
Abstract
In this study, newly synthesised compounds 6, 8, 10 and other compounds (1–5, 7 and 9) and their inhibitory properties against the human isoforms hCA I and hCA II were reported for the first time. Compounds 1–10 showed effective inhibition profiles with KI values in the range of 5.13–16.9 nM for hCA I and of 11.77–67.39 nM against hCA II, respectively. Molecular docking studies were also performed with Glide XP to get insight into the inhibitory activity and to evaluate the binding modes of the synthesised compounds to hCA I and II. More rigorous binding energy calculations using MM-GBSA protocol which agreed well with observed activities were then performed to improve the docking scores. Results of in silico calculations showed that all compounds obey drug likeness properties. The new compounds reported here might be promising lead compounds for the development of new potent inhibitors as alternatives to classical hCA inhibitors.
1. Introduction
Carbonic anhydrases (CAs, EC 4.2.1.1) are one of the metalloenzymes catalysing the hydration process of CO2 to HCO3− and H+. All living organisms contain CAs encoded by six phylogenetically gene families.Citation1,Citation2 Fifteen CA isoenzymes belonging to α-CA gene family have been characterised in human-beings. Some human CA (hCA) isoenzymes are cytosolic isoforms (hCA I, II, III, VII and XIII), some isoenzymes are membrane-bound isoforms (hCA IV, IX, XII and XIV), both hCA VA and VB are mitochondrial isoforms and hCA VI isoform is involved in saliva. Three hCA isoenzymes (CA VIII, X and XI) are characterised as acatalytic protein forms. Inhibition and activation studies on the catalytic activity of CAs are crucial for the treatment of numerous clinically important diseases.Citation2,Citation3 The inhibitors of CA isoenzymes (e.g., CA I and CA II) are used to design new class of drugs for epilepsy and glaucoma. Therefore, new CA inhibitors have been required to develop as therapeutic agents.Citation2–4 Several groups have studied the inhibition of hCAs with anions,Citation4 catecholamines,Citation5 thiourea derivatives,Citation6 uracil derivatives,Citation7 bromophenolsCitation8 and sulphonamides.Citation9 In addition, pyrazoles and chalcones have also been studied to inhibit hCAs as well.Citation10
Heterocyclic compounds have a vital role in medicine, pharmacy and agriculture.Citation11 Pyrazoles possess various important bio-medical features.Citation12 This type of derivatives exhibits several therapeutic activities such as insecticidal,Citation13 acaricidal,Citation14 anticonvulsantCitation15 antidepressant,Citation16 antiulcer, and anticancer features.Citation17 So far to date, many chalcone derivatives have been synthesised and their biological activities examined.Citation18 Several investigations have shown that chalcones possess important pharmacological characteristics including antitumor, anti-inflammatory, antifungal and antioxidant properties.Citation19 The development of effective CA inhibitors is limited by the lack of selectivity which could lead to serious side effects.Citation2 Hence, it has been of interest to us to develop not only potent hCA inhibitors but also with a promising selectivity for a specific isoform. We have previously carried out synthesis of various phenols and methoxyphenols in addition to derivatives of some natural products which possess different structures.Citation20 Some of our recently synthesised compounds were found to inhibit CAs in the milimolar to low nanomolar ranges.Citation21 In the current study, we focussed on the synthesis and inhibitory effects of some pyrazole derivatives against hCA I and II isoforms. Computational studies were also used to enlighten their activities based on the binding interactions with the target enzymes and their calculated molecular properties.
2. Methods and materials
2.1. Chemistry
1H and 13 C spectra were recorded on Bruker Ascend 400 (100)–MHz spectrometers and chemical shifts were reported (λ) relative to Me4Si as internal standard. The elemental analyses were performed on a Costech ESC 4010 instrument. The IR spectra were determined using a Perkin Elmer 1600 Fourier Transform-infrared (FT-IR) spectrophotometer on a KBr disc. Melting points were determined by using a Barnstead electrothermal 9200 series digital apparatus. Absorption spectra were recorded on a Shimadzu UV-1800 spectrophotometer. CA esterase activity was determined according to Verpoorte et al.Citation22
2.2. General methods for the synthesis of chalcones (1–5)
An aqueous solution of NaOH (60%, 10 ml) was added into the ethanol (6 ml) solution of substituted carbaldehyde (20.0 mmol) and a suitable acetophenone (20.0 mmol). The mixture was stirred for a day at room temperature and it was then poured on ice-water. The mixture was neutralised using 6 M hydrochloric acid. The yellow precipitate obtained was filtered and crystallized from ethanol-water. (E)-1-(4-aminophenyl)-3-(3,4,5-trimethoxyphenyl)prop-2-en-1-one (1), (E)-1-(4-bromophenyl)-3-(3,4,5-trimethoxyphenyl)prop-2-en-1-one (2), (E)-1-p-tolyl-3-(3,4,5-trimethoxyphenyl)prop-2-en-1-one (3), (E)-1-(4-aminophenyl)-3-(3,4-dimethoxyphenyl)prop-2-en-1-one (4), (E)-3-(4-(dimethylamino)phenyl)-1-(4-hydroxyphenyl)prop-2-en-1-one (5) were synthesised according to the literature,Citation23 respectively.
2.2.1. General methods for the synthesis of pyrazoles (6–10)
A mixture of (0.007 mol) chalcone and (0.014 mol) thiosemicarbazide were refluxed in ethanol (15 ml) while stirring vigorously. After complete dissolution of the reactants, a solution of (0.014 mol) of KOH in ethanol (15 ml) was added dropwise. The solution was refluxed for another 18 h, allowed to warm at room temperature and then stirred for 4 h. The crude product was refrigerated overnight. The precipitate formed was filtered off and crystallized from ethanol twice yielding yellow crystals.
2.2.2. 3-(4-Aminophenyl)-5-(3,4,5-trimethoxyphenyl)-4,5-dihydro-1H-pyrazole-1-carbothioamide (6)
IR (ATR), ν/cm−1: 3439, 3331, 1591, 1342. 1H-NMR (DMSO-d6), (δ:ppm): 3.10 (1H, dd, J = 3.2 and 3.2 Hz), 3.62 (3H, s), 3.70 (6H, s), 3.86 (1H, dd, J = 13.6 and 10.8 Hz), 5.80 (1H, dd, J = 3.2 and 2.8 Hz), 6.40 (2H, s), 6.56 (2H, d, J = 8.8 Hz), 7.53 (2H, d, J = 8.4 Hz). 13 C-NMR (DMSO-d6), (δ:ppm): 175.80, 156.52, 153.35, 151.91, 139.31, 136.82, 129.21, 118.08, 113.71, 103.07, 62.93, 60.38, 56.59, 42.95. Anal. calcd. for: C19H22N4O3S: C, 59.05; H, 5.74; N, 14.50; Found: C, 59.07; H, 5.71; N, 14.48.
2.2.3. 3-(4-Bromophenyl)-5-(3,4,5-trimethoxyphenyl)-4,5-dihydro-1H-pyrazole-1-carbothioamide (7)
IR (ATR), ν/cm−1: 3439, 3263, 1587, 1334. 1H-NMR (DMSO-d6), (δ:ppm): 3.21 (1H, dd, J = 3.6 and 3.2 Hz), 3.64 (3H, s), 3.71 (6H, s), 3.90 (1H, dd, J = 12.6 and 12.0 Hz), 5.89 (1H, dd, J = 2.8 and 2.8 Hz), 6.43 (2H, s), 7.65 (2H, d, J = 8.4 Hz), 7.82 (2H, d, J = 8.4 Hz). 13 C-NMR (DMSO-d6), (δ:ppm): 176.20, 154.52, 153.80, 139.64, 136.90, 132.07, 130.42, 129.54, 124.46, 104.37, 63.57, 59.99, 55.98, 42.75. Anal. calcd. for: C19H20BrN3O3S: C, 50.67; H, 4.48; N, 9.33; Found: C, 50.65; H, 4.47; N, 9.29.
2.2.4. 3-p-Tolyl-5-(3,4,5-trimethoxyphenyl)-4,5-dihydro-1H-pyrazole-1-carbothioamide (8)
IR (ATR), ν/cm−1: 3433, 3263, 1580, 1338. 1H-NMR (DMSO-d6), (δ:ppm): 2.35 (3H, s), 3.20 (1H, dd, J = 3.6 and 3.2 Hz), 3.64 (3H, s), 3.71 (6H, s), 3.92 (1H, dd, J = 12.0 and 11.0 Hz), 5.88 (1H, dd, J = 3.2 and 2.8 Hz), 6.43 (2H, s), 7.27 (2H, d, J = 4 Hz), 7.76 (2H, d, J = 4 Hz). 13 C-NMR (DMSO-d6), (δ:ppm): 176.76, 155.73, 153.40, 141.02, 139.18, 136.88, 129.74, 128.59, 127.59, 103.05, 63.34, 60.38, 56.63, 39.68, 21.50. Anal. calcd. for: C20H23N3O3S: C, 62.32; H, 6.01; N, 10.90; Found: C, 62.31; H, 6.02; N, 10.89.
2.2.5. 3-(4-Aminophenyl)-5-(3,4-dimethoxyphenyl)-4,5-dihydro-1H-pyrazole-1-carbothioamide (9)
IR (ATR), ν/cm−1: 3443, 3304, 1576, 1357. 1H-NMR (DMSO-d6), (δ:ppm): 3.06 (1H, dd, J = 2.8 and 2.8 Hz), 3.70 (3H, s), 3.71 (3H, s), 3.82 (1H, dd, J = 7.2 and 6 Hz), 5.73 (1H, s), 5.81 (1H, dd, J = 2.8 and 2.4 Hz), 6.57 (2H, d, J = 8.8 Hz), 6.77 (1H, d, J = 2), 6.85 (1H, d, J = 8.4), 7.53 (2H, d, J = 8.8 Hz). 13 C-NMR (DMSO-d6), (δ:ppm): 175.51, 156.48, 151.87, 149.08, 148.16, 136.04, 130.61, 129.17, 114.78, 113.73, 112.29, 110.21, 65.92, 55.99, 55.93, 42.93. Anal. calcd. for: C18H20N4O2S: C, 60.65; H, 5.66; N, 15.72; Found: C, 60.64; H, 5.63; N, 15.72.
2.2.6. 5-(4-(Dimethylamino)phenyl)-3-(4-hydroxyphenyl)-4,5-dihydro-1H-pyrazole-1-carbothioamide (10)
IR (ATR), ν/cm−1: 3430, 3260, 1580, 1346. 1H-NMR (DMSO-d6), (δ:ppm): 2.99 (6H, s), 3.06 (1H, dd, J = 2.8 and 2.0 Hz), 3.72 (1H, dd, J = 10.8 and 11.2 Hz), 5.78 (1H, dd, J = 2.4 and 2.4 Hz), 6.65 (2H, d, J = 8.8 Hz), 6.83 (2H, d, J = 8.8 Hz), 6.95 (2H, d, J = 8.4 Hz), 7.71 (2H, d, J = 8.4 Hz), 7.82 (br, –NH2), 11.20 (br, –OH). 13 C-NMR (DMSO-d6), (δ:ppm): 175.75, 160.32, 156.50, 151.91, 130.31, 129.21, 127.15, 119.01, 113.95, 63.81, 42.05, 41.78. Anal. calcd. for: C18H20N4OS: C, 63.50; H, 5.92; N, 16.46; Found: C, 63.54; H, 5.88; N, 16.44.
2.3. Biological activity
2.3.1. Inhibition studies of carbonic anhydrase I and II isoforms
Enzyme activity was determined spectrophotometrically by following the change in absorbance at 348 nm of 4-nitrophenylacetate to 4-nitrophenolate over a period of 3 min at 25 °C.Citation21 The enzymatic reaction contained 1.4 ml 0.05 M Tris-SO4 buffer (pH 7.4), 1 ml 3 mM 4-nitrophenylacetate, 0.5 ml H2O and 0.1 ml enzyme solution, in a total volume of 3.0 ml.Citation24 Inhibitory effects of compounds 1–10 were compared with acetazolamide (AZA). Different inhibitor concentrations were used and all compounds were tested in triplicate at each concentration used. Control cuvette activity was acknowledged as 100% in the absence of inhibitor. An Activity% – [Inhibitor] graph was drawn for each inhibitor.Citation25 The curve-fitting algorithm allowed for obtaining the IC50 values, working at the lowest concentration of substrate of 0.15 mM, from which Ki values were calculated.Citation20,Citation21 The catalytic activity of these enzymes was calculated from Lineweaver-Burk plots, as reported previously, and represent the mean from at least three different determinations. The hCA I and II isoenzymes used here were purified from human blood as previously described.Citation21
2.4. Computational section
2.4.1. Ligand and protein preparation
As a crucial step to meet minimum requirements for further computational calculations, all the studied ligands and target proteins were prepared. LigPrep toolCitation26 interfaced with Maestro module of SchrödingerCitation26 suite was used for the ligand preparation. The 3 D structures including all possible tautomers and ionisation states at pH 7.0 ± 2.0 of all the ligands 1–10 and the reference compound AZA were generated and geometrically minimised using optimised potential liquid simulations (OPLS3) force field.Citation27
Schrödinger’s multi-step Protein Preparation Wizard PrepWizard)Citation28 were used for the protein preparations. As an initial step, high-resolution protein crystal structures of CA I and II (PDB Ids: 2NMX and 3HS4, respectively), both in complexed with a native ligand, were retrieved from RCSB Protein Data Bank. Charges and bond orders were assigned, hydrogens were added to the heavy atoms, all water molecules and heteroatoms were then removed keeping the native ligands and zinc metals in the active site. The final structures were optimised and finally minimised using OPLS3 force field to avoid steric clashes between the atoms.
2.4.2. Molecular docking
A grid representing the binding pocket was generated using the centroid of co-crystallized native ligands. Default settings were kept in each case. Glide XP (extra precision)Citation29 module of Schrödinger Suite was used to dock the synthesised compounds into the active site of the crystal structures. The rescoring was performed to calculate and improved binding energy calculations with Prime’s Molecular Mechanics‐Generalized Born Surface Area (MM‐GBSA) protocol using VSGB solvation model.Citation26,Citation30
2.4.3. Calculation of physicochemical and ADME properties
QikPropCitation26 module of Schrodinger was used to calculate some commonly used molecular descriptors such as dipole moment, logarithm of octanol-water partition coefficient (QPlogPo/w), percent human oral absorption, polar surface area (PSA) and violations to the Lipinski’s rule of five.Citation30
3. Results and discussion
3.1. Chemistry
The new 4,5-dihydro-1H-pyrazole-1-carbothioamide derivatives (6–10) were prepared from the chalcones 1and 5 according to the reactions outlined in . Initially, chalcones (1–5) were prepared through Claisen-Schmidt condensation, which is the most important reaction in the formation of 1,3-diphenyl-2-propene-1-ones (chalcones), of various benzaldehydes with 4-amino, bromo, methyl and hydroxyl acetophenones. The reaction was carried out in 60% sodium hydroxide: ethanol for 24 h as stated in previous works.Citation23 Then, new 1-thiocarbamoyl-3,5-diaryl-4,5-dihydro-(1H)-pyrazole derivatives (6–10) were obtained by cyclisation of chalcones (1–5) with two equivalents of thiosemicarbazide with sodium hydroxide presence in ethanol. Compound 7–9 was reported in a previous study.Citation23 The synthesis of the compounds 6, 8 and 10 is being reported for the first time in this work. At the end of the synthesis, the crude product was purified by recrystallization two times from EtOH: H2O to obtain yellow crystals. The structures of all the compounds were confirmed by spectral FT-IR, 1H and 13 C NMR and elemental analyses. 1H NMR spectra of the title compounds were consistent with expected resonance signals in terms of chemical shifts and integrations. All the 13 C NMR findings confirmed the structures proposed. Selected 13 C NMR spectrum are given in .
Figure 1. (a) 60% Aq NaOH, EtOH, 24 h; (b) Thiosemicarbazide, KOH, EtOH, reflux 18 h.
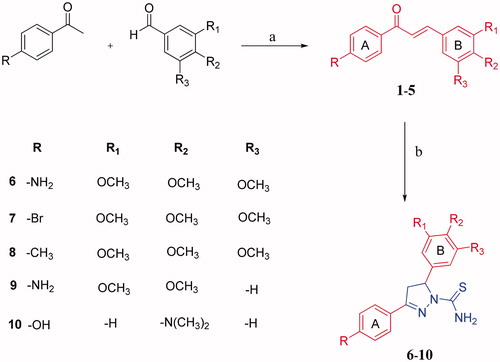
Figure 2. 13 C NMR spectrum of compound 6.
3.2. Biological evaluation
The inhibitory effects of all the synthesised compounds were investigated for the first time against hCA isozymes: hCA I and hCA II. Human CA I and II isozymes were purified by one step chromatography technique and the activity of the effluents was determined by the hydratase method, using CO2 as substrate and further kinetic studies were performed using the esterase activity method, using 4-nitrophenyl acetate (NPA) as substrate.Citation21 Inhibition characteristics of five pyrazoles, five chalcones and various reference compounds are given in . It is clear from the results that all molecules were found to act as low-nanomolar hCA I-II inhibitors. According to the experimental findings, all chalcone and new pyrazole compounds used in this study had better inhibition constants than the clinically used inhibitor acetazolamide (AZA) and other widely used inhibitors (11, 12, 13) and also comparable IC50 values with AZA. However, they possessed different selectivity against hCAI-II. The new compounds 6 and 10 with the IC50 values ranging between 23.87 and 24.37 nM showed promising powerful inhibitory profiles compared to the standard drug AZA and they all had comparable IC50 values against hCA I. The amino or hydroxyl substituents on phenyl ring could easily be predicted to be involved in making hydrogen bonds with the active site as observed in classical hCA I sulphonamide inhibitors. Changing the hydrophilic substituents, –NH2 (6) or –OH (10) with hydrophilic substituents, –Br (7) or –CH3 (8) on ring A, have negligible effect on the observed activity. It is clear that the lack of moieties which can make favourable hydrogen bonding are being compensated with hydrophobic groups that also enhance affinity by hydrophobic interactions. Furthermore, replacing methoxy substituents with N,N-dimethylamino substituent (10) does not seem to have much effect on the activity. It can be concluded that methyl groups in both cases have favourable contacts with hydrophobic sites of the active region. Oxygen atoms in metoxy groups could have extra interactions with the hydrophilic regions. However, methoxy substituents on ring B have large impact on the activity towards hCA II. The IC50 value diminishes more than two-folds when trimethoxy phenyl is being replaced with N,N-dimethy aniline (10) which results in a promising selectivity profile for this compound. As a result of those observations, we found it necessary to carry out in silico studies.
Table 1. The data of hCA I and II inhibition with compounds 1–10 and AZA.
3.3. Computational study
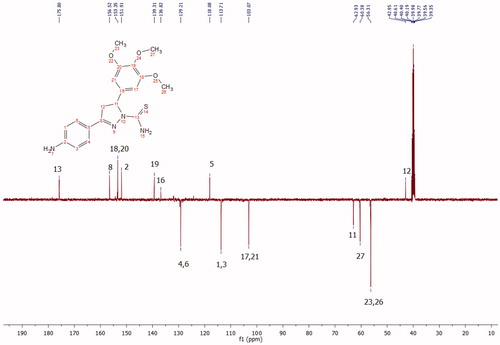
The active site of both hCAI and II, as in all other CAs, is lined with both hydrophobic and hydrophilic residues (). Hydrophobic part is believed to be responsible for entrapping the lipophilic CO2 molecule, and the other the part of the active site helps releasing the polar components after CO2 hydration reaction to the environment.Citation32 All the synthesised new compounds were found to have low IC50 values in the low nanomolar range (). Although it is well known that the commonly used docking software available at the market performs well in predicting the active conformations of the biologically active compounds but the present scoring functions are not expected to discriminate between active and inactive compounds.Citation33 We have obtained satisfying results using Glide in our recent studies.Citation34 In the current study, in order to improve the docking scores, we performed more rigorous binding energy calculations using MM-GBSA protocol. We have let the residues in 3 Å distance from the ligands to be relaxed during the computations. MM-GBSA ΔGbinding values substantially agreed well with the experimental inhibition data. According to the MM-GBSA calculations, compound 6 scored top in both hCA isoforms. It is noteworthy to mention that, according to the IC50 results listed in , the compound 10 showed a slight hCA I versus hCA II selectivity, with a selectivity ratio (SR) of 3.46. The 2 D and 3 D ligand interaction diagrams of both compounds with the hCA I and II isoforms are shown in . Compound 6 binds in a rather similar manner with the active site of both isoforms. The amino group of aniline part acts as a hydrogen bond donor and interacts with the TYR199 residue while the aromatic part establishes a π-π interaction with the residue HIS94. It also has favourable interactions with the polar residues at the active site entrance. Compound 10 has same type of interactions with the critical residues at the binding site of hCA I: phenolic -OH group participates in hydrogen bonding with THR199 and the same π-π interaction of the aromatic ring with the residue HIS94 is observed. Interestingly, the orientation is inverted in the active region of hCA II. The N,N-dimethyl aniline moiety is buried deep in the active site whereas phenol interacts with the negatively charged residue GLU69. The lack of contacts with the key residues HIS 94 and THR 199 could be responsible for the decreased activity towards hCA II.
Figure 3. A representative hydrophobic/philic surfaces of the active site of hCA I complexed with a native ligand. (PDB ID: 2NMX; hydrophilic surfaces cyan colour; hydrophobic surfaces: orange colour).
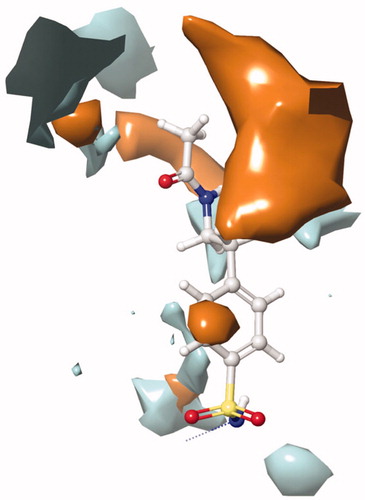
Figure 4. The 2 D and 3 D ligand interaction diagrams of hCA I-6 (R) (top) and hCA II-6 (R) (bottom) complexes obtained from prime MM-GBSA using Glide XP docked poses (In 3 D representation, hydrogen bonds are shown with yellow dashed lines, π-π interactions are shown with cyan colour. The distance between Zinc metal and interacting atom is shown with pink colour).
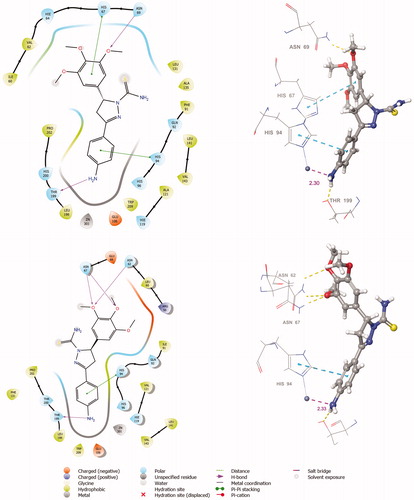
Figure 5. The 2 D and 3 D ligand interaction diagrams of hCA I-10 (S) (top) and hCA II-10 (S) (bottom) complexes obtained from prime MM-GBSA using Glide XP docked poses (In 3 D representation, hydrogen bonds are shown with yellow dashed lines, π-π interactions are shown with cyan colour. The distance between zinc metal and interacting atom is shown with pink colour).
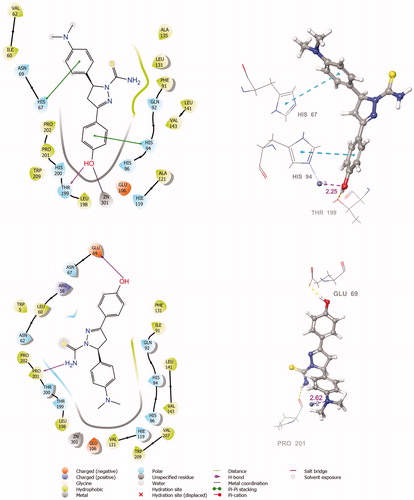
Table 2. Glide XP Docking scores, MM-GBSA ΔGbinding energy values and selected molecular properties of compounds 1–10 and AZA.
We have also calculated some molecular descriptors commonly used in absorption, distribution, metabolism and excretion (ADME) analysis (). As could be seen from the table, all of the new compounds obey Lipinski’s rule of five, which is an indication of the drug-likeness of a molecule, and PSA values are within the range that Veber et al. suggested.Citation35
4. Conclusion
In the current study, starting from some chalcones, design, synthesis and characterisation of new pyrazole derivatives were reported. All the synthesised compounds were then evaluated for their inhibitory properties against hCA I and hCA II isoenzymes. They exhibited significant inhibitory features at low nanomolar concentrations ranging between 21.98 and 25.14 nM. Molecular docking studies further supported observed inhibitory profiles. Compound 10 which had slight hCA I versus hCA II selectivity, binds with hCA I in similar orientations with other compounds but it adopts different conformation in the active site of hCA II. According to the in silico molecular properties calculations, all compounds also obeyed the drug likeness properties. The new compounds presented in this study, might be promising lead compounds for the development of more selective and potent inhibitors as alternatives to the classical CA inhibitors.
Disclosure statement
The authors declare that they have no competing interest.
Related Research Data
References
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81.
- (a) Arslan T, Celik G, Celik H. Synthesis and biological evaluation of novel bischalcone derivatives as carbonic anhydrase inhibitors. Arch Pharm 2016;349:741–8; (b) Vullo D, Del Prete S, Fisher GM. Sulfonamide inhibition studies of the η-class carbonic anhydrase from the malaria pathogen Plasmodium falciparum. Bioorg Med Chem 2015;23:526–31.
- (a) Senturk M, Alici HA, Beydemir S. In vitro and in vivo effects of some benzodiazepine drugs on human and rabbit erythrocyte carbonic anhydrase enzymes. J Enzyme Inhib Med Chem 2012;27:680–4; (b) De Simone G, Supuran CT. (In)organic anions as carbonic anhydrase inhibitors. J Inorg Biochem 2012;111:117–29; (c) Akin Kazancioglu E, Guney M, Senturk M, et al. Simple methanesulfonates are hydrolyzed by the sulfatase carbonic anhydrase activity. J Enzyme Inhib Med Chem 2012;27:880–5.
- Orhan F, Şentürk M, Supuran CT. Interaction of anions with a newly characterized alpha carbonic anhydrase from Halomonas sp. J Enzyme Inhib Med Chem 2016;31:1119–23.
- (a) Abdel-Aziz AA-M, El-Azab AS, Ekinci D, et al. Investigation of arenesulfonyl-2-imidazolidinones as potent carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2015;30:81–4; (b) Urcar H, Senturk E, Senturk M, et al. Investigation of effects of some catecholamines on the activity of carbonic anhydrase enzyme purified from bovine kidney tissue. Acta Physiol 2016;218:57; (c) Tas M, Senturk E, Ekinci D, et al. Comparison of blood carbonic anhydrase activity of athletes performing interval and continuous running exercise at high altitude. J Enzyme Inhib Med Chem 2019;34:219–25.
- Korkmaz N, Obaidi OA, Senturk M, et al. Synthesis and biological activity of novel thiourea derivatives as carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2015;30:75–80.
- (a) Guney M, Cavdar H, Senturk M, et al. Synthesis and carbonic anhydrase inhibitory properties of novel uracil derivatives. Bioorg Med Chem Lett 2015;25:3261–3; (b) Durdagi S, Senturk M, Guney M, et al. Design of novel uracil derivatives as inhibitors of carbonic anhydrase I & II, acetylcholinesterase, butyrylcholinesterase, and glutathione reductase using in silico, synthesis and in vitro studies. FEBS J 2016;283:106–106.
- (a) Balaydin HT, Senturk M, Goksu S, et al. Synthesis and carbonic anhydrase inhibitory properties of novel bromophenols and their derivatives including natural products: Vidalol B. Eur J Med Chem 2012;54:423–8; (b) Balaydin HT, Senturk M, Menzek A. Synthesis and carbonic anhydrase inhibitory properties of novel cyclohexanonyl bromophenol derivatives. Bioorg Med Chem Lett 2012;22:1352–7; (c) Balaydin HT, Durdaği S, Ekinci D, et al. Inhibition of human carbonic anhydrase isozymes I, II and VI with a series of bisphenol, methoxy and bromophenol compounds. J Enzyme Inhib Med Chem 2012;27:467–75.
- (a) Arslan T, Türkoğlu EA, Şentürk M, et al. Novel chalcone substituted benzenesulfonamides as inhibitors for human carbonic anhydrases. Bioorg Med Chem Lett 2016;26:5867–70; (b) Demirdag R, Comakli V, Senturk M, et al. Characterization of carbonic anhydrase from sheep kidney and effects of sulfonamides on enzyme activity. Bioorg Med Chem 2013;21:1522–5; (c) Yaseen R, Ekinci D, Senturk M, et al. Pyridazinone substituted benzenesulfonamides as potent carbonic anhydrase inhibitors. Bioorg Med Chem Lett 2016;26:1337–41; (d) Ekinci D, Senturk M, Senturk E. Purification and characterization of carbonic anhydrase enzyme from bovine heart tissue and investigation of inhibition effects of some sulphonamide derivative drugs. Acta Physiol 2015;215:99–99.
- (a) Cavusoglu K, Celebi F, Celik M, et al. Investigation of the effect of Shiga-toxin on rat serum carbonic anhydrase enzyme. Acta Physiol 2017;221:125–125; (b) Ekinci D, al-Rashida M, Abbas G, et al. Chromone containing sulfonamides as potent carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2012;27:744–7; (c) Karagoz L, Arslan T, Ekinci D, et al. Purification of carbonic anhydrase enzyme from bovine liver tissue and investigation of the inhibitory effects of bischalcone derivatives. Acta Physiol 2016;218:57–57.
- Reddy VG, Reddy TS, Nayak VL, et al. Design, synthesis and biological evaluation of N-((1-benzyl-1H-1,2,3-triazol-4-yl)methyl)-1,3-diphenyl-1H-pyrazole-4-carboxamides as CDK1/Cdc2 inhibitors. Eur J Med Chem 2016;122:164–77.
- Alam R, Wahi D, Singh R, et al. Design, synthesis, cytotoxicity, HuTopoIIα inhibitory activity and molecular docking studies of pyrazole derivatives as potential anticancer agents. Bioorg Chem 2016;69:77–90.
- Jiang D, Zheng X, Shao G, et al. Discovery of a novel series of phenyl pyrazole inner salts based on fipronil as potential dual-target insecticides. J Agric Food Chem 2014;62:3577–83.
- Furuya T, Suwa A, Nakano M, et al. Synthesis and biological activity of a novel acaricide, pyflubumide. J Pestic Sci 2015;40:38–43.
- Kaushik D, Khan SA, Chawla G, et al. N'-[(5-chloro-3-methyl-1-phenyl-1H-pyrazol-4-yl)methylene] 2/4-substituted hydrazides: synthesis and anticonvulsant activity. Eur J Med Chem 2010;45:3943–9.
- Abdel-Aziz M, Abuo-Rahma GE-DA, Hassan AA. Synthesis of novel pyrazole derivatives and evaluation of their antidepressant and anticonvulsant activities. Eur J Med Chem 2009;44:3480–7.
- Kumar S, Ceruso M, Tuccinardi T, et al. Pyrazolylbenzo[d]imidazoles as new potent and selective inhibitors of carbonic anhydrase isoforms hCA IX and XII. Bioorg Med Chem 2016;24:2907–13.
- (a) Arslan T. Synthesis and characterisation of new sulfonamide chalcones containing an azo group. J Chem Res 2018;42:267–70; (b) Arslan T. An efficient synthesis of novel type chalcones containing 8-hydroxyquinoline under green conditions. Erzincan University J Sci Technol 2018;11:321–7.
- Fu D-J, Zhang S-Y, Liu Y-C, et al. Design, synthesis and antiproliferative activity studies of novel dithiocarbamate-chalcone derivates. Bioorg Med Chem Lett 2016;26:3918–22.
- (a) Isik S, Vullo D, Durdagi S, et al. Interaction of carbonic anhydrase isozymes I, II, and IX with some pyridine and phenol hydrazinecarbothioamide derivatives. Bioorg Med Chem Lett 2015;25:5636–41; (b) Fidan I, Salmas RE, Arslan M, et al. Carbonic anhydrase inhibitors: design, synthesis, kinetic, docking and molecular dynamics analysis of novel glycine and phenylalanine sulphonamide derivatives. Bioorg Med Chem 2015;23:7353–8; (c) Ekinci D, Cavdar H, Durdagi S, et al. Structure-activity relationships for the interaction of 5,10-dihydroindeno[1,2-b]indole derivatives with human and bovine carbonic anhydrase isoforms I, II, III, IV and VI. Eur J Med Chem 2012;49:68–73.
- (a) Ozdemir ZO, Senturk M, Ekinci D. Carbonic anhydrase inhibitors: Inhibition of mammalian isoforms I, II and VI with thiamine and thiamine-like molecules. J Enzyme Inhib Med Chem 2013;28:316–9; (b) Yerlikaya E, Erdogan O, Demirdag R, et al. Expression of hCA IX isoenzyme by using sumo fusion partner and examining the effects of antitumor drugs. Turk J Biochem 2015;40:334–42; (c) Arslan T, Biyiklioglu Z, Şentürk M. The synthesis of axially disubstituted silicon phthalocyanines, their quaternized derivatives and firstly inhibitor effect on human cytosolic carbonic anhydrase isozymes hCAI and II. RSC Advances 2018;8:10172–8.
- Verpoorte JA, Mehta S, Edsall JT. Esterase activities of human carbonic anhydrases B and C. J Biol Chem 1967;242:4221–9.
- (a) Tomar V, Bhattacharjee G, Kamaluddin S, et al. Synthesis of new chalcone derivatives containing acridinyl moiety with potential antimalarial activity. Eur J Med Chem 2010;45:745–51; (b) Nepali K, Singh G, Turan A, et al. A rational approach for the design and synthesis of 1-acetyl-3,5-diaryl-4,5-dihydro(1H)pyrazoles as a new class of potential non-purine xanthine oxidase inhibitors. Bioorg Med Chem 2011;19:1950–8.
- Turkoglu EA, Senturk M, Supuran CT, et al. Carbonic anhydrase inhibitory properties of some uracil derivatives. J Enzyme Inhib Med Chem 2017;32:74–7.
- Lineweaver H, Burk D. The determination of enzyme dissociation constants. J Am Chem Soc 1934;56:658–66.
- Schrödinger Release 2017-3: Schrödinger Suite 2017-3 Protein Preparation Wizard; Epik, Schrödinger, LLC, New York, NY, 2017; Impact, Schrödinger, LLC, New York, NY, 2017; LigPrep, Schrödinger, LLC, New York, NY, 2017; Prime, Schrödinger, LLC, New York, NY, 2017; QikProp, Schrödinger, LLC, New York, NY, 2017.
- Harder E, Damm W, Maple J, et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J Chem Theory Comput 2016;12:281–96.
- Sastry GM, Adzhigirey M, Day T, et al. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des 2013;27:221–34.
- Friesner RA, Banks JL, Murphy RB, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 2004;47:1739–49.
- (a) Bashford D, Case DA. Generalized born models of macromolecular solvation effects. Annu Rev Phys Chem 2000;51:129–52; (b) Li J, Abel R, Zhu K, et al. The VSGB 2.0 model: a next generation energy model for high resolution protein structure modeling. Proteins 2011;79:2794–812; (c) Lipinski A, Lombardo F, Dominy BW, et al. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Delivery Rev 1997;23:3–26.
- Singasane N, Kharkar PS, Ceruso M, et al. Inhibition of carbonic anhydrase isoforms I, II, IX and XII with Schiff’s bases incorporating iminoureido moieties. J Enzyme Inhib Med Chem 2015;30:901–7.
- De Simone G, Alterio V, Supuran CT. Exploiting the hydrophobic and hydrophilic binding sites for designing carbonic anhydrase inhibitors. Expert Opin Drug Discov 2013;8:793–810.
- (a) Ece A, Sevin F. The discovery of potential cyclin A/CDK2 inhibitors: a combination of 3D QSAR pharmacophore modeling, virtual screening, and molecular docking studies. Med Chem Res 2013;22:5832–43; (b) Mascarenhas NM, Ghoshal N. An efficient tool for identifying inhibitors based on 3D-QSAR and docking using feature-shape pharmacophore of biologically active conformation—a case study with CDK2/cyclinA. Eur J Med Chem 2008;43:2807–18.
- (a) Er M, Ergüven B, Tahtaci H, et al. Synthesis, characterization, preliminary SAR and molecular docking study of some novel substituted imidazo [2, 1-b][1, 3, 4] thiadiazole derivatives as antifungal agents. Med Chem Res 2017;26:615–30; (b) Tahtaci H, Karacik H, Ece A, et al. Design, synthesis, SAR and molecular modeling studies of novel imidazo[2,1-b][1,3,4]thiadiazole derivatives as highly potent antimicrobial agents. Mol Inform 2018;37:83–92; (c) Türk S, Karakuş S, Ece A, et al. Synthesis, structure elucidation and biological activities of some novel 4 (3H)-quinazolinones as anti-biofilm agents. Lett Drug Des Disc 2019;16:313–321; (d) Ece A. Towards more effective acetylcholinesterase inhibitors: a comprehensive modelling study based on human acetylcholinesterase protein–drug complex. J Biomol Struct Dyn 2019;1–8; (e) Yamali C, Gul HI, Ece A, et al. Synthesis, biological evaluation and in silico modelling studies of 1, 3, 5-trisubstituted pyrazoles carrying benzenesulfonamide as potential anticancer agents and selective cancer-associated hCA IX isoenzyme inhibitors. Bioorg Chem 2019;92:103222.
- Veber DF, Johnson SR, Cheng HY, et al. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem 2002;45:2615–23.