
Abstract
A novel series of cyclin-dependent kinases (CDKs) inhibitors, which play critical roles in the cell cycle control and regulation of cell transcription, were synthesised. A systematic study of enzymatic and cellular assays led to the identification of compound X22 with a nanomolar potency against CDK4 and CDK9 and potent antiproliferative activities against a panel of tumour cell lines. X22 could induce cell cycle arrest and cell apoptosis in cancer cell lines. X22 dose-dependently inhibits signalling pathways downstream of CDKs in cancer cells. In vivo antitumor activity assays, oral administration of X22 led to significant tumour regression in mouse model without obvious toxicity. Superior anti-cancer efficacy in vitro and in vivo of X22 demonstrated combined depletion of cell cycle and transcriptional CDK all contributed to antitumor activity. Taken together, concomitant inhibition of cell cycle and transcriptional CDK activities provided valuable guide for further structural optimisation.
1. Introduction
Cancer is a multigenetic disease with the hallmark that multiple signalling pathways aberration, which often require multiple therapeutic interventionsCitation1,Citation2. Kinases mediate various cellular activities due to their critical roles in cellular signalling, such as proliferation, apoptosis, transcription, differentiation and so onCitation3,Citation4, which have been demonstrated as promising drug targets for the treatment of many diseases such as cancersCitation5–7. Kinase inhibitors are widely employed in clinical oncologyCitation8,Citation9. Simultaneous inhibition of different mechanisms using multi-kinase inhibitors could yield superior efficacy, such as synergy effects, avoiding drug resistance and so onCitation10.
Out of numerous kinases, cyclin-dependent kinases (CDKs) are protein kinases involved in important cellular processes due to the complexity of their roles. They regulate the cell division, apoptosis, transcription and differentiation, which involved in a number of pathological conditions such as human cancerCitation11,Citation12. CDKs are divided into two groups based on their roles in cell cycle progression and transcription regulationCitation13,Citation14. The first group including CDK subtypes 1, 2, 4 and 6 mainly is involved in cell cycle and the second group including CDK subtypes 7, 8 and 9 are associated with transcription controlCitation11. CDK4/6 binding to cyclin D forming CDK4/6-cyclin D complex, is an important component of cell cycle activation and mediates the transition from G1 to S phase, where cells grow and synthesise proteins in preparation for DNA synthesisCitation15. Among the transcriptional CDKs, CDK9 has attracted major interest of many groups. CDK9 forming heterodimeric complex with subunit cyclin T or cyclin K phosphorylates the COOH-terminal domain (CTD) of RNA polymerase II (RNAPII) to control the transcription progressionCitation16. As a component of a larger protein complex called positive transcription elongation factor b (P-TEFb), CDK9 stimulate transcription elongation of most protein coding genesCitation17. Phosphorylation of RNAPII is regarded as a marker involved in a variety of human pathological conditions, such as cancer. Inhibition of CDK9 results in reduced levels of antiapoptotic proteins of cancerCitation18,Citation19.
Dysregulation of CDKs-cyclin pathway resulting in uncontrolled proliferation have been observed in various cancersCitation20–22. Owing to the important role of CDKs in the control of cell division, numerous drugs targeting CDKs were designed and reported in the clinicCitation23. Several selective CDK4/6 inhibitors have been reported in the recent years. Pfizer’s palbociclib represents the first CDK inhibitor approved by the US FDA in February 2015 to treat HR+/HER2-breast cancerCitation24,Citation25. Novartis’ ribociclib and Lilly’s abemaciclib also got a fast approval by FDA in March 2017 and September 2017, respectively, to treat HR+/HER2-breast cancerCitation26,Citation27. While as reported, both palbociclib and ribociclib as selective CDK4/6 inhibitors need to combine with letrozole for the treatment of breast cancerCitation28,Citation29. Abemaciclib with additional kinase activities, such as CDK1, CDK2 and CDK9 shows unique single-agent activitiesCitation30,Citation31. These studies indicate that monotherapy targeting of individual cell cycle CDKs may be insufficient for cancer therapy.
Simultaneous regulating the cell cycle and transcription control could provide superior anticancer efficacyCitation32–34. In an effort to discover novel CDKs inhibitors with high inhibition potency against both cell cycle and transcriptional CDKs, we recently started a medicinal chemistry study based on ribociclib, a latest and selective CDK4 inhibitor and performed a rational drug design. According to the researches, many sulphur atom-containing compounds have immense importance in medicinal chemistry and exhibit anticancer activityCitation35. Moreover, isothiocyanate plays a prominent role in the field of pharmaceuticals with anticancer effects in various cancer typesCitation36. Sulphur atom or isothiocyanate-containing derivatives are biologically active agents for drugs used for the treatment of cancer. We reasoned to introduce sulphur atom and isothiocyanate moiety based on pyrrolo-[2,3-d] pyrimidines-2-amine skeleton to proceed structural optimisation on R, L and linker regions (Supplementary Figure S1). In our study, we synthesised a series of novel compounds simultaneously mediating the cell cycle and transcription control of CDKs with high potent inhibition against solid tumour. Compound X22 targeting CDK4 and CDK9 was discovered and exhibited potent antitumor efficacy in vitro and in vivo.
2. Materials and methods
2.1. General methods for chemistry
The commercially obtained chemicals were used directly without further purification. Solvents were purified and distilled following the standard procedures. All the reactions were monitored by thin-layer chromatography (TLC). The NMR spectra were taken on a Bruker AV-400 MHz spectrometer (400 MHz for 1H and 101 MHz for 13 C) and chemical shifts were expressed in ppm downfield using tetramethylsilane as the internal standard. High-resolution mass spectra (HRMS) were performed on a VG ZAB-HS mass spectrometer under electron spray ionisation (ESI). All the derivatives for testing bioactivity were purified to >95% purity which was determined by HPLC analysis on a Shimadzu Prominence-i LC-2030C 3D system (column, InertSustain C18, 4.6 × 250 mm, 5 μM; mobile phase, gradient elution of methanol/H2O (90:10); low rate, 1.0 ml/min; UV wavelength, 190 − 800 nm; temperature, 40 °C; injection volume, 10 μL). The detailed synthesis of compounds is presented in Supplementary Material.
2.2. Molecular docking
Docking studies were finished by Discovery Studio 3.1 to explore the predicted binding modes of compound X22 in CDK4, CDK6 (PDB code: 4EZ5) and CDK9 (PDB code: 4BCF) respectively. Hydrogen atoms were added by Gold (version 5.0). The images were created by PyMOL.
2.3. Cell lines and cell culture
All the human cancer cell lines were obtained from ATCC. Cells were cultured in RPMI-1640 (BioInd) medium according to the instructions from ATCC, with the medium containing 10% FBS (BioInd), 1% antibiotics (penicillin and streptomycin) at 37 °C in an atmosphere of 5% CO2.
2.4. Kinase inhibition assays
Kinase inhibition profiles were determined using KinaseProfiler services provided by Eurofins, and ATP concentrations used the Km of corresponding kinases.
2.5. Cytotoxicity assay
Cytotoxicity assay were conducted as described previously using a Cell Counting Kit-8 (CCK-8) assay (#CK04, Dojindo, Kumamoto, Japan)Citation37.
2.6. Cell cycle assay
Cells were plated on six-well culture plates at a density of 5 × 105 cells/mL and were treated with the indicated concentrations of X22 or ribociclib for 12, 24, 36 or 48 h after they adherence. Cells were harvested and washed with phosphate buffered saline (PBS) for three times and then fixed with ice cold 75% ethanol overnight. The fixed cells were then washed with PBS and stained with propidium iodide (50 mg/mL) in the presence of RNase A (0.5 mg) for 30 min at 37 °C. The stained cells were then subjected to flow cytometry (Modfit, BD) for cell cycle analysis.
2.7. Annexin V-FITC/PI apoptosis assay
Cells at a density of 3 × 105 cells/mL were seeded in six-well plates and treated with compounds at different concentrations for 48 h. The cells were then harvested and washed twice with cold PBS. Then the cells were subjected to an Annexin V/PI Apoptosis Detection kit (BD Biosciences) for staining according to manufacture’s instructions, and finally analysed by flow cytometry (Modfit, BD).
2.8. Western blotting
Protein extraction and western blotting methods were performed as described previouslyCitation37. The antibodies used in this study including anti-β-actin (Santa Cruz; sc-47778), anti-p53 (CST; #9282), anti-phospho-p53Ser15 (CST; #9284), anti-Bax (CST; #2772), anti-Bcl-2 (BD; #51-6511GR), anti-Rb (CST; #9309), anti-phospho-RbSer807/811 (CST; #8516), anti-phospho-RbSer780 (CST; #9307), anti-RNA polymerase II CTD (Abcam; ab817), anti-RNA polymerase II CTDphospho S5 (Abcam; ab5131), anti-RNA polymerase II CTDphospho S2 (Abcam; ab5095).
2.9. In vivo assay
The experimental procedures of the animal study were proved by the Animal Care and Use Committee at Nankai University. 4T1 breast cancer cells (5 × 104) were injected in the mammary fat pads of 6 − 8 weeks old female BALB/c mice. Once the tumours grew to a volume of approximately 100–150 mm3, they were placed into five treatment groups (n = 3, with a total tumour number of 15). The mice were treated daily for 18 days via oral gavage. Body weights and tumour size were determined every other day. Tumour measurements were found using a digital vernier calliper, and the volumes were determined using the following calculation: (short2) × long × 0.5. Experiments were performed under an approved IACUC protocol according to federal and institutional guidelines and regulations. Inhibition rate of tumour growth was calculated using the following formula: 100 × {1 − [(tumour volumefinal − tumour volumeinitial) for X22-treated group]/[(tumour volumefinal − tumour volumeinitial) for the vehicle-treated group]}.
2.10. Statistical analysis
Statistical analysis results were analysed values by GraphPad Prism version 6.0 software. For Student t test and ANOVA, p < 0.05 was considered statistically significant. Values were expressed as means ± SEM. Significance was determined by χ2 test, others were determined by Student’s t-test. A value of p < 0.05 was used as the criterion for statistical significance. ***indicates significant difference with p < 0.001, **indicates p < 0.01, *indicates p < 0.05.
3. Results and discussion
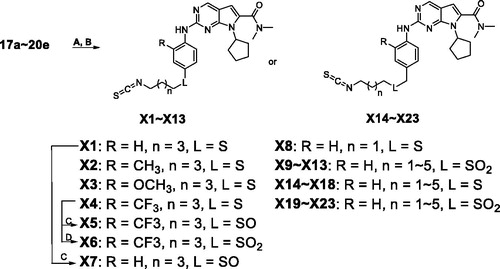
In our previous study, we found structural optimisation based on the pyrrolo-[2,3-d] pyrimidines-2-amine scaffold could improve the inhibitory activity against the cancer cell cycle as targeting CDK4/6 and transcription as targeting CDK9Citation38. As the noteworthy antitumor functions of sulphur atom and isothiocyanate as mentioned above, we introduced these moieties to form molecules to investigate the structure–activity relationship. We changed the substituent at R position on the phenyl ring, as well as the functional group L and length of spacer between the isothiocyanato and phenyl ring. The pathways adopted for the synthesis of compounds X1–X23 were carried out as shown in Schemes 1–3. The synthesis of the key intermediates 5a–5e was depicted in Scheme 1. Firstly, refluxing the commercial obtained potassium phthalimide 1 with various dibromoalkane in acetone yielded the mono-bromides 3a–3e bearing the phthalimide tail meeting the different length for SAR. Then, 3a–3e were refluxed with potassium thioacetate (KSAc) in THF to afford thioacetates 4a–4e, following acidic hydrolysis reaction in methanol to give the thiols 5a–5e. In Scheme 2, prepared 5a–5e were arylated by p-fluoronitrobenzene with various R substituents (6a–6d) to give the intermediates 7a–7h. On the other hand, 5a–5e reacted with 1-(bromomethyl)-4-nitrobenzene (11) in DMF under the alkaline condition of K2CO3 to obtain the 12a–12e. Especially emphasised in dotted line cage, thioether intermediates 7a–7e and 12a–12e, were oxidised absolutely to corresponding sulphone 8a–8e and 13a–13e. Subsequently, nitro group in intermediates 7a–8e and 12a–13e need to be reduced to primary amine using either iron powder activated by acetic acid or zinc powder (9a–9e, 10a–10e, 14a–14e, 15a–15e). After that, all the primary amine intermediates described above underwent Buchwald–Hartwig coupling reaction with commercially available 16 by catalytic amount of palladium acetate to create the key intermediates 17a–18e and 19a–20e. As shown in Scheme 3, the derivatives except X5–X7 were produced via two successive steps without intermediate purification. 17a–20e first were refluxed with 80% hydrazine hydrate in methanol to deprotect, following reacting with carbon disulphide and N,N′-dicylohexylcarbodiimide to afford the isothiocyanates derivatives X1–X4 and X8–X23. Finally, sulphone derivative X6 was obtained by oxidising X4 completely with an excess of m-CPBA. Sulfoxide derivatives X5 and X7 were obtained through incomplete oxidation from X4 and X1, respectively, which were accomplished through controlling strictly the amount of 1.1 equiv m-CPBA.
Scheme 1. Synthesis of intermediates 5a–5ea. aReagents and conditions: (A) Acetone, 70 °C, overnight-; (B) Potassium thioacetate (3.0 eq), THF, 75 °C, 5 h; (C) concentrated HCl, MeOH, 60 °C, 4 h.
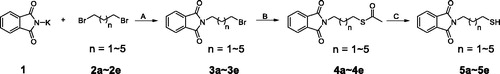
Scheme 2. Synthesis of intermediates 17a–18e, 19a–20ea. aReagents and conditions: (A) K2CO3 (2.0 eq), DMF, rt, 4.5 h; (B) m-CPBA (4.5 eq), DCM, 0 °C to rt, 2 h; (C) Fe/AcOH, MeOH, 60 °C, 5 h; (D) Zn, MeOH, rt, 1.5 h; (E) Pd(OAc)2 (0.10 eq), BINAP (0.06 eq), Cs2CO3 (2.0 eq), 1,4-dioxane, 105 °C, 7 h.
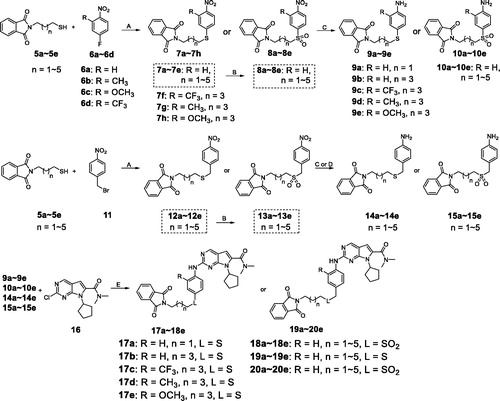
Scheme 3. Synthesis of derivatives X1–X23a. aReagents and conditions: (A) Hydrazine hydrate (6.0 eq), MeOH, rt, 3 h; (B) CS2 (20.0 eq), dicyclohexylcarbodiimide (DCC, 1.1 eq), THF, rt, overnight; (C) m-CPBA (1.5 eq) , DCM, −10 °C to 0 °C, 2 h; (D) m-CPBA (4.5 eq), DCM, 0 °C to rt, 2 h.
The results of the enzymatic-inhibition assays are listed in and with ribociclib as the positive control. We expected to explore an optimisation strategy for improving the inhibitory activity against cell cycle as targeting on CDK4/6 and simultaneously targeting CDK9 for cell transcription. The structure–activity relationships were discussed based on the optimisation at the R, L substituents and the carbon tail length (shown as Formula I and II in ).
Table 1. Structure and enzymatic inhibition activity evaluation of compounds X1–X23.Table Footnotea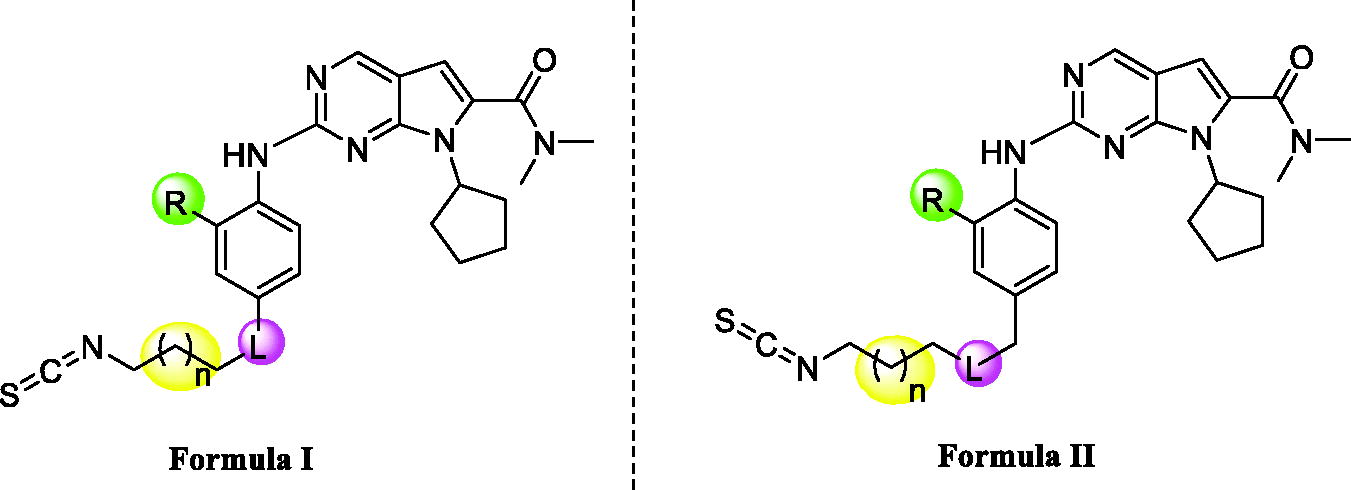
Table 2. Enzymatic inhibition activity evaluation of selected compounds.Table Footnotea
Firstly, we explored the R position with various substituents, such as H, CH3, OCH3, CF3 (X1–X4, ). The results showed that H and CH3 could only maintain the potent against CDK9 compared with ribociclib, while the OCH3 and CF3 groups were not tolerable at all because of the cliff loss potency on all the three kinases. In total, all the optimisation on R position would decrease enzymatic inhibitory activity on different levels, especially for CDK4/6. We presumed whether the joint position L between the isothiocyanato tail and phenyl ring affected the activity. Thus, X4 as a candidate for its poor potency on activity was selected to be oxidised to X5 and X6 structurally characterised as sulfoxide and sulphone, all of which were tested together to confirm the importance of the joint position L as shown in . As expected, X5–X6 displayed more notable increasing inhibitory activity than X4 against all of three kinases. Oxidation state of sulphur at the joint position L might be superior than sulphur, that was also be confirmed in the example of X1 versus X7 (). Further, the derivatives bearing oxidation state of sulphur were tested the IC50 value against CDK4 and CDK9 due to the high inhibition at 1 micromolar concentration (). Compared with ribociclib, X7 exhibited excellent potency against CDK4/9 (IC50: 7 and 26 nM, respectively). At the same time, a derivative (X8) with different space between isothiocyanato and phenyl ring was synthesised to determine the changing the activity produced by the length of the tail. The results indicated that the length of the tail could cause changing on the enzymatic inhibitory activity, but still need to be explored the rules of the changing in detail.
Based on preliminary research, we then adopted the H at R position to design several derivates (X9–X13, ) to focus on the effect of the sulphur and its oxidation state at L position and length of the tail on the enzyme test. Obviously, X9 with sulphone enhanced the inhibition against the three kinases, compared with X8. In the example of compound X9–X13, the inhibition against CDK9 was keeping at a relative high level, but the efficacy activity on CDK4/6 was declined with the increasing space between the isothiocyanato and sulphone structurally.
In view of the importance of the L position and the length of the tail, we attempt to insert a methylene between the L position and the phenyl leading to derivatives X14–X23 (). Excitingly, X19–X23 were proved high inhibition on all three kinases as well as no obvious difference for the enzymatic activity but X23. We summarised the structure–activity relationships as shown in Supplementary Figure S2.
Since our goal here is to discover CDKs inhibitors with cell cycle arrest and transcription blockade activities having high potency for cancer treatment, we screened compounds exhibiting better kinase inhibitory to do the cytotoxicity assays. The selected compounds, including X7, X19, X20 and X22 were carried out using standard CCK8 assay and the results are shown in . X8 and X12 with selective CDK9 inhibition and ribociclib as CDK4 inhibitor were assess the cancer cell inhibition assays as control. IC50 values were detected on compounds against breast cancer cell lines (4T1, T47D and MCF7) and lung cancer cell lines (A549, H1299 and H460). The best tumour cell potency of these analogues was obtained for molecule X22, which was also rational inhibitor of CDK9 with IC50 value of 10 nM and CDK4 with IC50 value of 30 nM. The CDK4 inhibitor ribociclib alone was not potent in these solid tumour cells. X22 displayed higher inhibition potency than selective CDK4 inhibitor ribociclib and selective CDK9 inhibitor X8 and X12. This result demonstrated that dual-target compound had more efficient inhibition at the cellular level. As the results of enzymatic and antitumor activity in cell lines showed that X22 is the best in this class, we performed further in-depth in vitro and in vivo biological studies using this compound.
Table 3. In vitro cell growth inhibition (IC50) of selected compounds against multiple cancer cell lines.a
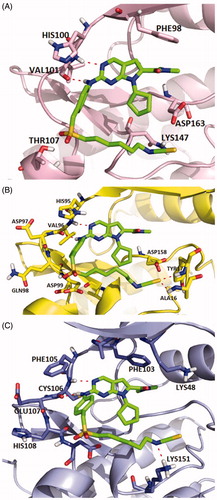
Molecular docking studies were carried out to investigate the binding modes of compound X22. X22 is a potent CDK4 and CDK9 inhibitor with IC50 value of 30 and 10 nM, respectively, and also has weak inhibition activity against CDK6 with IC50 value of 126 nM. As shown in , X22 binds to the ATP-binding sites of CDK6 with two key hydrogen bonds formed by aminopyrimidine and the backbone VAL101 residue, which retained the binding mode of the scaffold of ribociclib and CDK6 as reported. Given no 3D CDK4-ligand was reported, the structure of CDK6-ligand complex (PDB code: 4EZ5) was used as a template for homology modelling to generate the 3D structure of CDK4-ligand (). Importantly, two additional hydrogen bonds were formed because of the hydrophobic side chain containing isothiocyanate. The sulphur atom formed two hydrogen bonds with the backbone of ALA16 and TYR17 at the front of the ATP binding site of CDK4, respectively, which might explain the better inhibitory activity of the compound X22 against CDK4 (IC50 = 30 nM) than CDK6 (IC50=126 nM). In , the 2-aminopyrimidine formed two hydrogen bonds with the CYS106 residue of X22 in CDK9 (VAL101 in CDK6, VAL in CDK4). As well, the nitrogen atom formed a key hydrogen-bond with the residue of the LYS151 at the front of the ATP binding pocket. At the back of the ATP binding site, the pyrrolo-[2,3-d]-pyrimidine scaffold exploited the hydrophobic region close to the gatekeeper residue to form a favourable π–π interaction in all three kinases region.
Figure 1. Representation of the predicted binding modes of compound X22 with CDKs kinase domain. X22 is shown in green. Hydrogen bonds are shown in red. (A) Proposed binding mode of compound X22 with CDK6 (PDB code: 4EZ5), CDK6 backbone is shown in light pink. (B) Proposed binding mode of compound X22 with CDK4, which employed CDK6 (PDB entry: 4EZ5) as the template for homology modelling, CDK4 backbone is shown in yellow. (C) Proposed binding mode of compound X22 with CDK9 (PDB code: 4BCF), CDK9 backbone is shown in light blue.
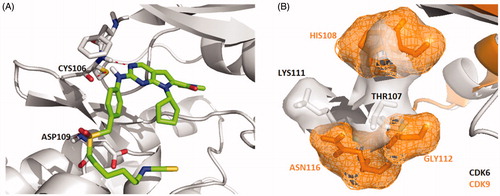
Interestingly, a hydrogen-bond was formed between the oxygen of the sulphonyl of X22 with the backbone of the ASP109 in another docking configuration (). At the front of the ATP binding pocket in CDK9, GLY112 possessing the smallest residue makes this hydrophobic site accommodate flexible side chain like compound X22, while THR107 at the same position in CDK6 shows a little crowded for flexible group (). The much bigger hydrophobic site permit the compound X22 exits in multiple configurations docking CDK9 forming hydrogen bond for binding tightly. The difference might offer an explanation the stronger inhibitory potency compound X22 against CDK9 (IC50= 10 nM). Thus, X22 as an ATP-competitive inhibitor potently binds in the ATP pockets of CDKs kinases.
Figure 2. (A) Representation of the predicted binding mode of compound X22 with CDK9 kinase domain (PDB code: 4BCF). CDK9 backbone is shown in grey. X22 is shown in green. Hydrogen bonds are shown in red. (B) Representation of the hydrophobic pocket difference at the front of the ATP-binding site by overlaying CDK6 and CDK9. CDK6 is shown in grey. CDK9 is shown in orange.
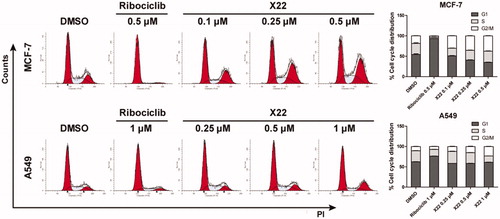
Given the strong inhibitory ability of X22 in MCF7 and A549 cells, we examined the effect of X22 on the cell cycle. Cells were treated with compound X22, ribociclib and DMSO for 24 h. Representative flow cytometry patterns are shown in . The results showed that X22 significantly blocked the cell cycle at G2/M phase in a dose-dependent manner accompanied by decreases in S phase in both cell lines, as compared to the cells incubated with DMSO. Ribociclib mainly blocked the cell cycle at G1 phase (). We also detected cell kinetics treated with X22, ribociclib and DMSO for 12, 36 and 48 h (Supplementary Figure S3). These results confirmed that X22 induced the cell cycle arrest, which are consistent with our observations aboveCitation39. Compared with CDK4 depletion alone, combined depletion CDK4 and CDK9 induced substantial G2/M arrest. These data indicated that X22 could induce G2/M phase arrest of the cell cycle in both breast and lung cancer cells.
Figure 3. X22 induced G2/M phase arrest in breast and lung cancer cells. Cells were harvested after treatment with different concentrations of X22 or ribociclib for 24 h. Cells were fixed by 70% ethanol and stained with propidium iodide. Data are represented as histogram on the left and the percentage of cell cycle distribution are shown on the right. The assays were performed in triplicate.
As reportedCitation40,Citation41, CDK4/6 preferentially bind D-type cyclins to form complexes. Activations of these complexes are responsible for the phosphorylation of retinoblastoma protein (Rb), which allows cell cycle to proceed from G1 to S phase and further results in cell proliferation. In contrast, CDK9 is not a typical Cdc-2-like kinase and does not participate in cell cycle regulation. It can form the P-TEFb complex, which is capable of phosphorylating the CTD of the largest subunit of RNAPII and regulate the RNA transcription elongation.
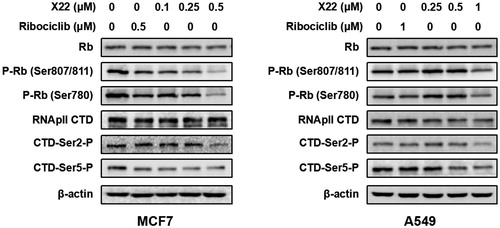
To further verify the inhibitory effect of compound X22 on CDKs, western blotting analysis was performed. We found that X22 treatment significantly suppressed the phosphorylation of Rb at CDK4/6 specific site Ser 780 and CDK9 specific sites Ser 807/811 in MCF7 and A549 cells respectively, which confirmed that X22 targeted on CDKs (). We also detected the level of phosphorylated Ser2 (Ser2-P), a well-established cellular target of CDK9 on the RNAPII CTD during transcription elongation. Compared to cells treated with DMSO, X22 effectively inhibited the phosphorylation of Ser2 and Ser5 at 0.5 µM in MCF7 cells and 1 µM in A549 cells (). Taken together, the results above revealed that X22 could specifically target CDKs, thus further regulating their downstream signalling proteins in cancer cells.
Figure 4. X22 suppressed the downstream signalling proteins of CDK4/9 in breast and lung cancer cells. Cells were incubated with the indicated concentrations of X22 or ribociclib for 24 h. Proteins were extracted and analysed by western blotting.
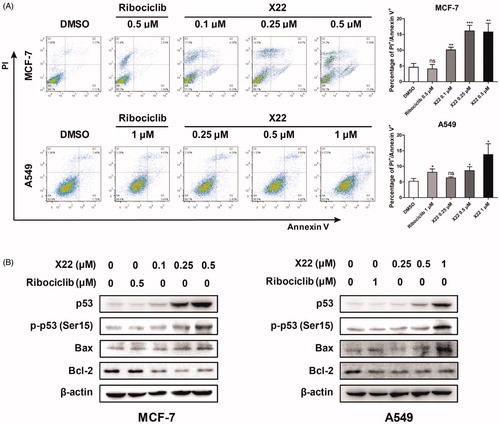
We next analysed whether X22 treatment promoted cell apoptosis by Annexin V-FITC/PI staining. After 48 h incubation, X22 dose-dependently increased the proportion of apoptotic cells (Q2 late apoptotic and Q3 early apoptotic) in A549 cells, compared to the cells treated with DMSO or ribociclib. Similar results were also observed in MCF7 cells. X22 at 0.1 to 0.5 µM increased the percentages of apoptotic cells by approximately 2.2 to 3.5-fold change, compared to DMSO-treated cells (). To further confirm the apoptosis induction of compound X22, we examined the expression of relevant proteins by Western blot. Cells were treated with or without compound X22 and control for 48 h and then lysed and analysed. Given the crucial role of p53 in regulating cell apoptosis, we detected whether compound X22 induced apoptosis depending on the expression of p53 and its downstream targets. As illustrated in , X22 treatment remarkably increased the expressions of p53 and promoted the phosphorylation of p53 at Ser15. We also demonstrated that X22-induced apoptosis in solid tumour cells, which reduced expression of the anti-apoptotic protein Bcl-2 and enhanced expression of Bax. These findings indicated that compound X22-induced apoptosis is associated with upregulating pro-apoptotic protein p53 and Bax and downregulating anti-apoptotic protein Bcl-2 expression. Collectively, X22 may induce cell apoptosis through p53/Bax/Bcl-2 pathway in breast and lung cancer cells.
Figure 5. X22 induced cell apoptosis in breast and lung cancer cells. (A) Cells were seeded in six-well plates and treated with the indicated concentrations of X22 or ribociclib for 48 h. Cells were then stained with the AnnexinV-FITC Apoptosis Detection Kit, followed by flow cytometry analysis. Quantitative data are expressed as mean ± SD of the percentages of apoptotic cells from three independent experiments. (B) Expressions of p53, phospho-p53 (Ser15), Bax, Bcl-2 in MCF-7 and A549 cells were detected by western blotting after the treatment with the indicated concentrations of ribociclib or X22 for 48 h. *p < 0.05 versus DMSO; **p < 0.01 versus DMSO; ***p < 0.001 versus DMSO.
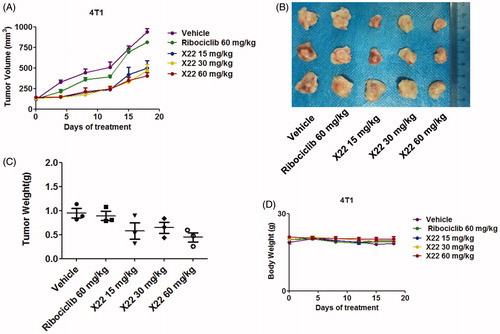
To better assess whether X22 could efficiently suppress tumour growth in vivo, homograft tumour models were established using 4T1 cells. Oral treatment with X22 significantly suppressed the tumour growth (). Representative photographs of excised tumours at day 18 were shown in . X22 treatment with 15 or 30 or 60 mg/kg substantially inhibited the tumour growth with tumour inhibition rates of 51.8 ± 17.5%, 65.4 ± 11.7% and 70.3 ± 4.4%, respectively. In contrast, tumour volume in ribociclib-treated group decreased slightly compared with vehicle group. Tumour growth inhibitions of 17.2 ± 5.2% were observed at doses of 60 mg/kg of ribociclib. The potencies of ribociclib are relatively weaker than those of X22. The average tumour weight of excised tumours in X22 groups was obviously lighter than those in other groups (). A preliminary toxicity evaluation for X22 was also carried out. Mouse weights were monitored twice per week over 18 days. There was no significant difference among the five groups of mice (). No obvious signs of toxicity were observed in the X22-treated groups, indicating that compound X22 is well tolerated in vivo.
Figure 6. X22 significantly inhibited growth of breast cancer in homograft mouse models. (A) Tumour growth curve of 4T1 tumour-bearing mice in response to different treatments. (B) Photographs of 4T1 homograft tumours. Mice were sacrificed 18 days after tumour implantation. (C) The average tumour weight of excised tumours at day 18. (D) Body weight curves of BALB/c mice in each group after injection.
4. Conclusions
To date, selective CDK inhibitors have produced only modest activity against solid tumours. In this article, we discovered and studied the medicinal chemistry of multi-CDKs inhibitors to assess their relative activities against the cell cycle and transcriptional CDKs. Several compounds inhibited CDK4 and CDK9 at low nanomolar levels and exhibited good antiproliferative activities in a panel of tumour cells. The most potent analogue X22 potently inhibited CDK4 and CDK9 with IC50 values of 30 and 10 nM, respectively, which showed marked antitumor activity. In in vitro cellar assays, X22 was capable of blocking the cell cycle at G2/M phase resulting in decreased S-phase populations. Compared with CDK4 depletion alone, combined depletion induced apoptosis in cancer cells in a dose-dependent manner. Western blot assays confirmed substantial apoptosis after X22-induced CDK4 and CDK9 depletion. Meanwhile, X22 reduced RNA polymerase II expression and CTD phosphorylation. In in vivo assay, oral administration of compound X22 once-daily at 15, 30 or 60 mg/kg for 18 days led to tumour regression without obvious toxicity. Thus, reduced CDK4 activity in concert with depleted CDK9 activity may enhance the antiproliferative effects. This study suggest combined depletion of cell cycle and transcriptional CDK activities may be worthy of clinical further development for solid tumour therapy.
Supplemental Material
Download PDF (1,008.9 KB)Disclosure statement
The authors report no declarations of interest.
Additional information
Funding
Related Research Data
References
- Ciceri P, Muller S, O'Mahony A, et al. Dual kinase-bromodomain inhibitors for rationally designed polypharmacology. Nat Chem Biol 2014;10:305–12.
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–74.
- Johnson LN, Lewis RJ. Structural basis for control by phosphorylation. Chem Rev 2001;101:2209–42.
- Adams JA. Kinetic and catalytic mechanisms of protein kinases. Chem Rev 2001;101:2271–90.
- Huang M, Shen AJ, Ding J, Geng MY. Molecularly targeted cancer therapy: some lessons from the past decade. Trends Pharmacol Sci 2014;35:41–50.
- Ma WW, Adjei AA. Novel agents on the horizon for cancer therapy. Ca Cancer J Clin 2009;59:111–37.
- Sun C, Bernards R. Feedback and redundancy in receptor tyrosine kinase signaling: relevance to cancer therapies. Trends Biochem Sci 2014;39:465–74.
- Wu P, Nielsen TE, Clausen MH. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol Sci 2015;36:422–39.
- Baker SJ, Reddy EP. Targeted inhibition of kinases in cancer therapy. Mt Sinai J Med 2010;77:573–86.
- Krug M, Hilgeroth A. Recent advances in the development of multi-kinase inhibitors. Mini Rev Med Chem 2008;8:1312–27.
- Asghar U, Witkiewicz AK, Turner NC, et al. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov 2015;14:130–46.
- Peyressatre M, Prevel C, Pellerano M, Morris MC. Targeting cyclin-dependent kinases in human cancers: from small molecules to peptide inhibitors. Cancers 2015;7:179–237.
- Deshpande A, Sicinski P, Hinds PW. Cyclins and cdks in development and cancer: a perspective. Oncogene 2005;24:2909–15.
- Kalra S, Joshi G, Munshi A, Kumar R. Structural insights of cyclin dependent kinases: Implications in design of selective inhibitors. Eur J Med Chem 2017;142:424–58.
- Harbour JW, Luo RX, Dei Santi A, et al. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell 1999;98:859–69.
- Romano G. Deregulations in the cyclin-dependent kinase-9-related pathway in cancer: implications for drug discovery and development. ISRN Oncol 2013;2013:1–14.
- Morales F, Giordano A. Overview of CDK9 as a target in cancer research. Cell Cycle 2016;15:519–27.
- Wang S, Fischer PM. Cyclin-dependent kinase 9: a key transcriptional regulator and potential drug target in oncology, virology and cardiology. Trends Pharmacol Sci 2008;29:302–13.
- Bose P, Simmons GL, Grant S. Cyclin-dependent kinase inhibitor therapy for hematologic malignancies. Expert Opin Investig Drugs 2013;22:723–38.
- Vijayaraghavan S, Moulder S, Keyomarsi K, et al. Inhibiting CDK in cancer therapy: current evidence and future directions. Targeted Oncol 2018;13:21–38.
- Sarosiek T. Inhibitors of cyclin-dependent kinases (CDK) – a new group of medicines in therapy of advanced breast cancer. Polski Merkuriusz Lekarski: organ Polskiego Towarzystwa Lekarskiego 2018;44:5–9.
- Heptinstall AB, Adiyasa I, Cano C, Hardcastle IR. Recent advances in CDK inhibitors for cancer therapy. Future Med Chem 2018;10:1369–88.
- Shapiro GI. Cyclin-dependent kinase pathways as targets for cancer treatment. J Clin Oncol 2006;24:1770–83.
- DiPippo AJ, Patel NK, Barnett CM. Cyclin-dependent kinase inhibitors for the treatment of breast cancer: past, present, and future. Pharmacotherapy 2016;36:652–67.
- Beaver JA, Amiri-Kordestani L, Charlab R, et al. FDA approval: palbociclib for the treatment of postmenopausal patients with estrogen receptor-positive, HER2-negative metastatic breast cancer. Clin Cancer Res 2015;21:4760–6.
- Shah A, Bloomquist E, Tang S, et al. FDA approval: ribociclib for the treatment of postmenopausal women with hormone receptor-positive, HER2-negative advanced or metastatic breast cancer. Clin Cancer Res 2018;24:2999–3004.
- FDA OKs abemaciclib for ER+, HER2-breast cancer. Cancer Discov 2017;7:OF1.
- Im SA, Mukai H, Park IH, et al. Palbociclib plus letrozole as first-line therapy in postmenopausal Asian women with metastatic breast cancer: results from the phase III, randomized PALOMA-2 study. J Glob Oncol 2019;5:1–19.
- Meattini I, Desideri I, Scotti V, et al. Ribociclib plus letrozole and concomitant palliative radiotherapy for metastatic breast cancer. Breast 2018;42:1–2.
- Single-agent abemaciclib active in breast cancer. Cancer Discov 2016;6:809–10.
- Hafner M, Mills CE, Subramanian K, et al. Multiomics profiling establishes the polypharmacology of FDA-approved CDK4/6 inhibitors and the potential for differential clinical activity. Cell Chem Biol 2019;26:1067–80.
- Lukasik PM, Elabar S, Lam F, et al. Synthesis and biological evaluation of imidazo[4,5-b]pyridine and 4-heteroaryl-pyrimidine derivatives as anti-cancer agents. Eur J Med Chem 2012;57:311–22.
- Singh U, Chashoo G, Khan SU, et al. Design of novel 3-pyrimidinylazaindole CDK2/9 inhibitors with potent in vitro and in vivo antitumor efficacy in a triple-negative breast cancer model. J Med Chem 2017;60:9470–89.
- Cai D, Latham VM, Jr., Zhang X, Shapiro GI. Combined depletion of cell cycle and transcriptional cyclin-dependent kinase activities induces apoptosis in cancer cells. Cancer Res 2006;66:9270–80.
- Surur AS, Schulig L, Link A. Interconnection of sulfides and sulfoxides in medicinal chemistry. Archiv Der Pharmazie 2019;352:e1800248.
- Gupta P, Kim B, Kim SH, Srivastava SK. Molecular targets of isothiocyanates in cancer: recent advances. Mol Nutr Food Res 2014;58:1685–707.
- Guo Q, Yu C, Zhang C, et al. Highly selective, potent, and oral mTOR inhibitor for treatment of cancer as autophagy inducer. J Med Chem 2018;61:881–904.
- Li Y, Luo X, Guo Q, et al. Discovery of N1-(4-((7-cyclopentyl-6-(dimethylcarbamoyl)-7 H-pyrrolo[2,3- d]pyrimidin-2-yl)amino)phenyl)-N8-hydroxyoctanediamide as a novel inhibitor targeting cyclin-dependent kinase 4/9 (CDK4/9) and histone deacetlyase1 (HDAC1) against malignant cancer. J Med Chem 2018;61:3166–92.
- Chiu HC, Huang WR, Liao TL, et al. Mechanistic insights into avian reovirus p17-modulated suppression of cell cycle CDK-cyclin complexes and enhancement of p53 and cyclin H interaction. J Biol Chem 2018;293:12542–62.
- Thangavel C, Boopathi E, Liu Y, et al. Therapeutic challenge with a CDK 4/6 inhibitor induces an RB-dependent SMAC-mediated apoptotic response in non-small cell lung cancer. Clin Cancer Res 2018;24:1402–14.
- Aleem E, Arceci RJ. Targeting cell cycle regulators in hematologic malignancies. Front Cell Dev Biol 2015;3:16.