
Abstract
A series of new carbohydrate-based sulphonamide derivatives were designed, synthesised by employing the so-call ‘sugar-tail’ approach. The compounds were evaluated in vitro against a panel of CAs. Compared to their parent compound p-sulfamoylbenzoic acid, these compounds showed nearly 100-fold improvement in their binding affinities against hCA II in vitro. All of compounds showed great water solubility and the pH value of their water solutions of compounds is 7.0. Such properties are advantageous to make them much less irritating to the eye when applied topical glaucomatous drugs, compared to the relatively highly acidic dorzolamide preparations (pH 5.5). Notably, compounds 7d, 7 g, 7 h demonstrated to topically lower intraocular pressure (IOP) in glaucomatous animals better than brinzolamide when applied as a 1% solution directly into the eye. Low cytotoxicity on human cornea epithelial cell was observed in the tested concentrations by the MTT assay.
Introduction
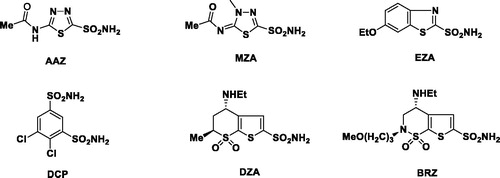
Glaucoma is an ophthalmologic disease featured by a high intraocular pressure (IOP), ultimately resulting in the irreversible loss of peripheral vision, and eventually into blindnessCitation1. Clinically, there are many treatments available for glaucoma such as surgery, laser, and pharmacologic therapies. For the majority of glaucoma patients, lowering IOP by pharmacologic therapy is the preferred choice. It is well-known that the systemic administration of inhibitors of carbonic anhydrase (CA) II, an enzyme present in ciliary epithelial cells located in the ciliary body, reduces aqueous humour secretion and lowers the IOPCitation2. Carbonic anhydrase inhibitors (CAIs) such as acetazolamide (AAZ), methazolamide (MZA), ethoxzolamide (EZA), and dichlorophenamide (DCP) can be used as pressure-lowering systemic drugs in the treatment of glaucomaCitation3–5. However, they may cause undesired side effects such as fatigue, augmented diuresis, or paresthesias due to inhibition of the carbonic anhydrase isoforms present in many other tissues and organs than the eyeCitation6,Citation7. To avoid the above-mentioned side effects, it is better for the inhibitors to have topical activity therefore can be applied directly into the eye. Currently, two topically effective CAIs have been developed: dorzolamide (DZA) and brinzolamide (BRZ)Citation8,Citation9. Both drugs can be applied topically as water solutions/suspensions and produce a prolonged reduction of IOPCitation10,Citation11. They showed fewer side effects as compared to the systemically applied sulphonamides. Clinically, they are used as hydrochloride salts in need of good water solubilityCitation12. However, in many cases it will lead to the lowering of the pH in such solutions. Consequently, the acidic solution will result in frequent stinging sensations, burning or reddening of the eye, blurred vision, pruritus, and other local side effects after the topical administration ()Citation13,Citation14.
Figure 1. Clinically used sulphonamides AAZ, MZA, EZA, DCP, DZA and BRZ.
To reduce this side effect, many efforts were made to design topical antiglaucoma sulphonamides with more balanced pKas. One successful approach for designing topical CAIs was termed “water-solubilizing tail approach”, which was a special version of ‘sugar-tail approach’. It has been applied for the synthesis of many classes of topical antiglaucoma sulphonamide CAIsCitation15–19. In this study, we employed this approach for designing and obtaining water-soluble, high-affinity sulphonamide CA inhibitors, which have great water solubility without the need to form hydrochloride salts. Herein, we report our work for obtaining carbohydrate-based CA II inhibitors with great water solubility in the neutral pH range, and potent and long-lasting in vivo properties as IOP lowering agents in glaucoma animal models.
Results and discussion
Sugar-tail approach to compound design
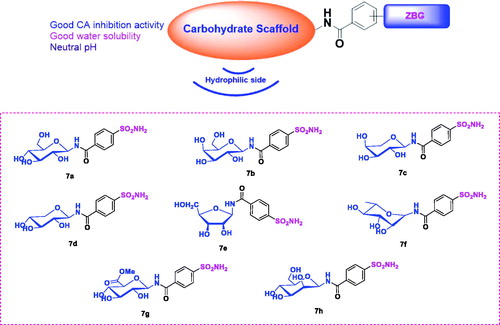
The main goal of this study was to obtain highly potent, water soluble CA inhibitors with potentially few side effects owing to the neutral pH of the eye drop solution when these agents were used in the treatment of glaucoma. Sugar-tail approach was employed for the following reasons (): (1) It was expected that compounds designed by this approach could form more interactions with the CA active site for the hydrophilic glycosyl moiety to interact with the hydrophilic half of the active site. (2) Due to the highly hydrophilic nature of sugar moieties, good water solubility could be achieved by incorporating them. (3) The pH of the water solutions of these compounds was in the neutral pH range therefore there was no need to form hydrochloride salts, making them much less irritating to the eyes. Target compounds were designed by attaching aminosaccharide group as the hydrophilic fragment via an amide bond linker to a p-sulfamoylbenzoic acid as the zinc binding group (ZBG). Different types of aminosaccharides were chosen to provide insight into the structure-activity relationship (SAR) (. Sugars used in this study were glucose, galactose, arabinose, xylose, ribose, rhamnose, glucuronic acid and mannose.
Figure 2. Structures of the sugar-tail compounds 7a–7h.
Chemistry
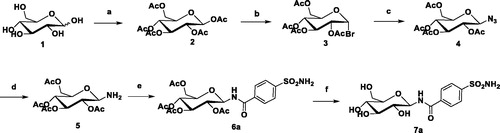
The general synthetic strategy for the synthesis of the target compounds is shown in Scheme 1. Monosaccharide was selected as the starting material, and acetylation with acetic anhydride to form intermediate 2 in pyridine. The terminal glycosyl bromide 3 was prepared by the reaction of 2 with HBr-AcOH in good yield. Intermediate 4 was formed by the reaction of a glycosyl bromide with an excess of sodium azide. Next, the intermediate 4 was reacted with hydrogen under the catalysis of Pd/C to form an intermediate 5. The condensation reaction of amino sugar with p-sulphonamide benzoic acid forms an intermediate 6 with an amide bond as a linker. Finally, deacetylation with a catalytic amount of NaOCH3 in CH3OH gave the target analogue 7Citation20. All compounds were extensively characterised (Supplementary Material).
Scheme 1. Synthesis of representative compound 7a. Reagents and Conditions: (a) Ac2O, pyridine, 5 h; (b) HBr-AcOH, CH2Cl2, r.t., 6 h, 62%; (c) NaN3, DMF, r.t. 5 h, 85%; (d) Pd/C, EA, 2 h, 89%; (e) EDCI, DCM, r.t., 2 h, 74%; (f) NaOMe, MeOH, r.t., 85%.
The different sugar derivatives were prepared by the same method. Direct condensation of monosaccharides, such as: glucose derivative 7a, galactose derivative 7b, xylcose derivative 7c, arabinesose derivative 7d, ribose derivative 7e, rhamnose derivative 7f, glucuronic acid derivative 7g, manose derivative 7h.
Biological activity
CA inhibition studies
To determine the enzymatic inhibitory activity, all the newly synthesised sugar derivatives 7a–7h were evaluated by enzyme assays in vitro on three physiologically relevant CA isoforms including CA I, CA II and CA IX (). AAZ was chosen as the control compound.
Table 1. The inhibitory activities of the compounds against hCA II and hCA I.
Inhibition data for sugar derivatives 7a–7h reported here, p-sulfamoylbenzoic acid and clinically used agent AAZ are reported in . The following structure-activity relationship (SAR) were summarised based on the CA inhibition data with these sugar derivatives investigated here:
The ubiquitously isoform hCA I in the body was weakly inhibited by the parent compound p-sulfamoylbenzoic acid (IC50 of 120 μM), as well as by sugar derivatives 7a–7h (IC50 1.8–12 μM). As CA I is considered an off-target CA isozyme, this weak CA I inhibition is desirable for the compounds designed to target the glaucoma-associated enzymes.
Against the physiologically dominant isoform hCA II, these sugar compounds showed IC50 values of 0.009–0.27 μM (). Compared to the parent compound, p-sulfamoylbenzoic acid, which has an IC50 value of 15 μM against hCA II, the sugar derivatives 7a–7h are at least two orders of magnitude more potent. Compound 7h, possessing manose fragment in its structure, was the best hCA II inhibitor (IC50 = 0.009 nM). The combination of both designed by the sugar-tail approach increased the inhibitory activity by two orders of magnitude, presumably due to simultaneously binding to the zinc ion and occupying the hydrophilic subpocket of the active site. Such modes of binding were further analysed and supported in detail in the molecular docking analysis section.
The SAR for the inhibition of the tumor-associated isoform hCA IX was similar to hCA II: Notably, compounds 7a–7h (IC50 = 0.54–0.95 μM) were also much better than p-sulfamoylbenzoic acid (IC50 = 24 μM) for hCA IX inhibition. This is also presumably due to the matching the hydrophilic half of the active site with the hydrophilic glucosyl moiety, in addition to the sulphonamide as the zinc binding group.
Then, the selectivity of compounds 7a–7h was explored for CA I, CA II and CA IX. The parent fragment p-sulfamoylbenzoic acid displayed weak selectivity for hCA II with respect to isozymes hCA I (8-fold) and hCA IX (1.6-fold). The compounds 7a–7h displayed high selectivity for the hCA II isozyme compared with both hCA I (400-fold) and IX (17-fold). Collectively, these results showed it was possible to generate hCA II selective inhibitors by tethering a sugar tail onto the CA anchor pharmacophore over the physiologically abundant hCA I and IX isozymes. These results also indicated that selective inhibition of carbonic anhydrase II of sugar-derivatives may reduce the side effects of current clinical drugs, and is expected to be a good candidate for treatment of antiglaucoma.
Solubility and pH assay
Generally, in order to have noticeable pharmacological effects, the majority of topical antiglaucoma drugs need to possess acceptable solubility. For example, dorzolamide must be dissolved as hydrochloride salt and as a consequence, the rather acidic solution (pH = 5.5) causing eye irritation. Therefore, some important physicochemical properties of new sulphonamides were investigated in detail (). Firstly, we explored the solubility of compounds 7a–7h in water. Compounds 7a–7h showed excellent water solubility which would be suitable for developing topical IOP lowering agents. In contrast, clinically used carbonic anhydrase inhibitors such as AAZ, DZA and BRZ possess lower water solubility. Next, we explored the pH values of compounds 7a–7h in water. It was shown that all the solutions had pH of 7 while the solutions of AAZ, DZA, and BRZ had pH lower than 7 (). Thus, it was easy to formulate them as eye-drops at neutral pH values. This also showed that due to the presence of the glycosyl tail in derivatives 7a–7h their water solubility was drastically increased while the pH of their solutions stayed at neutral pH, which constituted a very desirable pharmacological feature for topically acting antiglaucoma drugs.
Table 2. The solubility and pH of the solutions.
Topical antiglaucoma activity
The clinical typical symptom of glaucoma is IOP increase. Next, we investigated the IOP lowering ability of the most active compounds against the hCA II. An animal model of glaucoma, i.e., rabbits with high IOP, induced by the injection of 0.1 mL of hypertonic saline solution (5% in distilled water) into the vitreous of both eyesCitation21–23.
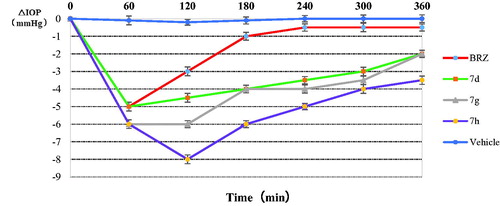
Three of the best in vitro CA inhibitors investigated here, 7d, 7g, 7h were formulated as 1% water solution and administered topically to hypertensive rabbits. The variation of IOP of hypertensive rabbits treated topically with one drop (50 μL) of 1% solution of inhibitors is shown in . The drug BRZ (1%, suspension) was also included in our experiments. It can be seen that BRZ was an efficient IOP-lowering agent, with a maximal effect (around 5 mm Hg) achieved at 1 h post-administration, and a return to basal IOP values after 3 h. However, the three sugar sulphanilamides investigated here were much more effective IOP lowering agents as compared to the clinically used drug BRZ. Especially, the arabinose derivative 7d was again maximally active after 1 h post-administration, producing an IOP lowering of around 5 mm Hg. A potent IOP lowering of 3.5–5 mm Hg was then maintained for the next 4–5 h. The glucuronic acid derivative 7g was slightly more effective than 7d. The maximal effect (6 mmHg) has been observed after 1 h post administration, and it lasted for up to 4–5 h. The best IOP lowering agent was the manose derivative 7h. In this case, the maximal IOP lowering (of around 8 mm Hg) has been achieved after 2 h post-administration, and this very potent effect was maintained for the next 5–6 h when pressure returned to the basal values. Thus, the three derivatives showed much more effective IOP lowering as compared to BRZ: both the magnitude of the effect as well as its duration of action were very much augmented, which constitute very desirable properties for topically acting antiglaucoma drugs. Moreover, no ocular discomfort or eye irritation of the experimental animals have been observed after administration of these sugar sulphonamide derivatives.
Figure 3. Effect of topically administered sulphonamide CA inhibitors (1% water solutions/suspensions) on the IOP of hypertensive rabbits. Errors were in the range of 3–5% of the reported values.
Cytotoxicity of tested compounds on mammalian cells
In order to ensure selectivity of the antiglaucoma effects, the cytotoxicity of compounds 7a–7h were evaluated by MTT assay using four mammalian cell lines, including HCEC (human corneal epithelial cell), HepG2 (human liver carcinoma), HT-29 (human colorectral cancer) and H9C2 (normal, rat myocytes). Meanwhile, we compared the effects of these sugar derivatives with those of the standard sulphonamide inhibitors AAZ and BRZ, working in the same conditions.
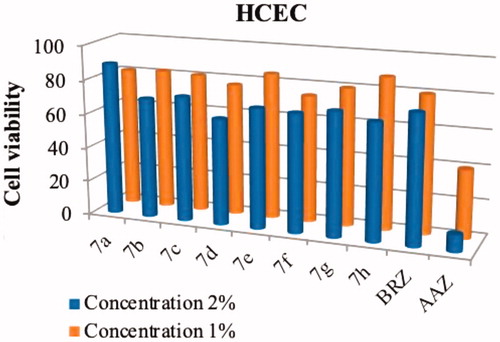
Compounds 7a–7h, AAZ and BRZ did not display any appreciable toxicity to the four mammalian cell lines when tested up to 100 μM (data not shown). However, our target compounds were designed as a topical antiglaucoma drug, which was usually made into an eye drop that concentration is 1–2%, so the topical concentration were large. With this in mind, we prepared compounds 7a–7h at concentrations ranging from 1% to 2% and tested the cytotoxicity on HCEC cell again. As shown in , compounds 7a–7h had low toxicity against the HCEC cell (74–88% cell viability) while the cell viability rates of AAZ and BRZ towards HCEC cell were 80% and 40% at 1% concentration. The increase of the concentration did significantly affect the compound toxicity. For instance, at 2% concentration AAZ showed a viability dropping (10%) and reduced cell viability was observed for compounds 7a–7h (62–89% cell viability).
Figure 4. Effects of compounds 7a–7h, BRZ and AAZ, at 1% and 2% concentrations, on the viability of HCEC cell line. Values are the average of three independent experiments. Relative errors are generally within 5–10%.
Molecular docking analyses
Molecular docking was employed to provide insight into the inhibitory activities against the CA I, II and IX enzymes listed in . The data showed that the inhibitory activities of the target compounds were more of the same magnitude towards the same enzyme. It was expected that the structural difference among the sugars were minor and did not cause big difference in the inhibitory activities. It was also observed that the inhibitory activities of the target compounds were more of the same magnitude between CA II and CA IX while about one order of magnitude more potent for CA II than CA I. It was also not surprising because sequence analysis showed that the amino acids composed of the active sites were almost identical between CA II and CA IX while there were several key differences between CA II and CA I. Consequently, the following docking studies were performed between compound 7c as the representative compound in the active sites of CA II and CA I.
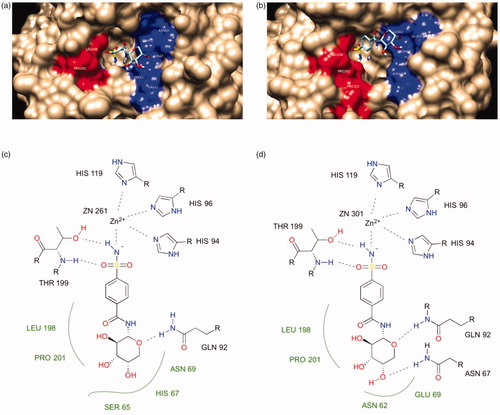
The docking studies started with a redocking experiment by AutoDock 4.2.6 with the AutoDock4Zn force field for AAZ with CA I and II (PDB: 1AZM and 3HS4). The redocking results showed that the experimental pose of AAZ was reproduced fairly well, especially with the interactions between the sulphonamide functional group of AAZ and the zinc ion in CA II. This is the characteristic of sulphonamides as CA inhibitors, which the compounds in will follow. Next, the same model was applied to the representative compound 7c. The results showed that the sulphonamide amine N atom of compound 7c binds directly to the active-site Zn atom along with the side chains of HIS94, HIS96 and HIS119 and the sulphonamide group forms a hydrogen bond to THR199. Furthermore, the sugar moiety of the compound preferably interacted with the hydrophilic region of the CA active site (highlighted in red in . It is consistent with the fact that sugars have many hydroxyl groups in their structures therefore they are highly hydrophilic and they tend to interact with the hydrophilic region of the active site favourably. The major difference in the active sites of CA I and II is the ASN 67 in CA II, which could form hydrogen bond with the nearby hydroxyl group in the sugar while the corresponding amino acid in CA I is HIS 67, which could not form hydrogen bond with the same hydroxyl group because it could not satisfy the criteria of distance and angle constraint. Such evidence explained that why compound 7c was about one order of magnitude more active against CA II than CA I.
Figure 5. The binding mode of compound 7c in the active site of CA I and II. (a) and (b) are surface representations for CA I and II, respectively. (c) and (d) are 2D representations for CA I and II, respectively.
Conclusion
Here, we report a novel class of water-soluble, topically very effective and long-acting antiglaucoma sulphonamides, obtained by attaching aminosaccharide moieties to well-known aromatic sulphonamides incorporating free carboxy group. Eight of the novel carbohydrate-based compounds exhibited excellent CA II inhibitory activity with 100-fold of the initial fragment p-sulfamoylbenzoic acid. In addition, all of the newly reported derivatives showed very good water solubility, in the range of 1–2.5% at neutral pH values. In particular, some of the most active inhibitors strongly lowered IOP in glaucomatous rabbits, showing a prolonged duration of action in comparison to brinzolamide. The cytotoxicity on human cornea epithelial cell from novel carbohydrate-based compounds 7a–7h was evaluated, and low cytotoxicity was observed. These compounds constitute valuable leading compounds for obtaining topically acting antiglaucoma CAIs of a new generation, with improved efficacy, longer duration of action, and reduced side effects.
Experimental section
Chemistry
Reagents and starting materials in current study were of analytic grade and used without further purification unless otherwise specified. Analytical thin layer chromatography was performed on silica gel HF254 plates. Preparative column chromatography was performed using silica gel H. Melting points were determined by a Büchi melting point B-540 apparatus. All 1H and 13C NMR spectra were recorded at 600 MHz and 150 MHz on a Bruker ARX 600 MHz model spectrometer in DMSO-d6 with TMS as the internal standard. NMR spectra were analysed and interpreted using MestReNova. ESI-MS spectra were recorded on an Agilent ESI-QTOF instrument. HR-MS were obtained on a Bruker micrOTOF_Q spectrometer.
General procedure for the synthesis of compounds (7a–7g)
To a solution of 1 (5 g, 27.8 mmol) in pyridine (50 mL) was added acetic anhydride (35 mL). The reaction mixture was stirred for 5 h and then H2O (100 mL) was added and stirring continued for 20 min. The reaction mixture was washed with 0.1 M HCl (15 mL), satd aq NaHCO3 (15 mL), and dried (MgSO4), Filtration, evaporation of solvent under diminished pressure to dryness to give 2. To a solution of 2 in dichloromethane (35 mL) was added HBr-HOAc (35 mL). The reaction mixture was stirred for 5 h and then H2O (50 mL) was added and stirring continued for 20 min. The reaction mixture was washed with satd aq NaHCO3 (25 mL), and dried (MgSO4). Filtration, evaporation of solvent under diminished pressure and chromatography of the residue (EtOAc-petroleum ether, 1:8) gave 3 as a white solid (4.4 g, 65%). To a solution of 3 (4.4 g, 10.8 mmol) in DMF (30 mL) was added NaN3 (3.5 g, 53.8 mmol). The reaction mixture was stirred for 4 h. Filtration, evaporation of solvent under diminished pressure and chromatography of the residue (EtOAc-petroleum ether, 1:5) gave 4 as a white solid (2.1 g, 53%). To a solution of 4 (2.1 g, 5.6 mmol) in EtOAc (20 mL) was added Pd/C(1.05 g) and add hydrogen. The reaction mixture was stirred for 2 h. Filtration, evaporation of solvent under diminished pressure to dryness to give 5 (1.6 g, 85%). To a solution of compound 5 (200 mg, 0.58 mmol) in dry CH2Cl2 (10 mL), p-sulfamoylbenzoic acid (116 mg, 0.58 mmol) and EDCI (111.2 mg,0.58 mmol) were added at room temperature and the reaction mixture was stirred for 2 h. The reaction mixture was washed with 0.1 M HCl (5 mL), satd aq NaHCO3 (5 mL), and dried (MgSO4), Filtration, evaporation of solvent under diminished pressure to dryness to give 6 (162 mg, 53%). To a solution of 6 (162 mg, 0.31 mmol) in CH2Cl2-MeOH (1:1, 5 mL), freshly prepared NaOMe in MeOH solution (1.0 mol/l, 1 mL) was added. After it was stirred overnight, the mixture was neutralised with Dowex H + resin to pH 7, then filtered. The filtrate was concentrated and purified by a silica gel column chromatography (5:1, CH2Cl2-MeOH) to afford 7a (52 mg, 47%) as a white solid.
4-Aminosulfonyl-N-(β-D-glucopyranosyl) benzamide (7a)
M.p. 112.2–114.2 °C; 1H NMR (400 MHz, DMSO) δ 9.04 (d, J = 8.8 Hz, 1H), 8.06 (d, J = 8.4 Hz, 2H), 7.91 (d, J = 8.4 Hz, 2H), 7.48 (s, 2H), 5.02 (d, J = 4.5 Hz, 1H), 4.99 (d, J = 5.3 Hz, 1H), 4.95 (t, J = 9.0 Hz, 1H), 4.91 (d, J = 5.3 Hz, 1H), 4.51 (t, J = 5.7 Hz, 1H), 3.68 (dd, J = 10.3, 5.6 Hz, 1H), 3.44 (dt, J = 11.5, 5.7 Hz, 1H), 3.37–3.32 (m, 1H), 3.28–3.23 (m, 1H), 3.22–3.17 (m, 1H), 3.14–3.07 (m, 1H). 13C NMR (151 MHz, DMSO) δ 166.11, 146.93, 137.47, 128.74, 126.00, 80.82, 79.31, 77.93, 72.58, 70.47, 61.42. ESI-MS (m/z): 363.1 [M + H]+; HRMS (ESI): Calcd for [M + H]+ C13H19N2O8S: 363.0817, Found 363.0858.
4-Aminosulfonyl-N-(β-D-galactopyranosyl) benzamide (7 b)
M.p. 115.7–117.3 °C; 1H NMR (600 MHz, DMSO) δ 9.10 (d, J = 8.9 Hz, 1H), 8.08 (d, J = 8.4 Hz, 2H), 7.89 (d, J = 8.3 Hz, 2H), 7.49 (s, 2H), 4.92 (t, J = 8.9 Hz, 1H), 4.88 (s, 2H), 4.62 (s, 1H), 4.50 (s, 1H), 3.73 (s, 1H), 3.65 (t, J = 9.2 Hz, 1H), 3.52 (s, 1H), 3.46 (s, 2H), 3.40 (s, 1H). 13 C NMR (151 MHz, DMSO) δ 166.12, 146.86, 137.52, 128.81, 125.92, 81.31, 77.48, 74.59, 69.83, 68.77, 61.02. ESI-MS (m/z): 363.1 [M + H]+; HRMS (ESI): Calcd for [M + H]+ C13H19N2O8S: 363.0817, Found 363.0850.
4-Aminosulfonyl-N-(β-L-arabinopyranosyl) benzamide (7c)
M.p. 114.7–116.6 °C; 1H NMR (400 MHz, DMSO) δ 9.04 (d, J = 8.7 Hz, 1H), 8.05 (d, J = 8.5 Hz, 2H), 7.91 (d, J = 8.5 Hz, 2H), 7.49 (s, 2H), 5.08 (d, J = 4.5 Hz, 1H), 5.04 (d, J = 5.3 Hz, 1H), 4.99 (d, J = 5.0 Hz, 1H), 4.86 (t, J = 8.9 Hz, 1H), 3.70 (dd, J = 11.1, 5.2 Hz, 1H), 3.36 (s, 1H), 3.30 (dd, J = 9.4, 5.2 Hz, 1H), 3.20 (td, J = 8.8, 4.5 Hz, 1H), 3.11 (t, J = 10.8 Hz, 1H). 13 C NMR (151 MHz, DMSO) δ 166.37, 146.96, 137.41, 128.74, 126.02, 81.62, 77.95, 72.33, 70.13, 68.01. ESI-MS (m/z): 333.1 [M + H]+; HRMS (ESI): Calcd for [M + H]+ C12H17N2O7S: 333.0712, Found 333.0748.
4-Aminosulfonyl-N-(β-D-xylopyranosyl) benzamide (7d)
M.p. 123.7–125.2 °C; 1H NMR (400 MHz, DMSO) δ 9.06 (d, J = 8.7 Hz, 1H), 8.05 (d, J = 8.4 Hz, 2H), 7.90 (d, J = 8.4 Hz, 2H), 7.48 (s, 2H), 4.90 (d, J = 5.1 Hz, 2H), 4.86 (d, J = 8.5 Hz, 1H), 4.60 (d, J = 2.9 Hz, 1H), 3.70 (s, 1H), 3.69–3.67 (m, 1H), 3.67–3.61 (m, 1H), 3.50 (d, J = 11.3 Hz, 1H), 3.45 (ddd, J = 8.5, 5.2, 3.2 Hz, 1H). 13C NMR (151 MHz, DMSO) δ 166.17, 146.92, 137.50, 128.72, 126.01, 81.35, 73.90, 69.75, 68.49, 67.82. ESI-MS (m/z): 333.1 [M + H]+; HRMS (ESI): Calcd for [M + H]+ C12H17N2O7S: 333.0712, Found 333.0748.
4-Aminosulfonyl-N-(β-D-ribofuranosyl) benzamide (7e)
M.p. 109.8–111.3 °C; 1H NMR (600 MHz, DMSO) δ 8.85 (d, J = 8.1 Hz, 1H), 7.94 (s, 4H), 7.51 (s, 2H), 5.64 (s, 1H), 5.34 (dd, J = 8.1, 4.1 Hz, 1H), 5.22 (d, J = 6.9 Hz, 1H), 4.93 (d, J = 5.8 Hz, 1H), 3.93 (s, 1H), 3.65–3.62 (m, 1H), 3.58 (dd, J = 5.3, 2.7 Hz, 2H), 3.37 (q, J = 9.0 Hz, 1H). 13C NMR (151 MHz, DMSO) δ 165.76, 147.18, 137.51, 128.09, 126.44, 77.36, 71.73, 67.49, 67.04, 60.94. ESI-MS (m/z): 333.1 [M + H]+; HRMS (ESI): Calcd for [M + H]+ C12H17N2O7S: 333.0712, Found 333.0737.
4-Aminosulfonyl-N-(β-L-rhamnosyl) benzamide (7f)
M.p. 116.6–118.2 °C; 1H NMR (600 MHz, DMSO) δ 8.47 (d, J = 8.6 Hz, 1H), 8.03 (d, J = 8.4 Hz, 2H), 7.89 (d, J = 8.3 Hz, 2H), 7.50 (s, 2H), 5.23 (d, J = 8.6 Hz, 1H), 5.05 (d, J = 4.5 Hz, 1H), 4.81 (d, J = 5.3 Hz, 1H), 4.74 (d, J = 5.9 Hz, 1H), 3.72 (s, 1H), 3.38 (ddd, J = 9.0, 5.9, 3.3 Hz, 1H), 3.25–3.20 (m, 1H), 3.20–3.15 (m, 1H), 1.17 (d, J = 5.9 Hz, 3H). 13C NMR (151 MHz, DMSO) δ 165.26, 147.03, 137.11, 128.74, 126.09, 78.93, 74.36, 74.02, 72.15, 71.09, 18.46. ESI-MS (m/z): 347.1 [M + H]+; HRMS (ESI): Calcd for [M + H]+ C13H19N2O7S: 347.0868, Found 347.0904.
4-Aminosulfonyl-N-(β-D-glucuronic acid methyl ester) benzamide (7g)
M.p. 114.5–117.1 °C; 1H NMR (600 MHz, DMSO) δ 8.78 (d, J = 8.6 Hz, 1H), 8.04 (d, J = 8.3 Hz, 2H), 7.91 (d, J = 8.4 Hz, 2H), 7.52 (s, 2H), 5.71 (dd, J = 8.5, 5.3 Hz, 1H), 5.42 (d, J = 5.2 Hz, 1H), 5.23 (d, J = 5.7 Hz, 1H), 5.12 (d, J = 4.7 Hz, 1H), 4.06 (d, J = 9.0 Hz, 1H), 3.82 (dd, J = 8.5, 4.6 Hz, 1H), 3.64 (s, 3H), 3.59–3.55 (m, 1H), 3.44 (td, J = 8.6, 5.3 Hz, 1H). 13C NMR (151 MHz, DMSO) δ 170.35, 167.31, 147.04, 137.45, 129.08, 125.87, 77.54, 73.51, 72.19, 72.04, 70.39, 52.24. ESI-MS (m/z): 391.1 [M + H]+; HRMS (ESI): Calcd for [M + H]+ C14H19N2O9S: 391.0767, Found 391.0784.
4-Aminosulfonyl-N-(β-D-mannopyranosyl) benzamide (7h)
M.p. 112.6–114.4 °C; 1H NMR (600 MHz, DMSO) δ 8.48 (d, J = 8.7 Hz, 1H), 8.03 (d, J = 8.3 Hz, 2H), 7.90 (d, J = 8.0 Hz, 2H), 7.50 (s, 2H), 5.27 (d, J = 8.6 Hz, 1H), 5.05 (s, 1H), 4.77 (s, 2H), 4.48 (s, 1H), 3.71 (s, 2H), 3.45–3.41 (m, 2H), 3.17 (d, J = 6.6 Hz, 2H). 13C NMR (151 MHz, DMSO) δ 165.18, 147.04, 137.09, 128.71, 126.12, 79.84, 78.90, 74.29, 71.02, 67.23, 61.76. ESI-MS (m/z): 363.1 [M + H]+; HRMS (ESI): Calcd for [M + H]+ C13H19N2O8S: 363.0817, Found 363.0840.
CA inhibition
A multifunctional enzyme marking instrument has been used for assaying CA isoenzyme activities according to the esterase assay method described previously by Verpoorte et al.Citation24. Initial rates of 4-nitrophenyl acetate hydrolysis catalysed by different CA isozymes were recorded on a Cary 3 instrument at 400 nm interfaced with a DELL-compatible PC. Solutions of substrate (4-nitrophenyl acetate) were prepared in DMSO; the substrate concentration was 2 mM at room temperature. A molar absorption coefficient of 400 M−1 cm−1 was used for the 4-nitrophenolate formed by hydrolysis in the conditions of the experiments (pH 7.5). Nonenzymatic hydrolysis rates were subtracted from the observed rates. The experiments were repeated three times at each inhibitor concentration. Stock solutions of the inhibitor (100 mM) were prepared and diluted up to 0.5 nM with analytical buffer. At least eight different inhibitor concentrations have been used, ranging from 5 mM, 0.5 mM, 50 mM, 5 mM, 0.5 mM, 50 nM, 5 nM, 0.5 nM. Inhibitor and enzyme solutions were preincubated together for 10 min at r.t. prior to the assay study, to allow for the formation of the E-I complex. Enzyme concentrations were 1 ng/L for CA IICitation25. Isoforms hCA IX and hCA I have a low esterase activity compared to hCA II. However, we also obtained a good test condition by constantly adjusting the dosage of hCA IX and hCA I enzyme.
MTT assay in vitro
Cell proliferation was evaluated using an MTT assay. Briefly, 5 × 104 cells per well were seeded into 96-well plates and grown at 37 °C for 24 h. Subsequently, compounds AAZ, BRZ and 7a–7g were added to each well at increasing concentrations, and then the cells were incubated for 96 h. The MTT solution was added to each well, and the cells were incubated at 37 °C for another 4 h. Formazan crystals were dissolved in 150 mL of DMSO. Cell viability was assessed by measuring the absorbance at 540 nm wavelength using a microplate reader (BioTek ELx800 USA).
Hypertensive rabbit IOP lowering studies
All animal experiments were conformed to the guidelines of the Animal Ethics Committee of Shenyang Pharmaceutical University. Rabbits were provided by the Laboratory Animal Centre of Shenyang Pharmaceutical University. Adult male rabbits weighing 2.0–2.5 kg were employed in our studies. Animals were anaesthetised using zoletil and injected with 0.1 mL of hypertonic saline solution (5% in distilled water) into the vitreous of both eyes. IOP was measured using a Tono-Pen tonometer (Tono-pen Avia tonometer, Reichhert Inc., Depew, NY 14043, USA) prior to hypertonic saline injection (basal) at 1, 2, 3, 4, 5 and 6 h after drug administration. Vehicle (phosphate buffer 7.00 plus DMSO 2%) or drugs were instilled immediately after the injection of hypertonic saline. Eyes were randomly assigned to different groups. Vehicle or drug (0.50 mL) was directly instilled into the conjunctive pocket at the desired doses (1 − 2%)Citation21,Citation22. Four different animals were used for each tested compound.
Molecular docking simulations
The experimental crystallographic structures of CA I and CA II complex were retrieved from the Protein DataBank (PDB: 1AZM and 3HS4). The molecular model of the sugar tail sulphonamide derivative 7c were built by Discovery Studio 3.5. Both the protein and the ligands were prepared by adding polar hydrogen atoms and partial charges with the assistance of AutoDockTools 1.5.6. AutoDockZN force field was used to handle the zinc ion in the binding site. Grid points were increased to 60 at x, y, z axes. The number of Dock runs was increased to 20. All other parameters were kept as their default values.
Supplemental Material
Download PDF (1.1 MB)Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- (a) Kolker AE. Visual prognosis in advanced glaucoma: a comparison of medical and surgical therapy for retention of vision in 101 eyes with advanced glaucoma. Trans Am Ophthalmol Soc 1977;75:539–45. (b) Werner EB, Drance SM, Schulzer M. Trabeculectomy and the progression of glaucomatous visual field loss. Arch Ophthalmol 1977;95:1374–7.
- Wiederholt M, Bielka S, Schweig F, et al. Regulation of outflow rate and resistance in the perfused anterior segment of the bovine eye. Exp Eye Res 1995;61:223–34.
- Krupin T, Wax M, Moolchandani J. Aqueous production. Trans Ophthalmol Soc UK 1986;105:156–61.
- Mincione F, Scozzafava A, Supuran CT. The development of topically carbonic anhydrase inhibitors as antiglaucoma agents. Curr Pharm Des 2008;14:649–54.
- (a) Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81. (b) Supuran CT, Scozzafava A. Carbonic anhydrase inhibitors and their therapeutic potential. Exp Opin Ther Pat 2000;10:575–600.
- Bartlett JD, Jaanus SD. Clinical ocular pharmacology. Boston, MA: Butterworth, 1989:254–63.
- Supuran CT, Altamimi ASA, Carta F. Carbonic anhydrase inhibition and the management of glaucoma: a literature and patent review 2013–2019. Exp Opin Ther Pat 2019;29:781–92.
- Supuran CT. The management of glaucoma and macular degeneration. Exp Opin Ther Pat 2019;29:745–7.
- (a) Supuran CT, Berrino E. Rho-kinase inhibitors in the management of glaucoma. Exp Opin Ther Pat 2019;29:817–27. (b) Supuran CT, Bua S. Diagnostic markers for glaucoma: a patent and literature review (2013–2019). Exp Opin Ther Pat 2019;29:829–39.
- (a) Supuran CT, Nocentini A. Adrenergic agonists and antagonists as antiglaucoma agents: a literature and patent review (2013–2019). Exp Opin Ther Pat 2019;29:805–15. (b) Supuran CT, Angeli A. Prostaglandin receptor agonists as antiglaucoma agents (a patent review 2013–2018). Exp Opin Ther Pat 2019;29:793–803.
- (a) Supuran CT, Scozzafava A, Casini A. Carbonic anhydrase inhibitors. Med Res Rev 2003;23:146–89. (b) Supuran CT, Scozzafava A. Carbonic anhydrase inhibitors and their therapeutic potential. Exp Opin Ther Patents 2000;10:575–601. (c) Supuran CT, Scozzafava A. Applications of carbonic anhydrase inhibitors and activators in therapy. Exp Opin Ther Patents 2002;12:217–42.
- Casini A, Scozzafava A, Mincione F, et al. Carbonic anhydrase inhibitors: water-soluble 4-sulfamoylphenylthioureas as topical intraocular pressure-lowering agents with long-lasting effects. J Med Chem 2000;43:4884–92.
- Maren TH. Role of carbonic anhydrase in aqueous humor and cerebrospinal fluid formation. In: Segal MB, ed. Barriers and fluids of the eye and brain. London: MacMillan Press; 1992:37–48.
- (a) Supuran CT. Carbonic anhydrase inhibitors and their potential in a range of therapeutic areas. Exp Opin Ther Pat 2018;28:709–12. (b) Supuran CT. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Exp Opin Drug Discov 2017;12:61–88.
- Scozzafava A, Menabuoni L, Mincione F, et al. Carbonic anhydrase inhibitors. Synthesis of water-soluble, topically effective, intraocular pressure-lowering aromatic/heterocyclic sulfonamides containing cationic or anionic moieties: is the tail more important than the ring? J Med Chem 1999;42:2641–50.
- Scozzafava A, Briganti F, Mincione G, et al. Carbonic anhydrase inhibitors: synthesis of water-soluble, amino acyl/dipeptidyl sulfonamides possessing long-lasting intraocular pressure-lowering properties via the topical route. J Med Chem 1999;42:3690–700.
- Borras J, Scozzafava A, Menabuoni L, et al. Carbonic anhydrase inhibitors: synthesis of water-soluble, topically effective intraocular pressure lowering aromatic/heterocyclic sulfonamides containing 8-quinoline-sulfonyl moieties: is the tail more important than the ring? Bioorg Med Chem 1999;7:2397–406.
- Renzi G, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors: topical sulfonamide antiglaucoma agents incorporating secondary amine moieties. Bioorg Med Chem Lett 2000;10:673–6.
- (a) Ilies M, Supuran CT, Scozzafava A, et al. Carbonic anhydrase inhibitors. Sulfonamides incorporating furan-, thiophene- and pyrrole-carboxamido groups possess strong topical intraocular pressure lowering properties as aqueous suspensions. Bioorg Med Chem 2000;8:2145–55. (b) Winum JY, Casini A, Mincione F, et al. Carbonic anhydrase inhibitors: N-(p-sulfamoylphenyl)-α-D-glycopyranosylamines as topically acting antiglaucoma agents in hypertensive rabbits. Bioorg Med Chem Lett 2004;14:225–9.
- Wei GF, Cui SS, Luan WJ, et al. Natural product-based design, synthesis and biological evaluation of Albiziabioside A derivatives that selectively induce HCT116 cell death. Eur J Med Chem 2016;113:92–101.
- Krauss AHP, Impagnatiello F, Toris CB, et al. Ocular hypotensive activity of BOL-303259-X, a nitric oxide donating prostaglandin F2α agonist, in preclinical models. Exp Eye Res 2011;93:250–5.
- Fabrizi F, Mincione F, Somma T, et al. A new approach to antiglaucoma drugs: carbonic anhydrase inhibitors with or without NO donating moieties. Mechanism of action and preliminary pharmacology. J Enzyme Inhib Med Chem 2012;27:138–47.
- Masini E, Carta F, Scozzafava A, Supuran CT. Antiglaucoma carbonic anhydrase inhibitors: a patent review. Exp Opin Ther Pat 2013;23:705–16.
- Verpoorte JA, Mehta S, Edsall JT. Esterase activities of human carbonic anhydrases B and C. J Biol Chem 1967;242:4221–9.
- Truppo E, Supuran CT, Sandomenico A, et al. Carbonic anhydrase VII is glutathionylated without loss of catalytic activity and affinity for sulfonamide inhibitors. Bioorg Med Chem Lett 2012;22:1560–4.