
Abstract
Two series of aminoguanidines containing an alkynyl moiety were designed, synthesised, and screened for antibacterial and anticancer activities. Generally, the series 3a–3j with a 1,2-diphenylethyne exhibited better antibacterial activity than the other series (6a–6k) holding 1,4-diphenylbuta-1,3-diyne moiety antibacterial activity. Most compounds in series 3a–3j showed potent growth inhibition against the tested bacterial strains, with minimum inhibitory concentration (MIC) values in the range 0.25–8 µg/mL. Compound 3g demonstrated rapid and persistent bactericidal activity at 2 × MIC. The resistance study revealed that resistance of the tested bacteria towards 3g is not easily developed. Molecular docking studies revealed that compounds 3g and 6e bind strongly to the LpxC and FabH enzymes. Moreover, excellent activity of selected compounds against the growth of cancer cell lines A549 and SGC7901 was also observed, with IC50 values in the range 0.30–4.57 µg/mL. These findings indicate that compounds containing the aminoguanidine moiety are promising candidates for the development of new antibacterial and anticancer agents.
Introduction
Multidrug-resistant (MDR) Gram-negative bacterial strains have arisen against all antibiotics in clinical use. Infections caused by these MDR bacteria threaten global public healthCitation1,Citation2 and are associated with high mortality rates. New antibacterial drugs with novel chemical scaffolds and targets are urgently required to combat infections due to drug-resistant strainsCitation3,Citation4.
One of the validated antibiotic targets against Gram-negative bacteria is UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase (LpxC), an essential enzyme catalysing the first committed step in the lipid A biosynthetic pathway. Because there is no gene homology in humans, inhibiting LpxC could result in the death of bacteria without causing side effects in the bodyCitation5. Based upon these facts, LpxC has become a promising target for developing novel therapeutics against MDR Gram-negative pathogensCitation6–8.
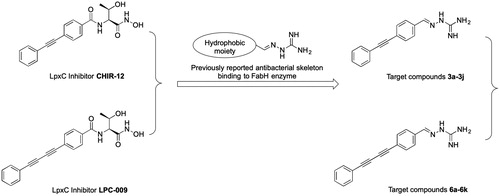
Studies of the enzymatic mechanism underlying LpxC have identified a hydrophobic tunnel that binds a myristate fatty acyl chain of the natural substrate and leads to a Zn2+ active site responsible for deacetylationCitation9,Citation10. Since the discovery of L-161240 as the first LpxC inhibitor in the 1990sCitation11, many LpxC inhibitors have been reported as antibacterial agentsCitation6–8. Most LpxC inhibitors share common structural features that mimic the natural Zn2+-binding substrate: (1) a hydroxamate head group, (2) a central linker, and (3) a lipophilic tailCitation12. Among the well-characterised compounds, threonyl-hydroxamate derivatives, such as CHIR-12 and LPC-009, are representative LpxC inhibitorsCitation13–15. The hydroxamate group of these compounds occupies the active site, and their diphenyl acetylene or phenyl-diyne group penetrates the hydrophobic passageCitation16,Citation17.
Aminoguanidine is functional group with a high polarity and capacity for hydrogen bonding with many critical amino acid residues as well as metal ions. Many aminoguanidine derivatives exhibit antitumor activity via the formation of complexes with metal ions. In our previous work, some aminoguanidine derivatives were reported as potent antibacterial agents against Gram-positive bacteria and Gram-negative bacteria including MDR clinical isolatesCitation18–20. The binding of these compounds involves a specific interaction with the β-ketoacyl-acyl carrier protein synthase III (FabH) enzymeCitation20. By analysing the SAR of these compounds, it can be concluded that aminoguanidine combined with a hydrophobic moiety in the form of azomethine imine is the common structural requirement for their antibacterial activity.
In this contribution, we built upon the above observations by replacing the threonyl-hydroxamate group in the lead compounds CHIR-12 and LPC-009 with aminoguanidine (), yielding two series of aminoguanidines 3a–3j and 6a–6k. We anticipated that this design would promote binding with both FabH and LpxC, resulting in high and broad-spectrum antibacterial activity.
Figure 1. The design of target compounds.
All the synthesised compounds were characterised by 1H-NMR, 13C-NMR, and high-resolution mass spectrometry, then evaluated for their antibacterial activity against 12 bacterial strains. To further characterise the antibacterial effects of compound 3g, the propensity for the development of bacterial resistance was determined and a bactericidal time-kill assay was carried out. Molecular docking studies of representative compounds 3g and 6e with LpxC and FabH were performed to understand the binding mechanism. Considering the reported anticancer activity of numerous compounds containing the guanidine moietyCitation21,Citation22, the anticancer activity was also evaluated for some selected compounds against two cancer cell lines (A549 and SGC7901). In this work, some aminoguanidines were discovered with promising antibacterial and anticancer activities.
Materials and methods
Instruments and reagents
All the reagents and solvents were purchased from Aladdin (Shanghai, China) or Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China), and were used as received. Melting points were determined in open capillary tubes and are uncorrected. Reaction courses were monitored by thin-layer chromatography (TLC) on silica gel-precoated F254 plates (Merck, Darmstadt, Germany). Developed plates were examined with UV lamps (254 nm). Nuclear magnetic resonance spectroscopy was performed on an AV-400 spectrometer (Bruker, Zurich, Switzerland) operating at 400 MHz for 1H and 100 MHz for 13C and using DMSO-d6 as the solvent and tetramethylsilane as the internal standard. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-MS) experiments were performed on a Bruker ultrafleXtreme MALDI-TOF/TOF mass spectrometer (Bruker Daltonik GmbH, Leipzig, Germany) equipped with a smartbeam II laser (1000 Hz).
Chemistry
Synthesis of 4-(substituted-phenylethynyl)benzaldehyde (2a–2j)
To a stirred solution of 4-ethynylbenzaldehyde (6.15 mmol, 1 eq) in THF (8 ml) was added substituted-iodobenzenes (6.76 mmol, 1 eq), TEA (0.4 ml), CuI (80 mg, 0.42 mmol, 0.07 eq) and Pd(PPh3)Cl2 (0.18 mmol, 0.03 eq) under N2. After stirring for 3 h at 45 °C, the resulting mixture was concentrated under vacuum. The residue was applied onto a silica gel column eluted with 1–4% ethyl acetate in petroleum ether to afford 4-(substituted-phenylethynyl)benzaldehyde (2a–2j) as a light yellow solid.
Synthesis of 2-(4-(substituted -phenylethynyl)benzylidene)hydrazine-1-carboximidamide (3a–3j)
To a stirred solution of hydrazinecarboximidamide carbonate (5.04 mmol, 1.3 eq) in water (8 ml) was added NaOAc (5.04 mmol, 1.3 eq). After stirring for 0.5 h at room temperature, a mixture of 4-(substituted-phenylethynyl)benzaldehyde (2a–2j) (3.88 mmol, 1 eq) in EtOH (8 ml) was added. Then the resulting solution was stirred at 70 °C for 6 h. The reaction mixture was diluted with water (16 ml) and then cooled to room temperature. After stirred for 3 h, large amount of solids was precipitated. The solids were collected by filtration, washed with EtOH (2 × 0.8 ml), and then dried in an oven under reduced pressure to afford 2-(4-(substituted-phenylethynyl)benzylidene)hydrazine-1-carboximidamide (3a–3j) as a light yellow solid.
2-(4-(Phenylethynyl)benzylidene)hydrazine-1-carboximidamide (3a)
Light yellow solid, m.p. 225–226 °C, yield 88%. 1H-NMR (DMSO-d6, 400 MHz): δ 5.65 (s, 2H, NH2), 6.04 (s, 2H, NH), 7.42–7.57 (m, 5H, Ar-H), 7.50 (d, 2H, J = 8.2 Hz, Ar-H), 7.72 (d, 2H, J = 8.2 Hz, Ar-H), 7.99 (s, 1H, N=CH). 13C-NMR (DMSO-d6, 100 MHz): δ 160.96, 142.06, 137.48, 131.43, 131.33, 128.80, 128.75, 126.32, 122.43, 120.88, 90.09, 89.84. MS m/z 263 (M + 1). ESI-HRMS calcd for C16H15N4+ ([M + H]+) 263.1291; found: 263.1287.
2-(4-((2-Fluorophenyl)ethynyl)benzylidene)hydrazine-1-carboximidamide (3b)
Light yellow solid, m.p. 189–190 °C, yield 75%. 1H-NMR (DMSO-d6, 400 MHz): δ 5.63 (s, 2H, NH2), 6.02 (s, 2H, NH2), 7.26–7.65 (m, 6H, Ar-H), 7.72 (d, 2H, J = 7.7 Hz, Ar-H), 7.99 (s, 1H, N=CH). 13C-NMR (DMSO-d6, 100 MHz): δ 162.20 (d, 1Jc-f = 247.72 Hz), 161.51, 142.38, 138.36, 133.86, 131.93, 131.43 (d, 3Jc-f = 7.92 Hz), 126.79, 125.32 (d, 4Jc-f = 3.4 Hz), 120.77, 116.22 (d, 2Jc-f = 20.8 Hz), 111.25 (d, 2Jc-f = 15.4 Hz), 95.25, 83.68. MS m/z 281 (M + 1). ESI-HRMS calcd for C16H14FN4+ ([M + H]+) 281.1197; found: 281.1193.
2-(4-((3-Fluorophenyl)ethynyl)benzylidene)hydrazine-1-carboximidamide (3c)
Light yellow solid, m.p. 223–224 °C, yield 76%. 1H-NMR (DMSO-d6, 400 MHz): δ 5.68 (s, 2H, NH2), 6.06 (s, 2H, NH2), 7.21–7.48 (m, 4H, Ar-H), 7.51 (d, 2H, J = 8.4 Hz, Ar-H), 7.74 (d, 2H, J = 8.4 Hz, Ar-H), 8.00 (s, 1H, N=CH). 13C-NMR (DMSO-d6, 100 MHz): δ 161.92 (d, 1Jc-f = 243.1 Hz), 161.00, 141.98, 137.79, 131.56, 130.89 (d, 3Jc-f = 8.9 Hz), 127.71 (d, 4Jc-f = 2.7 Hz), 126.33, 124.42 (d, 3Jc-f = 9.5 Hz), 120.39, 117.87 (d, 2Jc-f = 22.7 Hz), 115.96 (d, 2Jc-f = 20.9 Hz), 90.80, 88.78. MS m/z 281 (M + 1). ESI-HRMS calcd for C16H14FN4+ ([M + H]+) 281.1197; found: 281.1196.
2-(4-((4-Fluorophenyl)ethynyl)benzylidene)hydrazine-1-carboximidamide (3d)
Light yellow solid, m.p. 238–239 °C, yield 69%. 1H-NMR (DMSO-d6, 400 MHz): δ 5.62 (s, 2H, NH2), 6.01 (s, 2H, NH2), 7.26–7.64 (m, 4H, Ar-H), 7.49 (d, 2H, J = 8.3 Hz, Ar-H), 7.72 (d, 2H, J = 8.3 Hz, Ar-H), 7.99 (s, 1H, N=CH). 13C-NMR (DMSO-d6, 100 MHz): δ 161.99 (d, 1Jc-f = 246 Hz), 160.99, 142.02, 137.53, 133.65 (d, 3Jc-f = 8.5 Hz), 131.40, 126.30, 120.74, 118.92, 116.05 (d, 2Jc-f = 22.0 Hz), 89.56, 89.02. MS m/z 281 (M + 1). ESI-HRMS calcd for C16H14FN4+ ([M + H]+) 281.1197; found: 281.1199.
2-(4-((2-Chlorophenyl)ethynyl)benzylidene)hydrazine-1-carboximidamide (3e)
Light yellow solid, m.p. 200–203 °C, yield 81%. 1H-NMR (DMSO-d6, 400 MHz): δ 5.71 (s, 2H, NH2), 6.07 (s, 2H, NH2), 7.38–7.69 (m, 4H, Ar-H), 7.54 (d, 2H, J = 8.3 Hz, Ar-H), 7.75 (d, 2H, J = 8.3 Hz, Ar-H), 8.01(s, 1H, N=CH). 13C-NMR (DMSO-d6, 100 MHz): δ 161.01, 141.97, 137.91, 134.53, 133.31, 131.52, 130.28, 129.42, 127.42, 126.37, 122.14, 120.38, 94.86, 86.73. MS m/z 297 (M + 1). ESI-HRMS calcd for C16H14ClN4+ ([M + H]+) 297.0902; found: 297.0906.
2-(4-((3-Chlorophenyl)ethynyl)benzylidene)hydrazine-1-carboximidamide (3f)
Light yellow solid, m.p. 213–214 °C, yield 80%. 1H-NMR (DMSO-d6, 400 MHz): δ 5.62 (s, 2H, NH2), 6.02 (s, 2H, NH2), 7.44–7.65 (m, 6H, Ar-H), 7.73 (d, 2H, J = 8.4 Hz, Ar-H), 7.99 (s, 1H, N=CH). 13C-NMR (DMSO-d6, 100 MHz): δ 161.06, 141.90, 137.88, 133.35, 131.56, 130.74, 130.66, 129.99, 128.79, 126.30, 124.44, 120.29, 91.17, 88.55. MS m/z 297 (M + 1). ESI-HRMS calcd for C16H14ClN4+ ([M + H]+) 297.0902; found: 297.0900.
2-(4-((4-Chlorophenyl)ethynyl)benzylidene)hydrazine-1-carboximidamide (3g)
Light yellow solid, m.p. 232–234 °C, yield 77%. 1H-NMR (DMSO-d6, 400 MHz): 15.73 (s, 2H, NH2), 6.08 (s, 2H, NH2), 7.48–7.59 (m, 6H, Ar-H), 7.73 (d, 2H, J = 8.3 Hz, Ar-H), 8.00 (s, 1H, N=CH). 13C-NMR (DMSO-d6, 100 MHz): δ 160.88, 142.03, 137.62, 133.41, 133.03, 131.48, 128.95, 126.34, 121.32, 120.59, 90.91, 88.95. MS m/z 297 (M + 1). ESI-HRMS calcd for C16H14ClN4+ ([M + H]+) 297.0902; found: 297.0908.
2-(4-((2-Bromophenyl)ethynyl)benzylidene)hydrazine-1-carboximidamide (3h)
Light yellow solid, m.p(0).188–190 °C, yield 65%. 1H-NMR (DMSO-d6, 400 MHz): δ 5.71 (s, 2H, NH2), 6.07 (s, 2H, NH2), 7.33–7.68 (m, 4H, Ar-H), 7.53 (d, 2H, J = 8.2 Hz, Ar-H), 7.75 (d, 2H, J = 8.2 Hz, Ar-H), 8.02 (s, 1H, N=CH). 13C-NMR (DMSO-d6, 100 MHz): δ 160.98, 141.99, 137.88, 133.30, 132.51, 131.45, 130.37, 127.87, 126.35, 124.69, 124.31, 120.42, 94.16, 88.64. MS m/z 341 (M + 1). ESI-HRMS calcd for C16H14BrN4+ ([M + H]+) 341.0396; found: 341.0397.
2-(4-((3-Bromophenyl)ethynyl)benzylidene)hydrazine-1-carboximidamide (3i)
Light yellow solid, m.p. 223–225 °C, yield 73%. 1H-NMR (DMSO-d6, 400 MHz): δ 5.66 (s, 2H, NH2), 6.04 (s, 2H, NH2), 7.36–7.78 (m, 4H, Ar-H), 7.54 (d, 2H, J = 8.3 Hz, Ar-H), 7.73 (d, 2H, J = 8.3 Hz, Ar-H), 8.00 (s, 1H, N=CH). 13C-NMR (DMSO-d6, 100 MHz): δ 161.04, 141.91, 137.85, 133.55, 131.64, 131.56, 130.83, 130.30, 126.30, 124.69, 121.74, 120.31, 91.26, 88.46. MS m/z 341 (M + 1). ESI-HRMS calcd for C16H14BrN4+ ([M + H]+) 341.0396; found: 341.0391.
2-(4-((4-Bromophenyl)ethynyl)benzylidene)hydrazine-1-carboximidamide (3j)
Light yellow solid, m.p. 242–243 °C, yield 70%. 1H-NMR (DMSO-d6, 400 MHz): δ 5.65 (s, 2H, NH2), 6.03 (s, 2H, NH2), 7.49–7.52 (m, 4H, Ar-H), 7.64 (d, 2H, J = 8.4 Hz, Ar-H), 7.72 (d, 2H, J = 8.4 Hz, Ar-H), 7.99 (s, 1H, N=CH). 13C-NMR (DMSO-d6, 100 MHz): δ 160.98, 141.96, 137.71, 133.22, 131.84, 131.47, 126.31, 122.09, 121.67, 120.51, 91.06, 89.02. MS m/z 341 (M + 1). ESI-HRMS calcd for C16H14BrN4+ ([M + H]+) 341.0396; found: 341.0396.
Synthesis of 4-(iodoethynyl)benzaldehyde (4)
Ethynylbenzaldehyde (0.140 g, 1.076 mmol) was dissolved in acetone (10 ml) and AgNO3 (0.055 g, 0.323 mmol) was added. After 0.5 h Et2O (30 ml) and N-Iodosuccinimide (0.242 g, 1.076 mmol) were added. The mixture was stirred for 12 h after which time it was filtered and washed with ice-cold H2O (30 ml). The organic layer was separated. The aqueous layer was washed with Et2O (2 × 5 ml) and the combined organic parts were dried with Na2SO4. The solvent was removed under oil-pump vacuum and the residue was purified by chromatography on silica gel (20 cm; hexane/CH2Cl2 v/v, 1:1) to give 4-(iodoethynyl)benzaldehyde (4) as a yellow powder (Yield = 60%)Citation23.
Synthesis of 4-((substituted-phenyl)buta-1,3-diyn-1-yl)benzaldehyde (5a–5k)
To a stirred solution of 4-(iodoethynyl)benzaldehyde (800 mg, 3.12 mmol, 1 eq) in THF (8 ml) was added substituted-ethynylbenzene (3.32 mmol, 1. 07 eq), TEA (0.4 ml), CuI (42 mg, 0.22 mmol, 0.07 eq) and Pd(PPh3)Cl2 (65 mg, 0. 09 mmol, 0.03 eq) under N2. After stirring overnight at room temperature, the resulting mixture was concentrated under vacuum. The residue was applied onto a silica gel column eluted with 1–2% ethyl acetate in petroleum ether to afford a yellow solid.
Synthesis of 2-(4-((substituted-phenyl)buta-1,3-diyn-1-yl)benzylidene)hydrazine-1-carboximidamide (6a–6k)
To a stirred solution of hydrazinecarboximidamide carbonate (124.03 mg, 0.91 mmol, 1.4 eq) in water (5 ml) was added NaOAc (74.78 mg, 0.91 mmol, 1.4 eq). After stirring for 0.5 h at room temperature, a mixture of 4-((substituted-phenyl)buta-1,3-diyn-1-yl)benzaldehyde (0.65 mmol, 1 eq) in EtOH (5 ml) was added. Then the resulting solution was refluxed overnight. The reaction mixture was diluted with water (15 ml) and then cooled to room temperature. After stirred for 3 h, large amount of solids was precipitated. The solids were collected by filtration, washed with EtOH (2–0.5 ml), and then dried in an oven under reduced pressure to afford an off white solid.
2-(4-(Phenylbuta-1,3-diyn-1-yl)benzylidene)hydrazine-1-carboximidamide (6a)
Off white solid, m.p. 209–212 °C, yield 64%. 1H-NMR (DMSO-d6, 400 MHz): δ 5.66 (s, 2H, NH2), 6.05 (s, 2H, NH2), 7.43–7.63 (m, 5H, Ar-H), 7.54 (d, 2H, J = 8.4 Hz, Ar-H), 7.73 (d, 2H, J = 8.4 Hz, Ar-H), 7.98 (s, 1H, N=CH).13C-NMR (DMSO-d6, 100 MHz): δ 161.17, 141.63, 138.70, 132.48, 132.37, 129.96, 128.94, 126.30, 120.54, 118.69, 82.48, 82.26, 74.24, 73.76. MS m/z 287 (M + 1). ESI-HRMS calcd for C18H15N4+ ([M + H]+) 287.1291; found: 287.1286.
2-(4-((2-Fluorophenyl)buta-1,3-diyn-1-yl)benzylidene)hydrazine-1-carboximidamide (6 b)
Off white solid, m.p. 218–220 °C, yield 76%. 1H-NMR (DMSO-d6, 400 MHz): δ 5.83 (s, 2H, NH2), 6.17 (s, 2H, NH2), 7.27–7.71 (m, 6H, Ar-H), 7.75 (d, 2H, J = 8.4 Hz, Ar-H), 7.99 (s, 1H, N=CH). 13C-NMR (DMSO-d6, 100 MHz): δ 162.97 (d, 1Jc-f = 249.6 Hz), 160.90, 141.72, 138.72, 134.35, 132.57, 132.25 (d, 3Jc-f = 8.0 Hz), 126.37, 125.05 (d, 4Jc-f = 3.5 Hz), 118.50, 115.92 (d, 2Jc-f = 20.0 Hz), 109.14 (d, 2Jc-f = 15.2 Hz), 83.64, 78.40, 75.43, 73.85. MS m/z 305 (M + 1). ESI-HRMS calcd for C18H14FN4+ ([M + H]+) 305.1197; found: 305.1195.
2-(4-((3-Fluorophenyl)buta-1,3-diyn-1-yl)benzylidene)hydrazine-1-carboximidamide (6c)
Off white solid, m.p. 186–190 °C, yield 69%. 1H-NMR (DMSO-d6, 400 MHz): δ 5.84 (s, 2H, NH2), 6.13 (s, 2H, NH2), 7.34–7.55 (m, 4H, Ar-H), 7.58 (d, 2H, J = 8.4 Hz, Ar-H), 7.74 (d, 2H, J = 8.4 Hz, Ar-H), 7.99 (s, 1H, N=CH). 13C-NMR (DMSO-d6, 100 MHz): δ 161.79 (d, 1Jc-f = 243.8 Hz), 161.08, 141.59, 138.82, 132.58, 131.14 (d, 3Jc-f = 8.8 Hz), 128.86, 126.35, 122.52 (d, 3Jc-f = 9.8 Hz), 118.99 (d, 2Jc-f = 23.2 Hz), 118.48, 117.43 (d, 2Jc-f = 20.8 Hz), 83.12, 80.78, 74.58, 73.96. MS m/z 305 (M + 1). ESI-HRMS calcd for C18H14FN4+ ([M + H]+) 305.1197; found: 305.1199.
2-(4-((4-Fluorophenyl)buta-1,3-diyn-1-yl)benzylidene)hydrazine-1-carboximidamide (6d)
Off white solid, m.p. 206–210 °C, yield 78%. 1H-NMR (DMSO-d6, 400 MHz): δ 5.91 (s, 2H, NH2), 6.22 (s, 2H, NH2), 7.27–7.75 (m, 4H, Ar-H), 7.54 (d, 2H, J = 8.4 Hz, Ar-H), 7.74 (d, 2H, J = 8.4 Hz, Ar-H), 7.99 (s, 1H, N=CH). 13C-NMR (DMSO-d6, 100 MHz): δ 162.70 (d, 1Jc-f = 248.3 Hz), 160.76, 141.86, 138.45, 134.95 (d, 3Jc-f = 8.8 Hz), 132.51, 126.42, 118.90, 117.05(d, 4Jc-f = 3.3 Hz), 116.34 (d, 2Jc-f = 22.2 Hz), 82.36, 81.29, 74.26, 73.54. MS m/z 305 (M + 1). ESI-HRMS calcd for C18H15FN4+ ([M + H]+) 305.1197; found: 305.1189.
2-(4-((2-Chlorophenyl)buta-1,3-diyn-1-yl)benzylidene)hydrazine-1-carboximidamide (6e)
Off white solid, m.p. 207–210 °C, yield 75%. 1H-NMR (DMSO-d6, 400 MHz): δ 5.95 (s, 2H, NH2), 6.23 (s, 2H, NH2), 7.38–7.76 (m, 8H, Ar-H), 8.00 (s, 1H, N=CH). 13C-NMR (DMSO-d6, 100 MHz): δ 161.31, 142.23, 139.12, 136.04, 135.02, 133.06, 131.85, 130.02, 128.01, 126.86, 121.01, 119.05, 84.51, 79.03, 78.81, 74.43. MS m/z 321(M + 1). ESI-HRMS calcd for C18H14ClN4+ ([M + H]+) 321.0902; found: 321.0906.
2-(4-((3-Chlorophenyl)buta-1,3-diyn-1-yl)benzylidene)hydrazine-1-carboximidamide (6f)
Off white solid, m.p. 198–201 °C, yield 81%. 1H-NMR (DMSO-d6, 400 MHz): δ 5.83 (s, 2H, NH2), 6.12 (s, 2H, NH2), 7.45–7.50 (m, 8H, Ar-H), 7.99 (s, 1H, N=CH). 13C-NMR (DMSO-d6, 100 MHz): δ 161.09, 141.57, 138.83, 133.46, 132.57, 131.67, 131.15, 130.78, 130.08, 126.33, 122.58, 118.44, 83.23, 80.58, 74.88, 73.96. MS m/z 321 (M + 1). ESI-HRMS calcd for C18H14ClN4+ ([M + H]+) 321.0902; found: 321.0901.
2-(4-((4-Chlorophenyl)buta-1,3-diyn-1-yl)benzylidene)hydrazine-1-carboximidamide (6g)
Off white solid, m.p. 202–205 °C, yield 45%. 1H-NMR (DMSO-d6, 400 MHz): δ 5.92 (s, 2H, NH2), 6.21 (s, 2H, NH2), 7.51–7.56 (m, 4H, Ar-H), 7.64 (d, 2H, J = 8.3 Hz, Ar-H), 7.74 (d, 2H, J = 8.3 Hz, Ar-H), 7.99 (s, 1H, N=CH). 13C-NMR (DMSO-d6, 100 MHz): δ 159.84, 142.21, 137.82, 134.86, 134.18, 132.62, 129.19, 126.74, 119.44, 82.83, 81.22, 74.69, 74.33. MS m/z 321 (M + 1). ESI-HRMS calcd for C18H14ClN4+ ([M + H]+) 321.0902; found: 321.0909.
2-(4-((2-Bromophenyl)buta-1,3-diyn-1-yl)benzylidene)hydrazine-1-carboximidamide (6h)
Off white solid, m.p. 217–219 °C, yield 72%. 1H-NMR (DMSO-d6, 400 MHz): δ 5.69 (s, 2H, NH2), 6.06 (s, 2H, NH2), 7.39–7.78 (m, 6H, Ar-H), 7.58 (d, 2H, J = 8.4 Hz, Ar-H), 7.99 (s, 1H, N=CH). 13C-NMR (DMSO-d6, 100 MHz): δ 161.68, 142.02, 139.40, 135.20, 133.14, 133.05, 131.99, 128.50, 126.78, 125.79, 123.23, 118.82, 84.56, 80.73, 78.13, 74.41. MS m/z 365 (M + 1). ESI-HRMS calcd for C18H14ClN4+ ([M + H]+) 365.0396; found: 365.0394.
2-(4-((3-Bromophenyl)buta-1,3-diyn-1-yl)benzylidene)hydrazine-1-carboximidamide (6i)
Off white solid, m.p. 196–198 °C, yield 68%. 1H-NMR (DMSO-d6, 400 MHz): δ 5.95 (s, 2H, NH2), 6.24 (s, 2H, NH2), 7.38–7.83 (m, 4H, Ar-H), 7.56 (d, 2H, J = 8.4 Hz, Ar-H), 7.75 (d, 2H, J = 8.4 Hz, Ar-H), 7.99 (s, 1H, N=CH). 13C-NMR (DMSO-d6, 100 MHz): δ 160.70, 141.82, 138.57, 134.45, 132.94, 132.58, 131.50, 130.93, 126.43, 122.83, 121.81, 118.66, 83.21, 80.51, 74.95, 74.04. MS m/z 365 (M + 1). ESI-HRMS calcd for C18H14ClN4+ ([M + H]+) 365.0396; found: 365.0391.
2-(4-((4-Bromophenyl)buta-1,3-diyn-1-yl)benzylidene)hydrazine-1-carboximidamide (6j)
Off white solid, m.p. 227–230 °C, yield 50%. 1H-NMR (DMSO-d6, 400 MHz): δ 5.93 (s, 2H, NH2), 6.22 (s, 2H, NH2), 7.55–7.57 (m, 4H, Ar-H), 7.65 (d, 2H, J = 8.4 Hz, Ar-H), 7.74 (d, 2H, J = 8.4 Hz, Ar-H), 8.00 (s, 1H, N=CH). 13C-NMR (DMSO-d6, 100 MHz): δ 161.12, 142.37, 138.92, 134.69, 132.99, 132.49, 126.92, 124.05, 120.25, 119.27, 83.47, 81.66, 75.30, 74.64. MS m/z 365 (M + 1). ESI-HRMS calcd for C18H14ClN4+ ([M + H]+) 365.0396; found: 365.0395.
2-(4-(m-Tolylbuta-1,3-diyn-1-yl)benzylidene)hydrazine-1-carboximidamide (6k)
Off white solid, m.p. 202–204 °C, yield 63%. 1H-NMR (DMSO-d6, 400 MHz): δ 2.32 (s, 3H, CH3), 5.72 (s, 2H, NH2), 6.09 (s, 2H, NH2), 7.29–7.43 (m, 4H, Ar-H), 7.54 (d, 2H, J = 8.2 Hz, Ar-H), 7.72 (d, 2H, J = 8.2 Hz, Ar-H), 7.98 (s, 1H, N=CH). 13C-NMR (DMSO-d6, 100 MHz): δ 161.26, 142.30, 138.88, 138.83, 133.08, 132.94, 131.23, 129.99, 129.28, 126.85, 120.84, 119.42, 82.96, 82.73, 74.84, 73.91, 21.15. MS m/z 301 (M + 1). ESI-HRMS calcd for C19H16N4+ ([M + H]+) 301.1448; found: 301.1452.
Evaluation of antibacterial activity in vitro
Test bacteria were grown to mid-log phase in Mueller–Hinton broth (MHB) or Tryptone Soya Broth (TSB) and diluted 1000-fold in the same medium. Bacteria (105 CFU/mL) were inoculated into MHB or TSB and dispensed at 0.2 ml/well into a 96-well microtiter plate. As positive controls, gatifloxacin, moxifloxacin, norfloxacin, oxacillin, and penicillin were used. Test compounds were prepared in DMSO, the final concentration of which did not exceed 0.05%. The minimum inhibitory concentration (MIC) was defined as the concentration of a test compound that completely inhibited bacterial growth after 24 h incubation at 37 °C. Bacterial growth was determined by measuring the absorption at 630 nm using a microplate reader. All experiments were carried out three times.
Evaluation of bacterial resistance development
To evaluate the propensity for developing bacterial resistance, one of the compounds with high antibacterial activity (3g) was used. The initial MIC values of 3g were determined against bacteria Staphylococcus aureus CMCC 25923 and Escherichia coli CMCC 44568, using norfloxacin and colistin, respectively, as antibiotic controls. Subsequently, serial passaging was initiated by transferring bacterial suspension grown at the sub-MIC of the compound/antibiotics (MIC/2) to a new plate and subjecting it to another assay to determine the (new) MIC. After 22 h incubation, cells grown at the sub-MIC of the test compound/antibiotics were once again transferred and the MIC determined. The process was repeated for 20 or 30 passages for S. aureus and E. coli, respectively. The MIC for 3g, norfloxacin and colistin was plotted as a function of time in days (number of passages) to determine the propensity of bacterial resistance developmentCitation24.
Time-kill assay
Methicillin-resistant S. aureus ATCC 33591 grown in MHB was used to determine time-kill kinetics. Bacterial suspensions (105 CFU/mL) containing test compounds (norflocaxin, compound 3g) at final concentrations of 1 × MIC and 2 × MIC were incubated at 37 °C with shaking. Aliquots (10 μL) were removed from the cultures after 0, 0.5, 1, 2, 3, 4, 6, 8 and 12 h, serially diluted 1000-fold in nutrient solution, and plated onto sterile Mueller-Hinton agar medium. Plates were then incubated for 24 h at 37 °C, the number of CFU was counted, and the total bacterial log10 CFU/mL was determined.
Evaluation of cytotoxicity activity in vitro
A lung cancer cell line (A549), gastric cancer cell line (SGC7901) and human hepatocytes (L02) were used to test the anticancer activity and cytotoxicity of the new compounds. 5-Fluorouracil (5-FU) was used as the positive control against cancer cell lines. The A549, SGC7901 and L02 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% foetal bovine serum (FBS) and antibiotics (100 U/mL penicillin-streptomycin). Cells at 80–90% confluence were split by trypsin (0.25% in PBS; pH 7.4), and the medium was changed at 24 h intervals. The cells were cultured at 37 °C in a 5% CO2 incubator. The cells were passaged three times, then approximately 1 × 104 cells were seeded into each well of a 96-well plate and allowed to incubate to allow attachment of the cells to the surface. After 24 h, the medium was replaced with DMEM supplemented with 10% FBS containing various concentrations (0.1, 0.3, 1, 3, 10, and 30 μg/mL) of test compounds. Each concentration was tested in triplicate. After 48 h treatment, 20 µL of CCK-8 solution was added to each well and the optical density measured at 450 nm after 3 h using a microplate reader. The IC50 values were defined as the concentrations inhibiting 50% of cell growth.
Docking studies
Molecular docking of compounds 3g and 6e to Pseudomonas aeruginosa LpxC (PDB code: 3P3E), E. coli LpxC (PDB code: 3P3G) and the E. coli FabH-CoA complex structure (PDB code: 1HNJ) was carried out using the DS–CDOCKER protocol implemented through the graphical user interface of the Discovery Studio software (version 2019). The structures of 3P3E, 3P3G, and 1HNJ were downloaded from Protein Data Bank. The three–dimensional structures of 3g and 6e were constructed using Chem3D Ultra 12.0 software (Chemical Structure Drawing Standard; CambridgeSoft Corporation, Waltham, MA, 2010) and was energetically minimised using the MMFF94 force field with 5000 iterations and a minimum RMS gradient of 0.10. For protein preparation, the hydrogen atoms were added and water and impurities were removed. The 3D structure of 3g or 6e was placed during the molecular docking procedure. Types of docking interactions of the proteins with tested compounds were analysed and ranked, and those with maximum binding energy were selected to analyse the interaction patterns.
Results and discussion
Chemistry
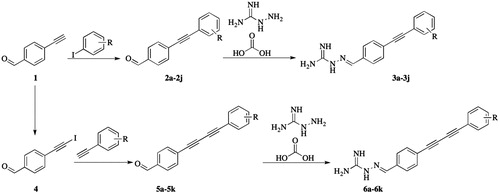
The synthesis of two series of aminoguanidine-linked alkynyl derivatives followed the general pathway outlined in Scheme 1 using 4-ethynylbenzaldehyde as starting material. The reaction of 4-ethynylbenzaldehyde with substituted-iodobenzenes in the presence of triethylamine (TEA), CuI and Pd(PPh3)Cl2 in tetrahydrofuran (THF) produced 4-(substituted-phenylethynyl)benzaldehyde (2a–2j) under the protection of nitrogen. The reaction of 4-ethynylbenzaldehyde with N-iodosuccinimide in the presence of AgNO3 in acetone produced 4-(iodoethynyl)benzaldehyde (4). The obtained 4-(iodoethynyl)benzaldehyde reacted with ethynylbenzenes in the presence of TEA, CuI and Pd(PPh3)Cl2 in THF yielding 4-((substituted-phenyl)buta-1,3-diyn-1-yl)benzaldehyde (5a–5k) under the protection of nitrogen. The target compounds 3a–3j and 6a–6k were prepared by the condensation of 2a–2j or 5a–5k with hydrazinecarboximidamide carbonate, respectively. Finally, the structures of the target compounds were characterised by 1H-NMR, 13C-NMR, and high-resolution mass spectrometry.
Scheme 1. The synthetic route of aminoguanidine-linked alkynyl derivatives.
Spectroscopic analyses of all synthetic compounds fully supported their depicted structures. Taking compound 3a as an example, the 1H-NMR spectrum yielded two singlets due to N-H of guanidyl at 5.65 and 6.04 ppm, which were assigned to two NH2 groups involved in the tautomerism of the guanidyl group. Peaks corresponding to the aromatic protons of terminal benzene ring were observed in the range 7.42–7.57 ppm. Two doublets (J = 8.2 Hz) due to aromatic protons of the para-substituted phenyl ring were observed at 7.50 and 7.72 ppm. The absorption peak of the C–H in imine was found at 7.99 ppm. The 13C-NMR spectra also identified 12 carbon nuclei in different chemical environments, which was also consistent with the structure of 3a. Moreover, high-resolution mass spectrometry of 3a displayed an [M + H]+ signal at m/z 263.1287, in agreement with its molecular weight of 263.1291.
In vitro antibacterial activity
The evaluation of antibacterial activity of all compounds (3a–3j and 6a–6k) was performed by a serial dilution method to determine the MIC against a panel of pathogens comprising five Gram-positive strains (S. aureus (CMCC(B) 26003 and CMCC 25923), Streptococcus mutans BNCC 336931, Enterococcus faecalis CMCC 29212, and Bacillus subtilis CMCC 63501), four Gram-negative strains (E. coli (CMCC 25922 and CMCC 44568) and P. aeruginosa (CMCC 27853 and CMCC 10104)), as well as three methicillin-resistant clinical isolates (S. aureus ATCC 43300 and ATCC 33591, P. aeruginosa ATCC BAA-2111). The results are described in and . Gatifloxacin, moxifloxacin, norfloxacin, oxacillin, and penicillin were used as positive controls.
Table 1. Inhibitory activity (MIC, μg/mL) of compounds 3a–3j and 6a–6k against Gram-positive bacteria and Gram-negative bacteria.
Table 2. Inhibitory activity (MIC, µg/mL) of compounds 3c–3e, 3g, 3i, 3j, 6a, 6b and 6e against clinical isolates of multidrug-resistant strains.
In general, the inhibitory activity of the new target compounds against Gram-positive strains was better than that against Gram-negative strains. Compared with compounds 6a–6k with a 1,4-diphenylbuta-1,3-diyne moiety, compounds 3a–3j containing a 1,2-diphenylethyne moiety exhibited good to excellent antibacterial activity against all strains except P. aeruginosa CMCC 27853. Pseudomonas aeruginosa are able to rapidly develop resistance to multiple classes of antibiotics, making the treatment of infectious diseases becomes more challenging. The outer membrane porin OprD and the multidrug efflux pumps of P. aeruginosa represent the main barriers for drug entry into the cell. The decrease or loss of antibacterial activity of most compounds synthesized against P. aeruginosa CMCC 27853 may be due to the above factsCitation25.
All compounds in series 3a–3j showed potent inhibitory effects against the five Gram-positive strains with MICs in the range 0.25–8 μg/mL, except compound 3j, which showed inhibitory activity at 128 μg/mL against B. subtilis CMCC 63501. In general, compounds 3e, 3f, 3g, 3j, containing 2-Cl, 3-Cl, 4-Cl, and 4-Br, respectively, had greater inhibitory activity against Gram-positive strains. Compounds 3a–3j were less active against the four Gram-negative strains, with MICs ranging from 1 µg/mL to more than 128 µg/mL. For E. coli CMCC 25922 and CMCC 44568, and P. aeruginosa CMCC 10104, the change of substituents had no obvious effect on the inhibitory activity, while for the P. aeruginosa CMCC 27853, the compounds with H, 3-F, 4-F groups had more potency than compounds with other substituents. Among the nine strains, compounds 3a–3j presented the most potent inhibitory activity against E. faecalis CMCC 29212 with a MIC of 0.5 or 1 μg/mL, which is comparable to gatifloxacin, moxifloxacin and norfloxacin (MIC = 1 µg/mL) and is 256 or 128-fold more potent than oxacillin and penicillin (MIC = 128 µg/mL). Compound 3g showed the most potent inhibitory activity against B. subtilis CMCC 63501 (MIC = 0.25 μg/mL).
Compounds in the series of 6a–6k displayed different degrees of inhibitory activity (MICs ranging from 0.25 µg/mL to more than 128 µg/mL) against the five Gram-positive strains and two Gram-negative strains (E. coli CMCC 25922 and P. aeruginosa CMCC 10104), while all of them were insensitive towards E. coli CMCC 44568 and P. aeruginosa CMCC 27853. The structure–activity relationship showed that the para-position substituents reduce the antibacterial activity, giving compounds 6d, 6g, 6i a MIC ≥128 against almost all of the tested strains. Compounds 6b and 6e, with F and Cl in the ortho position, showed the most potent inhibitory activity against S. aureus CMCC(B) 26003 (MIC = 0.25 μg/mL). Compound 6a, without substituents on the terminal phenyl group, was more effective against P. aeruginosa CMCC 10104 than other compounds in this series.
Based upon their superior performance in the above assays, compounds 3c, 3d, 3e, 3g, 3i, 3j, 6a, 6b and 6e were further evaluated for their inhibitory activity against the growth of several clinical isolates of MDR bacterial strains (methicillin-resistant and/or oxacillin-resistant S. aureus ATCC 43300 and ATCC 33591 and multi-drug resistant P. aeruginosa ATCC BAA-2111). As shown in , compounds 3c, 3d, 3e, 3g, 3i, 3j, 6a, 6b and 6e presented good activities (MIC = 1–8 μg/mL) against MDR S. aureus ATCC 43300, and showed the potent inhibitory activity against S. aureus ATCC 33591 (MIC = 0.5 or 1 μg/mL). They were slightly less active than gatifloxacin, moxifloxacin and norfloxacin (MIC = 0.25 µg/mL) but were more potent than oxacillin (MIC = 8 µg/mL) and penicillin (MIC > 32 µg/mL). Only compound 3g, with Cl atom in the para position, showed moderate inhibitory activity against multi-drug resistant P. aeruginosa ATCC BAA-2111. It was also the most potent compound against growth of S. aureus ATCC 43300 and ATCC 33591.
Propensity to develop bacterial resistance
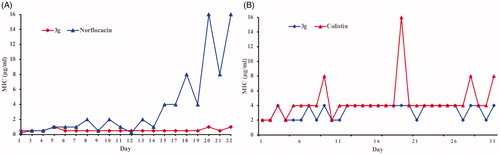
Bacterial resistance against most antibiotics presents a major problem of current timesCitation26–29. Therefore, it is crucial to evaluate the potential emergence of bacterial resistance against these biocides. The propensity for development of bacterial resistance of the synthesised compounds was evaluated by using the most active compound (3g) against both Gram-positive S. aureus and Gram-negative E. coliCitation30,Citation31. Norfloxacin, an antibiotic generally used to treat the Gram-positive infections, was used as a positive control for S. aureus, whereas colistin, a lipopeptide antibiotic active against Gram-negative bacteria, was used as a positive control for E. coli. These antibacterial agents were repeatedly challenged against bacteria at their sub-MIC values to allow bacteria to develop resistance. Resistance is usually defined as a > 4-fold increase in the original MICCitation32. Interestingly, little change in the MIC of compound 3g was observed over 20 generations. In comparison, an approximately 32-fold and 4 or 8-fold increase in MIC was observed for norfloxacin and colistin, respectively (). The above results indicated that bacteria do not develop resistance against this type of aminoguanidine within the experimental time period.
Figure 2. Propensity of the development of bacterial resistance towards compound 3g by (A) S. aureus and (B) E. coli.
Bactericidal time-kill kinetics
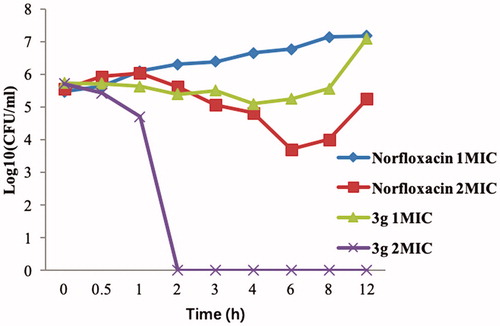
To study the bactericidal activity of the promising compound 3g, next we carried out in vitro time-kill assay against MRSA (starting bacterial concentration of 5.5 log10 CFU/mL) at two different concentrations (1 × MIC and 2 × MIC) using norfloxacin as a control (). Compound 3g was rapidly bactericidal at 2 × MIC (>5 log10 CFU/mL reduction) after 2 h and its bactericidal activity persisted for 12 h. In the case of norfloxacin, a concentration-dependent activity was seen at 1 × MIC to 2 × MIC, but its effects were bacteriostatic, not bactericidal (). These results clearly demonstrate the superiority of compound 3g over the commonly used antibiotic norfloxacin in killing MRSA bacteria.
Figure 3. Bactericidal activities of compound 3g and norfloxacin against MRSA.
Molecular modelling
LpxC is an essential enzyme in the lipid A biosynthetic pathway. Developing novel LpxC inhibitors has been an important approach to obtain new antibacterial drugs targeting Gram-negative pathogens. To gain insight into the molecular interactions of these compounds with LpxC, the co-crystal structures of 3g and 6e complexed with P. aeruginosa LpxC and E. Coli LpxC were obtained. Generally, the 1,2-diphenylethyne group in the series of 3a–3j and 1,4-diphenylbuta-1,3-diyne moiety in the series of 6a–6k functionally interacted with many of the hydrophobic residues in the lipophilic tunnel. The aminoguanidine was responsible for forming hydrogen bonds with several amino acid residues. The aminoguanidine of compound 3g bound to the active site Zn2+ ion of P. aeruginosa LpxC () and formed hydrogen bonds with ASP241, HIS264, MET62, and GLU77 that line this polar region. In the interaction of P. aeruginosa LpxC with compound 6e (), the phenyl ring of 1,4-diphenylbuta-1,3-diyne moiety showed interactions with critical amino acid residues LEU18, ILE197, and VAL216 via aromatic stacking and hydrophobic intermolecular forces. The guanidine group of compound 6e acted as a hydrogen bond donor in the interaction with the carbonyl group of ASP241, HIS264 and MET62.
Figure 4. Interactions of compound 3g and 6e with P. aeruginosa LpxC (A for 3g; B for 6e).
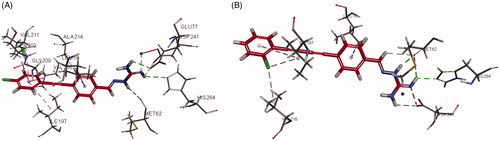
Interactions of compounds 3g and 6e with E. coli LpxC are shown in , respectively. The aminoguanidine of compound 3g was bound to the active site Zn2+ ion and forms hydrogen bond interactions with ASP242 and HIS265. The 1,2-diphenylethyne group was responsible for forming various aromatic stacking interactions and hydrophobic intermolecular forces with LEU18, ALA215, VAL217, GLY210, SER211, ALA215, and ILE198. In addition, the Cl atom attached to the terminal phenyl group showed hydrophobic interactions with amino acid residues PHE212 and MET195 that increased the binding force with E. coli LpxC. The aminoguanidine of compound 6e was also bound to the zinc atom of E. coli LpxC, as well as the amino acid residues ASP242, GLU78, and HIS265 via hydrogen bond interaction. Aromatic stacking interactions and hydrophobic force interactions were additionally formed between the hydrophobic 1,4-diphenylbuta-1,3-diyne moiety of 6e and LEU18, CYS207, ALA215, LEU62, MET195, GLY210, SER211, PHE212, and VAL217.
Figure 5. Interactions of (A) compound 3g and (B) 6e with E. coli LpxC.
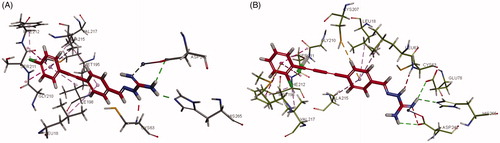
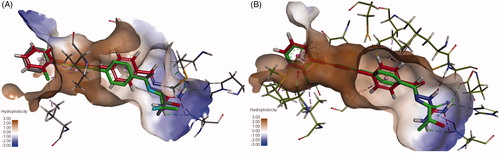
For comparison, the interactions of compound 6e with P. aeruginosa LpxC and E. coli LpxC were superimposed on the co-crystallised LPC-009. As seen in , compound 6e (red) showed similar interactions as LPC-009 (green). This suggests that binding of 6e to LpxC may be, at least in part, responsible for its antibacterial activity.
Figure 6. Overlay of 6e and LPC-009 binding to (A) P. aeruginosa LpxC and (B) E. coli LpxC.
It is likely that targets other than LpxC might be involved in the broad-spectrum antibacterial activity of the synthesised compounds against Gram-positive and Gram-negative bacteria. The FabH receptor is a condensing enzyme that plays key roles in fatty acid biosynthesisCitation33. The potential interaction of some hydrazine compounds with FabH prompted us to investigate the molecular interactions of the representative compounds 3g and 6e with FabH receptor (PDB ID: 1HNJ)Citation20,Citation34,Citation35.
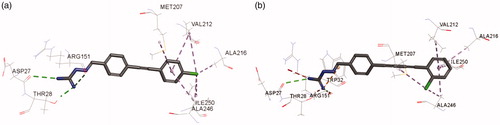
As shown in , residues including ASP27, THR28, ARG151, VAl212, ALA216, ILE250, and ALA246, were involved in the binding of 3g to the active site of E. coli FabH. The C = N and NH groups of compound 3g were involved in the interaction with ASP27, THR28, ARG151 via hydrogen bonding, while the terminal phenyl group and its attached chlorine atom formed hydrophobic interactions with VAl212, ALA216, ILE250, and ALA246. The binding of 6e with E. coli FabH resembled that of 3g. As shown in , residues ASP27, THR28, ARG151, TRP32, VAl212, ALA216, ILE250, and ALA246, were involved in the interactions with 6e in E. coli FabH enzyme.
Figure 7. Interactions of compound (A) 3g and (B) 6e with E. coli FabH.
Anticancer activity
The cytotoxic effects of compounds 3e, 3f, 3g, 3i, 6a, 6b, 6e and 6k were evaluated using two human cancer cell lines (A549and SGC7901) and one human normal cell line (L02). The IC50 values of the tested compounds and 5-FU are shown in . All the tested compounds showed excellent activity against the investigated cancer cells (IC50 = 0.30–4.57 µg/mL); however, no correlation between the substituents and the cytotoxic activity could be identifies. The highest activity against A549 cells was exhibited by compound 6b (IC50 = 2.22 µg/mL), although it is not comparable to that of 5-FU, which exhibited IC50 of 0.88 µg/mL. The highest activity against SGC7901 cells was exhibited by compound 3f (IC50 = 0.30 µg/mL), followed by compounds 3e and 6k (IC50 = 0.45, 0.55 µg/mL, respectively). All the tested compounds showed higher cytotoxic activity against SGC7901 cells than 5-FU. Compounds 3e, 3f, 3g, 3i, 6a, 6b, 6e and 6k showed IC50 values in the range of 10.25–20.85 µg/mL against the normal cell line L02, comparing to the IC50 value of 8.44 µg/mL of 5-FU. This result indicated that these compounds have low toxicity towards normal cells in comparison to cancer cells, suggesting potential for a good therapeutic index.
Table 3. The Inhibitory activity (IC50, µg/mL) of compounds 3e, 3f, 3g, 3i, 6a, 6b, 6e and 6k against cancer cell lines A549 and SGC7901 and normal cell lines L02.
Conclusion
In the present work, a series of novel aminoguanidines containing an alkynyl moiety were synthesised and characterised. The antibacterial and anticancer activities of these compounds were screened, with Gram-positive bacteria being more susceptible than Gram-negative ones. The aminoguanidine derivatives (3a–3j), having a 1,2-diphenylethyne, exhibited greater antibacterial activity than 6a–6j with a 1,4-diphenylbuta-1,3-diyne moiety. Compounds 3a–3j showed potent inhibitory activity against the selected bacterial strains with MIC values in the range of 0.25–8 μg/mL, including the multidrug resistant strains. Among them, compound 3g was the most promising, having superior activity to oxacillin and penicillin against the tested MDR strains. Resistance of the tested bacteria towards 3g was not easily developed and this compound was rapidly bactericidal. Furthermore, 3 g exhibited significant anticancer activity against lung (A549) and gastric cancer cells (SGC7901), with IC50 values of 4.42 and 1.01 µg/mL, respectively, and low-toxicity towards normal cells. To understand the binding pattern, molecular docking of representative compounds 3g and 6e was performed, demonstrating that they bind strongly to the LpxC enzyme and FabH enzyme. These findings indicate that compounds containing the aminoguanidine moiety are promising candidates for the development of new antibacterial and anticancer agents.
Acknowledgements
The School of Life Sciences, Jinggangshan University, was appreciated for providing the BIOVIA Discovery Studio software in this study. The authors would like to thank Editage (www.editage.cn) for English language editing.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Magiorakos AP, Srinivasan A, Carey RB, et al. Multidrug-resistant, extensively drug-resistant and pan drug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin Microbiol Infect 2012;18:268–81.
- Magiorakos AP, Suetens C, Monnet DL, et al. The rise of carbapenem resistance in Europe: just the tip of the iceberg?. Antimicrob Resist Infect Control 2013;2:6.
- Freire-Moran L, Aronsson B, Manz C, et al. Critical shortage of new antibiotics in development against multidrug-resistant bacteria-Time to react is now. Drug Resist Updat 2011;14:118–24.
- Payne DJ, Gwynn MN, Holmes DJ, et al. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov 2007;6:29–40.
- Whitfield C, Trent MS. Biosynthesis and export of bacterial lipopolysaccharides. Annu Rev Biochem 2014;83:99–128.
- Liu F, Ma S. Recent process in the inhibitors of UDP-3-O-(R-3-hydroxyacyl)-nacetylglucosamine deacetylase (LpxC) against Gram-negative bacteria. Mini Rev Med Chem 2018;18:310–23.
- Zhang J, Zhang L, Li X, Xu W. UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase (LpxC) inhibitors: a new class of antibacterial agents. Curr Med Chem 2012;19:2038–50.
- Erwin AL. Antibacterial drug discovery targeting the lipopolysaccharide biosynthetic enzyme LpxC. Cold Spring Harb Perspect Med 2016;6:a025304.
- Coggins BE, Li X, McClerren AL, et al. Structure of the LpxC deacetylase with a bound substrate-analog inhibitor. Nat Struct Biol 2003;10:645–51.
- Clayton GM, Klein DJ, Rickert KW, et al. Structure of the bacterial deacetylase LpxC bound to the nucleotide reaction product reveals mechanisms of oxyanion stabilization and proton transfer. J Biol Chem 2013;288:34073–80.
- Onishi HR, Pelak BA, Gerckens LS, et al. Antibacterial agents that inhibit lipid A biosynthesis. Science 1996;274:980–2.
- Lee PS, Lapointe G, Madera AM, et al. Application of virtual screening to the identification of new LpxC inhibitor chemotypes, oxazolidinone and isoxazoline. J Med Chem 2018;61:9360–70.
- Hale MR, Hill P, Lahiri S, et al. Exploring the UDP pocket of LpxC through amino acid analogs. Bioorg Med Chem Lett 2013;23:2362–7.
- Mansoor UF, Vitharana D, Reddy PA, et al. Design and synthesis of potent Gram-negative specific LpxC inhibitors. Bioorg Med Chem Lett 2011;21:1155–61.
- Liang X, Lee CJ, Chen X, et al. Syntheses, structures and antibiotic activities of LpxC inhibitors based on the diacetylene scaffold. Bioorg Med Chem 2011;19:852–60.
- Lee CJ, Liang X, Chen X, et al. Species-specific and inhibitor-dependent conformations of LpxC: implications for antibiotic design. Chem Biol 2011;18:38–47.
- Liang X, Lee CJ, Zhao J, et al. Synthesis, structure, and antibiotic activity of aryl-substituted LpxC inhibitors. J Med Chem 2013;56:6954–66.
- Gao ZM, Wang TT, LI SZ, et al. Synthesis and antibacterial activity evaluation of (2-chloroquinolin-3-yl)methyleneamino guanidine derivatives. Chin J Org Chem 2016;36:2484–8.
- Yu HH, Zhou SC, Guo TT, et al. Synthesis and antimicrobial activity evaluation of aminoguanidine derivatives containing a biphenyl moiety. Chin J Org Chem 2019;39:1497–502
- Song MX, Wang SB, Wang ZT, et al. Synthesis, antimicrobial and cytotoxic activities, and molecular docking studies of N-arylsulfonylindoles containing an aminoguanidine, a semicarbazide, and a thiosemicarbazide moiety. Eur J Med Chem 2019;166:108–18.
- Sidoryk K, Świtalska M, Rózga P, et al. An efficient synthesis of indolo[2,3-b]quinoline guanidine derivatives with their in vitro and in vivo study. Med Chem Res 2017;26:3354–66.
- Wei ZY, Chi KQ, Yu ZK, et al. Synthesis and biological evaluation of chalcone derivatives containing aminoguanidine or acylhydrazone moieties. Bioorg Med Chem Lett 2016;26:5920–5.
- Osowska K, Lis T, Szafert S. Protection/deprotection-free syntheses and structural analysis of (*Keto-aryl)diynes. Eur J Org Chem 2008;27:4598–606.
- Qiu X, Janson CA, Smith WW, et al. Refined structures of β-ketoacyl-acyl carrier protein synthase III. J Mol Biol 2001;307:341–56.
- Lister PD, Wolter DJ, Hanson ND. Antibacterial-resistant Pseudomonas aeruginosa: clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin Microbiol Rev 2009;22:582–610.
- Niu Y, Wang RE, Wu H, Cai J. Recent development of small antimicrobial peptidomimetics. Future Med Chem 2012;4:1853–62.
- Taubes G. The bacteria fight back. Science 2008;321:356–61.
- Walsh C. Molecular mechanisms that confer antibacterial drug resistance. Nature 2000;406:775–81.
- Alekshun MN, Levy SB. Molecular mechanisms of antibacterial multidrug resistance. Cell 2007;128:1037–50.
- Makovitzki A, Avrahami D, Shai Y. Ultrashort antibacterial and antifungal lipopeptides. Proc Natl Acad Sci U S A 2006;103:15997–6002.
- Hoque J, Akkapeddi P, Yadav V, et al. Broad spectrum antibacterial and antifungal polymeric paint materials: synthesis, structure-activity relationship, and membrane-active mode of action. ACS Appl Mater Interfaces 2015;7:1804–15.
- Kosowska-Shick K, Clark C, Pankuch GA, et al. Activity of telavancin against staphylococci and enterococci determined by MIC and resistance selection studies. Antimicrob Agents Chemother 2009;53:4217–24.
- Heath RJ, Rock CO. Inhibition of beta-ketoacyl-acyl carrier protein synthase III (FabH) by acyl-acyl carrier protein in Escherichia coli. J Biol Chem 1996;271:10996–1000.
- Chu W-C, Bai P-Y, Yang Z-Q, et al. Synthesis and antibacterial evaluation of novel cationic chalcone derivatives possessing broad spectrum antibacterial activity. Eur J Med Chem 2018;143:905–21.
- Zhang HJ, Qin X, Liu K, et al. Synthesis, antibacterial activities and molecular docking studies of Schiff bases derived from N-(2/4-benzaldehyde-amino) phenyl-N′-phenyl-thiourea. Bioorg Med Chem 2011;19:5708–15.