
Abstract
The inhibition of δ- and η-class carbonic anhydrases (CAs; EC 4.2.1.1) was poorly investigated so far. Only one δ-CA, TweCA from the diatom Thalassiosira weissflogii, and one η-CA, PfCA, from Plasmodium falciparum, have been cloned and characterised to date. To enrich δ- and η-CAs inhibition profiles, a panel of 22 phenols was investigated for TweCA and PfCA inhibition. Some derivatives showed effective, sub-micromolar inhibition of TweCA (KIs 0.81–65.4 µM) and PfCA (KIs 0.62–78.7 µM). A subset of compounds demonstrated a significant selectivity for the target CAs over the human physiologically relevant ones. This study promotes the identification of new potent and selective inhibitors of TweCA and PfCA, which could be considered as leads for finding molecular probes in the study of carbon fixation processes (in which TweCA and orthologue enzymes are involved) or drug candidates in the treatment of malaria.
1. Introduction
Carbonic anhydrases (CAs; EC 4.2.1.1) compose a superfamily of metalloenzymes that owe the role of speeding up the carbon dioxide hydration to bicarbonate and protonCitation1,Citation2. Crucial biological processes in most organisms of tree of life are related to such a reversible reaction: respiration, photosynthesis, pH regulation, CO2 and HCO3− transport, biosynthetic processes, production of body fluids, bone resorption, etcCitation3,Citation4. Eight evolutionarily unrelated CA classes have been identified to date, which are named as α-, β-, γ-, δ-, ζ-, η-, θ- and ι-CAsCitation4–8. The α-CAs are present in vertebrates, protozoa, algae, corals, bacteria and cytoplasm of green plantsCitation4. Human, in particular, encode only for α-class isozymesCitation3. The β-CAs have been identified in bacteria, fungi, Archaea, algae and chloroplasts of both mono- and dicotyledonsCitation4. The γ-CAs are encoded in Archaea, bacteria and plantsCitation4,Citation9. δ-CAs have been discovered in marine phytoplankton, such as haptophytes, dinoflagellates, diatoms and chlorophytic prasinophytes, while ζ-CAs appear to be present only in marine diatomsCitation6. A unique η-CA has been identified to date in the protozoa Plasmodium falciparumCitation7. θ-CAs have been recently discovered in the marine diatom Phaeodactylum tricornutumCitation10. A first specimen of ι-CAs was recently labelled from the marine diatom Thalassiosira pseudonanaCitation8.
A unique δ-CA, TweCA, from the diatom Thalassiosira weissflogii was cloned and characterised in detail to dateCitation11, though orthologues of this enzyme have been identified in most diatoms from natural phytoplankton assemblages and are responsible (along with other CAs) for CO2 fixation by marine organismsCitation12. TweCA is upregulated by low pCO2 and, under Zn-limited conditions, the zinc ion at the active site can be substituted by Co(II) in vivoCitation12,Citation13. TweCA is a protein of 281 amino acid residues. A subunit molecular mass of 32.0 kDa was estimated by SDS-PAGE, while the molecular mass of 32.2 kDa was calculated from the amino acid sequence. TweCA does not share any sequence homology to any other known CAs. The alignment of the amino acid sequence of TweCA with the polypeptide chain of the bovine α-CA (isoform bCA II) shows the low degree of identity with the mammalian α-CACitation11. Nonetheless, it was shown that the active site of TweCA is similar to that of mammalian α-CACitation11, with the metal coordination pattern formed by three histidines as found in α- and γ-CAs (). Unfortunately, no structural data are available on δ-CAs. A phylogenetic analysis carried out using α-, γ- and δ-CAs from different prokaryotic and eukaryotic organisms showed that the α-CAs appear closely related to the δ-CAs, but clustered in a branch distinct from that of γ-CAsCitation14. CA inhibitors, such as sulphonamides, inorganic anions, mono- and dithiocarbamates were screened as TweCA inhibitorsCitation14–16 with the aim to uncover molecular probes to investigate the role of this enzyme in the carbon fixation processes in marine diatoms that are responsible for removing large amounts of CO2 from the atmosphere.
Figure 1. Metal ion coordination in the different CA families: (A) α-, γ - and δ-CAs (in the α- and δ-classes the coordinating residues are from the same monomer, whereas in γ -CAs the third His is from an adjacent monomer). (B) β-CAs (ζ -CAs possess a Cd(II) bound within the active site and show analogue coordination pattern). (C) η-CAs.
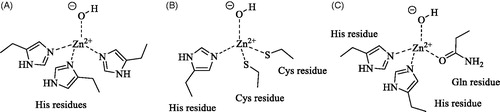
The η-class of CAs was firstly described in 2015 by analysis of the amino acid sequences of CAs from Plasmodia, parasitic protozoa responsible of malaria in humans and other animalsCitation7. The first and unique member of the family to be characterised in vitro to date was PfCA, a protein of 600 amino acid residues, identified in Plasmodium falciparumCitation17–20, one of the five species causing malaria in humans. Interestingly, PfCA was initially described as an α-CA enzyme, due to significant similarities with members of this class, but was subsequently reclassified into a new CA class, the η, due to some peculiar featuresCitation17. In particular, the zinc coordination pattern of PfCA is formed by two histidines and one glutamine, distinctly from α-CAs, and many insertions and deletions in the protozoan enzyme were identified with respect to common α-CAs: insertions were observed in PfCA at the N-terminus and in the middle of the protein (69 additional residues after residue 152 of hCA II, chosen as reference protein). A three-dimensional model of PfCA was built by homology using the structure of Thermovibrio ammonificans CA (TaCA) as templateCitation21. Because of low sequence homology only 267 residues (198–327 and 397–535) out of the 600 of the full-length protein could be modelled. A folding similar to that of α-CAs was found with the active site located in a large cavity with the zinc ion on the bottom (), coordinated by His299, His301 and Gln320. The 69 residues insertion was located at the edge of the active site cleft, being presumably implicated in the catalysisCitation17.
A significant interest is being dedicated to PfCA, because the enzyme has been recognised as possible target for the development of antimalarial drugs based on innovative mechanism of action. Indeed, a crucial role was suggested for PfCA in the Plasmodium parasites, being involved in the production of HCO3− necessary as precursor of the pyrimidine biosynthetic pathwayCitation22. Its targeting to block this pathway could thus represent an efficient strategy for the development of new pharmacological agents against malariaCitation23. In 1998, Sein and Aikawa showed that addition of CA inhibitors (CAIs) to a culture of P. falciparum provoked a remarkable reduction in parasitemiaCitation24. Successive reports illustrated that specific CA inhibition in P. falciparum and in the rodent parasite P. berghei produced the death of the parasite in in vitro culturesCitation22. Starting from these data, the search of new PfCA inhibitors has started with sulphonamides and inorganic anions, and, though encouraging results have been obtained, more efforts are still necessary to obtain candidate drug moleculesCitation18–20.
Here, a series of phenolic derivatives (1–22, ) was assessed for the inhibition of TweCA and PfCA to extend such isoforms inhibition profiles, in search of novel leads for drug candidates or molecular probes which show the selective modulation of CAs from diatoms and protozoa over human isozymes.
2. Methods
2.1. Chemistry
Phenols 1–22 were commercially available from Sigma-Aldrich (Milan, Italy) and were used without further purification (purity >95%). All other reagents, salts, buffers and solvents were the highest purity available ones from Sigma-Aldrich (Milan, Italy).
2.2. Carbonic anhydrase inhibition
An Sx.18Mv-R Applied Photophysics (Oxford, UK) stopped-flow instrument has been used to assay the catalytic activity of various CA isozymes for CO2 hydration reactionCitation25. Phenol red (at a concentration of 0.2 mM) was used as indicator, working at the absorbance maximum of 557 nm, with 20 mM TRIS (pH 8.3) as buffer, and 20 mM NaClO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalysed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor, at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (0.1 mM) were prepared in distilled-deionised water and dilutions up to 0.01 nM were done thereafter with the assay buffer. Inhibitor and enzyme solutions were preincubated together for 1 h at room temperature prior to assay, in order to allow for the formation of the E–I complex. The inhibition constants were obtained by nonlinear least-squares methods using PRISM 3 and the Cheng-Prusoff equation, as reported earlier, and represent the mean from at least three different determinationsCitation26–28. TweCAδ, PfCA, hCA I and II were recombinant proteins obtained in-house as reported earlierCitation29–31.
3. Results and discussion
3.1. Selection of δ- and η-class CAs and chemistry
The kinetic parameters of the CO2 hydration reaction catalysed by TweCA and PfCA are reported in in comparison with hCAs I and II. TweCA showed a significant catalytic activity with a kcat of 1.3 × 105 s−1 and a kcat/KM of 3.3 × 107 M−1 s−1. Similarly to β-, γ- and ζ-CAs, δ-CAs do not possess esterase activity.Citation14 TweCA is stable up to 80 °C with residual activity of 40%, when the incubation time did not exceed 30 min. In contrast, bCA is inactivated at temperatures higher than 60 °C Citation11, suggesting that the δ-CA from T. weissflogii probably possess a more compact 3 D structure than other mammalian α-CAs.
Table 1. Kinetic parameters for the CO2 hydration reaction catalysed by the human cytosolic isozymes hCA I and II (α-class CAs), TweCAδ and PfCA measured at 20 °CCitation14,Citation18.
Data of show that PfCA shows a significant catalytically activity for the CO2 hydration reaction, being the kcat 3.8 × 105 s−1 and the kcat/Km of 7.2 × 107 M−1 × s−1 Citation18. PfCA is more effective even compared to hCA I, and approximately 50% less effective compared to hCA II.
Phenolic compounds were shown to act as CAIs by a very distinct inhibition mechanism compared to primary sulphonamides, many of which are clinically used as diuretics, antiglaucoma, antiepileptic or in clinical trials for the management of advanced, hypoxic solid tumorsCitation30. In fact, whether sulphonamides directly coordinate the Zn(II) ion from the CA active site replacing the non-protein ligand, phenols were shown to anchor to the zinc-coordinated water molecule/hydroxide ion by a hydrogen bond networkCitation30. Up to now, phenolic derivatives, among which compounds 1–22 investigated here (), were assayed as inhibitors of the human CA I, II, IX and XIICitation31, of β-CAs, from the fungi Saccharomyces cerevisiae, Candida albicans, Cryptococcus neoformans and Malassezia GlobosaCitation32,Citation33 or the bacterium Mycobacterium tuberculosisCitation34 and γ-CAs from the pathogenic bacteria Burkholderia pseudomallei, Pseudomonas gingivalis, Vibrio cholerae and from the Antarctic bacteria Pseudoalteromonas haloplanktis and Colwellia psychrerythraeaCitation35.
Figure 2. Structures of phenolic compounds 1–22.
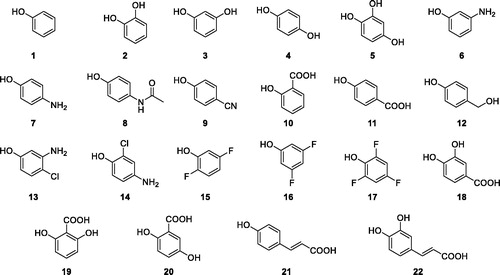
As the δ- and η-CAs active sites are narrower than those of α-CAs, only phenyl derivatives, and not complex natural polyphenols, were consideredCitation1. A large variety of electron donating and electron withdrawing groups were investigated as substituents on the phenolic scaffold to uncover on the role of acidity of the anchoring group in the inhibitory activity ().
Table 2. Inhibition data of TweCAδ and PfCA with phenols 1–22 and the standard sulphonamide inhibitor acetazolamide (AAZ) by a stopped flow CO2 hydrase assayCitation27.
3.2. δ- and η-class carbonic anhydrases inhibition
Phenols 1–22 were assayed as inhibitors of the unique δ- and η-class CAs identified to date, specifically from the marine diatom T. weissflogii and protozoan P. falciparum, respectively. A stopped flow CO2 hydrase assay was used including acetazolamide (AAZ) as standard inhibitorCitation25. The inhibition profiles against the human ubiquitous CAs I and II are displayed for comparisonCitation31. The following structure–activity relationships (SAR) can be drawn up from the inhibition data reported in .
As a general trend, it can be stated that phenolic compounds are able to interfere with the CO2 hydrase activity of δ- and η-class CAs in the micromolar range. Inhibition constants (KIs) spanned, in fact, between 0.81 and 65.4 µM against TweCA and 0.62 and 78.7 µM against PfCA, while compounds 13 and 14 did not show inhibition below 100 µM.
It is fair to immediately stress that even carboxylic acids can act as CAIs, and can do that by two distinct mechanisms of action: coordination of the metal(II) ion or anchorage to the zinc-bound nucleophile. As a result, one cannot exclude that compounds 10, 11, 18–22, which bear both phenolic and carboxylic groups, produce CA inhibition by the COOH function in place of the OH group.
Most substitutions at the phenol 1 scaffold produce enhancement in the inhibition of both TweCA and PfCA, with the exception of m-substituents of the amine type (6 and 13) and an o-chlorine atom (14), that presumably induce significant steric hindrance for the binding in the active site. Also a p-CN group at the phenol scaffold led to light worsening of inhibitory action of 9 against TweCA in comparison to the lead 1 (KIs of 52.3 and 56.9 µM, respectively).
As for TweCA, a consistent subset of derivatives showed KIs lower than 10 µM (KIs in the range 0.81–7.9 µM). In particular, 1,2-diols 1 and 5 exhibited the most potent TweCA inhibition (KIs of 4.5 and 2.0 µM) among those compounds possessing solely OH and not COOH groups. On the other hand, swapping the second aromatic OH group to the m- or p- position did not produce a consistent increase of TweCA inhibition which settled for 3 and 4 at 48.2 and 34.9 µM. The substitution of hydrogens with fluorine atoms on the phenol scaffold increased the inhibition of TweCA by 15–17 (KIs in the range 13.8–30.7 µM) with respect to the lead 1. In contrast, all benzoic derivatives reported low- to sub-micromolar activity. Precisely, the 2-hydroxy-benzoic acids 10 and 20 resulted to be the best TweCA inhibitors with submicromolar KIs of 0.95 and 0.81 µM. The presence of COOH group of the cinnamic acid type, such as in 21 or 22 did not elicit the same inhibition increased observed with benzoic acids, though the presence of a 1,2-diol portion in 22 drove its KI against TweCA below 10 µM. None of the assayed compounds provoked as inhibitory effect as the reference AAZ (KI of 83 nM).
As anticipated above, a superimposable inhibitory trend was measured for PfCA with phenols 1–22 (). The 1,2,4-triol 5 showed inhibition of the plasmodial CA in the submicromolar range (KI of 0.83 µM) reaching almost the same efficacy of benzoic acids 10, 11, 18–20 (KIs in the range 0.62–1.6 µM). Among the latter, the 2-hydroxybenzoic acid 20 stood out again as the best inhibitor here screened, also against PfCA, with a KI being less than the double of that shown by AAZ (KI of 360 nM). The incorporation of a p-olefin portion in the 1,2-diol scaffold of 2 such as in 22 worsened the inhibitory efficacy from 1.4 to 11.2 µM. Swapping the second aromatic OH group to the m- or p- position produced a more consistent increase of PfCA inhibition than that observed against TweCA, as the KIs of 3 and 4 settled at 26.9 and 21.0 µM. The parallelism observed in inhibition profile of TweCA and PfCA with phenolic and/or carboxylic compounds might suggest similar binding modes of these chemotypes within the active sites of δ- and η-class CAs that, as stated above, both resemble that of α-CAs.
reported the selectivity index (SI) calculated for TweCA and PfCA over the human off-target CAs I and II. First, the inhibition profiles of hCAs I and II with phenols 1–22 should be briefly summarised. KIs against CA I show a peculiar trend as the half compounds are inhibitors in a low micromolar range below 10 µM, 12, 14 and 16 exhibited KIs between 38.8 and 68.9 µM, whereas the remaining ones did not inhibit CA I below 100 µM. In contrast, a minor set of compounds did not inhibit hCA II (only 5, 7, 15 and 17). Again 12, 14 and 16 exhibited KIs above 10 µM (33.9–95.3 µM) and the most compounds effectively inhibited hCA II with KIs even reaching nanomolar such as in the cases of 4 and 9. The SI values in show that almost the half derivatives here screened exhibited selectivity of action against TweCA over hCAs I and II. The 1,2-diol 5 was the most selective among the screened compounds with SI over 50 against the diatom CA over off-target ones. Benzoic acids 10, 11 and 20 also reported a significant selectivity of action with SI settling between 5 and 10 over both CA I and II. Also the trifluorophenol 17 displayed an interesting selectivity against TweCA over human CAs (SI of 7).
Table 3. Selectivity index (SI) for target CA over the off-target hCA I and II.
Even higher SI were calculated against PfCA over both hCAs I and II (). 1,2-Diols 2 and 5 showed the most selective and promising inhibition of the target PfCA with respect to human CAs. While the SI of 2 settled at 70 and 4 over hCA I and II, respectively, those of 5 were even higher than 100 in both cases. The hCAs/PfCA SI values were also increased with most benzoic acid derivatives with respect to those observed for TweCA. As sole exceptions, carboxylates 18, 20 and 21 should be cited, since reported specificity of action for the human isozyme over the target ones (SI < 1). Analogue selectivity trend was observed for most other phenols (not showing COOH groups), such as 13 and 14, which were particularly selective against hCAs over the plasmodial and diatom isozymes.
4. Conclusions
CAs of δ- and η-classes have not been extensively characterised from the inhibitory standpoint in comparison to α- and β-class isozymes. A unique δ-CA, TweCA, from the diatom Thalassiosira weissflogii was cloned and characterised in detail to date, though orthologues of this enzyme have been identified in most diatoms from natural phytoplankton assemblages and are responsible, along with other CAs for CO2 fixation by marine organisms. The identification of selective inhibitors of these isozymes is of significant importance to uncover molecular probes to investigate the role of this enzyme in the carbon fixation processes of marine diatoms that are responsible for removing large amounts of CO2 from the atmosphere.
Meanwhile a significant interest has been dedicated to PfCA, the unique specimen of η-CA, which was identified in Plasmodium falciparum, one of the five species causing malaria in humans. The research of PfCA inhibitors has started with sulphonamides and inorganic anions, and, though encouraging results have been obtained, more efforts are still necessary to obtain candidate drug molecules.
To extend TweCA and PfCA inhibition profiles, in search of novel leads for drug candidates or molecular probes selectively modulating these CAs over human isozymes, a panel of 22 phenols was investigated for these isozymes’ inhibition. The exploration of the chemical space around the main functional group led to the discovery of a number of such derivatives showing effective, sometimes sub-micromolar, inhibition against TweCA (KIs 0.81 and 65.4 µM) and PfCA (KIs 0.62 and 78.7 µM). A subset of compounds even demonstrated a significant selectivity for the target CAs over the human physiologically relevant isoforms CA I and II. This study improves the knowledge on the modulation of CAs belonging to uncommon classes such as δ and η. As a result, it promotes the identification of new potent and selective inhibitors against diatom and plasmodial isoforms over human off-target CAs, which could be adopted as leads for finding molecular probes in the study of carbon fixation processes or drug candidates in the treatment of malaria.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- (a) Nocentini A, Supuran CT. Advances in the structural annotation of human carbonic anhydrases and impact on future drug discovery. Expert Opin Drug Discov 2019;14:1175–97. (b) Supuran CT. Carbon-versus sulphur-based zinc binding groups for carbonic anhydrase inhibitors? J Enzyme Inhib Med Chem 2018;33:485–95.
- (a) Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nature Rev Drug Discov 2008;7:168–81. b) Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31:345–60.
- (a) Alterio V, Di Fiore A, D’Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68. (b) De Simone G, Supuran CT. (In)organic anions as carbonic anhydrase inhibitors. J Inorg Biochem 2012;111:117–29.
- Supuran CT. Structure and function of carbonic anhydrases. Biochem J 2016;473:2023–32.
- Xu Y, Feng L, Jeffrey PD, et al. Structure and metal exchange in the cadmium carbonic anhydrase of marine diatoms. Nature 2008;452:56–61.
- Lane TW, Saito MA, George GN, et al. Biochemistry: a cadmium enzyme from a marine diatom. Nature 2005;435:42.
- Del Prete S, Vullo D, Fisher GM, et al. Discovery of a new family of carbonic anhydrases in the malaria pathogen Plasmodium falciparum-the η-carbonic anhydrases. Bioorg Med Chem Lett 2014;24:4389–96.
- Jensen EL, Clement R, Kosta A, et al. A new widespread subclass of carbonic anhydrase in marine phytoplankton. ISME J 2019;13:2094–06.
- Iverson TM, Alber BE, Kisker C, et al. A closer look at the active site of gamma-class carbonic anhydrases: high-resolution crystallographic studies of the carbonic anhydrase from Methanosarcina thermophila. Biochemistry 2000;39:9222–31.
- Kikutani S, Nakajima K, Nagasato C, et al. Thylakoid luminal θ-carbonic anhydrase critical for growth and photosynthesis in the marine diatom Phaeodactylum tricornutum. Pnas 2016;113:9828–33.
- Cox EH, McLendon GL, Morel FM, et al. The active site structure of Thalassiosira weissflogii carbonic anhydrase 1. Biochemistry 2000;39:12128–30.
- McGinn PJ, Morel FM. Expression and regulation of carbonic anhydrases in the marine diatom Thalassiosira pseudonana and in natural phytoplankton assemblages from Great Bay, New Jersey. Physiol Plant 2008;133:78–91.
- Lane TW, Morel FM. Regulation of carbonic anhydrase expression by zinc, cobalt, and carbon dioxide in the marine diatom Thalassiosira weissflogii. Plant Physiol 2000;123:345–52.
- Del Prete S, Vullo D, De Luca V, et al. Biochemical characterization of the δ-carbonic anhydrase from the marine diatom Thalassiosira weissflogii. TweCA. J Enzyme Inhib Med Chem 2014;29:906–11. Dec
- Vullo D, Del Prete S, Osman SM, et al. Sulfonamide inhibition studies of the δ-carbonic anhydrase from the diatom Thalassiosira weissflogii. Bioorg Med Chem Lett 2014;24:275–9.
- Bua S, Bozdag M, Del Prete S, et al. Mono- and di-thiocarbamate inhibition studies of the δ-carbonic anhydrase TweCAδ from the marine diatom Thalassiosira weissflogii. J Enzyme Inhib Med Chem 2018;33:707–13.
- De Simone G, Di Fiore A, Capasso C, Supuran CT. The zinc coordination pattern in the eta-carbonic anhydrase from Plasmodium falciparum is different from all other carbonic anhydrase genetic families. Bioorg Med Chem Lett 2015;25:1385–9.
- Del Prete S, De Luca V, De Simone G, et al. Cloning, expression and purification of the complete domain of the eta-carbonic anhydrase from Plasmodium falciparum. J Enzyme Inhib Med Chem 2016;31:54–9.
- Del Prete S, Vullo D, De Luca V, et al. Cloning, expression, purification and sulfonamide inhibition profile of the complete domain of the eta-carbonic anhydrase from Plasmodium falciparum. Bioorg Med Chem Lett 2016;26:4184–90.
- Del Prete S, Vullo D, De Luca V, et al. Anion inhibition profiles of the complete domain of the eta-carbonic anhydrase from Plasmodium falciparum. Bioorg Med Chem 2016;24:4410–4.
- James P, Isupov MN, Sayer C, et al. The structure of a tetrameric alpha-carbonic anhydrase from Thermovibrio ammonificans reveals a core formed around intermolecular disulfides that contribute to its thermostability. Acta Crystallogr D Biol Crystallogr 2014;70:2607–18.
- Krungkrai J, Prapunwatana P, Wichitkul C, et al. Molecular biology and biochemistry of malarial parasite pyrimidine biosynthetic pathway. Southeast Asian J Trop Med Public Health 2003;34:32–43.
- (a) Capasso C, Supuran CT. Bacterial, fungal and protozoan carbonic anhydrases as drug targets. Expert Opin Ther Targets 2015;19:1689–704. (b) Supuran CT. Carbonic anhydrases and metabolism. Metabolites 2018;8:25.
- Sein KK, Aikawa M. The pivotal role of carbonic anhydrase in malaria infection. Med Hypotheses 1998;50:19–23.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73.
- (a) Nocentini A, Bua S, Del Prete S, et al. Natural polyphenols selectively inhibit β-carbonic anhydrase from the dandruff-producing fungus malassezia globosa: activity and modeling studies. ChemMedChem 2018;13:816–23. (b) D'Ascenzio M, Guglielmi P, Carradori S, et al. Open saccharin-based secondary sulfonamides as potent and selective inhibitors of cancer-related carbonic anhydrase IX and XII isoforms. J Enzyme Inhib Med Chem 2017;32:51–9. (c) Nocentini A, Ceruso M, Bua S, et al. Discovery of β-adrenergic receptors blocker-carbonic anhydrase inhibitor hybrids for multitargeted antiglaucoma therapy. J Med Chem 2018;61:5380–94.
- (a) Vermelho AB, da Silva Cardoso V, Ricci Junior E, et al. Nanoemulsions of sulfonamide carbonic anhydrase inhibitors strongly inhibit the growth of Trypanosoma cruzi. J Enzyme Inhib Med Chem 2018;33:139–46. (b) Nocentini A, Carta F, Tanc M, et al. Deciphering the mechanism of human carbonic anhydrases inhibition with sulfocoumarins: computational and experimental studies. Chemistry 2018;24:7840–44. (c) Awadallah FM, Bua S, Mahmoud WR, et al. Inhibition studies on a panel of human carbonic anhydrases with N1-substituted secondary sulfonamides incorporating thiazolinone or imidazolone-indole tails. J Enzyme Inhib Med Chem 2018;33:629–38.
- (a) Ferraroni M, Gaspari R, Scozzafava A, et al. Dioxygen, an unexpected carbonic anhydrase ligand. J Enzyme Inhib Med Chem 2018;33:999–1005. (b) El-Gazzar MG, Nafie NH, Nocentini A, et al. Carbonic anhydrase inhibition with a series of novel benzenesulfonamide-triazole conjugates. J Enzyme Inhib Med Chem 2018;33:1565–74. (c) Nocentini A, Bonardi A, Gratteri P, et al. Steroids interfere with human carbonic anhydrase activity by using alternative binding mechanisms. J Enzyme Inhib Med Chem 2018;33:1453–59.
- (a) Nocentini A, Trallori E, Singh S, et al. 4-Hydroxy-3-nitro-5-ureido-benzenesulfonamides selectively target the tumor-associated carbonic anhydrase isoforms IX and XII showing hypoxia-enhanced antiproliferative profiles. J Med Chem 2018;61:10860–74. (b) Nocentini A, Moi D, Balboni G, et al. Discovery of thiazolin-4-one-based aromatic sulfamates as a new class of carbonic anhydrase isoforms I, II, IV, and IX inhibitors. Bioorg Chem 2018;77:293–99. (c) Entezari Heravi Y, Bua S, Nocentini A, et al. Inhibition of Malassezia globosa carbonic anhydrase with phenols. Bioorg Med Chem 2017;25:2577–82.
- (a) Supuran CT, Ilies MA, Scozzafava A. Carbonic anhydrase inhibitors. Part 29. Interaction of isozymes I, II and IV with benzolamide-like derivatives. Eur J Med Chem 1998;33:739–52. (b) Köhler K, Hillebrecht A, Schulze Wischeler J, et al. Saccharin inhibits carbonic anhydrases: possible explanation for its unpleasant metallic aftertaste. Angew Chem Int Ed Engl 2007;46:7697–9. (c) Scozzafava A, Menabuoni L, Mincione F, et al. Carbonic anhydrase inhibitors: perfluoroalkyl/aryl-substituted derivatives of aromatic/heterocyclic sulfonamides as topical intraocular pressure-lowering agents with prolonged duration of action. J Med Chem 2000;43:4542–51. (d) Sentürk M, Gülçin I, Daştan A, et al. Carbonic anhydrase inhibitors. Inhibition of human erythrocyte isozymes I and II with a series of antioxidant phenols. Bioorg Med Chem 2009;17:3207–11.
- (a) Durdagi S, Vullo D, Pan P, et al. Protein-protein interactions: inhibition of mammalian carbonic anhydrases I-XV by the murine inhibitor of carbonic anhydrase and other members of the transferrin family. J Med Chem 2012;55:5529–35. (b). Scozzafava A, Briganti F, Mincione G, et al. Carbonic anhydrase inhibitors: synthesis of water-soluble, aminoacyl/dipeptidyl sulfonamides possessing long-lasting intraocular pressure-lowering properties via the topical route. J Med Chem 1999;42:3690–700.
- (a) Davis RA, Hofmann A, Osman A, et al. Natural product-based phenols as novel probes for mycobacterial and fungal carbonic anhydrases. J Med Chem 2011;54:1682–92. (b) Sarikaya SBÖ, Topal F, Şentürk M, et al. In vitro inhibition of α-carbonic anhydrase isozymes by some phenolic compounds. Bioorg Med Chem Lett 2011;21:4259–62.
- (a) Bilginer S, Unluer E, Gul HI, et al. Carbonic anhydrase inhibitors. Phenols incorporating 2- or 3-pyridyl-ethenylcarbonyl and tertiary amine moieties strongly inhibit Saccharomyces cerevisiae β-carbonic anhydrase. J Enzyme Inhib Med Chem 2014;29:495–9. (b) Supuran CT, Clare BW. Carbonic anhydrase inhibitors. Part 57. Quantum chemical QSAR of a group of 1,3,4-thiadiazole and 1,3,4-thiadiazoline disulfonamides with carbonic anhydrase inhibitory properties. Eur J Med Chem 1999;34:41–50.
- (a) Cau Y, Mori M, Supuran CT, et al. Mycobacterial carbonic anhydrase inhibition with phenolic acids and esters: kinetic and computational investigations. Org Biomol Chem 2016;14:8322–30. (b) Supuran CT, Nicolae A, Popescu A. Carbonic anhydrase inhibitors. Part 35. Synthesis of Schiff bases derived from sulfanilamide and aromatic aldehydes: the first inhibitors with equally high affinity towards cytosolic and membrane-bound isozymes. Eur J Med Chem 1996;31:431–8.
- Nocentini A, Osman SM, Del Prete S, et al. Extending the γ-class carbonic anhydrases inhibition profiles with phenolic compounds. Bioorg Chem 2019;93:103336.