
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Cyclin-dependent kinase 2 (CDK2) is the family of Ser/Thr protein kinases that has emerged as a highly selective with low toxic cancer therapy target. A multistage virtual screening method combined by SVM, protein-ligand interaction fingerprints (PLIF) pharmacophore and docking was utilised for screening the CDK2 inhibitors. The evaluation of the validation set indicated that this method can be used to screen large chemical databases because it has a high hit-rate and enrichment factor (80.1% and 332.83 respectively). Six compounds were screened out from NCI, Enamine and Pubchem database. After molecular dynamics and binding free energy calculation, two compounds had great potential as novel CDK2 inhibitors and they also showed selective inhibition against CDK2 in the kinase activity assay.
1. Introduction
Cyclin-dependent kinases are the family of Ser/Thr protein kinases that are essential in regulating cell progression through cell cycle G/S and G2/MCitation1–3. The activities of cyclin-dependent kinases are regulated by the regulatory subunits of the complex cyclins and phosphorylation. Cyclin E binds G1 phase CDK2, which is required for the transition from G1 to S phase, while binding with Cyclin A is required to progress through the S phaseCitation2,Citation3. The precise regulation of CDK activity is essential for the stepwise execution of the many processes required for cell growth and division, including DNA replication and chromosome separationCitation4,Citation5. Abnormal CDK control of the cell cycle has been strongly linked to the molecular pathology of cancer, and CDK2 is now thought to be dispensable for tumour formation and maintenanceCitation5,Citation6. Additionally, increasing studies have shown that inhibition of CDK2 could induce cancer cell apoptosis with no damage to normal cellsCitation6. Therefore, CDK2 is an attractive target for the development of a novel anticancer agentCitation7.
CDK2 is activated by binding to cyclin A. The activation of CDK2 causes a change in the conformation of the ATP-binding region. Researchers have designed and synthesised a large number of CDK2 inhibitors, but most of them were designed to be based on the monomer structure of CDK2. Although some of these inhibitors such as R-roscovitine, SNS-032, MK7965, AT7519 and R-547 have entered clinical studies, they were terminated during phase II or phase III trials due to unexpected pharmracological effects and low specificity resulting in off-target interactions. Therefore, development of CDK2 selective inhibitiors would be valuable. But it is difficult to make CDK2-specific inhibitors that do not possess affinity for other kinases, especially for CDK4. The level of homology is 64% between CDK2 and CDK4 and that of sequence identity is 46%, which bring a huge challenge to design the CDK2 selective inhibitorsCitation8.
In this study, we introduced a multistage virtual screening method based on SVM to screen the CDK2 inhibitors from chemical database NCI (265,242 compounds), Enamine (1,960,321 compounds) and Pubchem (192,845,102 compounds). By deconstructing the CDK2 selective inhibitors, a molecular descriptor model based on SVM would be trained to screen out the compounds with selective inhibitory activity. PLIF pharmacophore and docking methods were introduced into the screening process to reduce the false positive rate and increase the isoform selectivityCitation9–12. Simultaneously, molecular dynamics and binding free energy calculation were used to verify the binding stability and affinity of the screened ligand to CDK2.
2. Material and method
2.1. Compounds collection and dataset construction
All the 743 CDK2 inhibitors were collected from The Binding Database (Target name: CDK2/Cyclin A, 671 from Binding DB with the IC50≤10 µM, http://www.bindingdb.org/bind/index.jsp) and the A Directory of Useful Decoys database (72 from DUD, http://dud.docking.org/), and 180 CDK2 non-inhibitors were collected from the Binding DB with the IC50≥100 µM (the structure of 743 CDK2 inhibitors and 180 CDK2 non-inhibitors was showed in Table S4). The two cut-off values (IC50≤10 µM and IC50≥100 µM) will minimise the risk of including potential CDK2 inhibitors in the negative group and reduce the number of false positives during virtual screening. The decoys were selected so that they would have similar physical properties with, but be chemically distinct from CDK2 inhibitorsCitation13–15. In detail, a parallel strategy to Shoichet’s and Garcia-Vallve’s was applied to develop the decoy sets from the NCI databaseCitation13, PubChem and the BD. First, Tanimoto coefficients between a set of 651 known CDK2 inhibitors from the BD (positives from the training set and positives from the validation set) and compounds to be sieved (265,242 from the NCI database, 192 845 102 from PubChem, and compounds from the BD) were calculated based on an ECFP (Extended-connectivity fingerprints) similarity analysisCitation16. The compounds with Tanimoto coefficients of less than 0.5 with any of the selected CDK2 inhibitors were selected. Second, the five physical properties including molecular weight (MW), number of hydrogen-bond donors and hydrogen-bond acceptors, number of rotatable bonds, Octanol-water partition coefficient (log P) were defined by DecoyFinder 2.0 for the CDK2 inhibitors collected from the BindingDB and DUD. Then, the compounds in NCI, Pubchem and BindingDB database with the five calculated physical properties similar to any of the CDK2 inhibitors were further selected in a decoys dataset. Then, 10,245 decoys from NCI, 41,250 decoys from Pubchem, and 114,278 decoys from BindingDB were utilised to build the training and independent validation dataset. Finally, the datasets included the training set (298 positive compounds together with the 10,245 decoys obtained from NCI), test set (70 positive compounds together with the 150 negative compounds (non-inhibitor) from binding database) and validation set (375 positive compounds together with 114278 decoys from BindingDB and 41,250 decoys from Pubchem).
2.2. Support vector machines (SVM) modelling
First, a total of 354 descriptors for the training compounds sets were calculated using the software MOE 2016 after energy minimisationCitation17. Then, the GA-SVMCitation18 was used to select optimised descriptors. The termination generation number was set to 600, the crossover rate was set to 0.6 and the mutation rate was set to 0.0033. LIBSVMCitation19 software was used to establish the SVM classification model based on descriptors selected from GA-SVM ().
Figure 1. The schematic hyperplane of SVM separating positive class (+1) and negative class (−1) with the maximum margin.
2.3. Pharmacophore modeling
All the CDK2 crystal structures were collected from the RSCB protein databank (http://www.rcsb.org/), the proteins with the inhibitor in the ATP competitive site and the same amino acid sequence were aligned over for putting into the protein database. The interaction fingerprints were then generated by the PLIF Setup function in Database Viewer panel, all the interactions with the min score 1 and score 2 were 0.5 kcal/mol and 1.5 kcal/mol respectively. Then, the Query Generator tool was carried out for generating pharmacophore queries based on the frequencies of protein-ligand contacts. The max Radius was set to 3, the Feature Coverage was set to 25%-75%, Excluded Volumes was set to ON, other parameters were kept at their default values.
2.4. Molecular docking study
The Genetic Optimisation of Ligand Docking (GOLD) module in MOE2016 software was used to perform the molecular docking study and analyse the interaction between the ligand and receptorCitation20,Citation21. The targeted protein was corrected, protonated, tethered, and stage minimised by QuickPrep function in MOE panel: the Structure, Protonate3D and ASN/GLN/HIS Filps were set to on and the water farther than 4.5 Å from Ligand or receptor were deleted. The binding pocket was defined near the ATP-competitive site within 6.0 Å to make sure that the space was large enough for accommodating the ligand scaleCitation20. During the case of selecting function scores, 50 inhibitors with IC50 values were docked into the binding pocket by using GoldScore, ChemScore, ASP and PLP function score, DCG algorithm to examine the consistency between the experimental IC50 and different score functions. The reli refers to the pIC50 and the IDCG refers to the ordered IC50 values. The closer the Normalised Discounted Cumulative Gain (NDCG) value is to 1, the better the consistency between the IC50 and function score is. Finally, the selected function score was used to evaluate the affinity between the ligand and compoundsCitation22.
2.5. Molecular dynamics
Preliminary MD simulations for the model protein was performed using the programme NAMD (NAnoscale Molecular Dynamics programme, v2.9), and all files were generated using visual molecular dynamics (VMD)Citation23,Citation24. NAMD is freely available software designed for high-performance simulation of large biomolecular systemsCitation24. During MD simulation, the minimisation and equilibration of the original and docked protein were in a 15 Å3 size water box, Amber 10 EHT force field file was applied for energy minimisation and equilibration together with Gasteiger-Huckel charge using the Boltzmann initial velocityCitation25,Citation26. The integrator parameters included 2fs/step for all rigid bonds and nonbonded frequencies were selected for 1 Å, and full electrostatic evaluations were conducted for 2 Å and used with ten steps for each cycleCitation26. The pressure was maintained at 101.325 kPa using the Langevin piston and temperature was controlled to 310 K using Langevin dynamics. The covalent interactions that exist between hydrogen and heavy atoms were observed to be in constrained as revealed by the SHAKE/RATTLE algorithmCitation25. Finally, a 10 ns MD simulation for docked protein was utilised to compare and verify the binding affinity and stability of the ligand-receptor complex.
2.6. Binding free energy calculation
The binding free energies (G-bind) were estimated using the molecular mechanics–Poisson–Boltzmann/surface area (MM/PBSA) method which is a reliable and successful method to investigate the interaction between the ligand and the proteinCitation18,Citation24,Citation27. The 300 snapshots from the latter 2 ns Molecular Dynamics trajectories of the ligand-receptor complexes were extracted for performing the calculation of the binding free energies by the software NAMD. The binding free energies were calculated by the following formula:
The and
refers to the van der Waals energy and electrostatic contribution respectively, and
and
refers to the polar and non-polar solvation energy respectively.
2.7. Cell-free detection of CDK2 activity
The inhibitory activity of the inhibitor on kinase was measured using the ADP-Glo Kinase Assay. The ADP and ATP provided by the kit were diluted to 40 µM with a kinase reaction buffer (40 mM Tris, 200 mM NaCl, 1 mM MgCl2). The test compound and the positive drug (Milciclib) were formulated into four concentration gradient solutions (6 × 10−2 M, 6 × 10−4 M, 6 × 10−6 M, 6 × 10−8 M). Ten microlitres kinase reaction system was shaken at 37 °C After 30 min, 10 µl of ADP-Glo reagent was added to react at room temperature for 40 min. Finally, 20 µl of kinase assay reagent was added to react at room temperature for 30 min to detect chemiluminescence values.
2.8. CDK2 inhibitors anti-tumour cell proliferation in vitro
The HCT116 and A549 cell lines were cultured using RPMI1640 (containing 10% foetal bovine serum) in standard humidified incubation condition at 37 °C in 5% CO2. The cancer cells (5000/well) were placed on 96-well plates in triplicate. After 6 h (cells adherent), the cells were treated with a series of concentrations of Compound 1 and Compound 3. The positive control contained the same concentrations of Milciclib, and the negative control received the same volume of DMSO. After 36 h incubation, cells were stained with 5% MTT (10 µL/well), and the optical density was recorded at an absorbance wavelength of 570 nm.
3. Result and discussion
3.1. Evaluation and validation the single SVM model
The constructed CDK2 inhibitor and non-inhibitor training set contained 298 inhibitors and 10245 decoys. Firstly, the 435 molecular descriptors of the training set compounds were calculated by the Calculate Descriptors function in MOE2016 software, which included geometrical, topological, and electronic properties. Then, the 435 descriptors were pre-processed by the data filter plugin in python to remove redundant data: (1) descriptors with large quantities of null, constant and zero values were eliminated, (2) descriptors with very small standard deviation values (stdev <0.5%) were eliminated, and (3) descriptors with high correlation to others (correlation coefficient > 0.95) were eliminated. Then the remained 142 molecular descriptors were normalised to a range of 0 to +1, which was necessary since the different ranges of descriptor values will influence the quality of the SVM model generatedCitation12. Then the GA-SVM method was utilised to perform a further feature selection process to these 142 normalised descriptors. Finally, 52 of 142 molecular descriptors were selected from the GA-SVM method and used for further evaluation and validation, and these molecular descriptors were divided into 10 groups in . The Grid Search result showed that the parameter C and g with the value of 32 and 0.125, the LibSVM model demonstrated the best accuracy.
Table 1. The 52 molecular descriptors filtered by GA-SVM method for building SVM model
The ten-fold cross-validation was utilised to evaluate the constructed SVM model by using the training set. The evaluation result was showed in , of the 298 CDK2 inhibitors, the 287 compounds (TP) were correctly predicted and 11 compounds (FN) were wrongly predicted, the sensitivity (SE) was 96.3%. As for the 10245 CDK2 non-inhibitors, 10224 compounds were correctly (TN) predicted and 21 compounds (FP) were incorrectly predicted with the specificity (SP) value of 99.69%. The overall accuracy (Q) was 99.79%, which demonstrated that the constructed SVM model was appropriate for the inhibitor and non-inhibitor compounds in training dataset. Then the independent test set was used to validate the predictive ability of the constructed SVM model to the dataset other than itself. The prediction and evaluation result was showed in . Of the 70 CDK2 inhibitors in test set, there were 61 compounds (TP) being correctly predicted with the SE value of 87.1%. As for the 150 CDK2 non-inhibitors, there were 143 compounds (TN) being correctly predicted with the SP value of 95.3%. Of all the 220 compounds in the test set, there were 204 compounds being correctly predicted with the Q value of 92.72%. Validation and evaluation results of the ten-fold cross-validation and independent test indicated that the constructed SVM model fit with both training set and test set, possessed high accuracy and predictive ability.
Table 2. The evaluation and validation result of the ten-fold cross-validation and independent test.
3.2. Validation and evaluation of the PLIF pharmacophore model
The 66 CDK2 crystal structures with the inhibitor were collected from PDB database. All the superposed ligand-receptor complexes were imported into the MOE database for calculating the PLIF rows (in Table S1), and then these PLIF rows were utilised for generating the amino acids interaction fingerprints. As depicted in , the single letter codes of the residues for which fingerprint bits were generated. The 66 CDK2 inhibitors formed with interaction with residues Ile10, Gly13, Val18, Lys33, Glu81, Phe82, Leu83, Gln85, Asp86, Lys89, Gln131 and Asp145. The residues Leu83 had the most single letter codes, which demonstrated that almost all the CDK2 inhibitors generate interaction with the Leu83. The moiety of residues Glu81, Lys33 and Asp86 also formed many interactions with the inhibitor in the ATP-competitive pocket. Most compounds exhibited the hydrogen bond interaction with Leu83 and Glu81, and the residues Asp83 formed the highest number of interaction types which included side-chain hydrogen bond donor, backbone hydrogen bond acceptor, ionic attraction and surface contact (Table S2). Based on these interaction fingerprints, four pharmacophore models were elicited and further evaluated their performance by the CDK2 inhibitor and non-inhibitor training set to select the best one. The result of the evaluation was showed in , the pharmacophore model 3 () with the highest values of yields showed that the 267 of the 298 CDK2 inhibitors were correctly predicted and 2127 of 10245 CDK2 non-inhibitors were also matched properly with this pharmacophore model. All the pharmacophore models had the ability to differentiate the CDK2 inhibitors from non-inhibitors to some degree. But these models also bring a large number of false positive prediction results, that is the reason we adopted the multistage virtual screening method.
Figure 2. (A) The barcodes and letter mode of the amino acids interaction fingerprint generated by MOE2016 software. (B) The best pharmacophore models evaluated by training set, the purple and cyan features indicted the hydrogen-bond donor and acceptor respectively. (C) The matching of the pharmacophore with the superposed structures indicates that the pharmacophore can describe the superposition characteristics of the substituent of the 66 CDK2 inhibitors well.
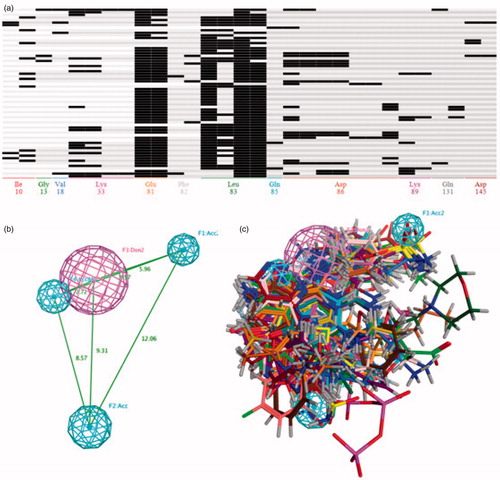
Table 3. The evaluation and validation result of the four Pharmacophore models generated by PLIF.
3.3. Determining docking parameter and validation
The optimisation of docking parameter and selection of the function score are considered important in the process of docking virtual screening because it has a great impact on the final results. The crystal structure of the kinase domain of CDK2 complexes (PDB ID: 3R9H) with 4-amino-N-(2,6-difluorophenyl)-2-[(4-sulfamoylphenyl)amino]-1,3-thiazole-5-carboxamide (RC-2–142) was chosen as the reference structure of the receptor since it has the highest resolution (2.1 Å) among all the CDK2 crystal structuresCitation28,Citation29. In order to determine the best docking parameters, eight active compounds that have been co-crystallized with CDK2 were docked back to the active site of CDK2Citation30–32. The parameters in GOLD panel in MOE2016 software were reconfigured to be as similar as possible to the original build-in inhibitor structures in the ATP-competitive pocket binding site of CDK2. The calculated values of root mean square deviation (RMSD) between the docked active compounds and the crystallized structures in CDK2 were showed in . All the RMSD values were less than 2.1 Å, which manifested that the GOLD plugin in MOE2016 software have the capability of getting the credible ligand-receptor CDK2 complexes in this series of configuration.
Table 4. The RMSD values of the eight active compounds between their docking conformation result and build-in ligand in CDK2 inhibitors.
In the case of selecting the function score, the 50 CDK2 inhibitors with defined IC50 values were docked into the active pocket of the CDK2 inhibitors. The results of NDCG were showed in Table S3: Four function score GoldScore, ChemScore, ASP and PLP values of the 50 inhibitors were calculated respectively for comparing the order to the values of IC50. The Values of ChemScore demonstrated the highest consistency to the experimental IC50 (with the NDCG values of 0.946). Finally, the ChemScore function score was utilised to evaluate the results of the Docking-Virtual Screening.
The training set was utilised to evaluate and validate the GOLD docking with setting parameters. With the threshold function value of 7.0, the 246 of 298 CDK2 inhibitors were correctly predicted with the yield of 82.5%, and 1120 of 10245 decoys were also wrongly predicted as the CDK2 inhibitors. Similar to the pharmacophore model virtual screening, the docking process also had the ability to distinguish the CDK2 inhibitors from the training set, but the multistage virtual screening method made sense because of the high TP (1120) suffered in the result of docking virtual screening.
3.4. Validation and evaluation the performance of the multistage virtual screening method
Each of the virtual screening methods has its advantages and disadvantages in terms of speed and prediction accuracy, and all of them suffer from a high false-positive rate. Multilevel virtual filtering is composed of SVM, pharmacophore and docking virtual screening method. The three filters were cascaded by using the fastest SVM filters first, then medium speed filters, and the slowest docking filters at the end. The validation set (with 375 CDK2 inhibitors and 155528 decoys) will be utilised for evaluating and validating the performance of this multistage virtual screening method.
During the process of validating and evaluating the multistage virtual screening method, the SVM, pharmacophore and docking virtual screening method were individually performed for screening the CDK2 inhibitors from the validation set first. To evaluate the prediction accuracy of virtual screening, the yield (percentage of predicted compounds in known inhibitors), hit rate (percentage of known inhibitors in predicted compounds), and enrichment factor (ratio of hit rate to the percentage of known inhibitors in validation set) were assessed in . For the SVM model, 313 of 375 CDK2 inhibitors were correctly predicted and the number of negatives was 153253 (of the 155528 decoys), the values of yield, hit rate and enrichment factor were 83.5%, 12.1% and 50.3% respectively. The time spent in the process of screening the validation set by SVM model was 0.2 h (on the desktop computer with 8 processors of IntelR XeonR CPU E5-1620@ 3.70 GHz). For the pharmacophore virtual screening method, the number of predicted positive was 28108. 313 of them were correctly predicted with a yield of 87.2%, the values of hit rate and enrichment factor were 1.17% and 4.86 respectively. The time used in the pharmacophore virtual screening was 6.95 h. For docking virtual screening, the 290 of 375 CDK2 inhibitors were correctly predicted and other 17211 decoys were also wrongly predicted as the positive inhibitors. The result of the hit rate, the enrichment factor and the value of yield was 1.66%, 6.90 and 77.3, the time cost in this process was 378.28 h. Hence, the SVM method was selected as the first filter, the pharmacophore and docking method were selected as the second and third one respectively.
Table 5. Validation and evaluation the various virtual screening method by the validation set that contains 375 known CDK2 inhibitors and 155528 decoys.
Then, SVM method was combined with pharmacophore method to investigate the speed and accuracy of screening validation set. After SVM virtual screening method, 313 of 375 CDK2 inhibitors were correctly predicted and 153253 of 155520 decoys were correctly predicted, so the set compose of 2275 decoys and 313 CDK2 inhibitors were selected as the initial set for combined virtual screening method. For the SVM-pharmacophore virtual screening method, 298 of 313 CDK2 inhibitors were correctly predicted with a yield of 79.4%. The value of hit rate and enrichment factor were 28.5% and 118.32 respectively, the time cost in this combined method was 0.93 h. The combination of the two methods was better than either SVM method or pharmacophore method alone in terms of both speed and accuracy (higher hit-rate and enrichment factor). This is because the false positive of SVM method (FP = 2275) in the screening process is far less than that of pharmacophore method (FP = 27454) and method 3 (FP = 17211). The introduction of SVM method as the first level filter will increase the proportion of correct positive results in all predicted positive results. Therefore, the values of hit-rate and enrichment factor will be increased greatly to facilitate distinguishing the positive and negative element.
Finally, the multistage virtual screening method based on SVM, pharmacophore and docking model was performed to screen the CDK2 inhibitors in validation set. The combined method got a set of 749 CDK2 non-inhibitors and 298 inhibitors, after performing the docking virtual screening method, the 277 CDK2 inhibitors and 680 non-inhibitors were correctly predicted. The value of yield, hit-rate, and enrichment factor were 73.8%, 80.1% and 332.83 respectively, the whole multistage virtual screening process took 1.62 h.
The multistage virtual screening method got the highest hit-rate among other methods which was up to 80.1%. With the addition of docking method, the accuracy of virtual screening has been greatly improved than the combined method, although it came with a slight loss of the yield (73.87%). Moreover, the screening time of the slower pharmacophore and docking method was significantly reduced because the fastest SVM method ignored too many negative results initially. We found that all the separate virtual screening methods were not as efficient and accurate as the multistage method as the following aspects: (1) all the separate approach virtual screening method suffer from the problem that positive results accounted for a high proportion of predicted positive results. This will result in a low hit-rate and enrichment factor (particularly in pharmacophore and docking method, the hit-rate and enrichment factor are less than 2% and which is untrustworthy and meaningless in virtual screening result; (2) in the case of screening the large chemical library containing millions or even tens of millions of compounds by performed the pharmacophore and docking method, in addition to the problem of time-consuming process, we should also consider that the ligand-based and structure-based method are often hard to find compounds with novel scaffolds. And using SVM method alone will result in over-fitting between the constructed SVM model and the training set which cannot be verified and avoided by subsequent methods.
3.5. Using the multistage virtual screening method for obtaining the CDK2 inhibitors from NCI database
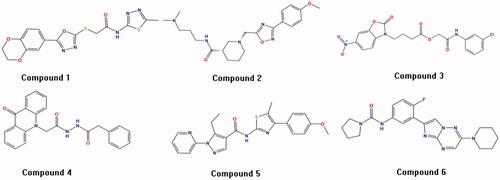
The multistage virtual screening method (SVM-Pharmacophore-Docking) was utilised to screen the CDK2 inhibitors from the chemical database NCI (265,242 compounds), Enamine (1,960,321 compounds) and Pubchem (192,845,102 compounds). The 30120 positive compounds obtained from the first SVM virtual screening approach. These compounds continued to be filtered using pharmacophore method, and then 5541 positive compounds were screened. After implementing the final GOLD docking approach, we ended up with 310 compounds in Table S4. Moreover, the fingerprint BIT-MACCS of all compounds was calculated by MOE2016 software, the 310 compounds were divided into 10 groups according to the similarity metric of tanimoto coefficient. One representative compound was selected in each group based on the number of interactions with important amino acids (Ile10, Gly13, Val18, Lys33, Glu81, Phe82, Leu83, Gln85, Asp86, Lys89, Gln131 and Asp145) and the degree of difficulty in synthesis. Finally, six compounds with novel scaffolds were selected for the further molecular dynamics experiments (in ). We further analysed the drug-like properties of the best hits and we observed that all of them satisfied the Lipinski's rule of five.
Figure 3. The six compounds with novel scaffolds obtained from the multistage virtual screening method.
3.6. Validation and comparison of the binding stability between the screened compounds and CDK2 receptor protein
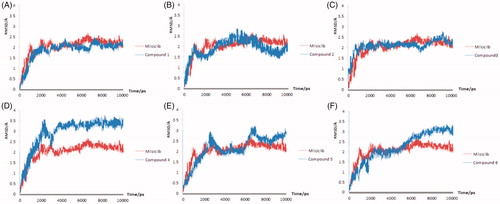
The molecular dynamics results of the Milciclib and the six hit compounds were showed in ). The RMSD plot of the Milciclib-CDK2 complex exhibited a sharp upward trend in the first 3 ns, then the value of the RMSD plot was flat around 2 Å until the end of the simulation. Out of all six compound-CDK2 complexes, only the Compound 1 and Compounds 3 showed the same stability to CDK2 receptor as Milciclib. In , the RMSD plot of Compound 1-CDK2 reached to 2 Å in 2.8 ns and it kept a little fluctuating around that value until the end of 10 ns. In , the RMSD plot of compound 3-CDK2 showed the same tendency to Milciclib-CDK2 plot in the first 7 ns. Although the values had a small fluctuation around 2 to 2.5 Å, it was still levelled off around 2 Å at the end of simulation. The other four compounds either had problems of serious fluctuation in CDK2 pocket (Compound 2-CDK2 and Compound 5-CDK2 in respectively) or demonstrated higher RMSD values than Milciclib-CDK2 complex (Compound 4-CDK2 and Compound 6-CDK2 in respectively), so it's unstable when it bound to the ATP competitive pocket of CDK2.
Figure 4. The molecular dynamics results of the Milciclib and six screened compounds, the RMSD of Milciclib-CDK2 complex was painted in red, the six screened compounds were painted in blue in (A–F).
3.7. The result of the binding free energy calculation
The result of the Binding free energy calculation (MM/PBSA) was depicted in . The van der Waals energy, electrostatic contribution and nonpolar energy were favourable for the stability of the binding pattern, while the polar energy was unfavourable. There was no significant difference in the polar energy and nonpolar energy values of the six compounds, so the main effect of the binding free energy were the van der Waals energy and electrostatic contribution. The Compound 1 and Compound 3 (with the binding free energy values of −258.98 KJ/mol and −252.94 KJ/mol respectively) exhibited the similar values of binding free energy to Milciclib (−260.30 KJ/mol). The other four screened compounds possessed higher binding free energy than Milciclib, indicated that these four compounds were not sufficiently stable to the active pocket of CDK2. The compound 2, 4, 5 and 6 exhibited the relatively high van der Waals energy, electrostatic contribution. The higher van der Waals energy and electrostatic contribution value were, the more likely it is that the compound will be solvated and will not interact with the residues in the active site of the CDK2.
Table 6. Binding free energy of the Milciclib and six potential CDK2 inhibitors.
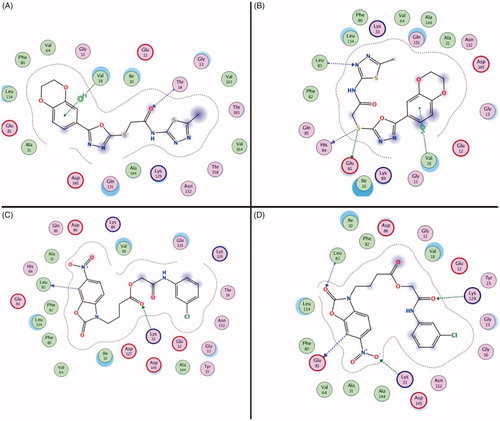
The result of the binding free energy calculation demonstrated that the compound 1 and compound 3 have the potent binding stability to CDK2. The binding mode of Compound 1 and Compound 3 at different simulation stages (0 ns and 10 ns) were showed in . Compound 1 formed the hydrogen bonds with Val18 and Thr14 at 0 ns when interacting with CDK2. When the simulation was over and the complex was stable, the interaction of Thr14 was lost but two new interactions (Glu81, His84 and Leu83) were generated. The benzdioxan moiety formed the aromatic ring interaction to the residue Val18, and the aromatic ring interaction and thioether chains also generated the hydrogen bonds interaction to the residues Glu81, His84 and Leu83. Almost all of these effects occur when an inhibitor binds to CDK2 (). Two hydrogen bonds were generated by interacting with Glu81 and Leu83 residues in the 0 ns of compound 3 & CDK2 molecular dynamics, the residues Lys129 and Glu81 formed new hydrogen bonds interaction with the compound in pocket. The nitryl and benzoxazole scaffold of the compound 3 formed many important aromatic interactions that can stabilise the ligand-receptor complex. The binding mode of the different stage revealed that the stabilisation of two compounds with the novel scaffold is due to the formation of hydrogen bonds interaction, which was similar to the existing CDK2 inhibitors.
Figure 5. The interaction between the compounds and the amino acid residues in the CDK2 active pockets during the molecular dynamics simulation (Compound 1 in 0 ns and 5 ns was showed in (A and B), Compound 3 in 0 ns and 5 ns was showed in (C and D).
3.8. Detection the inhibitory activities of compound 1 and compound 3
The inhibition of CDK2 and CDK4 kinase by Compound 1 and Compound 3 was determined by the chemiluminescence method, and Milciclib was used as a positive control. The inhibitory effects of Compound 1 and Compound 3 on CDK2 were comparable to those of the positive drug, and both of the two hits showed significant selectivity (). The above results were consistent with the molecular dynamics results. It also indicated that the multistage virtual screening method based on SVM was applicable to screen for the compounds with selective CDK2 inhibitory activity.
Table 7. The CDKs inhibition activity of Milciclib and two hit compounds
The proliferation experiment of Compound 1 and Compound 3 against HCT116 and A549 was also carried out. Compound 1 showed preferable inhibitory activity against both HCT116 and A549, especially against HCT116 with IC50 value of 7.2 ± 1.4, which was equivalent to the positive drug Milciclib, while Compound 3 only showed inhibitory activity against HCT116. Besides, the inhibitory activity was not as good as that of Compound 1 (). Comparing the structural characteristics of the two compounds, it was found that Compound 3 contained a nitro group. The group possessed strong polarity, its introduction made it difficult for small molecules to penetrate the cell membrane. This might be the reason that two compounds with similar inhibitory activity at the protein level differ greatly at the cell level.
4. Conclusion
A multistage virtual screening method combined by SVM, pharmacophore, and docking method was utilised for screening the CDK2 inhibitors from the NCI database. Initially, the SVM, pharmacophore, and docking methods were evaluated and optimised individually with the training and test set constructed by the BindingDB, Pubchem, and NCI database. After optimising and selecting the parameter and function score of docking method, the multistage virtual screening method with SVM as the first filter, pharmacophore as the second filter and docking as the last filter was established. The validation set was applied for evaluating the performance of this multistage method, the values of hit-rate and enrichment were higher than any other methods used alone. Then this multistage virtual screening method screened 6 compounds for further molecular dynamics, the result showed that Compound 1 and Compound 3 were stable in the ATP-binding site and exhibited similar binding modes to the existing CDK2 inhibitors. Finally, we used ADP-Glo luminescence to detect the inhibitory effect of the hit compounds on CDK2 in a cell-free system. The result showed that the compounds have good inhibitory activity and selectivity against CDK2. Our study proved that the multistage virtual screening method combined by SVM, pharmacophore, and docking methods was able to screen out compounds with potential as selective CDK2 inhibitors. Moreover, the further in vitro and in vivo experiments and structural modification of these two compounds will be carried out for investigating the mechanism and structure-function relationship of the two novel scaffolds and designing novel selective CDK2 inhibitors.
Supplemental Material
Download Zip (333.1 KB)Disclosure statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Funding
Related Research Data
References
- Sánchez-Martínez C, Gelbert LM, Lallena MJ, et al. Cyclin dependent kinase (CDK) inhibitors as anticancer drugs. Cheminform 2015;46:3420–35.
- Hydbring P, Malumbres M, Sicinski P. Non-canonical functions of cell cycle cyclins and cyclin-dependent kinases. Nat Rev Mol Cell Biol 2016;17:280–92.
- Han SH, Chung JH, Kim J, et al. New role of human ribosomal protein S3: regulation of cell cycle via phosphorylation by cyclin-dependent kinase 2. Oncol Lett 2017;13:3681–7.
- Chen JZ, Pan Y, Qian H, et al. Cyclin-dependent kinase-2 as a target for cancer therapy: progress in the development of CDK2 inhibitors as anti-cancer agents. Curr Med Chem 2014;22:237–63.
- Roskoski R. Cyclin-dependent protein kinase inhibitors including palbociclib as anticancer drugs. Pharmacol Res 2016;107:249–75.
- Wu L, Sun J, Su X, et al. A review about the development of fucoidan in antitumor activity: progress and challenges. Carbohydr Polym 2016;154:96–111.
- Javier HJ, Michael P, Lindsay S, et al. Giving drugs a second chance: overcoming regulatory and financial hurdles in repurposing approved drugs as cancer therapeutics. Front Oncol 2017;7:273.
- Kontopidis G, Mcinnes C, Pandalaneni SR, et al. Differential binding of inhibitors to active and inactive CDK2 provides insights for drug design. Chem Biol (Cambridge) 2006;13:201–11.
- Drwal MN, Griffith R. Combination of ligand- and structure-based methods in virtual screening. Drug Disc Today Technol 2013;10:e395.
- Kumar A, Zhang K. Hierarchical virtual screening approaches in small molecule drug discovery. Methods 2015;71:26–37.
- Lei DW, Li LL, Wang WJ, et al. Identification of CDK2 inhibitors with new scaffolds by a hybrid virtual screening approach based on Bayesian model; pharmacophore hypothesis and molecular docking. J Mol Graph Model 2012;36:42–7.
- Ren JX, Li LL, Zheng RL, et al. Discovery of novel Pim-1 kinase inhibitors by a hierarchical multistage virtual screening approach based on SVM model, pharmacophore, and molecular docking. J Chem Inf Model 2011;51:1364.
- Ceretomassagué A, Guasch L, Valls C, et al. DecoyFinder: an easy-to-use python GUI application for building target-specific decoy sets. Bioinformatics 2012;28:1661–2.
- Mysinger MM, Carchia M, Irwin JJ, et al. Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J Med Chem 2012;55:6582.
- Wei D, Zheng H, Su N, et al. Binding energy landscape analysis helps to discriminate true hits from high-scoring decoys in virtual screening. J Chem Inf Model 2010;50:1855–64.
- Rogers D, Hahn M. Extended-connectivity fingerprints. J Chem Inf Model 2010;50:742–54.
- Vilar S, Cozza G, Moro S. Medicinal chemistry and the molecular operating environment (MOE): application of QSAR and molecular docking to drug discovery. Curr Top Med Chem 2008;8:1555–72.
- Hou T, Wang J, Li Y, et al. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J Chem Inf Model 2011;51:69–82.
- Chang CC, Lin CJ. LIBSVM: a library for support vector machines. ACM Trans Intell Syst Technol 2011;2:1.
- Cotesta S, Giordanetto F, Trosset JY, et al. Virtual screening to enrich a compound collection with CDK2 inhibitors using docking, scoring, and composite scoring models. Proteins Struct Funct Genet 2005;60:629–43.
- Verdonk ML, Cole JC, Hartshorn MJ, et al. Improved protein-ligand docking using GOLD. Proteins Struct Funct Genet 2003;52:609–23.
- Ullah MZ, Aono M, Seddiqui MH. Estimating a ranked list of human hereditary diseases for clinical phenotypes by using weighted bipartite network. Conf Proc IEEE Eng Med Biol Soc 2013;2013:3475–8.
- Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph 1996;14:33–8.
- Phillips JC, Braun R, Wang W, et al. Scalable molecular dynamics with NAMD. J Comput Chem 2005;26:1781–802.
- Bártová I, Otyepka M, Kríz Z, et al. The mechanism of inhibition of the cyclin-dependent kinase-2 as revealed by the molecular dynamics study on the complex CDK2 with the peptide substrate HHASPRK. Protein Sci 2010;14:445–51.
- Jiang W, Phillips JC, Huang L, et al. Generalized scalable multiple copy algorithms for biological molecular dynamics simulations in NAMD. Comput Phys Commun 2014;185:908–16.
- Rastelli G, Del Rio A, Degliesposti G, et al. Fast and accurate predictions of binding free energies using MM-PBSA and MM-GBSA. J Comput Chem 2010;31:797–810.
- Schonbrunn E, Betzi S, Alam R, et al. Development of highly potent and selective diaminothiazole inhibitors of cyclin-dependent kinases. J Med Chem 2013;56:3768–82.
- Richardson CM, Nunns CL, Williamson DS, et al. Discovery of a potent CDK2 inhibitor with a novel binding mode, using virtual screening and initial, structure-guided lead scoping. Bioorg Med Chem Lett 2007;17:3880–5.
- Betzi S, Alam R, Martin M, et al. Discovery of a potential allosteric ligand binding site in CDK2. ACS Chem Biol 2011;6:492–501.
- Kryštof V, McNae IW, Walkinshaw MD, et al. Antiproliferative activity of olomoucine II, a novel 2,6,9-trisubstituted purine cyclin-dependent kinase inhibitor. Cell Mol Life Sci 2005;62:1763–71.
- Su YW, Mcnae I, Kontopidis G, et al. Discovery of a novel family of CDK inhibitors with the program LIDAEUS: structural basis for ligand-induced disordering of the activation loop. Structure 2003;11:399–410.