growth medium and then induced to differentiate for further 48h either. Non-treated cells were used as a control (CTRL). Representative images of immunofluorescence for Myosin Heavy Chain (MyHC, green) are shown. Nuclei were counterstained with DAPI (blue). (b) Graph shows the quantification of the percentage of nuclei inside MyHC positive myotubes (Differentiation index). Data are presented as the average of n=3 replicates +/- SD. Significance has been assessed by t-test (*p < 0.05).
4. Discussion
Statin intolerance is often a consequence of statin-associated myopathyCitation38,Citation39; hence, it is fundamental to understand the molecular effect of statin in the cells. Yeast is recognised as a good model to study the cellular mechanisms of mitochondrial human diseases and S. cerevisiae is unique among yeasts because it is able to survive with non-functional mitochondriaCitation17. We confirmed here that statins act as cholesterol lowering drugs in S. cerevisiae as was previously publishedCitation14; mass spectrometry analysis revealed a lower ergosterol content in statins treated cells than the wild type strain (Supplementary Figure 1); in yeast cells treated with statins, the Erg27 enzyme localised in the endoplasmic reticulum indicating low level of ergosterol, because in cells with normal ergosterol content, the enzyme is localised in lipid dropletsCitation19 (. Of note, statin treatments resulted in W303 growth curves rates intermediate between the non-treated strain (W303) and the strain devoid of mitochondrial DNA (W303 rho°), while the supply of ergosterol in the medium restores a wild type growth in statin treated cells and in W303 rho° (Supplementary Figure 3(B)). Also, the growth curves of the yeast K. lactis, which has a metabolism more similar to human cells, revealed that statins slightly impair the growth, as in S. cerevisiae (Supplementary Figure 6). Finally, we treated with statins a yeast model of human MELAS diseaseCitation18. Statins are not administered to MELAS patients because they precipitate myopathic symptomsCitation40; indeed, we found an effect on C25T strain growth, indicating an additive negative affect of stations if associated with a MELAS mutation (Supplementary Figure 7).
Taking together these results indicate that S. cerevisiae is a suitable model to investigate the mitochondrial effects of statins and also to enlighten the regulation of ergosterol pathway based on the dynamic localisation of Erg enzymes in the cell. Another case of relocalization of an enzyme of the ergosterol biosynthetic pathway was previously described: Erg1 shows a dual localisation both in the ER and LDs, but it is redistributed from LDs to ER in low iron condition in the cellsCitation41. The cross talk between the mitochondrial respiration and the ergosterol biosynthetic pathway is still poorly understoodCitation19.
Statins induce a petite phenotype ( and Supplementary Figure 2), caused by the loss of mitochondrial DNA (); S. cerevisiae is able to survive without mitochondrial DNA (rho° cells) but can also maintain rearrangements of mitochondrial DNA (rho− cells), induced by chemical agents or by genetic mutationsCitation42. The lack of mitochondrial DNA in statin treated cells, suggested an effect of statins in the stability of mitochondrial DNA and not in mitochondrial DNA transcription/replication/recombination. The percentage of treated cells showing cells devoid of mitochondrial DNA () fits well with the percentage of petite colonies ().
The supplementation of ergosterol in the growth medium was able to suppress the mitochondrial DNA loss ( and ), demonstrating that a correct amount of ergosterol is essential for mitochondrial function.
To date, a demonstration of how mitochondrial DNA molecules are attached to the inner mitochondrial membrane is still lacking, but lipid rafts, membrane microdomains enriched in cholesterol and sphingolipidsCitation43 are essential to maintain mitochondrial DNA in human cellsCitation44. Furthermore, the maintenance of mitochondrial nucleoids is coordinated with the mitochondrial fusion and fission machinery to ensure the distribution of mitochondrial DNA in the cellsCitation45; of note, the mitochondrial protein ATAD3, which binds cholesterol in the inner mitochondrial membrane, has been proposed to have a central role in nucleoid organisationCitation46–49.
The statins investigated in this work showed different behaviours: pravastatin produced less petites, it is less toxic, but the ergosterol content is comparable to other statins, and the respiration is more similar to the wild type strain. Rosuvastatin produced the higher percentage of petites and it is more toxic, and the respiration is similar to rho° cells. Atorvastatin is intermediate between pravastatin and rosuvastatin. The same behaviour is observed when K. lactis was treated with the statins.
An extensive literature reports defects in respiration in mammalian cells treated with statins, by measuring the activity of respiratory chain complexesCitation50. We hypothesise that the mitochondrial defects described in literature can be reconciled/explained by the low levels of mitochondrial DNA copy number, that imbalance the ratio between mitochondrial and nuclear-encoded subunits of the respiratory complexes, resulting in an altered respiration rate. Indeed, yeast cells treated with statins showed a respiration rate lower than the wild type strain but, when the same cultures were grown in YPD medium were supplemented with ergosterol, the respiration rate was higher (. Of note, the complex II is the only to be entirely nuclear encoded (i.e. this complex does not contain mitochondrial encoded subunits); indeed, the complex II resulted unaffected by simvastatin treatment on skeletal muscleCitation51,Citation52; on the contrary, the other respiratory complexes contain nuclear and mitochondrial- encoded subunits are affected by statinsCitation42; we hypothesise that a reduced level of mitochondrial DNA could results in an imbalance of the nuclear and mitochondrial encoded subunits, causing an altered respiration rate.
Of note, skeletal muscle biopsies from patients diagnosed with myopathies induced by statins, were used to quantify mitochondrial DNA relative to nuclear DNA (mitochondrial DNA content) by qPCR, and it was found that skeletal muscle mitochondrial DNA is decreased in patients with statin-induced myopathyCitation53,Citation54. In agreement with this, we observed a small but significant reduction of mitochondrial DNA in C2C12 myoblasts after statin treatment. This reduction could explain why muscle cells are more sensitive to statins than other cells, resulting in muscle-related side effects. Indeed, mitophagy has been reported to be essential for proper muscle differentiationCitation55 and during muscle differentiation, 4 times more DNA and 8 times more respiratory complexes are needed compared to other cellsCitation56. Our results support the notion that the amount of mitochondrial DNA is critical in muscle differentiation because after degradation of mitochondria, new mitochondrial DNA synthesis and new membranes are needed to maintain functional myofibers. The statins-induced reduction of the amount of cholesterol necessary to maintain mitochondrial DNA, and for membrane construction, might be the reason why chronic statins treatment induces muscle-related side effects. Accordingly, in presence of statins the differentiation from myoblast to myotubes is dramatically reduced (.
In conclusion, we have used S. cerevisiae as a model to study the mechanism of statin side effects on mitochondria, and we confirm that a relevant effect of low cholesterol level induced by statins is on mitochondrial DNA stability, explaining why muscle cells are more prone to statins side effects.
Supplemental Material
Download PDF (793.5 KB)Acknowledgements
We thank Silvia Francisci and Arianna Montanari for providing the C25T strain and Dr. Luigi Gatta and Dr. Andrea Morelli for scientific discussion. T.R. thanks Laura Frontali for constant support on mitochondrial research and for sharing the invaluable experience on this field.
Disclosure statement
The authors report no declarations of interest.
Additional information
Funding
Related Research Data
References
- Buhaescu I, Izzedine H. Mevalonate pathway: a review of clinical and therapeutical implications. Clin Biochem 2007;40:575–84.
- Friesen JA, Rodwell VW. The 3-hydroxy-3-methylglutaryl coenzyme-A (HMG-CoA) reductases. Genome Biol 2004;5:248.
- Burg JS, Espenshade PJ. Regulation of HMG-CoA reductase in mammals and yeast. Prog Lipid Res 2011;50:403–10.
- Istvan ES. Structural mechanism for statin inhibition of 3-hydroxy-3-methylglutaryl coenzyme A reductase. Am Heart J 2002;144:S27–S32.
- Golomb BA, Evans MA. Statin adverse effects. Am J Cardiovasc Drugs 2008;8:373–418.
- Tomaszewski M, Stępień KM, Tomaszewska J, Czuczwar SJ. Statin-induced myopathies. Pharmacol Rep 2011;63:859–66.
- du Souich P, Roederer G, Dufour R. Myotoxicity of statins: mechanism of action. Pharmacol Ther 2017;175:1–16.
- Matsubara A, Oda S, Akai S, et al. Establishment of a drug-induced rhabdomyolysis mouse model by co-administration of ciprofloxacin and atorvastatin. Toxicol Lett 2018;291:184–93.
- Knauer MJ, Urquhart BL, Meyer zu Schwabedissen HE, et al. Human skeletal muscle drug transporters determine local exposure and toxicity of statins. Circ Res 2010;106:297–306.
- Tournadre A. Statins, myalgia, and rhabdomyolysis. Joint Bone Spine 2019;1–6.
- Kaufmann P, Török M, Zahno A, et al. Toxicity of statins on rat skeletal muscle mitochondria. Cell Mol Life Sci 2006;63:2415–25.
- Apostolopoulou M, Corsini A, Roden M. The role of mitochondria in statin‐induced myopathy. Eur J Clin Invest 2015;45:745–54.
- Bolotin‐Fukuhara M, Fairhead C. Candida glabrata: a deadly companion? Yeast 2014;31:279–88.
- Lorenz RT, Parks LW. Effects of lovastatin (mevinolin) on sterol levels and on activity of azoles in Saccharomyces cerevisiae. Antimicrob Agents Chemother 1990;34:1660–5.
- Westermeyer C, Macreadie IG. Simvastatin reduces ergosterol levels, inhibits growth and causes loss of mtDNA in Candida glabrata. FEMS Yeast Res 2007;7:436–41.
- Barrientos A. Yeast models of human mitochondrial diseases. IUBMB Life 2008;55:83–95.
- Rinaldi T, Dallabona C, Ferrero I, et al. Mitochondrial diseases and the role of the yeast models. FEMS Yeast Res 2010;10:1006–22.
- Francisci S, Montanari A. Mitochondrial diseases: yeast as a model for the study of suppressors. Biochim Biophys Acta Mol Cell Res 2017;1864:666–73.
- Cirigliano A, Macone A, Bianchi MM, et al. Ergosterol reduction impairs mitochondrial DNA maintenance in S. cerevisiae. Biochim Biophys Acta Mol Cell Res 2019;1864:290–303.
- Ottaviano D, Montanari A, De Angelis L, et al. Unsaturated fatty acids-dependent linkage between respiration and fermentation revealed by deletion of hypoxic regulatory KlMGA2 gene in the facultative anaerobe-respiratory yeast Kluyveromyces lactis. FEMS Yeast Res 2015; 15:fov028.
- De Luca C, Zhou Y, Montanari A, et al. Can yeast be used to study mitochondrial diseases? Biolistic tRNA mutants for the analysis of mechanisms and suppressors. Mitochondrion 2009;9:408–17.
- De Angelis L, Rinaldi T, Cirigliano A, et al. Functional roles of the fatty acid desaturases encoded by KlOLE1, FAD2 and FAD3 in the yeast Kluyveromyces lactis. Microbiology 2016;162:1435–45.
- Kang C, Goodman CA, Hornberger TA, Ji LL. PGC-1a overexpression by in vivo transfection attenuates mitochondrial deterioration of skeletal muscle caused by immobilization. FASEB J. 2015;29(10):4092–106.
- Bard M, Lees ND, Burnett AS, Parker RA. Isolation and characterization of mevinolin resistant mutants of Saccharomyces cerevisiae. Microbiology 1988;134:1071–8.
- Fowler DM, Cooper SJ, Stephany JJ, et al. Suppression of statin effectiveness by copper and zinc in yeast and human cells. Mol BioSyst 2011;7:533–44.
- Maciejak A, Leszczynska A, Warchol I, et al. The effects of statins on the mevalonic acid pathway in recombinant yeast strains expressing human HMG-CoA reductase. BMC Biotechnol 2013;13:68.
- Davies BS, Rine J. A role for sterol levels in oxygen sensing in Saccharomyces cerevisiae. Genetics 2006;174:191–201.
- Serratore ND, Baker KM, Macadlo LA, et al. A novel sterol-signaling pathway governs azole antifungal drug resistance and hypoxic gene repression in Saccharomyces cerevisiae. Genetics 2018;208:1037–55.
- Dimmer KS, Fritz S, Fuchs F, et al. Genetic basis of mitochondrial function and morphology in Saccharomyces cerevisiae. Mol Biol Cell 2002;13:847–53.
- Altmann K, Westermann B. Role of essential genes in mitochondrial morphogenesis in Saccharomyces cerevisiae. Mol Biol Cell 2005;16:5410–7.
- Chen XJ, Clark-Walker GD. Mutations in MGI genes convert Kluyveromyces lactis into a petite-positive yeast. Genetics 1993;133:517–25.
- Bianchi MM, Brambilla L, Protani F, et al. Efficient homolactic fermentation by Kluyveromyces lactis strains defective in pyruvate utilization and transformed with the heterologous LDH gene. Appl Environ Microbiol 2001;67:5621–5.
- Ramachandran R, Wierzbicki A. Statins, muscle disease and mitochondria. J Clin Med 2017;6:75.
- Tay SK, DiMauro S, Pang AY, et al. Myotoxicity of lipid-lowering agents in a teenager with MELAS mutation. Pediatr Neurol 2008;39:426–8.
- Vladutiu GD, Simmons Z, Isackson PJ, et al. Genetic risk factors associated with lipid‐lowering drug‐induced myopathies. Muscle Nerve 2006;34:153–62.
- Feuermann M, Francisci S, Rinaldi T, et al. The yeast counterparts of human ‘MELAS’mutations cause mitochondrial dysfunction that can be rescued by overexpression of the mitochondrial translation factor EF‐Tu. EMBO Rep 2003;4:53–8.
- Montanari A, Besagni C, De Luca C, et al. Yeast as a model of human mitochondrial tRNA base substitutions: investigation of the molecular basis of respiratory defects. RNA 2007;14:275–83.
- Blau HM, Pavlath GK, Hardeman EC, et al. Plasticity of the differentiated state. Science 1985;230:758–66.
- Katz DH, Intwala SS, Stone NJ. Addressing statin adverse effects in the clinic: the 5 Ms. J Cardiovasc Pharmacol Ther 2014;19:533–42.
- Sirvent P, Fabre O, Bordenave S, et al. Muscle mitochondrial metabolism and calcium signaling impairment in patients treated with statins. Toxicol Appl Pharmacol 2012;259:263–8.
- Shakoury-Elizeh M, Protchenko O, Berger A, et al. Metabolic response to iron deficiency in Saccharomyces cerevisiae. J Biol Chem 2010;285:14823–33.
- Chariot P, Abadia R, Agnus D, et al. Simvastatin-induced rhabdomyolysis followed by a MELAS syndrome. Am J Med 1993;94:109.
- Ferguson LR, Von Borstel RC. Induction of the cytoplasmic ‘petite’mutation by chemical and physical agents in Saccharomyces cerevisiae. Mutat Res 1992;265:103–48.
- Sezgin E, Levental I, Mayor S, Eggeling C. The mystery of membrane organization: composition, regulation and roles of lipid rafts. Nat Rev Mol Cell Biol 2017;18:361.
- Gerhold JM, Cansiz-Arda Ş, Lõhmus M, et al. Human mitochondrial DNA-protein complexes attach to a cholesterol-rich membrane structure. Sci Rep 2015;5:15292.
- Murley A, Lackner LL, Osman C, et al. ER-associated mitochondrial division links the distribution of mitochondria and mitochondrial DNA in yeast. Elife 2013;2:e00422.
- He J, Cooper HM, Reyes A, et al. Mitochondrial nucleoid interacting proteins support mitochondrial protein synthesis. Nucleic Acids Res 2012;40:6109–21.
- Gilkerson R, Bravo L, Garcia I, et al. The mitochondrial nucleoid: integrating mitochondrial DNA into cellular homeostasis. Cold Spring Harb Perspect Biol 2013;5:a011080.
- Peralta S, Goffart S, Williams SL, et al. ATAD3 controls mitochondrial cristae structure in mouse muscle, influencing mtDNA replication and cholesterol levels. J Cell Sci 2018;131:jcs217075.
- De Pinieux G, Chariot P, Ammi‐Said M, et al. Lipid‐lowering drugs and mitochondrial function: effects of HMG‐CoA reductase inhibitors on serum ubiquinone and blood lactate/pyruvate ratio. Br J Clin Pharmacol 2003;42:333–7.
- Nadanaciva S, Dykens JA, Bernal A, et al. Mitochondrial impairment by PPAR agonists and statins identified via immunocaptured OXPHOS complex activities and respiration. Toxicol Appl Pharmacol 2007;223:277–87.
- Sirvent P, Bordenave S, Vermaelen M, et al. Simvastatin induces impairment in skeletal muscle while heart is protected. Biochem Biophys Res Commun 2005;338:1426–34.
- Larsen S, Stride N, Hey-Mogensen M, et al. Simvastatin effects on skeletal muscle: relation to decreased mitochondrial function and glucose intolerance. J Am Coll Cardiol 2013;61:44–53.
- Schick BA, Laaksonen R, Frohlich JJ, et al. Decreased skeletal muscle mitochondrial DNA in patients treated with high‐dose simvastatin. Clin Pharmacol Ther 2007;81:650–3.
- Stringer HA, Sohi GK, Maguire JA, Côté HC. Decreased skeletal muscle mitochondrial DNA in patients with statin-induced myopathy. J Neurol Sci 2013;325:142–7.
- Sin J, Andres AM, Taylor DJ, et al. Mitophagy is required for mitochondrial biogenesis and myogenic differentiation of C2C12 myoblasts. Autophagy 2016;12:369–80.