
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
A urease inhibitor with good in vivo profile is considered as an alternative agent for treating infections caused by urease-producing bacteria such as Helicobacter pylori. Here, we report a series of N-monosubstituted thioureas, which act as effective urease inhibitors with very low cytotoxicity. One compound (b19) was evaluated in detail and shows promising features for further development as an agent to treat H. pylori caused diseases. Excellent values for the inhibition of b19 against both extracted urease and urease in intact cell were observed, which shows IC50 values of 0.16 ± 0.05 and 3.86 ± 0.10 µM, being 170- and 44-fold more potent than the clinically used drug AHA, respectively. Docking simulations suggested that the monosubstituted thiourea moiety penetrates urea binding site. In addition, b19 is a rapid and reversible urease inhibitor, and displays nM affinity to urease with very slow dissociation (koff=1.60 × 10−3 s−1) from the catalytic domain.
Introduction
Helicobacter pylori, a gram-negative and microaerophilic bacterium, colonises the gastric mucosa of over 50% of the global populationCitation1. In some individuals, chronic infection can induce a significant inflammatory response, which triggers a loss of gastric epithelial cells, resulting in gastric, duodenal ulcers and approximately 90% of cases of intestinal-type gastric carcinomaCitation2. With the help of urease (EC 3.5.1.5), a nickel dependent metalloenzyme with an ability to catalyse the hydrolysis of urea to ammonia and carbamates, H. pylori creates a local neutral environment for survival by continuously releasing ammonia into succus gastricusCitation3. In addition, Eaton et al. and Karita et al. demonstrated that urease-negative mutant of the H. pylori strain was unable to colonise the gastric mucosa under the acidic conditions of the stomachCitation4,Citation5. Therefore, urease is considered as a virulence factor playing an essential role for establishment of H. pylori colonisation in human. Therefore, urease inhibitors could serve as drugs for treating H. pylori caused disease such as gastritis and peptic ulcersCitation6.
In the past decades, thousands of urease inhibitors have been reported, and they were designed exclusively by either attacking the metallocenter or mimicking the substrate of ureasesCitation7–10. However, urease has a highly specific substrate urea, which makes it very challenging for the development of urease inhibitors. In spite of enormous efforts being made, only acetohydroxamic acid (AHA) has so far been approved by FDA for the treatment of urinary tract infections. Novel urease inhibitors with high potency are urgently needed. We therefore have focussed our efforts on this field for several years, and some potent urease inhibitors with structural diversity were reported such as catechols, diarylethylenes, flavonoids, arylamides, and hydroxamic acidsCitation11–15.
Thiourea derivatives, substrate analogues of urease, were reported as potential urease inhibitorsCitation16–18. In chemical structure, they are all N,N′-disubstituted thioureas, resulting in a different binding mode from that of ureaCitation18,Citation19, which may be caused by the high hindrance around thiourea score. Considering that the urea binding pocket is narrow and small, we believe that the thiourea moiety of N-monosubstituted thioureas could reach the bottom of the urea-binding pocket and chelate the two Ni atoms due to a tinny head. Based on this hypothesis and as a continuation of urease inhibitor screening, a series of N-arylacetothioureas were designed and synthesised. Some resulted compounds showed excellent potency against H. pylori urease.
Materials and methods
Biology materials
Protease inhibitors (Complete, Mini, EDTA-free) were purchased from Roche Diagnostics GmbH (Mannheim, Germany) and Brucella broth was from Becton Dickinson and Company (Sparks, MD). Sheep sterile and defibrinated blood were from Hyclone (Logan, UT).
Bacteria
H. pylori (ATCC 43504; American Type Culture Collection, Manassas, VA) was grown in Brucella broth supplemented with 10% sheep sterile and defibrinated blood for 24 h at 37 °C under microaerobic conditions (5% O2, 10% CO2, and 85% N2), as our previously described literatureCitation13,Citation14.
Preparation of H. pylori urease
For urease inhibition assays, 50 mL broth cultures (2.0 × 108 CFU/mL) were centrifuged (5000×g, 4 °C) to collect the bacteria, and after washing twice with phosphate-buffered saline (pH 7.4), the H. pylori precipitation was stored at –80 °C for 8 h, and then was returned to room temperature, and after addition of 3 mL of distilled water and protease inhibitors, sonication was performed for 60 s. Following centrifugation (15,000×g, 4 °C), the supernatant was desalted through Sephadex G-25 column (PD-10 columns, Amersham Pharmacia Biotech, Uppsala, Sweden). The resultant crude urease solution was added to an equal volume of glycerol and stored at 4 °C until use in the experiment.
Measurement of urease inhibitory activity
Urease activity was determined in the 96-well assay plate by measuring ammonia production using the indophenol method as described by WeatherburnCitation20. Briefly, to each well, 50 µL of urea (10 mM) in phosphate buffer solution was added in a mixture of 25 µL (10 U) of H. pylori urease and 25 µL of the test compound, which was incubated at 37 °C for 0.5 h. Fifty µL of phenol reagent (containing 127 mM phenol and 0.168 mM sodium nitroprusside) and 50 µL of alkali reagent (containing 125 mM NaOH and 0.168 mM NaOCl) were added in turn. The resulted mixture was incubated at 37 °C for another 0.5 h for colouration developing. The increasing absorbance was measured at 630 nm after 50 min using a microplate reader (Molecular Devices, San Jose, CA). Percentage inhibitions were calculated from the following formula (EquationEquation (1)(1)
(1) ). Experiments were performed in triplicate and AHA was used as reference drug, and the IC50 value was determined as the concentration of compound that give 50% inhibition of maximal activity. As for the urease assay of intact cells, 25 µL (10 U) of H. pylori urease was replaced by 25 µL of cell suspension (4.0 × 107 CFU/mL).
Determination of minimal inhibitory concentrations
The minimum inhibitory concentration (MIC) values were determined using the broth microdilution protocol according to the methods of the Clinical and Laboratory Standards Institute (CLSI)Citation21.
Ligand affinity study
The binding kinetics of selected compounds were assayed via surface plasmon resonance (SPR) using an OPEN SPR instrument (Nicoya Lifesciences, Kitchener, Canada). First, urease dissolved (50 µg/mL) in PBS buffer (1 mM KH2PO4, 155 mM NaCl, 3 mM Na2HPO4-12H2O, pH 7.4), was immobilised to a CM5 chip using a standard amine coupling procedureCitation22. Then, SPR measurements were carried out in PBS, and stock solutions were diluted in the same buffer. Data were collected with OpenSPR control software. Experiments were performed by monitoring the refractive index changes as a function of time under constant flow rate of 20 µL/min. The relative amount of inhibitor bound to the urease was determined by measuring the net increase in refractive index over time compared to control running buffer. There is an inline subtraction of reference surface during the run. This change is usually reported in response units (RU). Sensograms were processed and analysed using TraceDrawer software. The binding curves were fit to determine the equilibrium dissociation constant (KD).
Enzyme kinetic study
Based on the indophenol method, the velocity of ammonia production (V) was measured in the presence of concentration gradients of urea ([S]) for every specific concentration of selected compounds ([I]). Nonlinear fitting curves to data of V and [S] were used to determine the type of enzyme inhibition based on the general kinetics equation (EquationEquation (1)(1)
(1) ). Subsequently, the resulted fitting constants (
or
) and [I] were linearly fitted to give kinetic parameters
and
respectively. The experimental assay was performed against a pure urease (Jack bean urease) for the consideration of precision.
(1)
(1)
Protocol of docking study
Molecular docking of compounds b19 into the structure of H. pylori urease complex structure was carried out using SYBYL-X version 2.1.1 software suite (Tripos, Inc., St. Louis, MO)Citation23. The X-ray structure of urease from H. pylori was downloaded from the Protein Data Bank (PDB code: 1e9y)Citation24 and was modified by adding hydrogen atoms and removing water as well as cocrystallised substrate (AHA). The active site was defined as all the amino acid residues confined within a 5 Å radius sphere centred about AHA, and the composite structure without original ligand was utilised as the in silico model for docking studies. Default parameters and values within the minimisation dialogue were used except where otherwise mentioned. The docked conformations of ligands were evaluated and ranked using Surflex-dock and four scoring functions implemented in the CSCORE software module within the SYBYL-X environment. The CSCORE module allowed consensus scoring that integrated multiple well-known scoring functions such as ChemScore, D-Score, G-Score, and PMF-Score to evaluate docked ligand conformations.
Molecular dynamic simulations protocol
Molecular dynamics was performed using Desmond 4.2 with the standard RESPA integration and 2 fs time step. The TIP3P water model was used and exhibited well when combining with the OPLS force field. Each simulation system including the prepared protein, the inhibitor, and several Cl– added to achieve charge neutrality was immersed in a cubic box (10 Å). First, all the prepared systems were minimised until a gradient threshold (25 kcal/mol/Å) was reached by the steepest-descent (SD) method, and then coupled to the Berenson thermostat with 300 K reference temperature and the Berendsen barostat with 1.01325 bar reference pressure. The calculation of long-range electrostatics was based on the Particle-Mesh Ewald method. The cut-off for coulomb interaction was set at 9.0 Å. After equilibrated, all the systems began to run in the NPT ensemble for 6 ns. The relative RMSD and root mean square fluctuation (RMSF) were calculated based on the analysis of the MD trajectories by the VMD (version 1.9.1) and xmgrace tool.
Cytotoxicity assay
The stock solutions of the selected compounds (250 µg/mL, in PBS) were prepared in medium. L-02 cells and P69 cells were grown in medium (90% RPMI-1640 medium, 10% foetal bovine serum, 1% penicillin/streptomycin) and maintained at 37 °C in a humidified atmosphere containing 5% CO2, respectively. Cells were seeded in 96-well cell culture plates. On the day when seeding, the cells were exposed to 250 µg/mL of compounds and further cultured for 72 h at 37 °C. Cell proliferation was then determined using the thiazolyl blue tetrazolium bromide (MTT) assay.
Chemistry
All chemicals (reagent grade) used were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Melting points (uncorrected) were determined on a XT4 MP apparatus (Taike Corp., Beijing, China). EI mass spectra were obtained on Agilent 6120 mass spectrometer, and 1H NMR spectra were recorded on a Bruker AV-600 spectrometer at 25 °C with TMS and solvent signals allotted as internal standards. Chemical shifts were reported in ppm (δ). Elemental analyses were performed on a Foss Heraeus CHN-O-Rapid instrument and were within ±0.4% of the theoretical values.
General procedure for the preparation of compounds b
A selected arylacetic acid (10 mmol) was solved in SOCl2 (50 mL), which was heated to 90 °C and stirred for 1–2 h until the reaction was completed. The crude product (a1–a29) was furnished after removal of SOCl2 under reduced pressure. To the resulting residue, 50 mL of toluene was added and stirred for 30 min at room temperature. Thiourea (40 mmol) was subsequently added, and the reaction solution was heated to 100 °C for 1.5–2 h. After the end of the reaction was established by TLC, the solvent was removed under vacuum, and excess saturated NaHCO3 solution was added. The resulted mixture was extracted with ethyl acetate, dried over MgSO4, filtered, and concentrated under vacuum. The product was purified by a silica gel column using ethyl acetate and petroleum ether as eluent to afford compound b (b1–b29) in moderate to high yield as white powder (all the 1H NMR and 13C NMR Spectra of Compoundsb1-29 in Supporting Information).
N-Phenylacetourea (b1). White powder, 47%, mp: 110.9–112.1 °C, 1H NMR (600 MHz, CDCl3) δ (ppm): 3.71 (s, 2H, CH2); 7.28 (d, J = 6.8 Hz, 2H, Ar); 7.35 (t, J = 7.3 Hz, 1H, Ar); 7.39 (t, J = 7.3 Hz, 2H, Ar); 7.44 (s, 1H, NH2); 9.29 (s, 1H, NH2); 9.88 (s, 1H, NH); 13C NMR (150 MHz, CDCl3): 43.97; 128.06; 129.22; 129.38; 132.19; 171.72; 182.01; MS (ESI) m/z 195 (M + H)+; Anal. Calcd for C9H10N2OS: C, 55.65; H, 5.19; N, 14.42; S, 16.51; found: C, 55. 59; H, 5.19; N, 14.44; S, 16.53.
N-(3-Bromophenylaceto)urea (b2). White powder, 53%, mp: 207.4–208.8 °C, 1H NMR (600 MHz, CDCl3) δ (ppm): 3.58 (s, 2H, CH2); 6.98 (s, 1H, NH); 7.13 (dt, J = 7.3 Hz, J = 1.3 Hz, 1H, Ar); 7.20 (t, J = 7.8 Hz, 1H, Ar); 7.37 (t, J = 1.9 Hz, 1H, Ar); 7.42 (ddd, J = 8.0 Hz, J = 2.0 Hz, J = 1.1 Hz, 1H, Ar); 8.63 (s, 1H, NH2); 9.70 (s, 1H, NH2); 13C NMR (150 MHz, CDCl3): 42.21; 121.94; 129.04; 130.25; 130.95; 132.69; 137.54; 172.26; 182.04.
N-(3,4-Dichlorophenylaceto)urea (b3). White powder, 58%, mp: 202.1–203.4 °C, 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.75 (s, 2H, CH2); 7.28 (dd, J = 2.1 Hz, J = 10.3 Hz, 1H, Ar); 7.57 (d, J = 2.0 Hz, 1H, Ar); 7.59 (d, J = 8.0 Hz, 1H, Ar); 9.44 (s, 1H, NH2); 9.51 (s, 1H, NH2); 11.33 (s, 1H, NH); 13C NMR (150 MHz, DMSO-d6): 41.61; 130.12; 130.46; 130.87; 131.21; 132.08; 135.91; 171.9; 181.99.
N-(3-Hydroxyphenylaceto)urea (b4). White powder, 51%, mp: 160.3–161.5 °C, 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.60 (s, 2H, CH2); 6.65 (d, J = 7.1 Hz, 1H, Ar); 6.69 (d, J = 8.5 Hz, 1H, Ar); 6.70 (s, 1H, Ar); 7.10 (t, J = 7.8 Hz, 1H, Ar); 9.38 (s, 1H, OH); 9.41 (s, 1H, NH2); 9.58 (s, 1H, NH2); 11.26 (s, 1H, NH); 13C NMR (150 MHz, DMSO-d6): 42.87; 114.35; 116.60; 120.34; 129.80; 136.16; 157.77; 172.85; 182.18.
N-(3-Methoxyphenylaceto)urea (b5). White powder, 59%, mp: 184.5–186.1 °C, 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.67 (s, 2H, CH2); 3.73 (s, 3H, OCH3); 6.83 (dd, J = 8.3 Hz, J = 2.8 Hz, 1H, Ar); 6.85 (d, J = 7.5 Hz, 1H, Ar); 6.88 (t, J = 1.8 Hz, 1H, Ar); 7.23 (td, J = 7.9 Hz, J = 1.6 Hz, 1H, Ar); 9.42 (s, 1H, NH2); 9.57 (s, 1H, NH2); 11.28 (s, 1H, NH); 13C NMR (150 MHz, DMSO-d6): 42.89; 55.44; 112.73; 115.63; 121.99; 129.87; 136.31; 159.66; 172.71; 182.16.
N-(2-Fluorophenylaceto)urea (b6). White powder, 60%, mp: 157.2–159.0 °C, 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.75 (s, 2H, CH2); 7.14 (t, J = 9.1 Hz, 1H, Ar); 7.19 (t, J = 7.5 Hz, 1H, Ar); 7.24 (s, 1H, NH2); 7.29 (t, J = 7.7 Hz, 1H, Ar); 7.36 (dd, J = 14.1 Hz, J = 7.2 Hz, 1H, NH2); 9.04 (s, 1H, NH2); 9.81 (s, 1H, NH); 13C NMR (150 MHz, DMSO-d6): 37.53; 115.88 (d, J = 21.5 Hz); 119.49 (d, J = 15.7 Hz); 124.80 (d, J = 3.7 Hz); 130.22 (d, J = 8.2 Hz); 131.55 (d, J = 3.6 Hz); 160.94 (d, J = 246.8 Hz); 170.31; 181.98.
N-(3-Chlorophenylaceto)urea (b7). White powder, 59%, mp: 136.6–137.4 °C, 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.74 (s, 2H, CH2); 7.25 (dt, J = 7.2 Hz, J = 1.6 Hz, 1H, Ar); 7.32–7.36 (m, 2H, Ar); 7.38 (t, J = 1.8 Hz, 1H, Ar); 9.44 (s, 1H, NH2); 9.54 (s, 1H, NH2); 11.32 (s, 1H, NH); 13C NMR (150 MHz, DMSO-d6): 42.26; 127.36; 128.66; 129.82; 130.64; 133.30; 137.26; 172.25; 182.05.
N-(4-Hydroxyphenylaceto)urea (b8). White powder, 57%, mp: 177.1–178.3 °C, 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.56 (s, 2H, CH2); 6.70 (d, J = 8.0 Hz, 1H, Ar); 7.07 (d, J = 8.0 Hz, 1H, Ar); 9.32 (s, 1H, OH); 9.39 (s, 1H, NH2); 9.58 (s, 1H, NH2); 11.20 (s, 1H, NH); 13C NMR (150 MHz, DMSO-d6): 40.03; 115.62; 124.96; 130.74; 156.77; 173.39; 182.23.
N-(3-Trifluoromethylphenylaceto)urea (b9). White powder, 51%, mp: 204.3–205.8 °C, 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.86 (s, 2H, CH2); 7.57 (d, J = 7.5 Hz, 1H, Ar); 7.60 (d, J = 7.8 Hz, 1H, Ar); 7.64 (d, J = 7.0 Hz, 1H, Ar); 7.68 (s, 1H, Ar); 9.44 (s, 1H, NH2); 9.53 (s, 1H, NH2); 11.36 (s, 1H, NH); 13C NMR (150 MHz, DMSO-d6): 42.28; 124.12 (q, J = 3.8 Hz); 124.67 (q, J = 272.2 Hz); 126.63 (q, J = 3.8 Hz); 129.46 (q, J = 31.5 Hz); 129.82; 134.18; 136.23; 172.22; 182.03.
N-(3,4-Dimethoxyphenylaceto)urea (b10). White powder, 50%, mp: 146.3–147.6 °C, 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.61 (s, 2H, CH2); 3.72 (s, 3H, OCH3); 3.73 (s, 3H, OCH3); 6.81 (dd, J = 8.2 Hz, J = 2.0 Hz, 1H, Ar); 6.89 (d, J = 8.3 Hz, 1H, Ar); 6.90 (d, J = 2.0 Hz, 1H, Ar); 9.40 (s, 1H, NH2); 9.58 (s, 1H, NH2); 11.23 (s, 1H, NH); 13C NMR (150 MHz, DMSO-d6): 42.44; 55.89; 55.96; 112.26; 113.65; 121.87; 127.15; 148.29; 148.98; 173.10; 182.19.
N-(4-Methoxyphenylaceto)urea (b11). White powder, 56%, mp: 143.5–144.7 °C, 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.62 (s, 2H, CH2); 3.73 (s, 3H, OCH3); 6.89 (d, J = 8.6 Hz, 2H, Ar); 7.21 (d, J = 8.6 Hz, 2H, Ar); 9.40 (s, 1H, NH2); 9.58 (s, 1H, NH2); 11.25 (s, 1H, NH); 13C NMR (150 MHz, DMSO-d6): 41.98; 55.49; 114.28; 126.76; 130.84; 158.72; 173.22; 182.21.
N-(4-Bromophenylaceto)urea (b12). White powder, 53%, mp: 174.9–176.2 °C, 1H NMR (600 MHz, CDCl3) δ (ppm): 3.56 (s, 2H, CH2); 6.99 (s, 1H, NH); 7.07 (d, J = 8.3 Hz, 2H, Ar); 7.44 (d, J = 8.3 Hz, 2H, Ar); 8.67 (s, 1H, NH2); 9.70 (s, 1H, NH2); 13C NMR (150 MHz, CDCl3): 43.41; 122.37; 131.04; 131.14; 132.42; 170.59; 181.93.
N-(2-Chlorophenylaceto)urea (b13). White powder, 55%, mp: 148.8–151.2 °C, 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.91 (s, 2H, CH2); 7.29–7.32 (m, 2H, Ar); 7.37–7.40 (m, 1H, Ar); 7.42–7.45 (m, 1H, Ar); 9.42 (s, 1H, NH2); 9.52 (s, 1H, NH2); 11.38 (s, 1H, NH); 13C NMR (150 MHz, DMSO-d6): 40.84; 127.60; 129.44; 129.47; 132.92; 133.04; 134.17; 171.73; 182.01; MS (ESI) m/z 229 (M + H)+; Anal. Calcd for C9H9ClN2OS: C, 47.27; H, 3.97; Cl, 15.50; N, 12.25; S, 14.02; found: C, 47.21; H, 3.97; Cl, 15.52; N, 12.25; S, 14.03.
N-(3-Fluorophenylaceto)urea (b14). White powder, 58%, mp: 126.0–127.4 °C, 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.75 (s, 2H, CH2); 6.95–7.23 (m, 3H, Ar); 7.30–7.52 (m, 1H, Ar); 9.43 (s, 1H, NH2); 9.54 (s, 1H, NH2); 11.32 (s, 1H, NH); 13C NMR (150 MHz, DMSO-d6): 42.37 (d, J = 1.8 Hz); 114.20 (d, J = 20.8 Hz); 116.74 (d, J = 21.4 Hz); 126.07 (d, J = 2.7 Hz); 130.67 (d, J = 8.3 Hz); 137.52 (d, J = 8.0 Hz); 162.45 (d, J = 243.2 Hz); 172.28; 182.07.
N-(2-Bromophenylaceto)urea (b15). White powder, 54%, mp: 179.0–179.5 °C, 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.92 (s, 2H, CH2); 7.22 (td, J = 7.7 Hz, J = 1.7 Hz, 1H, Ar); 7.35 (td, J = 7.4 Hz, J = 1.8 Hz, 1H, Ar); 7.38 (dd, J = 7.6 Hz, J = 1.9 Hz,1H, Ar); 7.60 (dd, J = 8.6 Hz, J = 2.3 Hz, 1H, Ar); 9.41 (s, 1H, NH2); 9.52 (s, 1H, NH2); 11.38 (s, 1H, NH); 13C NMR (150 MHz, DMSO-d6): 43.28; 125.06; 128.14; 129.63; 132.72; 133.00; 134.83; 171.69; 182.01.
N-(4-Methylphenylaceto)urea (b16). White powder, 55%, mp: 171.2–172.5 °C, 1H NMR (600 MHz, DMSO-d6) δ (ppm): 2.27 (s, 3H, CH3); 3.65 (s, 2H, CH2); 7.12 (d, J = 7.9 Hz, 2H, Ar); 7.17 (d, J = 7.9 Hz, 2H, Ar); 9.40 (s, 1H, NH2); 9.57 (s, 1H, NH2); 11.26 (s, 1H, NH); 13C NMR (150 MHz, DMSO-d6): 21.13; 42.45; 129.41; 129.66; 131.84; 136.44; 173.04; 182.19.
N-(2-Methoxyphenylaceto)urea (b17). White powder, 61%, mp: 135.4–136.9 °C, 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.69 (s, 2H, CH2); 3.75 (s, 3H, OCH3); 6.89 (td, J = 7.4 Hz, J = 1.0 Hz, 1H, Ar); 6.97 (dd, J = 8.2 Hz, J = 2.8 Hz, 1H, Ar); 7.17 (dd, J = 7.5 Hz, J = 1.7 Hz, 1H, Ar); 7.25 (td, J = 7.8 Hz, J = 1.5 Hz, 1H, Ar); 9.36 (s, 1H, NH2); 9.57 (s, 1H, NH2); 11.17 (s, 1H, NH); 13C NMR (150 MHz, DMSO-d6): 37.94; 55.90; 111.16; 120.66; 123.22; 128.94; 131.69; 157.66; 172.95; 182.10.
N-(2-Methylphenylaceto)urea (b18). White powder, 60%, mp: 135.6–137.1 °C, 1H NMR (600 MHz, DMSO-d6) δ (ppm): 2.23 (s, 3H, CH3); 3.76 (s, 2H, CH2); 7.11–7.15 (m, 1H, Ar); 7.15–7.17 (m, 2H, Ar); 7.19 (dd, J = 6.7 Hz, J = 1.7 Hz, 1H, Ar); 9.41 (s, 1H, NH2); 9.57 (s, 1H, NH2); 11.29 (s, 1H, NH2); 13C NMR (150 MHz, DMSO-d6): 19.74; 40.69; 126.30; 127.52; 130.41; 130.68; 133.67; 137.20; 172.80; 182.11.
N-(4-Chlorophenylaceto)urea (b19). White powder, 57%, mp: 184.1–186.6 °C, 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.72 (s, 2H, CH2); 7.31 (d, J = 8.4 Hz, 2H, Ar); 7.39 (d, J = 8.4 Hz, 2H, Ar); 9.43 (s, 1H, NH2); 9.54 (s, 1H, NH2); 11.31 (s, 1H, NH); 13C NMR (150 MHz, DMSO-d6): 42.02; 128.77; 131.77; 132.12; 133.88; 172.48; 182.09.
N-(2-Nitrophenylaceto)urea (b20). White powder, 59%, mp: 177.6–178.4 °C, 1H NMR (600 MHz, DMSO-d6) δ (ppm): 4.17 (s, 2H, CH2); 7.55 (dd, J = 7.6, J = 1.4 Hz, 1H, Ar); 7.59 (td, J = 7.8, J = 1.5 Hz, 1H, Ar); 7.73 (td, J = 7.5, J = 1.3 Hz, 1H, Ar); 8.10 (dd, J = 8.2, J = 1.3 Hz, 1H, Ar); 9.41 (s, 2H, NH2); 11.41 (s, 1H, NH); 13C NMR (150 MHz, DMSO-d6): 41.21; 125.24; 129.32; 129.88; 134.41; 134.45; 148.92; 171.52; 181.93.
N-(2,4-Dichlorophenylaceto)urea (b21). White powder, 59%, mp: 166.6–167.2 °C, 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.92 (s, 2H, CH2); 7.41 (dd, J = 8.3 Hz, J = 2.0 Hz, 1H, Ar); 7.43 (d, J = 8.2 Hz, 1H, Ar); 7.61 (d, J = 2.2 Hz, 1H, Ar); 9.43 (s, 1H, NH2); 9.48 (s, 1H, NH2); 11.40 (s, 1H, NH); 13C NMR (150 MHz, DMSO-d6): 40.23; 127.74; 128.94; 132.23; 133.08; 134.19; 135.17; 171.32; 181.93.
N-(4-Nitrophenylaceto)urea (b22). White powder, 58%, mp: 199.1–200.9 °C, 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.91 (s, 2H, CH2); 7.57 (d, J = 8.7 Hz, 2H, Ar); 8.20 (d, J = 8.7 Hz, 2H, Ar); 9.46 (s, 1H, NH2); 9.50 (s, 1H, NH2); 11.40 (s, 1H, NH); 13C NMR (150 MHz, DMSO-d6): 42.43; 123.86; 131.38; 142.81; 147.04; 171.72; 181.99.
N-(4-Fluorophenylaceto)urea (b23). White powder, 57%, mp: 165.4–166.5 °C, 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.71 (s, 2H, CH2); 7.15 (t, J = 8.9 Hz, 2H, Ar); 7.32 (dd, J = 8.5 Hz, J = 5.7 Hz, 2H, Ar); 9.42 (s, 1H, NH2); 9.55 (s, 1H, NH2); 11.30 (s, 1H, NH); 13C NMR (150 MHz, DMSO-d6): 41.87; 115.57 (d, J = 21.1 Hz); 131.04 (d, J = 3.2 Hz); 131.78 (d, J = 8.0 Hz); 161.74 (d, J = 242.7 Hz); 172.75; 182.12.
N-(2-Naphthylaceto)urea (b24). White powder, 52%, mp: 202.6–204.4 °C, 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.90 (s, 1H); 7.45 (dd, J = 8.4 Hz, J = 1.7 Hz, 1H, Ar); 7.49 (dd, J = 6.8 Hz, J = 1.6 Hz, 1H, Ar); 7.51 (dd, J = 7.0 Hz, J = 1.8 Hz, 1H, Ar); 7.81 (d, J = 1.8 Hz, 1H, Ar); 7.88 (d, J = 8.1 Hz, 2H, Ar); 7.89 (d, J = 8.7 Hz, 1H, Ar); 9.45 (s, 1H, NH2); 9.60 (s, 1H, NH2); 11.40 (s, 1H, NH); 13C NMR (150 MHz, DMSO-d6): 42.96; 126.28; 126.68; 127.94; 127.98; 128.18; 128.29; 128.34; 132.41; 132.59; 133.38; 172.85; 182.17.
N-(Diphenylaceto)urea (b25). White powder, 54%, mp: 169.5–172.7 °C, 1H NMR (600 MHz, DMSO-d6) δ (ppm): 5.40 (s, 1H, CH); 7.23–7.32 (m, 6H, Ar); 7.33–7.38 (m, 4H, Ar); 9.52 (s, 1H, NH2); 9.61 (s, 1H, NH2); 11.49 (s, 1H, NH); 13C NMR (150 MHz, DMSO-d6): 56.77; 127.70; 129.03; 129.04; 139.00; 173.35; 182.12.
N-(Biphenyl-4-yl)acetourea (b26). White powder, 52%, mp: 187.3–189.6 °C, 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.76 (s, 2H); 7.36 (t, J = 7.4 Hz, 1H, Ar); 7.39 (d, J = 8.1 Hz, 2H, Ar); 7.46 (t, J = 7.7 Hz, 2H, Ar); 7.62 (d, J = 8.1 Hz, 2H, Ar); 7.65 (d, J = 6.8 Hz, 2H, Ar); 9.44 (s, 1H, NH2); 9.59 (s, 1H, NH2); 11.35 (s, 1H, NH); 13C NMR (150 MHz, DMSO-d6): 42.49; 127.07; 127.18; 127.85; 129.38; 130.41; 134.15; 139.31; 140.33; 172.83; 182.18.
N-(1-Naphthylaceto)urea (b27). White powder, 57%, mp: 195.5–197.9 °C, 1H NMR (600 MHz, DMSO-d6) δ (ppm): 4.24 (s, 2H, CH2); 7.46–7.49 (m, 2H, Ar); 7.53 (td, J = 7.4 Hz, J = 1.2 Hz, 1H, Ar); 7.57 (td, J = 8.3 Hz, J = 1.5 Hz, 1H, Ar); 7.84–7.89 (m, 1H, Ar); 7.94 (dd, J = 8.1, J = 1.4 Hz, 1H); 8.02 (d, J = 7.8 Hz, 1H); 9.43 (s, 1H, NH2); 9.54 (s, 1H, NH2); 11.48 (s, 1H, NH); 13C NMR (150 MHz, DMSO-d6): 40.20; 124.51; 125.99; 126.24; 126.74; 128.07; 128.74; 128.95; 131.49; 132.32; 133.81; 172.78; 182.12.
N-(2-(benzo[d][1,3]dioxol-5-yl)aceto)urea (b28). White powder, 56%, mp: 153.5–155.0 °C, 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.60 (s, 2H, CH2); 5.99 (s, 2H, OCH2O); 6.75 (dd, J = 8.0 Hz, J = 1.7 Hz, 1H, Ar); 6.85 (d, J = 7.7 Hz, 1H, Ar); 6.86 (d, J = 1.5 Hz, 1H, Ar); 9.41 (s, 1H, NH2); 9.56 (s, 1H, NH2); 11.23 (s, 1H, NH); 13C NMR (150 MHz, DMSO-d6): 42.43; 101.34; 108.60; 110.21; 122.95; 128.40; 146.64; 147.63; 172.95; 182.16.
N-(3,4-Dihydroxyphenylaceto)urea (b29). White powder, 56%, mp: 153.5–155.0 °C, 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.48 (s, 2H, CH2); 6.53 (dd, J = 8.1 Hz, J = 2.1 Hz, 1H, Ar); 6.65 (d, J = 8.0 Hz, 1H, Ar); 6.68 (d, J = 2.1 Hz, 1H, Ar); 8.79 (s, 1H, OH); 8.88 (s, 1H, OH); 9.39 (s, 1H, NH2); 9.60 (s, 1H, NH2); 11.17 (s, 1H, NH); 13C NMR (150 MHz, DMSO-d6): 42.29; 115.90; 117.04; 120.52; 125.52; 144.77; 145.53; 173.40; 182.25.
Results and discussion
Chemistry
A typical synthetic route towards arylacetothioureas is described in Scheme 1. The arylacetyl chloride intermediate was synthesised from an appropriate arylacetic acid by reaction with SOCl2. Amidation was followed with thiourea to afford the corresponding compound b.
Scheme 1. Synthesis of arylacetothioureas.
Inhibitory activity against cell-free urease
All synthesised arylacetothioureas were evaluated for their inhibitory activity against extracted H. pylori urease. As shown in , a compound bearing naphthyl (b24 and b27), benzo-1,3-dioxole (b28) or biphenyl (b26) moiety exhibited very low potency, even showed inactivity. In contrast, some compounds from phenylacetothioureas showed excellent potency with IC50 values lower than that of the positive control AHA. Out of these compounds, b19 was the most active inhibitor with IC50 of 0.16 ± 0.05 µM, showing 158-fold more potency than AHA. Replacement of the chloro group of compound b19 by other substitutions such as fluoro, bromo, methyl, and methoxy attenuated urease inhibition by 27- to 968-fold, which suggested that only suitable steric volume is tolerated. Movement of the chlorine from the para position (b19) to the meta position (b7) produced a 397-fold decrease in potency. In the case of meta-substituted derivatives, a different result was observed. Replacement of chloro group with fluoro (b14), bromo (b2), or methoxy (b5) group gives a comparable activity. However, hydroxyl-substituted analogue (b4) showed an about fourfold increase in potency in comparison with b7, which may be attributed to a possible hydrogen-bond building ability of the hydroxyl group. In comparison with the meta-substituted derivatives, the ortho-analogous resulted in no significant change in potency. It is to be noted that compound containing a strong electron-withdrawing nitro group (b20) resulted in a significant decrease in potency with IC50 value over 180 µM. In the case of double substituent on the phenyl ring, compound with 3,4-dihyroxyl group resulted in IC50 values of 18.4 µM, being the most active in this series and showing more potent than the positive control AHA.
Table 1. Structure, inhibitory activity (IC50), antibacterial activity (MIC50) against H. pylori urease of compounds b1–b29.
Inhibitory activity against urease in intact cell
Encouraged by the results of the extracted urease assay, compounds showing higher potency than AHA were selected to determine inhibitory activities against urease in intact H. pylori cell, and the results are shown in . A 5- to 24-fold increase of IC50 values against urease in intact cell was observed in comparison with those of extracted urease. Three compounds (b8, b16, and b19) were found to be much more potent than AHA, showing IC50 of 52.5 ± 3.9, 35.7 ± 1.7, and 3.86 ± 0.10 µM. Not surprisingly, the most potent compound b19 found in the extracted urease assay was also exhibited the highest activity against urease in intact H. pylori cell.
Antibacterial activity
To get an insight of the possibility of the identified urease inhibitors for further drug developing, compounds showing higher potency in enzyme assays than AHA were selected to test the potential growth inhibition against H. pylori, and the results are shown in . The assayed compounds showed no or very weak antibacterial activity against H. pylori at neutral pH. However, at low pH (about 4.0), compounds with significant inhibition against urease in intact H. pylori cell showed impressive antibacterial effect against H. pylori. This is consistent with that revealed by Eaton et al. and Karita et al. as above mentionedCitation4,Citation5. In general, these data indicated a potential for in vivo efficacy of compounds such as b19 and b16 in clearing H. pylori infection.
Ligand affinity
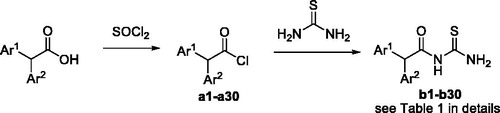
Compounds with IC50 lower than 20 µM against extracted H. pylori urease were selected for ligand-enzyme binding interaction based on SPR, which allows for the determination of affinities and kinetic parameters in a single experimentCitation25. In the present paper, the pure Jack bean urease from commercial was used for this assay because of non-availability of pure H. pylori urease. The binding affinities are shown in and the representative SPR plots are shown in . The most active compound b19 displayed the highest binding affinity to urease with a KD value of 4.50 ± 0.16 nM. For efficient and tight ligand binding, the rate constant koff is of particular interest, because koff has a potential to differentiate indistinguishable compounds with similar affinitiesCitation26. The results revealed that b19 displayed very slow dissociation from the catalytic domain of urease with a koff 1.73 × 10−3 s−1, indicating that b19 could be prioritised for further optimisation.
Figure 1. Sensograms of interactions between b19 and urease.
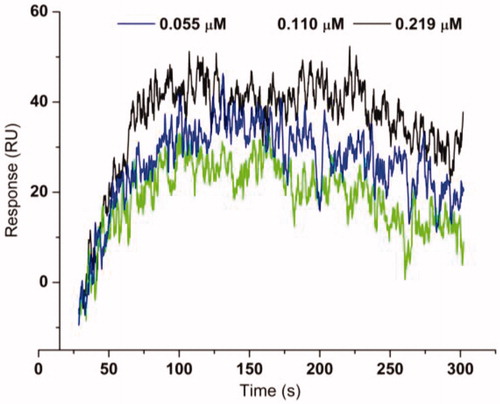
Table 2. The binding affinity data for urease–thioureas interactions.
Inhibition kinetics
Compounds with IC50 lower than 20 µM against extracted H. pylori urease were selected to perform kinetic assay for further insight of the inhibition mechanism. Mazzei et al. confirmed that urease shared the mechanism of catalysis and inhibition regardless of the biological sourcesCitation27,Citation28. For excluding the possible interference, a pure urease (Jack bean urease) was therefore used for kinetic assays. As an example, describes the preincubation-time dependence of urease inhibition by b19. The suppressed urease activity maintained at relatively constant values with the increasing preincubation time under different concentrations of b19, resulting in nearly equal IC50 values. The obtained results indicate that b19 can rapidly bind to the active site and inhibit urease in a time-independent manner.
Figure 2. Characterisation of urease inhibition by compound b19 for different preincubation time.
shows the nonlinear fitting curves (V vs. [S]) of the most potential urease inhibitor b19 based on the general kinetics equation (EquationEquation (1)(1)
(1) , and the corresponding plots by linearly fitting the fitting constants from V–[S] curves against [I], which provided the kinetic parameters
and
of b19. Herein,
is the dissociation constant for “urease-inhibitor → urease and inhibitor” and
is the dissociation constant for “urease-urea-inhibitor → urease-urea and inhibitor”Citation1. The calculated
and
of b19 are 0.040 and 0.16 µM, respectively, suggesting: (1) b19 is a reversible urease inhibitor (
and
indicate an irreversible inhibition); (2) b19 has a mixed competitive mechanism (
and
are not
and
). Similarly, kinetic parameters (
and
) and inhibition types of other compounds were also determined and are shown in . The values of
are larger than the corresponding
for all tested compounds, suggesting that the complex of urease-urea-inhibitor is less stable than that of the urease-inhibitor and competitive inhibition has relatively higher weight in the mixed competitive mechanism.
Figure 3. A velocity (V) was nonlinearly fitted against the concentrations of urea [S] in the presence of a specific concentration of compound b19. (B) (1) the fitted constants (
) from the corresponding V–S plot were plotted against concentrations of b19 ([I]); (2)
the fitted constants (
) from the corresponding V–S plot were plotted against concentrations of b19.
![Figure 3. A velocity (V) was nonlinearly fitted against the concentrations of urea [S] in the presence of a specific concentration of compound b19. (B) (1) Ki: the fitted constants (KmVmax(1+[I]Ki)) from the corresponding V–S plot were plotted against concentrations of b19 ([I]); (2) Ki': the fitted constants (1Vmax(1+[I]Ki′)) from the corresponding V–S plot were plotted against concentrations of b19.](/cms/asset/cbdcdec4-28e7-4e50-bece-e902474d1941/ienz_a_1706503_f0003_c.jpg)
Table 3. Data of inhibition mechanism.
Molecular docking
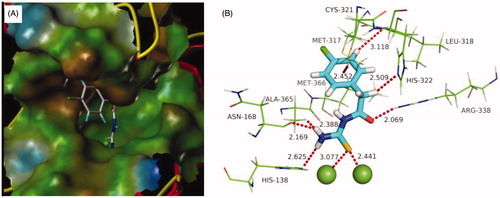
With the aim to explore the structural determinants of the guidance for further SAR studies, molecular docking of the most potent inhibitor b19 into urea binding site was performed, and the binding model is depicted in . This model revealed that the monosubstituted thiourea moiety is nicely bound to urea binding site (), and is of primary importance for its network of interactions (): it coordinates the nickel ion and establishes hydrogen bonds with N of His138, O of Asn168, and Me of Ala365, respectively. On the other hand, the benzene ring of b19 establishes appropriate hydrophobic contacts with the hydrophobic gap (Met317, Leu318, and Met366) under the movable flap, a helix-turn-helix motif composed of residues α313–α346. Moreover, benzene ring is further solid by an S–H···π with Cys321, and a C–H···N with Leu318. In addition, this model also suggested that the aceto moiety as a donor as well as an acceptor forms C–H···N and N–H···O hydrogen bonds with His322 and Arg338, respectively. The enzyme assay data and the molecular docking results indicated that compound b19 is a potential inhibitor of urease.
Figure 4. Predicted binding mode of ligand-urease (PDB code: 1e9y): (A) compound b19 shown as white sticks and the enzyme shown as surface. (B) Compound b19 shown as cyan sticks and enzyme shown as lines; Hydrogen bonds shown as red dashed lines.
Molecular dynamics
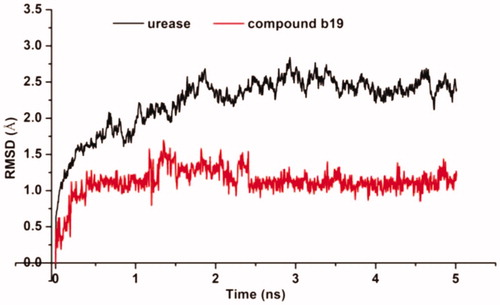
Molecular dynamic (MD) simulations were performed to understand the dynamic properties of urease–b19 complexes, and to provide some evidence for the suppression site identified by molecular dockings. shows the evolutions of the root mean square deviation (RMSD) values versus time in reference to the energy minimised complex structure. RMSD change suggested that simulation attains an equilibrium position within 1 ns, and the average RMSD fluctuation values of b19 were 1.1 Å after equilibrium was reached. The very low deviation from docked position indicated that molecular docking results of b19 are reliable, which was also evidenced by the high binding affinity observed in enzyme and SPR assays.
Figure 5. The RMSD values obtained during 6 ns of molecular dynamic simulation for urease and b19.
Cytotoxicity
The highly bioactive compounds with IC50 value against urease lower than that of AHA were further evaluated for its toxicity profile against human normal hepatic cell line L-02 and normal prostate cell line P69 using the colorimetric cell proliferation MTT assay. As shown in , most of the assayed compounds showed low cytotoxicity against these human normal cell lines with viability over 90% at concentration of 25 µg/mL. It is to be noted that the cell viability against the most potent compound b19 of the two cell lines was more than 93%, suggesting the low toxicity to mammalian cells. On the whole, the new identified urease inhibitor b19 showed lower cell toxicity than the positive control AHA.
Table 4. Cell Viability of selected compounds on L-02 and P69 at concentration of 25 μg/mL.
Conculsions
In summary, we have developed a series of mono-substituted thioureas as potent H. pylori urease inhibitors. The most potent compound b19 was identified as a reversible urease inhibitor, and significantly inhibits extracted urease and urease in intact cell with IC50 values of 0.16 ± 0.05 and 3.86 ± 0.10 µM, being 170- and 44-fold more potent than clinically used drug AHA, respectively. The SPR assay thus revealed that b19 exhibits very high urease affinities in the low nM range, probably binding to the urea site and showing very slow dissociation from the catalytic domain. Urease inhibition assay, SPR assay, molecular docking studies and cell proliferation assay suggested that the mono-substituted thioureas are a new kind of urease inhibitors acting at the urea site, with b19 having potential for further development as an agent to treat H. pylori caused diseases.
Supplemental Material
Download PDF (11.1 MB)Acknowledgements
Authors thank Dr Dong-Dong Li for molecular dynamics.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Liu Q, Ni W-W, Li Z, et al. Resolution and evaluation of 3-chlorophenyl-3-hydroxy-propionylhydroxamic acid as antivirulence agent with excellent eradication efficacy in Helicobacter pylori infected mice. Eur J Pharm Sci 2018;121:293–300.
- Valenzuela-Valderrama M, Cerda-Opazo P, Backert S, et al. The Helicobacter pylori urease virulence factor is required for the induction of hypoxia-induced factor-1 in gastric cells. Cancers 2019;11:799.
- Liu Q, Shi W-K, Ren S-Z, et al. Arylamino containing hydroxamic acids as potent urease inhibitors for the treatment of Helicobacter pylori infection. Eur J Med Chem 2018;156:126–36.
- Eaton KA, Brooks CL, Morgan DR, et al. Essential role of urease in pathogenesis of gastritis induced by Helicobacter pylori in gnotobiotic piglets. Infect Immun 1991;59:2470–5.
- Karita M, Tsuda M, Nakazawa T. Essential role of urease in vitro and in vivo Helicobacter pylori colonization study using a wild-type and isogenic urease mutant strains. J Clin Gastroenterol 1995;21:S160–S163.
- Rizvi F, Khan M, Jabeen A, et al. Studies on isoniazid derivatives through a medicinal chemistry approach for the identification of new inhibitors of urease and inflammatory markers. Sci Rep 2019;9:1–14.
- Shi W-K, Deng R-C, Wang P-F, et al. 3-Arylpropionylhydroxamic acid derivatives as Helicobacter pylori urease inhibitors: synthesis, molecular docking and biological evaluation. Bioorg Med Chem 2016;24:4519–27.
- Kanwal, Khan M, Arshia, et al. Syntheses, in vitro urease inhibitory activities of urea and thiourea derivatives of tryptamine, their molecular docking and cytotoxic studies. Bioorg Chem 2019;83:595–610.
- Rauf A, Shahzad S, Bajda M, et al. Design and synthesis of new barbituric- and thiobarbituric acid derivatives as potent urease inhibitors: structure activity relationship and molecular modeling studies. Bioorg Med Chem 2015;23:6049–58.
- Yu H-Y, Guo S-H, Cheng J-Y, et al. Synthesis and crystal structures of cobalt(III), copper(II), nickel(II) and zinc(II) complexes derived from 4-methoxy-N′-(pyridin-2-ylmethylene)benzohydrazide with urease inhibitory activity. J Coord Chem 2008;61:1212–20.
- Ni W-W, Liu Q, Ren S-Z, et al. The synthesis and evaluation of phenoxyacylhydroxamic acids as potential agents for Helicobacter pylori infections. Bioorg Med Chem 2018;26:4145–52.
- Xiao Z-P, Shi W-K, Wang P-F, et al. Synthesis and evaluation of N-analogues of 1,2-diarylethane as Helicobacter pylori urease inhibitors. Bioorg Med Chem 2015;23:4508–13.
- Wang X-D, Wei W, Wang P-F, et al. Synthesis, molecular docking and biological evaluation of 3-arylfuran-2(5H)-ones as anti-gastric ulcer agent. Bioorg Med Chem 2015;23:4860–5.
- Xiao Z-P, Peng Z-Y, Dong J-J, et al. Synthesis, structure–activity relationship analysis and kinetics study of reductive derivatives of flavonoids as Helicobacter pylori urease inhibitors. Eur J Med Chem 2013;63:685–95.
- Xiao Z-P, Ma T-W, Fu W-C, et al. The synthesis, structure and activity evaluation of pyrogallol and catechol derivatives as H. pylori urease inhibitors. Eur J Med Chem 2010;45:5064–70.
- Sivapriya K, Suguna P, Banerjee A, et al. Facile one-pot synthesis of thio and selenourea derivatives: a new class of potent urease inhibitors. Bioorg Med Chem Lett 2007;17:6387–91.
- Taha M, Wadood A. Synthesis and molecular docking study of piperazine derivatives as potent urease inhibitors. Bioorg Chem 2018;78:411–7.
- Bano B, Kanwal, Khan KM, et al. Synthesis, in vitro urease inhibitory activity, and molecular docking studies of thiourea and urea derivatives. Bioorg Chem 2018;80:129–44.
- Amtul Z, Atta-ur-Rahman BSP, Siddiqui R, et al. Chemistry and mechanism of urease inhibition. Curr Med Chem 2002;9:1323–48.
- Weatherburn MW. Phenol–hypochlorite reaction for determination of ammonia. Anal Chem 1967;39:971–4.
- Methods for antimicrobial susceptibility testing of anaerobic bacteria, M11. 7th ed. Wayne (PA): CLSI; 2007.
- Patil S, Sistla S, Jadhav J. Screening of inhibitors for mushroom tyrosinase using surface plasmon resonance. J Agric Food Chem 2014;62:11594–601.
- SYBYL molecular modeling software, version SYBYL-X 2.1. St. Louis (MO): Tripos Inc.; 2015.
- Ha NC, Oh ST, Sung JY, et al. Supramolecular assembly and acid resistance of Helicobacter pylori urease. Nat Struct Biol 2001;8:505–9.
- Frostell A, Vinterbäck L, Sjöbom H. Protein-ligand interactions using SPR systems. Methods Mol Biol 2013;1008:139–65.
- Guo D, Hillger JM, IJzerman AP, Heitman LH. Drug-target residence time—a case for G protein-coupled receptors. Med Res Rev 2014;34:856–92.
- Mazzei L, Cianci M, Musiani F, et al. Inactivation of urease by catechol: kinetics and structure. J Inorg Biochem 2017;166:182–9.
- Mazzei L, Cianci M, Contaldo U, et al. Insights into urease inhibition by N-(n-butyl) phosphoric triamide through an integrated structural and kinetic approach. J Agric Food Chem 2019;67:2127–38.