
Abstract
Overexpression of protein tyrosine phosphatase 1B (PTP1B) induces insulin resistance in various basic and clinical research. In our previous work, a synthetic oleanolic acid (OA) derivative C10a with PTP1B inhibitory activity has been reported. However, C10a has some pharmacological defects and cytotoxicity. Herein, a structure-based drug design approach was used based on the structure of C10a to elaborate the smaller tricyclic core. A series of tricyclic derivatives were synthesised and the compounds 15, 28 and 34 exhibited the most PTP1B enzymatic inhibitory potency. In the insulin-resistant human hepatoma HepG2 cells, compound 25 with the moderate PTP1B inhibition and preferable pharmaceutical properties can significantly increase insulin-stimulated glucose uptake and showed the insulin resistance ameliorating effect. Moreover, 25 showed the improved in vivo antihyperglycaemic potential in the nicotinamide–streptozotocin-induced T2D. Our study demonstrated that these tricyclic derivatives with improved molecular architectures and antihyperglycaemic activity could be developed in the treatment of T2D.
Graphical Abstract
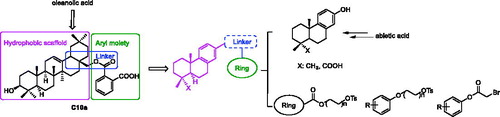
1. Introduction
The predominant pathophysiological factor of type 2 diabetes (T2D) is insulin resistanceCitation1–4. Though many anti-T2D drugs have emerged in the past decadeCitation5,Citation6, traditional anti-T2D agents such as repaglinide, metformin, dipeptidyl peptidase-IV inhibitors, α-glucosidase inhibitors and glucagon-like peptide-1 agonists, and thiazolidinediones are still the most efficacious oral drugs in the first-line monotherapy of T2DCitation7. It is undoubted that new insulin sensitisers will meet great needs of T2D patientsCitation8,Citation9.
Protein tyrosine phosphatase 1B (PTP1B) dephosphosphorylates the tyrosine-phosphorylated insulin receptor (IR) and the downstream insulin receptor substrate (IRS) to down regulate insulin transductionCitation10–15. PTP1B inhibitors could potentially improve insulin sensitivity and normalise glucose levels and therefore could be a promising therapeutic strategy in the T2D patients. Recent studies also identified the involvement of intra-islet PTP1B in the regulation of insulin release and reinforce the potential of PTP1B inhibitors for the treatment of beta-cell secretory failure in the pathogenesis of T2DCitation16,Citation17. Besides, PTP1B-mediated dephosphorylation has been implicated in the development of diabetesCitation18, cancerCitation19, hepatic fibrosisCitation20, bacterial infectionCitation21, rheumatoid arthritisCitation22 and hypertensionCitation23. Many PTP1B inhibitors have been reported, but the discovery of PTP1B inhibitors with superior cell permeability and in vivo potency is difficult and so far there is no PTP1B inhibitors entered III phase clinical trialCitation18,Citation24.
Hundreds of natural products have been isolated and identified as PTP1B inhibitors, and natural products with interesting structural diversity have potential to develop the new PTP1B inhibitorsCitation25–27. In our previous work, some oleanolic acid (OA) derivatives with modified A-ring, C-ring, and C17 moiety were designed and synthesizedCitation28–33. Within these OA derivatives, compound C10a () exhibited the most PTP1B inhibition (IC50: 3.12 μM), which was 7.6-fold more than the parent compound OACitation28. However, the triterpenoid derivative C10a has too large molecular weight (>500) and some pharmacological defects, such as weak cell permeability, poor bioavailability and improper lipid/water partition coefficient. C10a also showed the considerable cytotoxicity. Therefore, the structure of C10a needs to be optimised to develop the potent PTP1B inhibitors with favourable pharmacological properties.
Figure 1. The chemical structures of OA and lead compound C10a.
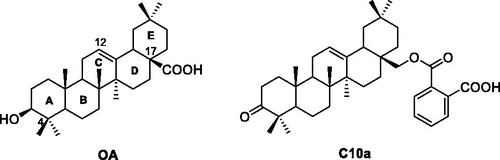
The structural optimisation strategy is shown in . The structure of C10a contains hydrophobic scaffold, linker and aryl moiety. As displayed in , the molecular docking has demonstrated the hydrophobic interactions between the terpenoid scaffold of C10a and the surrounding amino residues of PTP1B are critical for the complex stability, but the pentacyclic core of C10a is too complicated. We assumed this scaffold could be simplified to the smaller tricyclic fragment containing the same stereo-conformation of fused A/B ring junction, such as the tricyclic terpenoid scaffold of compound 15 as shown in . One of the methyl group at 4-position also was retained, because it was beneficial for interaction with Arg24Citation28, which is an important residue at the second site of PTP1B for substrate specificity (the second site of PTP1B is a noncatalytic cleft-like binding pocket, which is not conserved among all PTPs)Citation35. As shown in , C ring was replaced with the substituted benzene ring, which could provide opportunities to form more hydrophobic and π–π interactions. D ring and E ring were simplified to the linker from C ring to the aryl moiety. Insertion of polar group (e.g. carboxyl, ether) into this linker would be beneficial for the favourable balance between hydrophilicity and hydrophobicity. The overlapping figure of C10a and 15 indicated these compounds have similar docking modes with amino residues of PTP1B (). Only two hydrogen-bond interactions between C10a and PTP1B were observed (Tyr46 and Lys120), so the aryl moiety of C10a was replaced with various substituted rings in order to enhance inhibition, since the aryl moiety was important for the substrate recognitionCitation19.
Figure 2. The structural optimisation strategy.
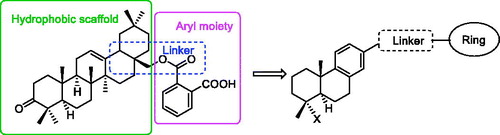
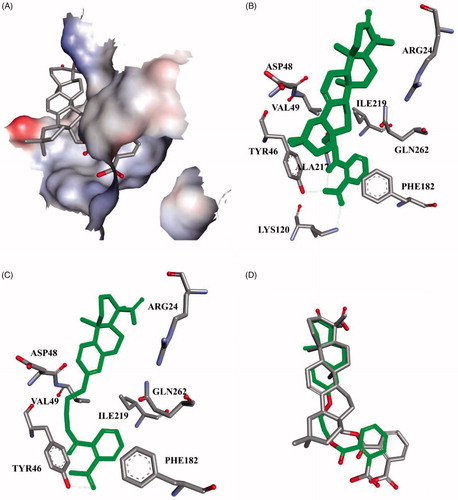
Figure 3. C10a and 15 docked in the PTP1B active site (PDB ID: 2B07Citation34). (A) Only the active site was shown, displaying the protein in surface representation and ligand C10a in stick representation; (B) C10a, Coloured green and displayed in stick representation, bound to these important residues in the interior of the active site. All hydrogen atoms are omitted for clarity; (C) 15 bound to the important residues in the interior of the active site; (D) The overlapping docking modes of C10a and 15.
2. Results and discussion
2.1. Chemistry
The synthesis procedure to achieve 15-hydroxydehydroabietic acid (3) from abietic acid (AA) involved addition, elimination, and oxidation. However, according to the literaturesCitation36–38, alcohol 3 was obtained in only 10% yield in our laboratory. We therefore improved the synthetic method and 3 was finally obtained in 70% overall yield (Scheme 1). According to the improved synthetic procedure, AA (1) was treated with 33% HBr/AcOH and the resulting 8, 15-dibromo derivative was heated in the presence of LiOH/DMF to afford diene (2), with four methyl groups of all singlets by 1HNMR. Oxidative rearrangement of 2 with SeO2 provided 15-hydroxydehydroabietate (3) in 80% yield. 3 was esterified by treatment with EtI (or BnBr) to give ester 4a (4 b). 4a was then reduced with LiAlH4 to give alcohol 5. We found 15-hydroxydehydroabietic derivatives are not suitable synthesis intermediates because of high steric hindrance of C-15 position, and thus hydroperoxide rearrangement of esters 4a–b with t-BuOOH and H2SO4/AcOH gave the 13-hydroxy-8,11,13-podocarpatriene derivatives (6a–b)Citation39. Finally, ester 6a was saponified with aq.NaOH to give acid 7.
Scheme 1. Synthesis of the compounds 2–7.
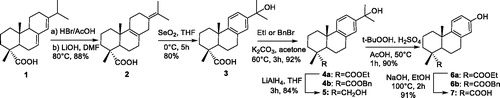
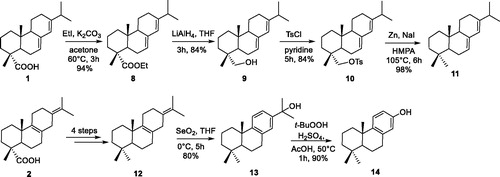
A couple of methyl groups at 4-position is most frequently found in the terpenoid scaffold. As shown in Scheme 2, the conversion of the COOH at 4-position to CH3 was also investigated. Acids 1 and 2 were firstly esterified by treatment with EtI and K2CO3 to give corresponding esters, which were subsequently reduced to afford alcohols. Conversion of alcohols to its tosylates in pyridine proceeded in satisfactory yield. Zn/NaI reductionCitation40 of abietane-tosylates afforded dienes 11–12 in satisfactory yield, respectively. Then, diene 12 was oxidative rearranged with SeO2 to obtain alcohol 13, which was converted to phenol 14 by hydroperoxide rearrangement.
Scheme 2. Synthesis of the compounds 8–14.
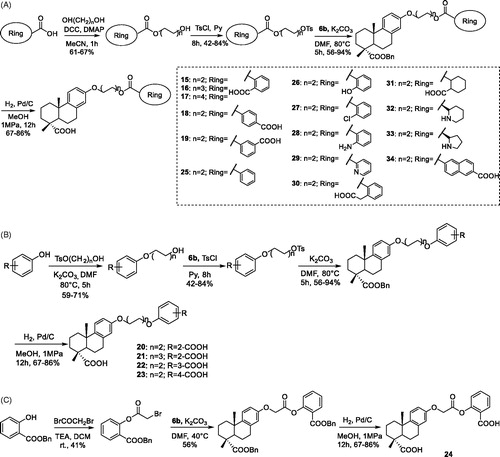
The final products 15–34 were synthesised via nucleophilic substitution of 6 b and corresponding tosylate or alkyl bromide, and subsequent deprotection of O-protected intermediates (Scheme 3(A–C)). As shown in Scheme 3(A), the tosylate intermediates of 15–19 and 25–34 were provided from corresponding alcohols, which were synthesised via condensation of the acids with different rings and diols with different length in the presence of DCC/DMAP. As shown in Scheme 3(B), the tosylate intermediates of 20–23 were provided from corresponding alcohols, which were synthesised via nucleophilic substitution. As shown in Scheme 3(C), the alkyl bromide intermediate of 24 was synthesised from the phenol and bromoacetyl bromide in alkaline condition.
Scheme 3. Synthesis of the compounds 15–34.
2.2. Enzymatic hPTP1B inhibitory activities and SAR analysis
As shown in , the PTP1B inhibitory activities of abietic derivatives with two types of terpenoid scaffolds (A and B) were firstly evaluated. Structure–activity relationship (SAR) was also discussed. The carboxyl group at 4-position was beneficial for PTP1B inhibition. Esterification of the carboxyl group to give corresponding ethyl ester (3 vs 4a), or reduction to the corresponding alcohol (1 vs 9, 3 vs 5) all resulted in loss of activity. Replacing the carboxyl group at 4-position with methyl group resulted in significant drop in activity (1 vs 11). Besides, transformation of hydroxy isopropyl group at 13-position of 8,11,13-podocarpatriene derivatives (scaffold B) to hydroxy group (3 vs 7, 13 vs 14) resulted in loss of activity.
Table 1. The PTP1B inhibitory activities of abietic derivatives 1–14.
Among tricyclic derivatives 15–34, the compounds 15 (2.9 μM), 28 (4.8 μM) and 34 (8.2 μM) exhibited the most PTP1B enzymatic inhibitory activities, which were equal to that of the lead compound C10a (3.1 μM), and they have smaller molecular weight (). SAR analysis demonstrated that the derivatives with different linker between terpenoid scaffold and ring moiety displayed different PTP1B inhibition activity. The inhibitory activity of PTP1B decreased along with an increase of the length of linker (15 > 16 > 17). Also, the structure of the linker influenced PTP1B inhibition. Compared with 20 and 24, 15 displayed higher PTP1B inhibitory activity. Besides, the derivatives containing a benzene or naphthalene at the ring moiety (15, 18–19, 25–28 and 34) showed higher inhibitory activity relative to the analogues bearing the heteroaromatic ring (29) or alicyclic ring (31–33). At the ring moiety, a clear preference was identified for the polar groups, with an increase in activity for the carboxyl group (15 and 34) and amino group (28), whereas substitution with carboxylmethyl group (30) led to significant loss of activity. The benzene ring with a carboxyl group at the o-position (15) increased the potency relative to those at the p-position (18) or m-position (19). Overall, our data demonstrated these compounds with tricyclic diterpene moiety are potent PTP1B inhibitors.
Table 2. The PTP1B inhibitory activities of tricyclic derivatives 15–34.
2.3. Cellular glucose uptake in insulin-resistant HepG2 cells
To assess the insulin sensitisation, the cellular effect of these compounds on insulin-resistant human hepatoma HepG2 cells was performed. The concentration of insulin and induction time were screened to induce insulin resistance in HepG2 cells according to the reported methodCitation41,Citation42 with some modification. As shown in , treatment with above 1 × 10−5 mmol of insulin reduced glucose uptake, whereas treatment with 6 × 10−6 mmol of insulin enhanced glucose uptake. Insulin resistance can be obviously observed when the HepG2 cells were treatment with 1.5 × 10−5 mmol of insulin, and an insulin sensitiser (rosiglitazone) can significantly decrease the insulin resistance and improve the glucose uptake. Treatment with higher concentration (>2 × 10−5 mmol) of insulin inhibited cell growth, and microscopic examination revealed atrophy of the cells. As shown in , insulin resistance was induced in HepG2 cells at 36 h, and rosiglitazone can decrease the insulin resistance. The cell growth was accelerated after 36 h and insulin resistance was decreased along with induction time. Therefore, the cells were treated with 1.5 × 1 0 −5 mmol of insulin for 36 h to induce the insulin resistance and produce the most obvious difference of glucose consumption between insulin resistance group (M group) and rosiglitazone group (Y group).
Figure 4. The effect of the compounds on glucose consumption in insulin-resistant HepG2 cells. (A) The HepG2 cells were induced with different concentrations of insulin; (B) The HepG2 cells were induced for different induction times; (C) HepG2 cells were induced with 1.5 × 10−5 mmol of insulin for 36 h to afford insulin-resistant cells, and then treated with rosiglitazone (Y group) or the compounds for 24 h. After incubation, glucose content in the culture medium was measured by glucose oxidase method. M group: insulin-resistant HepG2 cells without rosiglitazone and the compounds; K group: HepG2 cells without induction by insulin.
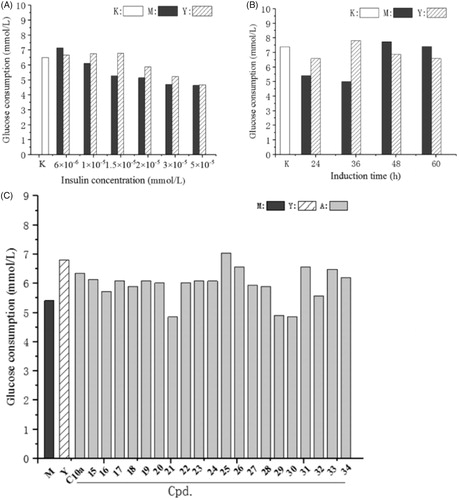
As shown in , most compounds can enhance the glucose consumption in insulin-resistant HepG2 cells, and the insulin stimulatory effects on glucose uptake were similar to that of C10a, but inferior to that of rosiglitazone. Cell viability was evaluated with the MTT assay and the result demonstrated that the cytotoxicities of most compounds were acceptable, while C10a showed the considerable cytotoxicity (see Figure 1S, Supplementary material). Compared with 15, which exhibited the most PTP1B enzymatic inhibitory potency, compound 25 has lower PTP1B inhibition (26.2 μM), but it can more significantly increase insulin-stimulated glucose uptake in cellular assay. This result was unsurprising, because most active site-directed PTP1B inhibitors are phosphotyrosine mimetics with weak cell permeabilityCitation43. We have previously reported that there is a trade-off between enzymatic inhibition and cell permeabilityCitation18. We calculated the pharmaceutical properties of 15, 25 and C10a using the Swiss ADME. As shown in , the MW and cLogP value of C10a are too large, as well as the HB acceptors of 15. Both of these compounds could not fulfil Lipinski RuleCitation44. 25 fulfilled Lipinski Rule of Five and has the lowest TPSA, which means more acceptable oral absorption and cell permeability. These results demonstrated in silico preferable pharmaceutical properties of 25 were beneficial for its cellular effect.
Table 3. Pharmaceutical properties of 15, 25 and C10a.
2.4. The antihyperglycaemic effect in NIDDM mice
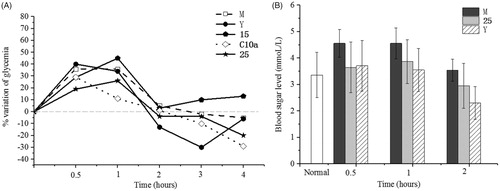
The antihyperglycaemic effect of the compounds 15, 25 and C10a were evaluated on non-insulin dependent diabetes mellitus (NIDDM) mice. Nicotinamide and streptozotocin were administrated intraperitoneally to induce hyperglycaemia and the plasma glucose of mice raised over 4.5 mmol/LCitation45. Rosiglitazone has not shown effect during the first hour, but it showed the significant antihyperglycaemic effect after 1 h (). Though C10a showed the antihyperglycaemic effect, some mice treated with C10a appeared the hydroabdomen, thrombus and tail necrosis, suggesting the acute toxicity of C10a. Compared with 15, 25 showed in vivo antihyperglycaemic effect, correlating with insulin-resistant cell assay.
Figure 5. The antihyperglycaemic effect in NIDDM mice (n = 8). (A) Diminution of plasmatic glucose concentration over NIDDM mice treated with 15, 25, C10a and rosiglitazone, respectively; (B) The acute antidiabetic effect of 25 were observed with respect to positive control. M group: NIDDM mice were treated with saline alone. Y group: NIDDM mice were treated with rosiglitazone.
3. Conclusions
In this article, a series of tricyclic derivatives containing various hydrophobic scaffold, linker and aryl moiety were designed and synthesised from 13-hydroxy-8,11,13-podocarpatriene, and their inhibitory activity against the PTP1B enzyme was assessed. Among these compounds, some tricyclic derivatives were described as potent inhibitors of PTP1B. Compound 25 showed the most effect on glucose consumption in insulin-resistant HepG2 cells. Moreover, 25 showed the most antihyperglycemic effect in NIDDM mice. The therapeutic potential of PTP1B inhibition, as well as the insulin sensitising and in vivo antihyperglycemic effect of these compounds, made this discovery an important opportunity.
4. Experimental section
4.1. Chemistry
1H NMR spectra were recorded on a Bruker Avance 400 spectrometer. 13C NMR spectra were recorded on a Bruker DPX 300 spectrometer. TMS was the internal standard. Chemical shifts as δ values were recorded in ppm and coupling constants as J values were recorded in Hz. HRMS spectra were recorded on a micrOTOF-Q II 10203 spectrometer. TLC were performed on the HSGF254 silica gel and UV detection at 254 nm and 365 nm, and appropriate chromogenic agents.
4.1.1. Synthesis of abieta-8(9),13(15)-dien-18-oic acid (2)
To a solution of 1 (20.00 g, 66.40 mmol) in 64 ml of HOAc was added to 33% HBr/HOAc (64 ml). The mixture was stirred at rt for 6 h, and then was filtered and the filter cake was washed with HOAc, dried in vacuum overnight to give corresponding crude dibromo derivative. To a solution of the crude 8,15-dibromo derivative was added LiOH (2.64 g, 64.72 mmol) in 200 ml of DMF, and the mixture was stirred at 80 °C for 7 h. Solvent was removed under reduced pressure, and the pH was adjusted to 3 with HCl (aq) and concentrated to give a brown oil 2 without further purification.
4.1.2. General procedure 1: SeO2-mediated oxidative rearrangement
To a solution of 1 equiv. diene in 10 ml/mmol of dry THF was added 4 equiv. SeO2, and the mixture was stirred at 0 °C for 5 h under an atmosphere of Ar. The solid was removed by filtration, and the filtrate was extracted with EtOAc. Solvent was removed under reduced pressure, and the resulting residue was purified by flash column chromatography to give product.
15-Hydroxydehydroabieta-18-oic acid (3). Yellow oil; mp: 92–93 °C; 1H NMR (400 MHz, CDCl3) δ 7.24 (m, 2H), 7.18 (s, 1H), 2.96 (m, 1H), 2.43–2.00 (m, 3H), 1.92–1.85 (m, 2H), 1.85–1.77 (m, 2H),1.77–1.70 (s, 3H), 1.58 (s, 6H), 1.46 (s, 3H), 1.31 (s, 3H). 13C NMR (300 MHz, CDCl3): δ 184.5, 147.8, 145.9, 134.7, 124.9, 124.1, 121.9, 120.2, 72.4, 60.4, 47.3, 44.5, 37.8, 36.9, 36.7, 31.6, 30.1, 25.0, 18.5, 16.2. HRMS (AP-ESI) Calcd. For C20H28NaO3: 339.2038 [M + Na]+. Found: 339.2087.
15-Hydroxydehydroabietane (13). Yellow oil; 1H NMR (400 MHz, CDCl3) δ 7.80 (d, J = 8.2 Hz, 1H), 7.37 (d, J = 8.0 Hz, 1H), 7.22 (s, 1H), 5.32 (s, 1H), 3.85 (d, J = 9.3 Hz, 1H), 3.62 (d, J = 9.4 Hz, 1H), 2.93–2.73 (m, 2H), 2.48 (s, 3H), 1.71 (m, 2H), 1.60 (s, 3H), 1.58 (s, 3H), 1.51–1.30 (m, 6H), 1.20 (s, 3H), 0.91 (s, 3H). 13C NMR (300 MHz, CDCl3) δ 129.8, 127.9, 124.8, 124.2, 121.9, 77.6, 72.2, 43.5, 38.0, 37.3, 37.1, 35.0, 31.6, 29.9, 21.6, 18.8, 18.3, 17.1. HRMS (AP-ESI) Calcd. for C20H30NaO: 309.2195 [M + Na]+. Found: 309.2185.
4.1.3. General procedure 2: K2CO3-mediated esterification
To a solution of 1 equiv. abieta-18-oic acid in acetone was added 3 equiv. K2CO3, and the mixture was stirred for 15 min. The solution was dropwise added 2 equiv. EtI (for 4a and 8) or BnBr (for 4 b) and the mixture was stirred at 60 °C for 3 h. The mixture was extracted with EtOAc. Solvent was removed under reduced pressure, and the resulting residue was purified by flash column chromatography to give product.
Ethyl 15-hydroxydehydroabietate (4a). Yellow solid; mp: 51–52 °C; 1H NMR (400 MHz, CDCl3) δ 7.26 (s, 2H), 7.19 (s, 1H), 4.11–4.18 (m, 2H), 3.05–2.61 (m, 2H), 2.44–2.25 (m, 2H), 1.91–1.83 (m, 2H), 1.83–1.70 (m, 3H),1.31–1.27 (m, 5H), 1.26 (s, 3H), 1.24 (s, 3H). 13C NMR (300 MHz, CDCl3) δ 178.4, 147.8, 145.9, 134.6, 126.8, 124.8, 124.1, 121.9, 72.1, 60.4, 47.3, 44.6, 37.8, 36.9, 36.4, 31.5, 30.2, 29.6, 25.0, 23.9, 21.5, 18.5, 16.4, 14.1. HRMS (AP-ESI) Calcd. for C22H32KO3: 283.1988 [M + K]+. Found: 383.1933.
Benzyl 15-hydroxydehydroabietate (4 b). Yellow solid; mp: 112–113 °C; 1HNMR (400 MHz, CDCl3) δ 7.42–7.31 (m, 1H), 7.26–7.15 (m, 1H), 7.06–7.00 (m, 1H), 6.89 (s, 1H), 5.19 (dd, J = 12.4, 3.4 Hz, 2H), 5.10 (s, 1H), 5.07 (s, 1H), 2.91–2.75 (m, 1H), 2.36–2.25 (m, 1H), 1.86–1.61 (m, 2H), 1.33–1.31 (m, 1H), 1.29 (s, 1H), 1.24 (dd, J = 6.4, 3.8 Hz, 1H). 13C NMR (300 MHz, CDCl3) δ 177.9, 146.9, 145.7, 136.1, 134.7, 128.9, 127.6, 127.1, 124.9, 124.3, 123.0, 73.9, 67.0, 51.8, 43.8, 38.4, 37.6, 31.6, 30.0, 28.3, 24.7, 20.1, 19.7. HRMS (AP-ESI) Calcd. for C27H35O3: 407.2581 [M + H]+. Found: 407.2585.
4.1.4. General procedure 3: LiAlH4- participated reduction
To a suspension of 4 equiv. LiAlH4 in THF was added 1 equiv. ester in 10 ml/mmol of THF at 0 °C. The mixture was stirred at rt for 3 h, and then was quenched by addition of aq.NaOH. The resulting solid was filtered off. Solvent of the filtrate was removed under reduced pressure, and the resulting residue was purified by flash column chromatography to give product.
1-Hydroxylmethyl-15-hydroxydehydroabietate (5). Colourless oil; 1H NMR (400 MHz, d6-DMSO) δ 12.17 (br, 1H), 8.99 (br, 1H), 7.03 (d, J = 8.6 Hz, 1H), 6.53 (dd, J = 8.5, 2.5 Hz, 1H), 6.37 (d, J = 2.4 Hz, 1H), 4.12 (br, 1H), 3.17 (d, J = 2.0 Hz, 2H), 2.73–2.67 (m, 2H), 2.25 (d, J = 12.4 Hz, 1H), 2.00 (dd, J = 12.3, 1.6 Hz, 1H), 1.75–1.55 (m, 8H), 1.38–1.24 (m, 4H), 1.15 (s, 3H), 1.09 (s, 3H). 13C NMR (300 MHz, d6-DMSO) δ 179.9, 155.0, 140.5, 135.8, 125.5, 114.9, 113.6, 53.7, 49.0, 46.7, 45.3, 38.4, 36.7, 36.5, 30.0, 25.4, 21.6, 18.6, 16.7, 14.4. HRMS (AP-ESI) Calcd. for C20H31O2: 303.2246 [M + Na]+. Found: 303.2221.
1-Hydroxymethylabietane (9). Yellow solid; mp: 125–126 °C; 1HNMR (400 MHz, CDCl3) δ 5.81 (s, 1H), 5.49–5.42 (m, 1H), 4.25 (s, 1H), 3.54–3.45 (m, 2H), 2.37–1.92 (m, 6H), 1.91–1.77 (m, 4H), 1.67–1.52 (m, 5H), 1.53–1.40 (m, 4H), 1.34–1.26 (m, 6H), 1.26–1.20 (s, 3H), 1.20–1.15 (m, 6H), 1.09–0.98 (m, 8H), 0.98–0.86 (m, 11H), 0.81 (s, 4H). 13C NMR (300 MHz, CDCl3) δ 146.0, 145.9, 134.7, 124.9, 124.1, 121.9, 75.7, 72.4, 48.3, 44.5, 37.8, 36.9, 36.7, 31.6, 30.2, 25.0, 21.7, 19.5, 14.2. HRMS (AP-ESI) Calcd. for C20H33O: 289.2526 [M + Na]+. Found: 289.2531.
4.1.5. General procedure 4: t-BuOOH-mediated hydroperoxide rearrangement
To a solution of 1 equiv. ester 5 in 5 ml/mmol AcOH was added 3 equiv. t-BuOOH followed by addition of 0.1 equiv. H2SO4. The mixture was stirred at 50 °C for 1 h. The mixture was diluted with water and the aqueous phase was extracted with DCM. The organic phase was washed with Na2CO3 (aq), water and dried. Solvent was removed under reduced pressure, and the resulting residue was purified by flash column chromatography to furnish the product.
Ethyl 13-hydroxy-8,11,13-podocarpatriene-1-carboxylate (6a). White solid; mp: 96–97 °C; 1H NMR (400 MHz, CDCl3) δ 7.12 (d, J = 8.6 Hz, 1H), 6.66 (dd, J = 8.5, 2.6 Hz, 1H), 6.53 (d, J = 2.4 Hz, 1H), 5.25 (br, 1H), 4.35–3.96 (m, 2H), 2.85 (dd, J = 10.8, 5.2 Hz, 2H), 2.44–2.17 (m, 2H), 1.90–1.62 (m, 7H), 1.29–1.24 (m, 6H), 1.21 (s, 3H). 13C NMR (300 MHz, CDCl3) δ 178.8, 153.2, 142.0, 136.6, 125.5, 114.8, 113.1, 60.6, 47.4, 44.9, 38.1, 36.5, 30.1, 25.2, 23.6, 21.5, 18.6, 16.4, 14.2. ESI-MS: 303.1 [M + H]+.
Benzyl 13-hydroxy-8,11,13-podocarpatriene-1-carboxylate (6 b). White solid; mp: 102–103 °C; 1H NMR (400 MHz, CDCl3) δ 9.26 (s, H),7.44–7.31 (m, 6H), 7.27–7.16 (m, 2H), 2.88–2.78 (m, 2H), 2.35–2.20 (m, 2H), 1.85 –1.67 (m, 2H), 1.58 (s, 3H), 1.23 (d, J = 3.3 Hz, 3H). 13C NMR (300 MHz, CDCl3) δ 178.5, 153.1, 142.0, 136.6, 136.3, 128.5, 128.1, 128.1, 125.6, 114.9, 113.1, 66.4, 47.7, 45.0, 38.1, 36.4, 36.5, 30.0, 25.3, 21.5, 18.6, 16.6. HRMS (AP-ESI) Calcd. for C24H29O3: 365.2111 [M + H]+. Found: 365.2119.
13-Hydroxy-8,11,13-podocarpatriene (14). Yellow solid. mp: 145–146 °C; 1H NMR (400 MHz, CDCl3) δ 7.14 (d, J = 8.6 Hz, 1H), 6.64 (dd, J = 8.5, 2.7 Hz, 1H), 6.53 (d, J = 2.6 Hz, 1H), 4.77 (s, 1H), 3.02–2.65 (m, 2H), 1.88 (m, 1H)1.81–1.56 (m, 4H), 1.54–1.33 (m, 2H), 1.33–1.23 (m, 2H),1.19 (s, 3H), 0.97 (s, 3H), 0.95 (s, 3H). 13C NMR (300 MHz, CDCl3) δ 148.1, 138.1, 132.2, 120.9, 110.1, 108.2, 45.8, 37.0, 34.3, 32.6, 28.7, 28.6, 25.7, 20.2, 16.9, 14.6, 14.3. HRMS (AP-ESI) Calcd. for C17H25O: 245.1878 [M + H]+. Found: 245.1895.
4.1.6. Synthesis of 13-hydroxy-8,11,13-podocarpatriene-1-carboxylic acid (7)
To a solution of 6 (0.10 g, 0.33 mmol) in 4 ml EtOH/H2O (1:1) was added NaOH (13.1 mg, 0.99 mmol), and the mixture was stirred at 100 °C for 2 h. The mixture was cooled to rt and pH was adjusted to 6 with HCl(aq). The resulting solid was filtered off to give the product 7 (82.5 mg, 91%) as a white solid. mp: 101–102 °C; 1H NMR (400 MHz, d6-DMSO) δ 12.17 (br, 1H), 8.99 (br, 1H), 7.03 (d, J = 8.6 Hz, 1H), 6.51 (dd, J = 8.5, 2.5 Hz, 1H), 6.38 (d, J = 2.4 Hz, 1H), 2.82–2.60 (m, 1H), 2.25 (d, J = 12.4 Hz, 1H), 2.00 (dd, J = 12.3, 1.6 Hz, 1H), 1.78–1.67 (m, 2H), 1.67–1.56 (m, 2H), 1.56–1.45 (m, 2H), 1.44–1.18 (m, 2H), 1.16 (s, 3H), 1.10 (s, 3H). 13C NMR (300 MHz, d6-DMSO) δ 180.0, 155.1, 140.5, 135.9, 125.5, 115.0, 113.6, 46.8, 45.4, 40.6, 39.3, 36.5, 30.1, 25.5, 21.6, 18.7, 16.8. HRMS (AP-ESI) Calcd. for C17H22NaO3: 297.1467 [M + Na]+. Found: 297.1461.
4.1.7. General procedure 5: TsCl-participated sulfonylation
The solution of 1 equiv. alcohol and 5 equiv. tolylsulfonyl chloride in 3 ml/mmol of pyridine was stirred at rt for 6 h. After removal of solvent by evaporation, the mixture was dissolved in DCM, followed by washed and dried. The mixture was then concentrated and subjected to flash column chromatography to furnish the product.
1-Hydroxymethyl-abietane-p-methylbenzenesulfonate (10). White solid; mp:67–68 °C; 1H NMR (400 MHz, CDCl3) δ 7.74–7.66 (d, 2H), 7.40–7.51 (d, 2H),5.81 (s, 1H), 5.58–5.34 (m, 1H), 2.53 (s, 3H),2.32–1.94 (m, 4H), 1.93–1.77 (m, 2H), 1.67–1.54 (m, 3H), 1.50–1.42 (m, 1H), 1.34–1.16 (m, 6H), 1.03 (dd, J = 6.8, 4.0 Hz, 6H), 0.94 (q, J = 6.5 Hz, 3H), 0.92–0.84 (m, 3H). 13C NMR (300 MHz, CDCl3) δ 146.0, 144.4, 140.4, 133.5, 130.5, 128.3, 122.4, 121.3, 73.1, 51.7, 51.1, 39.5, 37.0, 35.4, 34.9, 27.7, 24.2, 22.7, 21.0, 20.3, 18.4, 14.7. HRMS (AP-ESI) Calcd. for C27H39O3S: 443.2614 [M + H]+. Found: 443.2619.
4.1.8. General procedure 6: Zn/NaI-mediated reduction
The solution of 1 equiv. p-toluenesulfonate, 10 equiv. zinc and 5 equiv. sodium iodide in 10 ml/mmol of HMPA was stirred at 105 °C for 7 h. The mixture was dissolved in water and the aqueous layer was extracted with EtOAc. The combined organic layers were washed with brine and dried. Solvent was removed under reduced pressure, and the resulting residue was purified by flash column chromatography to furnish the product.
7-Isopropyl-1,1,4-trimethyl-decahydrophenanthrene (11). White oil;1H NMR (400 MHz, CDCl3) δ 5.81 (s, 1H), 5.58–5.34 (m, 1H), 2.32–1.94 (m, 2H), 1.93–1.77 (m, 2H), 1.67–1.54 (m, 3H), 1.50–1.42 (m, 1H), 1.34–1.16 (m, 6H), 1.03 (dd, J = 6.8, 4.0 Hz, 6H), 0.94 (q, J = 6.5 Hz, 3H), 0.92–0.84 (m, 3H), 0.81 (s, 3H). 13C NMR (300 MHz, d6-DMSO) δ 142.6, 137.5, 123.5, 120.6, 52.2, 50.4, 41.8, 40.4, 39.6, 35.05, 33.5, 31.6, 22.7, 20.8, 19.1, 18.4, 15.2, 14.3. HRMS (AP-ESI) Calcd. for C20H31: 271.2426 [M – H]−. Found: 271.2242.
4.1.9. General procedure 7: DCC-promoted condensation
To a solution of 1 equiv. carboxylic acid in 1.5 equiv. diol was added to a solution of 1.5 equiv. DCC and 0.05 equiv. DMAP in 5 ml/mmol of acetonitrile. The mixture was stirred at 0 °C for 1 h, then transferred to rt and stirred for 5 h. The mixture was extracted with EtOAc. The combined organic layers were washed with brine and dried. Solvent was removed under reduced pressure, and the resulting residue was purified by flash column chromatography to furnish the product.
4.1.10. General procedure 8: K2CO3-mediated nucleophilic substitution
A solution of 3 equiv. ester in 2 ml/mmol of DMF was added to a solution of 1 equiv. 6b and 5 equiv. K2CO3 in 2 ml/mmol of DMF. The mixture was stirred at 80 °C for 5 h. The mixture was extracted with EtOAc. The combined organic layers were washed with brine and dried. Solvent was removed under reduced pressure, and the resulting residue was purified by flash column chromatography to furnish the product.
4.1.11. General procedure 9: Pd/C-catalyzed hydrogenolysis of the O-protected group
Pd/C (10%, 1 mg/mg) was added to the solution of 1 equiv. benzyl ester in 10 ml/mmol of methanol. The mixture was hydrogenolised under 1 MPa at rt for 12 h. The reaction mixture was filtered. Solvent of the filtrate was removed under reduced pressure, and the resulting residue was purified by flash column chromatography to furnish the product.
Compound 15. White solid; mp: 72–73 °C; 1HNMR (400 MHz, CDCl3) δ 7.91 (d, J = 7.0 Hz, 1H), 7.67 (d, J = 7.1 Hz, 1H), 7.63–7.44 (m, 2H), 7.12 (t, J = 8.8 Hz, 1H), 6.73 (dd, J = 8.5, 1.9 Hz, 1H), 6.58 (s, 1H), 4.64 (s, 2H), 4.23 (s, 1H), 4.15 (q, J = 7.1 Hz, 1H), 2.84 (d, J = 5.4 Hz, 2H), 2.35–2.11 (m, 2H), 2.08 (s, 1H), 1.92–1.65 (m, 6H), 1.29 (t, J = 7.1 Hz, 5H), 1.19 (d, J = 7.4 Hz, 3H). 13C NMR (300 MHz, CDCl3) δ 185.4, 179.8, 173.7, 171.3, 156.1, 142.4, 136.3, 130.9, 129.9, 128.8, 125.5, 125.4, 115.0, 114.1, 113.1, 112.9, 65.5, 60.5, 47.4, 44.7, 38.0, 36.6, 30.2, 25.2, 21.7, 18.5, 16.2, 14.2. HRMS (AP-ESI) Calcd. for C27H29O7: 465.1919 [M – H]−. Found: 465.1921.
Compound 16. White solid; mp: 109–110 °C; 1HNMR (400 MHz, CDCl3) δ 7.92 (d, J = 6.8 Hz, 1H), 7.70 (d, J = 7.1 Hz, 1H), 7.59 (dt, J = 13.2, 7.2 Hz, 2H), 7.14 (d, J = 8.7 Hz, 1H), 6.71 (dd, J = 8.6, 2.8 Hz, 1H), 6.56 (dd, J = 11.1, 2.6 Hz, 1H), 5.32 (s, 2H), 4.64 (dd, J = 5.8, 2.8 Hz, 1H), 4.52 (t, J = 6.3 Hz, 1H), 4.29 (d, J = 5.2 Hz, 1H), 4.06 (t, J = 6.1 Hz, 1H), 4.00 (t, J = 6.2 Hz, 1H), 2.86 (s, 3H), 2.25 (dd, J = 30.0, 10.2 Hz, 4H), 2.11–2.02 (m, 1H), 1.91–1.78 (m, 4H), 1.78–1.68 (m, 3H), 1.35–1.24 (m, 5H), 1.20 (s, 3H).13C NMR (300 MHz, CDCl3) δ 185.6, 172.5, 168.2, 142.0, 136.3, 133.5, 132.3, 130.8, 129.9, 129.9, 128.7, 125.3, 113.9, 112.7, 64.2, 63.0, 47.5, 44.7, 38.0, 36.7, 36.6, 30.2, 28.4, 25.2, 21.7, 18.5, 16.2. HRMS (AP-ESI) Calcd. for C28H31O7: 479.2075 [M – H]−. Found: 479.2070.
Compound 17. White solid; mp: 113–114 °C; 1HNMR (400 MHz, CDCl3) δ 7.69 (d, J = 6.2 Hz, 1H), 7.57 (d, J = 4.8 Hz, 2H), 7.12 (t, J = 9.5 Hz, 2H), 6.67 (dd, J = 21.8, 7.4 Hz, 2H), 6.52 (d, J = 19.8 Hz, 2H), 4.40 (s, 1H), 3.95 (s, 1H), 2.84 (s, 3H), 2.25 (dd, J = 26.4, 12.1 Hz, 4H), 1.97–1.67 (m, 12H), 1.29 (s, 6H), 1.20 (s, 6H). 13C NMR (300 MHz, CDCl3) δ 156.4, 141.9, 136.6, 136.3, 131.9, 130.9, 128.7, 125.5, 125.3, 115.0, 113.9, 113.1, 112.6, 67.1, 65.7, 47.4, 44.8, 38.1, 36.8, 36.6, 30.0, 25.2, 25.2, 21.7, 18.5, 16.2. HRMS (AP-ESI) Calcd. for C28H33O7: 493.2232 [M – H]−. Found: 493.2226.
Compound 18. White solid; mp: 112–113 °C; 1HNMR (400 MHz, CDCl3) δ 8.12 (dt, J = 21.5, 10.8 Hz, 1H), 7.13 (d, J = 8.6 Hz, 1H), 6.65 (dd, J = 8.6, 2.4 Hz, 1H), 6.52 (d, J = 2.4 Hz, 1H), 5.33 (s, 1H), 4.70 (dd, J = 8.9, 4.3 Hz, 1H), 4.33 (t, J = 4.3 Hz, 1H), 2.96–2.79 (m, 2H), 2.34–2.17 (m, 2H), 1.93–1.80 (m, 3H), 1.80–1.70 (m, 2H), 1.54 (q, J = 6.9 Hz, 2H), 1.31 (s, 3H), 1.22 (d, J = 6.7 Hz, 3H). 13C NMR (300 MHz, CDCl3) δ 185.0, 165.7, 156.1, 153.1, 142.0, 136.6, 130.1, 129.8, 125.5, 115.0, 114.4, 113.1, 112.7, 65.8, 64.2, 47.4, 44.7, 38.1, 36.7, 36.6, 30.0, 25.2, 21.7, 18.5, 16.2. HRMS (AP-ESI) Calcd. for C27H29O7: 465.1919 [M – H]−. Found: 465.1928.
Compound 19. White solid; mp: 113–114 °C; 1HNMR (400 MHz, d6-DMSO) δ 8.50 (s, 1H), 8.20 (d, J = 7.6 Hz, 1H), 8.14 (d, J = 7.6 Hz, 1H), 7.65 (t, J = 7.7 Hz, 1H), 7.16 (d, J = 8.7 Hz, 1H), 6.75 (dd, J = 8.6, 2.1 Hz, 1H), 6.63 (s, 1H), 4.62 (s, 2H), 4.30 (s, 2H), 2.87–2.66 (m, 3H), 2.28 (d, J = 12.4 Hz, 1H), 2.01 (d, J = 11.7 Hz, 1H), 1.45–1.19 (m, 4H), 1.16 (s, 3H), 1.1 (s, 3H). 13 C NMR (300 MHz, CDCl3) δ 183.0, 169.7, 165.1, 153.1, 142.0, 136.6, 130.1, 129.8, 125.5, 118.0, 117.4, 116.1, 115.7, 65.8, 64.2, 47.4, 45.7, 38.1, 36.7, 36.6, 30.0, 29.2, 28.7, 24.7, 19.7. HRMS (AP-ESI) Calcd. for C27H29O7: 465.1919 [M – H]−. Found: 465.1926.
Compound 20. White solid; mp: 102–103 °C; 1HNMR (400 MHz, CDCl3) δ 8.21 (dd, J = 7.8, 1.4 Hz, 1H), 7.63–7.51 (m, 1H), 7.21–7.03 (m, 3H), 6.78 (dd, J = 8.6, 2.5 Hz, 1H), 6.63 (d, J = 2.3 Hz, 1H), 4.63–4.51 (m, 2H), 4.42–4.30 (m, 2H), 3.01–2.78 (m, 3H), 2.27 (dd, J = 31.5, 12.4 Hz, 3H), 1.95–1.81 (m, 4H), 1.81–1.70 (s, 3H), 1.28 (d, J = 13.2 Hz, 4H), 1.24–1.16 (m, 4H). 13 C NMR (300 MHz, CDCl3) δ 184.8, 166.0, 157.3, 155.5, 143.0, 136.6, 134.9, 133.9, 125.6, 122.7, 118.5, 114.2, 113.4, 112.7, 68.5, 65.3, 47.4, 44.7, 38.0, 36.7, 30.2, 25.2, 21.6, 18.5, 16.2. HRMS: m/z calcd for C26H31O6 [M + H]+ 439.2115, found 439.2123.
Compound 21. White solid; mp: 112–113 °C; 1HNMR (400 MHz, CDCl3) δ 8.19 (dd, J = 7.8, 1.7 Hz, 1H), 7.62–7.52 (m, 1H), 7.20–7.03 (m, 3H), 6.77 (dd, J = 8.7, 2.6 Hz, 1H), 6.61 (d, J = 2.5 Hz, 1H), 5.32 (s, 1H), 4.44 (t, J = 6.0 Hz, 2H), 4.17 (t, J = 5.4 Hz, 2H), 2.97–2.84 (m, 3H), 2.43–2.34 (m, 2H), 2.30 (d, J = 12.6 Hz, 1H), 2.23 (d, J = 12.3 Hz, 1H), 1.92–1.80 (m, 4H), 1.80–1.69 (m, 3H),1.54–1.28 (m, 3H),1.25 (s, 3H),1.25 (s, 3H). 13 C NMR (300 MHz, CDCl3) δ 184.9, 166.3, 157.7, 156.1, 142.4, 136.4, 135.0, 133.8, 125.5, 122.0, 117.8, 113.8, 112.7, 112.4, 67.5, 64.5, 47.4, 44.7, 38.1, 36.7, 36.6, 30.2, 29.1, 25.2, 21.6, 18.5, 16.2. HRMS: m/z calcd for C27H33O6 [M + H]+ 453.2272, found 453.2286.
Compound 22. White solid; mp: 156–157 °C; 1HNMR (400 MHz, CDCl3) δ 7.96–7.90 (m, 1H), 7.76–7.68 (m, 1H), 7.60 (tt, J = 13.6, 6.8 Hz, 2H), 5.27 (dd, J = 25.2, 11.3 Hz, 2H), 4.39 (d, J = 11.0 Hz, 1H), 3.92 (d, J = 11.1 Hz, 1H), 2.96 (d, J = 29.7 Hz, 1H), 2.64–2.50 (m, 1H), 2.40 (dd, J = 6.5, 3.5 Hz, 1H), 2.36 (dd, J = 6.4, 3.5 Hz, 1H), 2.15–2.07 (m, 1H), 2.02–1.87 (m, 6H), 1.77 (dt, J = 13.0, 8.3 Hz, 3H), 1.69–1.60 (m, 2H), 1.45 (s, 4H), 1.29 (t, J = 12.0 Hz, 5H), 1.23–1.15 (m, 6H). 13 C NMR (300 MHz, CDCl3) δ 185.2, 171.8, 158.7, 156.1, 142.5, 136.5, 130.6, 129.6, 125.4, 123.0, 121.2, 115.2, 114.3, 112.7, 66.8, 66.3, 53.4, 47.4, 44.7, 38.1, 36.6, 30.2, 25.2, 21.7, 18.5, 16.2. HRMS: m/z calcd for C26H31O6 [M + H]+ 439.2115, found 439.2120.
Compound 23. White solid; mp: 158–159 °C; 1HNMR (400 MHz, CDCl3) δ 7.74 (d, J = 7.6 Hz, 1H), 7.68 (s, 1H), 7.40 (t, J = 7.9 Hz, 1H), 7.25–7.16 (m, 2H), 6.82–6.76 (m, 1H), 6.65 (s, 1H), 5.33 (s, 1H), 4.36 (dd, J = 13.3, 3.8 Hz, 3H), 3.02–2.83 (m, 2H), 2.29 (dd, J = 23.2, 12.4 Hz, 2H), 1.87 (dd, J = 21.6, 11.8 Hz, 2H), 1.77 (t, J = 11.0 Hz, 2H), 1.57 (dd, J = 12.7, 6.6 Hz, 1H), 1.48–1.32 (s, 3H), 1.32–1.25 (s, 3H). 13 C NMR (300 MHz, CDCl3) δ 185.2, 171.8, 158.7, 156.1, 142.5, 136.5, 130.6, 129.6, 125.4, 123.0, 121.2, 115.2, 114.3, 112.7, 66.8, 66.3, 47.4, 44.7, 38.1, 36.7, 30.2, 25.2, 21.7, 18.5, 16.2. HRMS: m/z calcd for C26H31O6 [M + H]+ 439.2115, found 439.2118.
Compound 24. Yellow oil; mp: 145–146 °C; 1H NMR (400 MHz, CDCl3) δ 8.21 (dd, J = 7.8, 1.8 Hz, 1H), 7.67–7.56 (m, 1H), 7.28 (t, J = 4.4 Hz, 1H), 7.22 (t, J = 7.2 Hz, 1H), 7.04 (d, J = 8.3 Hz, 1H), 6.92 (dd, J = 8.6, 2.5 Hz, 1H), 6.83 (d, J = 2.5 Hz, 1H), 5.06 (s, 2H), 1.85–1.76 (m, 2H),1.76–1.68 (m, 2H),1.68–1.62 (m, 2H), 1.66–1.40 (m, 3H), 1.31 (s, 3H), 1.23 (s, 3H). 13 C NMR (300 MHz, CDCl3) δ 184.6, 166.5, 166.1, 156.5, 147.8, 147.3, 137.0, 134.9, 134.1, 125.7, 123.1, 120.9, 119.0, 118.2, 113.1, 66.4, 47.30, 44.3, 37.9, 37.1, 36.7, 29.9, 25.1, 21.4, 18.4, 16.2. HRMS: m/z calcd for C26H28O7 [M + H]+ 454.1908, found 454.1902.
Compound 25. White solid; mp: 138–139 °C; 1H NMR (400 MHz, CDCl3) δ 8.13–8.05 (m, 1H), 7.59 (t, J = 7.4 Hz, 1H), 7.46 (t, J = 7.7 Hz, 1H), 7.19 (d, J = 8.7 Hz, 1H), 6.78 (dd, J = 8.7, 2.6 Hz, 1H), 6.64 (d, J = 2.5 Hz, 1H), 4.67 (t, J = 9.2 Hz, 1H), 4.30 (t, J = 9.6 Hz, 1H), 3.04–2.83 (m, 2H), 2.39–2.14 (m, 2H), 1.95–1.69 (m, 5H), 1.65–1.48 (m, 2H), 1.31 (s, 3H), 1.23 (s, 3H). 13 C NMR (300 MHz, CDCl3) δ 185.0, 166.6, 156.2, 142.4, 136.5, 133.1, 129.9, 129.8, 128.4, 125.4, 125.4, 114.2, 112.7, 65.9, 63.6, 47.4, 44.7, 38.1, 36.7, 30.2, 26.9, 25.2, 21.7, 18.5, 16.2. HRMS: m/z calcd for C26H30O5 [M + H]+ 424.2166, found 224.2199.
Compound 26. White solid; mp: 111–112 °C; 1H NMR (400 MHz, CDCl3) δ 10.69 (s, 1H), 7.86 (dd, J = 8.0, 1.7 Hz, 1H), 7.51–7.43 (m, 1H), 7.19 (d, J = 8.7 Hz, 1H), 7.00 (d, J = 7.7 Hz, 1H), 6.92–6.85 (m, 1H), 6.77 (dd, J = 8.7, 2.7 Hz, 1H), 6.63 (d, J = 2.7 Hz, 1H), 4.74–4.63 (m, 2H), 4.33–4.26 (m, 2H), 2.31 (d, J = 12.7 Hz, 1H), 2.23 (dd, J = 12.4, 1.9 Hz, 1H), 1.97–1.68 (m, 5H), 1.60–1.41 (m,3H), 1.31 (s, 3H), 1.22 (s, 3H). 13C NMR (300 MHz, CDCl3) δ 184.8, 170.0, 161.7, 156.1, 142.6, 136.5, 135.9, 130.2, 125.5, 119.2, 117.6, 114.3, 112.8, 112.3, 65.7, 63.8, 60.4, 47.4, 44.7, 38.1, 36.8, 36.7, 30.2, 25.2, 21.7, 18.5, 16.2. HRMS: m/z calcd for C26H30O6 [M + H]+ 440.2115, found 440.2146.
Compound 27. White solid; mp: 128–129 °C; 1H NMR (400 MHz, CDCl3) δ 8.09–8.06 (m, 1H), 7.58 (t, J = 7.9 Hz, 1H), 7.48–7.43 (m, 1H), 7.19 (d, J = 8.8 Hz, 1H), 6.78 (dd, J = 8.7, 2.8 Hz, 1H), 6.66–6.64 (m, 1H), 4.67 (t, J = 9.2 Hz, 1H), 4.30 (t, J = 9.2 Hz, 1H), 1.90–1.83 (m, 4H), 1.83–1.73 (m, 3H),1.66–1.42 (m, 3H), 1.31 (s, 3H), 1.22 (s, 3H). 13 C NMR (300 MHz, CDCl3) δ 183.0, 166.0, 154.2, 140.5, 134.5, 133.3, 131.4, 130.0, 126.8, 113.8, 111.4, 66.4, 63.9, 51.5, 43.9, 38.4, 37.6, 37.4, 30.1, 28.1, 24.7, 20.1, 19.7. HRMS: m/z calcd for C26H29ClO5 [M + H]+ 457.1776, found 457.1774.
Compound 28. White solid; mp: 126–127 °C; 1H NMR (400 MHz, CDCl3) δ 7.88 (dd, J = 8.1, 1.4 Hz, 1H), 7.33–7.25 (m, 1H), 7.18 (d, J = 8.7 Hz, 1H), 6.77 (dd, J = 8.7, 2.7 Hz, 1H), 6.71–6.60 (m, 3H), 4.62 (t, J = 9.6, 2H), 4.28 (t, J = 9.6, 2H), 3.01–2.83 (m, 2H), 2.36–2.18 (m, 2H), 1.96–1.82 (m, 4H), 1.82–1.73 (m, 2H), 1.31 (s, 3H), 1.22 (s, 3H). 13 C NMR (300 MHz, CDCl3) δ 168.0, 156.2, 150.5, 142.4, 136.4, 134.2, 131.5, 125.4, 116.7, 116.3, 114.3, 112.8, 110.6, 66.0, 62.9, 47.4, 44.8, 38.1, 36.8, 36.7, 30.2, 25.2, 21.7, 18.5, 16.2. HRMS: m/z calcd for C26H31NO5 [M + H]+ 439.2275, found 439.2271.
Compound 29. White solid; mp: 158–159 °C; 1H NMR (400 MHz, CDCl3) δ 8.79 (d, J = 4.7 Hz, 1H), 8.14 (d, J = 7.8 Hz, 1H), 7.86 (t, J = 7.8 Hz, 1H), 7.56–7.47 (m, 1H), 7.16 (d, J = 8.8 Hz, 1H), 6.74 (s, 1H), 6.61 (d, J = 7.9 Hz, 1H), 4.76 (t, J = 9.2 Hz, 1H), 4.33 (t, J = 9.2 Hz, 1H), 3.01–2.83 (m, 2H), 2.36–2.18 (m, 2H), 1.95–1.83 (m, 4H), 1.83–1.70 (m, 2H), 1.31 (s, 3H), 1.22 (s, 3H). 13 C NMR (300 MHz, CDCl3) δ 168.7, 155.9, 142.7, 136.5, 125.5, 114.2, 112.6, 65.4, 64.6, 56.4, 53.5, 47.3, 44.8, 43.8, 38.2, 36.8, 36.7, 30.2, 25.7, 25.2, 21.7, 18.6, 16.4. HRMS: m/z calcd for C25H29NO5 [M + H]+ 424.2118, found 424.2115.
Compound 30. White solid; mp: 161–162 °C; 1H NMR (400 MHz, CDCl3) δ 8.04 (d, J = 8.7 Hz, 1H), 7.52 (t, J = 7.5 Hz, 1H), 7.38 (t, J = 7.3 Hz, 1H), 7.30 (d, J = 8.4 Hz, 1H), 7.16 (d, J = 8.8 Hz, 1H), 6.75 (dd, J = 8.7, 2.6 Hz, 1H), 6.61 (d, J = 2.5 Hz, 1H), 4.68–4.61 (m, 2H), 4.33–4.22 (m, 2H), 4.06 (s, 2H), 2.94–2.70 (m, 2H), 2.29 (d, J = 12.5 Hz, 1H), 2.20 (d, J = 13.9 Hz, 1H), 1.94–1.84 (m, 2H), 1.84–1.68 (m, 3H), 1.60–1.41 (m, 2H), 1.28 (s, 3H), 1.20 (s, 3H). 13 C NMR (300 MHz, CDCl3) δ 185.1, 177.0, 167.3, 156.1, 142.5, 136.4, 135.4, 132.8, 132.4 131.3, 129.4 127.7, 125.4, 114.2, 112.8, 65.7, 63.8, 47.4, 44.7, 40.7, 38.1, 36.7, 36.6, 30.1, 25.2, 21.6, 18.5, 16.2. HRMS: m/z calcd for C28H32O7 [M + Na]+ 504.2221, found 504.2071
Compound 31. White solid; mp: 116–117 °C; 1H NMR (400 MHz, CDCl3) δ 7.15 (d, J = 8.8 Hz, 1H), 6.71 (dd, J = 8.7, 2.6 Hz, 1H), 6.57 (s, 1H), 4.48–4.31 (m, 2H), 4.20–3.89 (m, 2H), 3.03–2.73 (m, 4H), 2.25 (dd, J = 32.2, 11.8 Hz, 2H), 2.04 (s, 1H), 1.88–1.82 (m, 2H), 1.82–1.76 (m, 2H), 1.76–1.71 (m, 2H), 1.55–1.36 (m, 5H), 1.28 (s, 3H), 1.20 (s, 3H). 13 C NMR (300 MHz, CDCl3) δ 185.1, 179.9, 173.6, 156.1, 142.4, 136.3, 125.3, 114.3, 114.2, 112.8, 112.7, 65.8, 63.0, 60.4, 47.3, 44.7, 42.5, 42.4, 38.1, 36.7, 36.6, 30.1, 25.2, 21.6, 21.1, 18.5, 16.2, 14.2. HRMS: m/z calcd for C27H36O7 [M + H]+ 474.2534, found 474.2570.
Compound 32. White solid; mp: 109–110 °C; 1H NMR (400 MHz, CDCl3) δ 7.15 (d, J = 8.7 Hz, 1H), 6.69 (d, J = 6.9 Hz, 1H), 6.55 (s, 1H), 4.54 (s, 2H), 4.15 (s, 2H), 3.51 (s, 1H), 2.84 (s, 2H), 2.37–1.89 (m, 5H), 1.75 (m, 6H), 1.59–1.36 (m, 3H), 1.28 (s, 3H), 1.18 (s, 3H). 13 C NMR (300 MHz, CDCl3) δ 168.7, 155.9, 142.7, 136.5, 125.5, 114.2, 112.6, 65.4, 64.6, 56.4, 53.5, 47.3, 44.8, 43.8, 38.2, 36.8, 36.7, 30.2, 25.7, 25.2, 21.7, 18.6, 16.4. HRMS: m/z calcd for C24H33NO5 [M + H]+ 417.2431, found 417.2462.
Compound 33. White solid; mp: 48–49 °C; 1H NMR (400 MHz, CDCl3) δ 7.16 (d, J = 10.0 Hz, 1H), 6.72 (d, J = 8.6 Hz, 1H), 6.58 (s, 1H), 4.39–3.66 (m, 6H), 3.34–3.29 (m, 2H), 2.99–2.78 (m, 2H), 2.38–2.05 (m, 4H), 1.80–1.63 (m, 4H), 1.54–1.38 (m, 2H), 1.26 (s, 3H), 1.20 (s, 2H). 13C NMR (300 MHz, CDCl3) δ 183.4, 156.2, 142.6, 136.5, 125.4, 114.2, 112.5, 69.1, 61.4, 53.1, 47.3, 45.8, 44.9, 38.2, 36.8, 36.7, 30.2, 25.2, 21.6, 18.6, 16.4. HRMS: m/z calcd for C25H35NO5 [M + H]+ 431.2588, found 431.2615.
Compound 34. White solid; mp: 189–190 °C; 1H NMR (400 MHz, d6-DMSO) δ 12.72 (s, 2H), 8.67 (d, J = 4.9 Hz, 2H), 8.23 (dd, J = 8.4, 4.8 Hz, 2H), 8.04 (dd, J = 14.0, 8.7 Hz, 2H), 7.14 (d, J = 8.7 Hz, 1H), 6.76 (d, J = 10.5 Hz, 1H), 6.64 (s, 1H), 4.66 (s, 2H), 4.34 (s, 2H), 2.25 (d, J = 12.0 Hz, 1H), 2.01 (d, J = 11.6 Hz, 1H), 1.78–1.48 (m, 2H), 1.15 (s, 3H), 1.09 (s, 3H). 13 C NMR (300 MHz, d6-DMSO) δ 179.9, 167.6, 166.0, 156.3, 142.6, 136.3, 134.8, 134.5, 130.9, 130.7, 130.6, 130.4, 130.3, 129.3, 126.5, 126.0, 125.8, 114.5, 113.2, 66.1, 64.4, 46.8, 45.3, 38.3, 36.7, 30.2, 25.3, 21.5, 18.6, 16.8. HRMS: m/z calcd for C31H32O7 [M + H]+ 517.2221, found 517.2231.
4.2. Biological assays
4.2.1. PTP1B inhibition assay
The inhibitory assay was performed using human PTP1B (purchased from Sigma) and p-nitrophenylphosphate (pNPP) as substrate by UV absorption. 400 nmol pNPP were dissolved in H2O and 48 μL of the solution was added to a 100 μL reaction. The indicated amount of synthetic compounds was diluted in DMSO at a concentration of 5 mM. The PTP1B was diluted in pH 7.2 buffer contained 50 mM HEPES, 3 mM dithiothreitol, 2 mM EDTA and 100 mM NaCl. About 50 μL PTP1B dilution was added, and the mixture incubated at 37 °C for 30 min. 1N NaOH was added to terminate the reaction. UV–Vis absorption was detected at 410 nm using a Tecan Infinite M1000 plate reader.
4.2.2. Glucose uptake assay
HepG2 cells were grown in DMEM with 10% foetal bovine serum, 100 mg/mL of streptomycin, and 100 U/mL of penicillin, in a humidified atmosphere of 5% CO2 at 37 °C. HepG2 cells were cultured in 96-well tuft plates. After overnight incubation, the cells were serum starved for 12 h. Then, the cells were treated with insulin (6 × 10−6–5 × 10−5 mmol/L) for 0–60 h to induce insulin resistance. HepG2 cells were then cultured for another 24 h in the compound (40 μM) or rosiglitazone (10 μM, available from Bied Pharmaceutical Technology Co., Ltd., Shanghai, China) containing DMEM. After cultivation, the glucose content was determined by the glucose oxidase method (the glucose analysis kit was purchased from Rongsheng Biopharmaceutical Co., Ltd., Shanghai, China). The sample and the kit working solution were thoroughly mixed at a ratio of 1:100, and placed in a 37 °C water bath for 15 min. The absorbance was measured at 506 nm using a microplate reader (SpectraMax i3x, Molecular Devices). The residual glucose concentration is expressed as AX/A0 × 5.55 mmol/L (AX: OD value of sample; A0: OD value of glucose standard solution).
4.2.3. Animal experiment
During the experiment, the male Kunming mice with 25–30 g body weight were fasted but received water prior to induction of diabetes. 110 mg/kg of nicotinamide and 65 mg/kg of streptozotocin (Macklin in Biochemical Technology Co., Ltd., Shanghai, China) were dissolved in pH 4.5 citrate buffer and intraperitoneally administrated to induce NIDDM mice model. The mice were intraperitoneally administered with a compound or rosiglitazone (100 mg/kg) dissolved in saline containing 1% DMSO and 10% Tween 80. Hyperglycaemia was confirmed by a plasma glucose increase of more than 4.5 mmol/L measured by a blood glucose metre (Sinocare GA-3, Changsha, China). Glucose concentrations from the tail of the mice were measured at 0, 0.5, 1, 2, 3 and 4 h by the blood glucose metre. Calculate the percentage of blood glucose by using the following formula: variation of glycaemia% = (AX − A0)/A0 × 100% (AX: blood glucose at selected time; A0: initial blood glucose).
Supplemental Material
Download PDF (2.8 MB)Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Chen L, Magliano DJ, Zimmet PZ. The worldwide epidemiology of type 2 diabetes mellitus-present and future perspectives. Nat Rev Endocrinol 2012;8:228–36.
- van Dieren S, Beulens JWJ, van der Schouw YT, et al. The global burden of diabetes and its complications: an emerging pandemic. Eur J Cardiov Prev R 2010;17:S3–S8.
- Zinman B, Wanner C, Lachin JM, et al. Investigators, empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. New Engl J Med 2015;373:2117–28.
- Catanzaro R, Lorenzetti A, Allegri F, et al. Inhibiting insulin resistance mechanisms by DTS phytocompound: an experimental study on metabolic syndrome-prone adipocytes. Acta bio-medica Atenei Parmensis 2012;83:95–102.
- Bonner C, Kerr-Conte J, Gmyr V, et al. Inhibition of the glucose transporter SGLT2 with dapagliflozin in pancreatic alpha cells triggers glucagon secretion. Nat Med 2015;21:512–U139.
- Green JB, Bethel MA, Armstrong PW, et al. Effect of sitagliptin on cardiovascular outcomes in type 2 diabetes. New Engl J Med 2015;373:232–42.
- Jia Y, Lao Y, Zhu H, et al. Is metformin still the most efficacious first-line oral hypoglycaemic drug in treating type 2 diabetes? A network meta-analysis of randomized controlled trials. Obes Rev 2019;20:1–12.
- Gouni-Berthold I, Berthold HK. Pharmacologic therapy for cardiovascular risk reduction in patients with the metabolic syndrome. Curr Pharm Des 2014;20:5025–38.
- Rajasurya V, Anjum H, Surani S. Metformin use and metformin-associated lactic acidosis in intensive care unit patients with diabetes. Cureus 2019;11:UNSP e4739.
- Ji WJ, Chen XL, Lv J, et al. Liraglutide exerts antidiabetic effect via PTP1B and PI3K/Akt2 signaling pathway in skeletal muscle of KKAy mice. Int J Endocrinol 2014;9:312452.
- Panzhinskiy E, Hua Y, Culver B, et al. Endoplasmic reticulum stress upregulates protein tyrosine phosphatase 1B and impairs glucose uptake in cultured myotubes. Diabetologia 2013;56:598–607.
- Hsu MF, Meng TC. Enhancement of insulin responsiveness by nitric oxide-mediated inactivation of protein-tyrosine phosphatases. J Biol Chem 2010;285:7919–28.
- Elchebly M, Payette P, Michaliszyn E, et al. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science 1999;283:1544–8.
- Delibegovic M, Zimmer D, Kauffman C, et al. Liver-specific deletion of protein-tyrosine phosphatase 1B (PTP1B) improves metabolic syndrome and attenuates diet-induced endoplasmic reticulum stress. Diabetes 2009;58:590–9.
- Bence KK, Delibegovic M, Xue B, et al. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat Med 2006;12:917–24.
- Fernandez-Ruiz R, Vieira E, Garcia-Roves PM, et al. Protein tyrosine phosphatase-1B modulates pancreatic beta-cell mass. PLoS One 2014;9:e90344.
- Liu SM, Xi YN, Bettaieb A, et al. Disruption of protein-tyrosine phosphatase 1B expression in the pancreas affects beta-cell function. Endocrinology 2014;155:3329–38.
- Qian S, Zhang M, He Y, et al. Recent advances in the development of protein tyrosine phosphatase 1B inhibitors for type 2 diabetes. Fut Med Chem 2016;8:1239–58.
- Yu M, Liu Z, Liu Y, et al. PTP1B markedly promotes breast cancer progression and is regulated by miR-193a-3p. Febs J 2019;286:1136–53.
- Garcia-Ruiz I, Blanes Ruiz N, Rada P, et al. Protein tyrosine phosphatase 1B deficiency protects against hepatic fibrosis by modulating NADPH oxidases. Redox Bio 2019;26:101263.
- Liu H, Sun D, Du H, et al. Synthesis and biological evaluation of tryptophan-derived rhodanine derivatives as PTP1B inhibitors and anti-bacterial agents. Eur J Med Chem 2019;172:163–73.
- Yu X, Sun JP, He Y, et al. Structure, inhibitor, and regulatory mechanism of Lyp, a lymphoid-specific tyrosine phosphatase implicated in autoimmune diseases. Proc Natl Acad Sci USA 2007;104:19767–772.
- Olivier M, Hsiung CA, Chuang LM, et al. Single nucleotide polymorphisms in protein tyrosine phosphatase 1 beta (PTPN1) are associated with essential hypertension and obesity. Hum Mol Genet 2004;13:1885–92.
- Combs AP. Recent advances in the discovery of competitive protein tyrosine phosphatase 1B inhibitors for the treatment of diabetes, obesity, and cancer. J Med Chem 2010;53:2333–44.
- Jiang CS, Liang LF, Guo YW. Natural products possessing protein tyrosine phosphatase 1B (PTP1B) inhibitory activity found in the last decades. Acta Pharmacol Sin 2012;33:1217–45.
- Li J, Bai L, Wei F, et al. Therapeutic mechanisms of herbal medicines against insulin resistance: a review. Front Pharmacol 2019;10:661.
- Wang LJ, Jiang B, Wu N, et al. Natural and semisynthetic protein tyrosine phosphatase 1B (PTP1B) inhibitors as anti-diabetic agents. RSC Adv 2015;5:48822–834.
- Qian S, Li H, Chen Y, et al. Synthesis and biological evaluation of oleanolic acid derivatives as inhibitors of protein tyrosine phosphatase 1B. J Nat Prod 2010;73:1743–50.
- Qian S, Chen QL, Guan JL, et al. Synthesis and biological evaluation of raddeanin A, a triterpene saponin isolated from Anemone raddeana. Chem Pharm Bull 2014;62:779–85.
- Qian S, Wu Y, He YX, et al. Synthesis and biological evaluation of oleanane triterpenoid with gamma-lactone functionality in ring C. Chem J Chin Univ 2012;33:969–75.
- Pei SC, Wu JB, Lei F, et al. Synthesis of oleanolic disaccharide derivatives. Chin Chem Lett 2012;23:403–6.
- Chen L, Wu JB, Lei F, et al. Synthesis and biological evaluation of oleanolic acid derivatives as antitumor agents. J Asian Nat Prod Res 2012;14:355–63.
- Qian S, Li JH, Zhang YW, et al. Synthesis and alpha-glucosidase inhibitory activity of oleanolic acid derivatives. J Asian Nat Prod Res 2010;12:20–29.
- Moretto AF, Kirincich SJ, Xu WX, et al. Bicyclic and tricyclic thiophenes as protein tyrosine phosphatase 1B inhibitors. Bioorg Med Chem 2006;14:2162–77.
- Puius YA, Zhao Y, Sullivan M, et al. Identification of a second aryl phosphate-binding site in protein-tyrosine phosphatase 1B: a paradigm for inhibitor design. Proc Natl Acad Sci USA 1997;94:13420–25.
- Abad A, Arno M, Domingo LR, et al. Synthesis of (+)-podocarp-8(14)-en-13-one and methyl-(+)-13-oxo-podocarp-8(14)-en-18-oate from abietic acid. Tetrahedron 1985;41:4937–40.
- Alvarez-Manzaneda EJ, Chahboun R, Guardia JJ, et al. New route to 15-hydroxydehydroabietic acid derivatives: application to the first synthesis of some bioactive abietane and nor-abietane type terpenoids. Tetrahedron Lett 2006;47:2577–80.
- Gonzalez MA, Correa-Royero J, Agudelo L, et al. Synthesis and biological evaluation of abietic acid derivatives. Eur J Med Chem 2009;44:2468–72.
- Boger DL, Coleman RS. Benzylic hydroperoxide rearrangement - observations on a viable and convenient alternative to the Baeyer-Villiger rearrangement. J Org Chem 1986;51:5436–39.
- Lee HJ, Ravn MM, Coates RM. Synthesis and characterization of abietadiene, levopimaradiene, palustradiene, and neoabietadiene: hydrocarbon precursors of the abietane diterpene resin acids. Tetrahedron 2001;57:6155–67.
- Liu ZQ, Liu T, Chen C, et al. Fumosorinone, a novel PTP1B inhibitor, activates insulin signaling in insulin-resistance HepG2 cells and shows anti-diabetic effect in diabetic KKAy mice. Toxicol Appl Pharmacol 2015;285:61–70.
- Xu F, Wang F, Wang Z, et al. Glucose uptake activities of bis (2, 3-dibromo-4,5-dihydroxybenzyl)ether, a novel marine natural product from red alga Odonthalia corymbifera with protein tyrosine phosphatase 1B inhibition, in vitro and in vivo. PLoS One 2016;11:e0147748.
- Cui DS, Lipchock JM, Brookner D, et al. Uncovering the molecular interactions in the catalytic loop that modulate the conformational dynamics in protein tyrosine phosphatase 1B. J Am Chem Soc 2019;141:12634–47.
- Hann MM, Oprea TI. Pursuing the leadlikeness concept in pharmaceutical research. Curr Opin Chem Biol 2004;8:255–63.
- Jose Ramirez-Espinosa J, Yolanda Rios M, Paoli P, et al. Synthesis of oleanolic acid derivatives: In vitro, in vivo and in silico studies for PTP-1B inhibition. Eur J Med Chem 2014;87:316–27.