
Abstract
The enzymes acetylcholinesterase (AChE) and butyrylcholinesterase (BChE) are primary targets in attenuating the symptoms of neurodegenerative diseases. Their inhibition results in elevated concentrations of the neurotransmitter acetylcholine which supports communication among nerve cells. It was previously shown for trans-4/5-arylethenyloxazole compounds to have moderate AChE and BChE inhibitory properties. A preliminary docking study showed that elongating oxazole molecules and adding a new NH group could make them more prone to bind to the active site of both enzymes. Therefore, new trans-amino-4-/5-arylethenyl-oxazoles were designed and synthesised by the Buchwald-Hartwig amination of a previously synthesised trans-chloro-arylethenyloxazole derivative. Additionally, naphthoxazole benzylamine photoproducts were obtained by efficient photochemical electrocyclization reaction. Novel compounds were tested as inhibitors of both AChE and BChE. All of the compounds exhibited binding preference for BChE over AChE, especially for trans-amino-4-/5-arylethenyl-oxazole derivatives which inhibited BChE potently (IC50 in µM range) and AChE poorly (IC50≫100 µM). Therefore, due to the selectivity of all of the tested compounds for binding to BChE, these compounds could be applied for further development of cholinesterase selective inhibitors.
Series of oxazole benzylamines were designed and synthesised
The tested compounds showed binding selectivity for BChE
Naphthoxazoles were more potent AChE inhibitors
HIGHLIGHTS
1. Introduction
Acetylcholinesterase (AChE, EC 3.1.1.7) and butyrylcholinesterase (BChE, EC 3.1.1.8) are two related enzymes present in vertebrates and plants. In humans, these enzymes are products of different genes but share about 54% of their amino acid sequenceCitation1. The major difference in their active site are the 14 aromatic amino acid residues in AChE which correspond to 8 aromatic and 6 aliphatic residues in BChECitation2. This enables BChE to hydrolyse larger substrates and ligands than AChE and is accountable for the binding selectivity of cholinesterasesCitation3–6.
AChE has an essential physiological role in the body as it controls the transmission of nerve impulses in the cholinergic synapses of the central and peripheral nervous system by hydrolysis of the neurotransmitter acetylcholine. It also has a role in neuritogenesis, cell adhesion, proliferation and cell interactions, synaptogenesis, dopamine neuronal activation, the formation of amyloid fibres characteristic for Alzheimer's disease, haematopoiesis and thrombopoiesisCitation7–9. The role of BChE is not physiologically essential but it could be assigned to the detoxification of xenobiotics (organophosphates and carbamate pesticides, cocaine, aspirin, succinyldicholine, etc.) and bioactivation of drugs (bambuterol, heroin, etc.)Citation10,Citation11. Also, BChE serves as a co-regulator of cholinergic neurotransmission and is capable of catalysing the hydrolysis of acetylcholineCitation12. It was found that high BChE levels are associated with neuritic plaques and neurofibrillary tangles, the neuropathologic hallmarks of Alzheimer’s disease (AD)Citation13,Citation14. Therefore, both cholinesterases are pharmacologically relevant targets in neurodegenerative disorders, and today’s treatment includes cholinesterase inhibitors like donepezil, galantamine, physostigmine, rivastigmine, ect.Citation15. Many other compounds acting as inhibitors of cholinesterase are therefore considered as potential AD therapeuticsCitation16–18.
Recently we have shown that 4/5-arylethenyloxazoles possess a moderate potency to inhibit AChE and BChECitation19. In this study, we designed new trans-amino-5-arylethenyl-oxazole derivatives where the oxazole molecule has an NH group on one of the substituents. For the synthesis of styryl-oxazoles, the Van Leusen reaction was utilised. Styryl-oxazole that has chlorine as a substituent was then N-alkylated by Buchwald-Hartwig type reaction to give oxazole benzylamines which were tested as cholinesterase inhibitors. All of the styryl-oxazole amines were also photochemically cyclized to give naphthoxazole benzylaminesCitation20. Naphthoxazoles synthesised by this manner were also tested. This gave a wide range of molecules with either oxazole or the naphthoxazole moiety to evaluate their impact on the cholinesterase inhibitory activity.
2. Results and discussion
2.1. Synthesis and photochemistry of novel oxazole benzylamines
Using the reaction of N-alkylation on the previously synthesised trans-chloro-arylethenyloxazole 1Citation20, new trans-amino-5-arylethenyl-oxazole derivatives trans-2–18 were synthesised (Scheme 1) with an aim to add a new functional group at the end of the oxazole derivative that resembles acetylcholine, the substrate of cholinesterase. The Buchwald-Hartwig reactionCitation21 was utilised with two catalysts and the reaction was optimised for best conditions to enhance the yield. Change of base was crucial for the optimisation of this reaction. Sodium tert-butoxide was previously used as a base but the dehalogenation of the starting material was observed. Caesium carbonate improved yield and conversion. Temperature, solvent and catalyst used were independently varied to give the best conversion. The best conditions found are given in Scheme 1. The catalysed N-alkylation reaction is a complex coupling reaction and it gave a vast array of yields. Some of the substrates were optimised to excellent yields, while in the example of others only moderate to low yields were obtained. There is still some room for optimisation in the future with additional catalysts but at this time this was sufficient.
Scheme 1. Synthesis of targeted compounds trans-2–18 by Buchwald-Hartwig reaction.
A vast number of new compounds was synthesised and spectroscopically characterised (See experimental and Supplementary Figures S1–S19). Compounds with a pyridine ring 15 and 16 and that bearing 2-chlorophenyl substituent 13 were synthesised only in trace amounts and these compounds were not further investigated. Pyridine derivatives 15 and 16 could not be obtained probably because of the influence of basicity of the heteroaromatic ring containing nitrogen on the complex reaction steps of Buchwald-Hartwig amination reaction. Only some of the compounds (trans-2, trans-6 and trans-18) were successfully photochemically cyclized into novel polycyclic derivatives 19–21 (Scheme 2). Other starting amines did not react in the electrocyclization reaction and remained unreacted in the reaction solution, some of them as mixture of configurational isomers. The photochemically reactive anilines showed cis-trans photoisomerization during the photoreaction and as the consequence of that gave photocyclization products 19–21 as only cis-configuration is suitable for electrocyclization. Only the cis-isomer of the amine 18 was isolated from the photomixture after the cyclisation reaction, and spectroscopically characterised. It does not mean that the photostationary state is not established in the photomixture in the case of other amines, but without further electrocyclization reaction. During the photocyclization of 2-thienyl (trans-18) derivative, the competitive cleavage of the heteroaromatic moiety occurred resulting in the isolation of the cis-22 and its electrocyclization product 23. The same products were seen also in the 1H NMR spectrum after photoreaction of trans-17. The difference between these two heteroaromatic amines is that trans-17 does not cyclize to the corresponding electrocyclization product and gave these products only in traces. The formation of the same product 23 can be also explained as the consequence of the cleavage of the heteroaromatic moiety from 21 (Scheme 2) and this pathway of formation of 23 cannot be excluded as both pathways can occur as competitive processes in the same photoreaction.
Scheme 2. Photochemical reactivity of amino-5-arylethenyl-oxazoles trans-2,6,18 into naphtho[1,2-d]oxazoles, 19,20 and 21, respectively.
![Scheme 2. Photochemical reactivity of amino-5-arylethenyl-oxazoles trans-2,6,18 into naphtho[1,2-d]oxazoles, 19,20 and 21, respectively.](/cms/asset/a3d58b7f-f1f9-4fab-8e86-903a50253e70/ienz_a_1707197_sch0002_c.jpg)
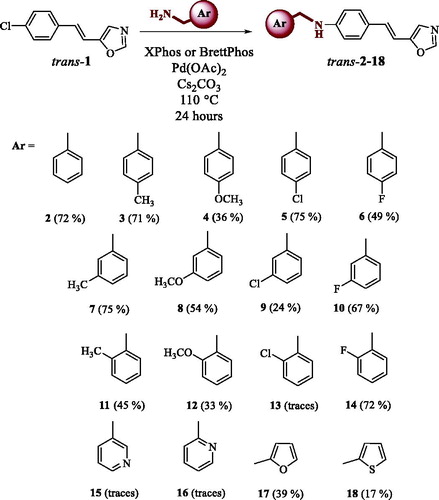
All compounds were completely spectroscopically characterised. On , UV spectra are given as they are used in the determination of wavelength in cyclisation reactions. Absorption maxima of all of the starting trans-isomers of compounds 2–18 are in the area between 340 nm and 350 nm and that is the reason why irradiation at 350 nm wavelength was used for cyclisation.
Figure 1. UV spectra of compounds trans-2 and trans-17 (a), para-substituted synthesised compounds trans-3–6 (b), meta-substituted synthesised compounds trans-7–10 (c) and ortho-substituted synthesised compounds trans-11, trans-12 and trans-14 (d).
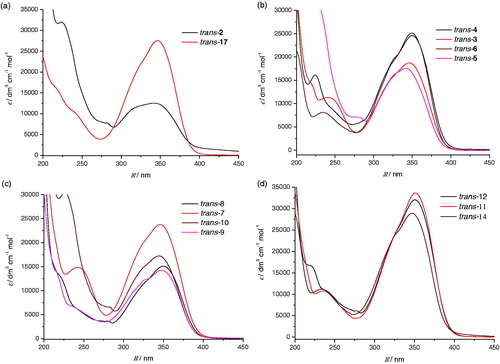
All isolated compounds exhibited in the 1H NMR spectrum a singlet in the range of 7.78–7.81 ppm, which was attributed to the proton on the position 2 of the oxazole ring due to the influence of nitrogen and oxygen found in its immediate vicinity unshaded and shifted to a lower field ( and Supplementary Figures S1–S19). The protons located at position 4 of the oxazole ring showed a singlet in the range of 6.95–6.99 ppm, the ethylenic protons are visible as doublets in the range of 6.99–7.08 ppm with coupling constants between 16 Hz and 17 Hz, characteristic for trans-isomers. For compounds trans-17 and trans-18, the characteristic signals for the furan or thiophene ring are also visible with characteristic coupling constants (See experimental and Supplementary Figure S16). In the spectra of electrocyclization products 19–21, two new doublets with cis coupling constants appeared, characteristic for the central ring of the cyclized naphthoxazole. The structure and purity of the synthesised amines were also confirmed by 13 C NMR and two-dimensional NMR techniques as well as HRMS analyses (See experimental and Supplementary Figures S1–S19).
Figure 2. Partial 1H NMR spectra of starting amines trans-2 and trans-17 and of the photocyclization product 19.
2.2. Inhibition of cholinesterases by novel oxazole benzylamines
The eleven new synthetised trans-amino-5-arylethenyl-oxazole derivatives (trans-2-trans-12, and trans-17) were tested in a wide concentration range as BChE inhibitors to evaluate the inhibitor concentration that inhibits 50% of enzyme activity (IC50), presented in . The most potent BChE inhibitors were compounds trans-12, trans-10 and trans-8 with an IC50 of about 30 µM. BChE had the lowest binding affinity for compound trans-11 which was 5.5-fold lower than the most potent inhibitor trans-12. It is interesting to note that the binding affinity of BChE for compounds trans-12, trans-10 and trans-8 was similar as reported for cholinesterase inhibitors BW284C51, huperzine or rivastigmine (IC50 30 – 54 µM)Citation22.
Table 1. Inhibition of BChE and AChE by tested trans-amino-5-arylethenyl-oxazole derivatives (trans-2-trans-17), naphtho[1,2-d]oxazoles (19–21 and 23), and amino-4/5-arylethenyl-oxazoles (cis-18 and cis-22), expressed as IC50 ± SE.
Generally, although these compounds, with the exception of compound trans-17, systematically differ only by their substituent on the benzyl group and its substituent position (ortho-, meta-, para-), this structural variation cannot be easily related to IC50 values. The most potent inhibitors, compounds trans-12, trans-10 and trans-8 have ortho-methoxy, meta-fluoro and meta-methoxy substituents at the phenyl rings, respectively. Moreover, ortho- (trans-12) and meta- (trans-8) analogues had about a 3 times lower IC50 than their para-methoxy analogue (trans-4). Similarly, the meta-fluoro substituted compound, trans-10, exhibited a 2.5 times lower IC50 than its para-fluoro analogue, trans-6. In case of the methyl substituent, the position of the substituent was not relevant because all three compounds (trans-3, trans-7 and trans-11) had a similar IC50 and were the weakest inhibitors among the tested compounds (). Nevertheless, the para-substitution and methyl-substitution led to inactive compounds, while the ortho/meta-methoxy and the meta-fluoro were active derivatives. It is not surprising that the activity was noticeably different between the most potent (trans-12) and the weakest inhibitor (trans-11) differed in the methoxy and methyl substituent at position 2, respectively.
Eleven trans-amino-5-arylethenyl-oxazole derivatives inhibited maximally 20% of AChE activity (Supplementary Figure S20) and IC50 values were not determined. Since higher concentrations than 100 µM could not be used due to AChE inhibition by solvent DMSOCitation23, the IC50 values for AChE were presumably much higher than 100 µM.
Four of the polycyclic naphtho[1,2-d]oxazoles compounds (19, 20, 21, 23) and two isolated cis-isomers of amino-5-arylethenyl-oxazole derivatives (cis-18, cis-22) were also tested as potential inhibitors of cholinesterases. All compounds, except 23, inhibited both enzymes more than 50% with concentrations in µM range and the evaluated IC50 values are given in . The IC50 for BChE and compound cis-18 was the lowest IC50 value evaluated in this study. cis-18 was about 5 times more potent inhibitor of BChE than amino-5-arylethenyl-oxazoles trans-12, trans-10 and trans-8 (). It is also interesting to note that cis-18 and cis-22 exhibited a higher inhibition effect for BChE than their electrocyclic products 21 and 23, respectively, while polycyclic derivative 20 had about 8-fold higher potency for BChE than its counterpart trans-6 ().
In the case of AChE, it seems that the electrocyclization and trans-cis isomerisation of amino-5-arylethenyl-oxazole derivatives enabled additional interactions in the active site improving the inhibition potency. The electrocyclization of cis-18 and cis-6 resulted in the thienyl-naphtho[1,2-d]oxazole, 21, and p-fluorophenyl-naphtho[1,2-d]oxazole, 20, respectively. Both compounds 21 and 20 are potent inhibitors of AChE. The naphtho[1,2-d]oxazole without aminoalkyl substituent 23 was the poorest inhibitor out of the six tested compounds (). Again, except for 23, there was a slight binding preference of BChE. Therefore, generally, the obtained results indicate that the tested oxazole amines as well as naphtho[1,2-d]oxazole derivatives may be classified as selective inhibitors of BChE.
3. Conclusions
New amino-5-arylethenyl-oxazoles trans-2–18, and cis-18 and cis-22, as well as naphthoxazole benzylamines 19–23 were successfully synthesised using the reaction of N-alkylation on previously synthesised trans-chloro-arylethenyl-oxazole 1. The IC50 values evaluated for BChE classified the tested compounds as moderate BChE inhibitors. Naphtho[1,2-d]oxazoles showed to be more potent AChE inhibitors than the trans-amino-5-arylethenyl-oxazole derivatives, which inhibited 20% of AChE activity at most at the highest concentration possible to test. Due to the selectivity of tested oxazole benzylamines for binding to BChE, the scaffold of these compounds could be used for further development of cholinesterase selective inhibitors.
4. Experimental section
4.1. Chemistry
4.1.1. General procedures
Reactions that required the use of anhydrous, inert atmosphere techniques were carried out under an atmosphere of nitrogen. Petroleum ether, bp 40–60 °C, was used. Solvents were purified by distillation. Column chromatography was carried out on columns with silica gel (Fluka 0.063–0.2 nm and Fluka 60 Å, technical grade). TLC was carried out using plates coated with silica gel (0.2 mm, 0.5 mm, 1.0 mm, Kieselgel 60 F254). Organic layers were routinely dried with anhydrous MgSO4 and evaporated using a rotary evaporator. 1H and 13 C NMR spectra were recorded on a spectrometer at 300 and 600 MHz. All NMR spectra were measured in CDCl3 using tetramethylsilane as reference. The assignment of signals was based on 2 D-CH correlation and 2 D-HH-COSY experiments. The following abbreviations are used: s, singlet; d, doublet; t, triplet; q, quartette, dd, doublet of doublets; m, multiplet and br, broad. UV spectra were measured on a UV/VIS spectrophotometer. IR spectra were recorded on a FTIR. Mass spectra were obtained on a GC-MS system. Melting points were obtained using a microscope equipped apparatus and are uncorrected. HRMS analyses were carried out on a mass spectrometer. The LC-MS system consisted of an Agilent 1290 LC coupled with an Agilent 6550 iFunnel quadrupole time-of-flight mass spectrometer (Agilent Technologies, Santa Clara, CA, USA). The LC-MS system equipped with a quaternary gradient pump, temperature-controlled column compartment, refrigerated autosampler component, diode array detector (DAD) and MS with electrospray ionisation were used for the identification. Chromatographic separations were performed using Acquity UPLC BEH C18 column, 50 × 2.1 mm, 1.7 µm (Agilent Technologies, Santa Clara, CA, USA). Gradient elution with mobile phase containing solvent A (0.1% formic acid) and solvent B (acetonitrile) was used. The mobile-phase flow rate was 0.4 mL/min and the column temperature was maintained at 50 ± 1 °C. Substances were analysed in positive electrospray ionisation mode. Nitrogen was a nebuliser and curtain gas. The capillary voltage was 3500 V. Gas temperature was 200 °C, gas flow was 14 L/min, nebuliser was 35 psi, sheath gas temperature was 350 °C and sheath gas flow was 11 L/min. Data acquisition and processing were performed on a MassHunter Data Acquisition for Q-TOF B.06.01 (B6157) software (Agilent Technologies). Irradiation experiments were performed in a closed quartz vessel in toluene solution in a photochemical reactor equipped with 360 nm lamps. The solvents were removed on the rotatory evaporator under reduced pressure in a ventilated hood.
4.1.2. Synthesis of (E)-5–(4-chlorostyryl)oxazole (trans-1)
Compound trans-1 was synthesised from (E)-3–(4-chlorophenyl)acryladehyde (6.00 mmol, 1 eq) by Van Leusen reaction,Citation24 with tosylmethylisocyanide (TosMIC) (5.76 mmol, 0.96 eq) reagent and potassium carbonate as base (5.79 mmol, 0.96 eq) in methanol. (E)-5–(4-chlorostyryl)oxazole (trans-1) of compoundCitation17 was isolated as yellow powder (0.950 g; 77.24%): mp 78–81 °C; Rf (PE/E, 20%) = 0.59; UV (EtOH) λmax/nm (ε/dm3mol−1 cm−1): 299 (27977), 312 (29351), 326 (20895); IR νmax/cm−1 (NaCl): 1697, 1610, 1491, 1089; 1H NMR (CDCl3, 600 MHz): δ/ppm 7.84 (s, 1H, H-2), 7.40 (dd, Jar= 8.5 Hz, Jar= 6.6 Hz, 2H, H-ar), 7.33 (dd, Jar= 8.5 Hz, Jar= 6.6 Hz, 2H, H-ar), 7.08 (s, 1H, H-4), 7.04 (d, Jet = 16.2 Hz, 1H, H-et), 6.88 (d, Jet = 16.2 Hz, 1H, H-et); 13 C NMR (CDCl3, 150 MHz): δ/ppm 150.63 (d), 149.23 (s), 147.54 (s), 137.03 (s), 132.58 (s), 129.92 (d), 128.33 (d), 128.12 (d), 127.52 (d), 125.52 (d), 122.11 (d), 112.48 (d), 108.52 (d), 46.88 (t); 13 C NMR (CDCl3, 150 MHz): δ/ppm 149.9 (d, C-2), 149.7 (s), 134.2 (s), 133.5 (s), 128.5 (d), 128.4 (d), 127.2 (d), 124.0 (d), 112.9 (d); MS m/z (EI) = 205 (100, M+); HRMS(Q-TOF) for C11H8ClNO: (M + H)+calcd = 206.0294, (M + H)+found = 206.0369.
4.1.3. Synthesis of new (E)-N-benzyl-4–(2-(oxazol-5-yl)vinyl)anilines
4.1.3.1. Synthesis with BrettPhos
BrettPhos (0.024 mmol 0.1 eq), Pd(OAc)2 (0.012 mmol, 0.05 eq) were suspended in 2 mL of dioxane with 0.01 mL water and heated to 120 °C. (E)-5–(4-chlorostyryl)oxazole (0.243 mmol, 1 eq) Cs2CO3 (0.365 mmol, 1,5 eq) and different benzyl-amines were added (0.486 mmol, 2 eq). The reaction mixture was heated in a pressure tube to 110 °C for 24 h. Solvent was evaporated under pressure and the compound purified by column chromatography on silicagel using petroleumether/dichloromethane (20–100%) as eluent.
4.1.3.2. Synthesis with XPhos
(E)-5–(4-chlorostyryl)oxazole (0.243 mmol, 1 eq), XPhos (0.049 mmol, 0.2 eq), Pd(OAc)2 (0.012 mmol, 0.05 eq) and Cs2CO3 (0.365 mmol, 1.5 eq) were dissolved in 2 mL of dioxane and benzyl-amines (0.486 mmol, 2 eq) were added. The reaction mixture was purged with argon and heated to 110 °C in a pressure tube for 24 h. Solvent was evaporated under pressure and the compound purified by column chromatography on silica gel using petroleumether/dichloromethane (20–100%) as eluent.
(E)-N-benzyl-4–(2-(oxazol-5-yl)vinyl)aniline (trans-2) was isolated (0.097 g (72.22%)) as yellow powder: mp 112–118 °C; Rf (DCM, 100%) = 0.32; UV (EtOH) λmax/nm (ε/dm3mol−1 cm−1): 224 (32046), 319 (Sh, 11687), 343 (12469); IR νmax/cm−1 (NaCl): 3421, 2924, 1748, 1607, 1524, 1490, 1453, 955, 817, 744, 638; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.79 (s, 1H, H-Ox2), 7.37–7.28 (m, 8H, H-Ar, H-NH), 7.00 (d, 1H, JEt1,Et2 = 16.07 Hz, H-Et1), 6.97 (s, 1H, H-Ox4), 6.69 (d, 1H, JEt2,Et1 = 16.42 Hz, H-Et2), 6.63 (d, 2H, JAr2a,Ar1a = 8.74 Hz, H-Ar2a), 4.37 (s, 2H, H-CH2); 13 C NMR (CDCl3, 75 MHz): δ/ppm 149.22 (s), 147.88 (s), 138.46 (s), 130.04 (d), 128.21 (d), 127.51 (d), 126.97 (d), 126.88 (d), 125.27 (s), 122.02 (d), 112.38 (d), 108.33 (d), 47.59 (t); HRMS(Q-TOF) for C18H16N2O: (M + H)+calcd = 277.1335, (M + H)+found = 277.1325.
(E)-N-(4-methylbenzyl)-4–(2-(oxazol-5-yl)vinyl)aniline (trans-3) was isolated (0.099 g (70.92%)) as yellow powder mp 128–131 °C; Rf (DCM, 100%) = 0.43; UV (EtOH) λmax/nm (ε/dm3mol−1 cm−1): 242 (11249), 325 (Sh,15804), 346 (18719); IR νmax/cm−1 (NaCl): 3416, 2925, 1612, 1577, 1558, 1519, 1476, 953, 820, 637; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.81 (s, 1H,H-Ox2), 7.32 (d, 2H, JAr1aAar2a = 8.58 Hz, H-Ar1a), 7.27 (d, 2H, JAr3b,Ar4b = 7.82 Hz, H-Ar3b), 7.18 (d, 2H, JAr4b,Ar3b = 7.82 Hz, H-Ar4), 7.02 (d, 1H, JEt1,Et2 = 16.65 Hz, H-Et1), 6.98 (s, 1H, H-Ox4), 6.70 (d, 1H, JEt2,Et1 = 16.40 Hz, H-Et2), 6.63 (d, 2H, JAr2a,Ar1a = 8.58 Hz, H-Ar2a), 4.34 (s, 2H, H-CH2), 4.21 (s, 1H, H-NH), 2.36 (s, 3H, H-CH3); 13 C NMR (CDCl3, 75 MHz): δ/ppm 147.95 (s), 136.56 (s), 135.38 (s), 130.10 (d), 128.88 (d), 127.51 (d), 126.93 (d), 125.16 (s), 122.01 (d), 112.36 (d), 108.25 (d), 47.34 (q), 20.58 (t); HRMS(Q-TOF) for C19H18N2O: (M + H)+calcd = 291.1492, (M + H)+found = 291.1487.
(E)-N-(4-methoxybenzyl)-4–(2-(oxazol-5-yl)vinyl)aniline (trans-4) was isolated (0.054 g (36.29%)) as yellow powder mp 153–159 °C; Rf (DCM, 100%) = 0.38; UV (EtOH) λmax/nm (ε/dm3mol−1 cm−1): 224 (16148), 328 (Sh, 19991), 350 (24578); IR νmax/cm−1 (NaCl): 3376, 2925, 1601, 1514, 1468, 952, 822, 638; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7,81 (s, 1H, H-Ox2), 7.34–7.29 (m, 4H, H-Ar), 7.02 (d, 1H, JEt1,Et2 = 16.51 Hz, H-Et1), 6.98 (s, 1H, H-Ox4), 6.90 (d, 2H, JAr1a,Ar2a = 8.77 Hz, H-Ar1a), 6.70 (d, 1H, JEt2,Et1 = 16.26 Hz, H-Et2), 0.63 (d, 2H, JAr2a,Ar1a = 8.51 Hz, H-Ar2a), 4.31 (s, 1H, H-CH2), 4.17 (s, 1H, H-NH), 3.82 (s, 3H, H-OCH3); 13 C NMR (CDCl3, 75 MHz): δ/ppm 158.50 (s), 147.93 (s), 130.43 (s), 130.07 (d), 128.24 (d), 127.17 (s), 125.17 (s), 121.99 (d), 113.62 (d), 112.36 (d), 108.26 (d), 54.80 (q), 47.06 (t); HRMS(Q-TOF) for C19H18N2O2: (M + H)+calcd = 307.1441, (M + H)+found = 307.1439.
(E)-N-(4-chlorobenzyl)-4–(2-(oxazol-5-yl)vinyl)aniline (trans-5) was isolated (0.114 g (75.49%)) as yellow powder mp 120–125 °C; Rf (DCM, 100%) = 0.43; UV (EtOH) λmax/nm (ε/dm3mol−1 cm−1): 242 (14854), 325 (Sh, 20225), 346 (23776); IR νmax/cm−1 (NaCl): 3325, 2924, 1605, 1520, 1473, 953, 818, 638; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.81 (s, 1H, H-Ox2), 7.33–7.28 (m, 6H, H-Ar), 7.01 (d, 1H, JEt1,Et2 = 16.15 Hz, H-Et1), 6.99 (s, 1H, H-Ox4), 6.70 (d, 1H, JEt2,Et1 = 16.15 Hz, H-Et2), 6.60 (d, 2H, JAr2a,Ar1a = 8.39 Hz, H-Ar2), 4.36 (s, 2H, H-CH2), 4.28 (s, 1H, H-NH); 13 C NMR (CDCl3, 75 MHz): δ/ppm 150.63 (d), 149.23 (s), 147.54 (s), 137.03 (s), 132.58 (s), 129.92 (d), 128.33 (d), 128.12 (d), 127.52 (d), 125.52 (s), 122.11 (d), 112.48 (d), 108.52 (d), 46.88 (t); HRMS(Q-TOF) for C18H15ClN2O: (M + H)+calcd = 311.0946, (M + H)+found = 311.0938.
(E)-N-(4-fluorobenzyl)-4–(2-(oxazol-5-yl)vinyl)aniline (trans-6) was isolated (0.070 g (48.95%)) as yellow powder mp 137–142 °C; Rf (DCM, 100%) = 0.43; UV (EtOH) λmax/nm (ε/dm3mol−1 cm−1): 234 (8159), 326 (Sh, 19305), 349 (25076); IR νmax/cm−1 (NaCl): 3325, 2925, 1606, 1520, 1509, 1470, 953, 818, 638; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.81 (s, 1H, H-Ox2), 7.36 (d, 2H, JAr1a,Ar2a = 8.56 Hz, H-Ar1a), 7.34 (d, 2H, JAr4b,Ar3b = 8.83 Hz, H-Ar4b), 7.05 (d, 1H, JEt1,Et2 = 16.86 Hz, H-Et1), 7.06 (d, 2H, JAr3b,Ar4b = 8.83 Hz, H-Ar3), 6.99 (s, 1H, H-Ox4), 6.71 (d, 1H, JEt2,Et1 = 16.32 Hz, H-Et2), 6.62 (d, 2H, JAr2a,Ar1a = 8.56 Hz, H-Ar2), 4.35 (s, 2H, H-CH2), 4.24 (s, 1H, H-NH); 13 C NMR (CDCl3, 75 MHz): δ/ppm 162.44 (s), 150.67 (s), 149.23 (s), 147.66 (s), 134.16 (s), 129.95 (d), 128.48 (d), 128.42 (d), 127.52 (d), 125.43 (d), 122.08 (d), 115.11 (d), 114.97 (d), 112.42 (d), 108.46 (d), 46.88 t); HRMS(Q-TOF) for C18H15FN2O: (M + H)+calcd = 295.1241, (M + H)+found = 295.1234.
(E)-N-(3-methylbenzyl)-4–(2-(oxazol-5-yl)vinyl)aniline (trans-7) was isolated (0.105 g (75.18%)) as yellow powder mp 75–80 °C; Rf (DCM, 100%) = 0.31; UV (EtOH) λmax/nm (ε/dm3mol−1 cm−1): 242 (14732), 325 (Sh, 19824), 346 (23783); IR νmax/cm−1 (NaCl): 3413, 3325, 2921, 1605, 1520, 1489, 1469, 953, 817, 638; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.81 (s, 1H, H-Ox2), 7.34–7.13 (m, 6H, H-Ar), 7.08 (d, 1H, JEt1,Et2 = 16.54 Hz, H-Et1), 6.98 (s, 1H, H-Ox4), 6.70 (d, 1H, JEt2,Et1 = 16.54 Hz, H-Et2), 6.63 (d, 2H, JAr2a,Ar1a = 8.14 Hz, H-Ar2a), 4.34 (s, 2H, H-CH2), 4.23 (s, 1H, H-NH), 2.37 (s, 3H, H-CH3); 13 C NMR (CDCl3, 75 MHz) δ/ppm: 151.23 (d, C-Ox2), 149,73 (s), 148,48 (s), 138, 92 (s), 138,42 (s), 130,57 (d, C-Et1), 128,63 (d, C-Ar), 128,23 (d, C-Ar), 128,16 (d, C-Ar), 128,04 (d, C-Ar), 125,65 (s), 124,53 (d, C-Ar), 122,52 (d, C-Ox4), 112,86 (d, C-Ar1a), 108,75 (d, C-Et2), 48,09 (t, C-CH2), 21,45 (q, C-CH3); HRMS(Q-TOF) for C19H18N2O: (M + H)+calcd = 291.1492, (M + H)+found = 291.1485.
(E)-N-(3-methoxybenzyl)-4–(2-(oksazol-5-yl)vinyl)aniline (trans-8) was isolated (0.080 g (53.76%)) as yellow oil: Rf (DCM, 100%) = 0.27; UV (EtOH) λmax/nm (ε/dm3mol−1 cm−1): 219 (Sh, 13312), 326 (Sh, 11808), 349 (15095); IR νmax/cm−1 (NaCl): 3307, 2927, 1605, 1522, 1489, 1465, 953, 817, 639; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.81 (s, 1H, H-Ox2), 7.33 (d, 2H, JAr1a,Ar2a = 8.56 Hz, H-Ar1), 7.02 (d, 1H, JEt1,Et2=16.51 Hz, H-Et1), 6.98 (s, 1H, H-Ox4), 6.97–6.90 (m, 1H, H-Ar5b), 6.93 (s, 1H, H-Ar2b), 6.84 (d, 2H, JAr4b,6b,Ar5b = 8.00 Hz, H-Ar4b, H-Ar6b), 6.70 (d, 1H, JEt2,Et1 = 16.38 Hz, H-Et2), 6.63 (d, 2H, JAr2a,Ar1a = 8.38 Hz, H-Ar2), 4.36 (s, 2H, H-CH2), 4.25 (s, 1H, H-NH), 3.82 (s, 3H, H-OCH3); 13 C NMR (CDCl3, 75 MHz) δ/ppm:133.05 (d), 131.30 (d), 130.05 (d), 129.23 (d), 127.50 (d), 125.47 (d), 125.26 (s), 119.72 (d), 119.13 (d), 112.56 (d), 112.40 (d), 112.19 (d), 108.31 (d), 47.55 (t); HRMS (Q-TOF) for C19H18N2O2: (M + H)+calcd = 307.1441, (M + H)+found = 307.1434.
(E)-N-(3-chlorobenzyl)-4–(2-(oxazol-5-yl)vinyl)aniline (trans-9) was isolated (0.036 g (23.83%)) as yellow oil: Rf (DCM, 100%) = 0.27; UV (EtOH) λmax/nm (ε/dm3mol−1 cm−1): 216 (Sh, 13861), 234 (6874), 329 (Sh, 12349), 347 (14260); IR νmax/cm−1 (NaCl): 3325, 2925, 1606, 1520, 1473, 953, 818, 638; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.81 (s, 1H, H-Ox2), 7.41–7.31 (m, 6H, H-Ar), 7.02 (d, 1H, JEt1,Et2 = 16.22 Hz, H-Et1), 6.99 (s, 1H, H-Ox4), 6.71 (d, 1H, JEt2,Et1 = 16.22 Hz, H-Et2), 6.62 (d, 2H, JAr2a,Ar1a = 8.48 Hz, H-Ar2a), 4.38 (s, 2H, H-CH2), 4,33 (s, 1H, H-NH); 13 C NMR (CDCl3, 75 MHz) δ/ppm: 158.5 (s), 147.9 (s), 130.4 (s), 130.1 (d), 128.5 (d), 128.4 (s), 128.3 (d), 128.2 (2d), 127.5 (d), 125.2 (s), 122.0 (d), 113.6 (d), 112.5 (d), 112.4 (2d), 108.2 (d), 47.1 (t); HRMS (Q-TOF) for C18H15ClN2O: (M + H)+calcd = 311.0946, (M + H)+found = 311.0939.
(E)-N-(3-fluorobenzyl)-4–(2-(oxazol-5-yl)vinyl)aniline (trans-10) was isolated (0.096 g (67.13%)) as yellow powder mp 92–95 °C;Rf (DCM, 100%) = 0.32; UV (EtOH) λmax/nm (ε/dm3mol−1 cm−1): 226 (30378), 325 (Sh, 14962), 345 (17263); IR νmax/cm−1 (NaCl): 3419, 2926, 1606, 1520, 1487, 1448, 953, 817, 638; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.81 (s, 1H, H-Ox2), 7.32 (d, 2H, JAr1a,Ar2a = 8.19 Hz, H-Ar1), 7.18–6.99 (m, 4H, H-Ar), 7.02 (d, 1H, JEt1,Et2 = 16.38 Hz, H-Et1), 6.99 (s, 1H, H-Ox4), 6.71 (d, 1H, JEt2,Et1 = 16.38 Hz, H-Et2), 6.61 (d, 2H, JAr2a,Ar1a = 8.19 Hz, H-Ar2), 4.39 (s, 2H, H-CH2), 4,32 (s, 1H, H-NH). 13 C NMR (CDCl3, 75 MHz) δ/ppm; 163.5 (s), 161.8 (s), 147.5 (s), 141.3 (s), 129.9 (d), 127.5 (2d), 125.5 (s), 122.2 (d), 122.1 (d), 113.8 (d), 113.7 (d), 113.6 (d), 113.5 (d), 112.4 (2d), 108.5 (d), 47.0 (t); HRMS(Q-TOF) for C18H15FN2O: (M + H)+calcd = 295.1241, (M + H)+found = 295.1237.
(E)-N-(2-methylbenzyl)-4–(2-(oxazol-5-yl)vinyl)aniline (trans-11) was isolated (0.064 g (45.35%)) as yellow oil: Rf (DCM, 100%) = 0.30; UV (EtOH) λmax/nm (ε/dm3mol−1 cm−1): 236 (10757), 329 (Sh, 25544), 350 (33604); IR νmax/cm−1 (NaCl): 3413, 3324, 2923, 1604, 1520, 1495, 1462, 953, 817, 638; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.81 (s, 1H, H-Ox2), 7.37–7.31 (m, 1H, H-Ar3), 7.35 (d, 2H, JAr1a,Ar2a = 8,81 Hz, H-Ar1a), 7.26–7.18 (m, 1H, H-Ar4b), 7.24 (d, 2H, JAr2b,Ar3b = JAr5b,Ar4b = 1,70 Hz, H-Ar2, H-Ar5), 7.04 (d, 1H, JEt1,Et2 = 16.48 Hz, H-Et1), 6.99 (s, 1H, H-Ox4), 6.72 (d, 1H, JEt2,Et1 = 16.48 Hz, H-Et2), 6.64 (d, 2H, JAr2a,Ar1a = 8.81 Hz, H-Ar2a), 4.33 (s, 2H, H-CH2), 4,10 (s, 1H, H-NH), 2,40 (s, 3H, H-CH3); 13 C NMR (CDCl3, 75 MHz) δ/ppm 150.72 (s), 149.19 (s), 148.03 (s), 136.08 (s), 135.80 (s), 130.09 (d), 130.01 (d), 127.68 (d), 127.54 (d), 127.09 (d), 125.72 (d), 125.17 (d), 122.00 (d), 112.22 (d), 108.28 (d), 45.67 (q), 18.41 (t); HRMS(Q-TOF) for C19H18N2O: (M + H)+calcd = 291.1492, (M + H)+found = 291.1481.
(E)-N-(2-methoxybenzyl)-4–(2-(oxazol-5-yl)vinyl)aniline (trans-12) was isolated (0.050 g (33.58%)) as yellow oil: Rf (DCM, 100%) = 0.18; UV (EtOH) λmax/nm (ε/dm3mol−1 cm−1): 221 (16630), 327 (Sh, 24690), 350 (32084); IR νmax/cm−1 (NaCl): 3413, 2930, 1605, 1521, 1490, 1463, 953, 817, 638; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.78 (s, 1H, H-Ox2), 7.32–7.23 (m, 1H, H-Ar3b), 7.29 (d, 2H, JAr1aAar2a = 8.45 Hz, H-Ar1a), 6.99 (d, 1H, JEt1,Et2 = 16.18 Hz, H-Et1), 6.96–6.88 (m, 3H, H-Ar2b,3b,4b), 6.95 (s, 1H, H-Ox4), 6.67 (d, 1H, JEt2,Et1 = 16.18 Hz, H-Et2), 6.63 (d, 2H, JAr2a,Ar1a = 8.45 Hz, H-Ar2a), 4.36 (s, 2H, H-CH2), 4.33 (s, 1H, H-NH), 3.87 (s, 3H, H-OCH3); 13 C NMR (CDCl3, 75 MHz) δ/ppm 149.14 (s), 148.24 (s), 130.16 (d), 128.31 (d), 127.98 (d), 127.46 (d), 124.96 (d), 121.90 (d), 120.06 (d), 112.51 (d), 109.84 (d), 108.08 (d), 54.83 (q), 42.75 (t)); HRMS (Q-TOF) for C19H18N2O2: (M + H)+calcd = 307.1441, (M + H)+found = 307.1444.
(E)-N-(2-fluorobenzyl)-4–(2-(oxazol-5-yl)vinyl)aniline (trans-14) was isolated (0.102 g (71.52%)) as yellow powder mp 119–122 °C; Rf (DCM, 100%) = 0.26; UV (EtOH) λmax/nm (ε/dm3mol−1 cm−1): 233 (11204), 326 (Sh, 24144), 346 (28843); IR νmax/cm−1 (NaCl): 3398, 2926, 1605, 1520, 1487, 1455, 952, 815, 758, 638; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.81 (s, 1H, H-Ox2), 7.40–7.25 (m, 1H, H-Ar3b), 7.34 (d, 2H, JAr1a,Ar2a = 8.15 Hz, H-Ar1a), 7.14–6.97 (m, 3H, H-Ar2b,3b,4b), 7.02 (d, 1H, JEt1,Et2 = 16.30 Hz, H-Et1), 6.99 (s, 1H, H-Ox4), 6.70 (d, 1H, JEt2,Et1 = 16.30 Hz, H-Et2), 6.64 (d, 2H, JAr2a,Ar1a = 8.15 Hz, H-Ar2a), 4.45 (s, 2H, H-CH2), 4.29 (s, 1H, H-NH); 13 C NMR (CDCl3, 75 MHz) δ/ppm 149.37 (s), 147.58 (s), 129.98 (d), 131.84 (d), 128.40 (d), 127.51 (d), 125.46 (d), 123.74 (d), 122.07 (d), 114.88 (d), 112.47 (d), 108.46 (d), 41.17 (t); HRMS(Q-TOF) for C18H15FN2O: (M + H)+calcd = 295.1241, (M + H)+found = 295.1236.
(E)-N-(furan-2-ylmethyl)-4–(2-(oxazol-5-yl)vinyl)aniline (trans-17) was isolated (0.050 g (38.63%)) as yellow powder mp 97–102 °C; Rf (DCM, 100%) = 0.22; UV (EtOH) λmax/nm (ε/dm3mol−1 cm−1): 215 (Sh, 16540), 241 (Sh, 10289), 325 (Sh, 22270), 346 (27578); IR νmax/cm−1 (NaCl): 3407, 2926, 1609, 1519, 1492, 964, 951, 820, 738, 637; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.79 (s, 1H, H-Ox2), 7.37 (d, 1H, JFur5,Fur4 = 1.80 Hz, H-Fur5), 7.32 (d, 2H, JAr1,Ar2 = 8.48 Hz, H-Ar1), 7.00 (d, 1H, JEt1,Et2 = 16.60 Hz, H-Et1), 6.97 (s, 1H, H-Ox4), 6.69 (d, 1H, JEt2,Et1 = 16.60 Hz, H-Et2), 6.65 (d, 2H, JAr2,Ar1 = 8.48 Hz, H-Ar2), 6.32 (dd, 1H, JFur4,Fur5 = 1.81 Hz, JFur4,Fur3 = 3.16 Hz, H-Fur4), 6,24 (d, 1H, JFur3,Fur4 = 3.16 Hz, H-Fur3), 4.35 (s, 2H, H-CH2), 4.21 (s, 1H, H-NH); 13 C NMR (CDCl3, 75 MHz) δ/ppm 151.52 (s), 150.64 (s), 149.23 (s), 147.37 (s), 141.55 (d), 129.97 (d), 127.46 (d), 125.66 (d), 122.10 (d), 112.63 (d), 109.87 (d), 108.55 (d), 106.65 (d), 40.69 (t).
(E)-4–(2-(oxazol-5-yl)vinyl)-N-(thiophen-2-ylmethyl)aniline (trans-18) was isolated (11.1 mg (17.10%)) as yellow oil; Rf (DCM) = 0.42; UV (EtOH) λmax/nm: 274 (12817), 291 (13098); IR νmax/cm−1 (NaCl): 3359, 2811, 1708, 1665; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.62 (s, 1H, H-Ox2), 7.20–7.09 (m, 4H), 7.07 (d, 1H, J = 5.1 Hz), 6.98 (d, 1H, J = 3.2 Hz), 6.79 (dd, 1H, J = 5.1, 3.2 Hz), 6.63 (s, 1H), 6.55 (d, 1H, J = 12.3 Hz), 6.23 (d, 1H, J = 12.3 Hz), 4.62 (s, 2H), 4.31 (s, 1H); MS m/z (%, fragment) (EI): 282 (M+, 100); HRMS (m/z): [M + H]+ calcd for 283.0827; found for 283.0848.
4.1.4. Photochemistry of the new (E)-N-benzyl-4(2-(oxazol-5-yl)vinyl)anilines
A quartz vessel was charged with (E)-N-aryl-4(2-(oxazol-5-yl)vinyl)anilines (trans-2–8, trans-10–12 and trans-17) in 50 ml of toluene (0.003 mmol/mL) with the addition of a small amount of iodine and irradiated at 350 nm in the Rayonet reactor for 2 h, 3 h, 4 h and 8 h. The conversion was followed by thin-layer chromatography. After irradiation, the solvent was removed in vacuum and the residue chromatographed on a silica gel column using dichloromethane as eluent. In the first fractions, the photoelectrocyclized products 19–21 were isolated, and in the last fractions the unreacted started amines and photolyzed products cis-18, cis-22 and 23.
N-benzylnaphtho[1,2-d]oxazol-8-amine (19) was isolated (25.0 mg (37.40%)) as yellow oil; Rf (DCM) = 0.53; UV (EtOH) λmax/nm: 247 (15132), 299 (5098), 344 (4899); IR νmax/cm−1 (NaCl): 3396, 2922, 2849, 1735, 1635, 1535; 1H NMR (CDCl3, 300 MHz) δ/ppm: 8.15 (s, 1H, H-Ox2), 7.75 (d, 1H, J = 8.8 Hz), 7.66 (d, 1H, J = 8.8 Hz), 7.52 (d, 1H, J = 2.4 Hz), 7.43 (t, 3H, J = 7.2 Hz), 7.35 (t, 2H, J = 7.3 Hz), 7.31–7.28 (m, 1H), 6.94 (dd, 1H, J = 8.7; 2.5 Hz), 4.52 (s, 2H); 13 C NMR (CDCl3, 75 MHz) δ/ppm: 149.7 (s), 146.2 (s), 138.2 (s), 129.3 (2d), 128.2 (d), 128.7 (s), 127.7 (s), 127.3 (2d), 126.9 (d), 126.4 (d), 125.9 (d), 124.2 (s), 116.1 (d); 105.9 (d), 98.8 (d), 47.8 (t); MS m/z (%, fragment) (EI): 274 (M+, 100); HRMS (m/z): [M + H]+ calcd for 275.1106; found for 275.1119.
N-(4-fluorobenzyl)naphtho[1,2-d]oxazol-8-amine (20) was isolated (18.6 mg (26.4%)) as yellow oil; Rf (DCM) = 0.55; UV (EtOH) λmax/nm: 247 (5711), 310 (4677), 339 (2013); IR νmax/cm−1 (NaCl): 3414, 2922, 1637, 1601, 1533, 1508, 1255, 1222; 1H NMR (CDCl3, 300 MHz) δ/ppm: 8.11 (s, 1H, H-Ox2), 7.75 (d, 1H, J = 8.5 Hz), 7.66 (d, 1H, J = 8.8 Hz), 7.49 (d, 1H, J = 2.5 Hz), 7.43 (d, 2H, J = 8.3 Hz), 7.42–7.39 (m, 2H), 7.04 (d, 2H, J = 8.1 Hz), 6.94 (dd, 1H, J = 8.8; 2.5 Hz), 4.50 (s, 2H), 4.39 (s, 1H, NH); 13 C NMR (CDCl3, 75 MHz) δ/ppm: 163.7 (s), 150.8 (s), 148.2 (s), 146.9 (s), 134.5 (s), 129.8 (d), 129.3 (d), 129.2 (d), 126.4 (d), 124.7 (s), 116.5 (d), 115.7 (d), 115.4 (d), 106.6 (d), 99.3 (d); 47.5 (t); MS m/z (%, fragment) (EI): 292 (M+, 100); HRMS (m/z): [M + H]+ calcd for 293.1012; found for 293.1004.
N-(thiophen-2-ylmethyl)naphtho[1,2-d]oxazol-8-amine (21) was isolated (8.5 mg (13.0%)) as yellow oil; Rf (DCM) = 0.67; UV (EtOH) λmax/nm: 231 (19138); IR νmax/cm−1 (NaCl): 3393, 2917, 2310, 1731, 1539, 1460; 1H NMR (CDCl3, 300 MHz) δ/ppm: 8.16 (s, 1H, H-Ox2), 7.76 (d, 1H, J = 8.8 Hz), 7.67 (d, 1H, J = 8.8 Hz), 7.56 (d, 1H, J = 2.3 Hz), 7.44 (d, 1H, J = 8.9 Hz), 7.24 (d, 1H, J = 5.0 Hz), 7.10 (d, 1H, J = 3.5 Hz), 6.99 (dd, 1H, J = 5.0, 3.5 Hz), 6.95 (dd, 1H, J = 8.8; 2.5 Hz), 4.71 (s, 2H), 4.40 (s, 1H); 13 C NMR (CDCl3, 75 MHz) δ/ppm: 153.7 (s), 149.3 (s), 136.9 (s), 129.9 (d), 128.7 (d), 128.1 (d), 127.5 (s), 127.0 (d), 126.6 (d), 125.3 (d), 124.4 (s), 116.4 (d), 106.2 (d), 99.5 (d), 50.1 (t); MS m/z (%, fragment) (EI): 280 (M+, 100); HRMS (m/z): [M + H]+ calcd for 281.0670; found for 281.0691.
(Z)-4–(2-(oxazol-5-yl)vinyl)-N-(thiophen-2-ylmethyl)aniline (cis-18) was isolated (11.1 mg (17.10%)) as yellow oil; Rf (DCM) = 0.42; UV (EtOH) λmax/nm: 274 (12817), 291 (13098); IR νmax/cm−1 (NaCl): 3359, 2811, 1708, 1665; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.62 (s, 1H, H-Ox2), 7.20–7.09 (m, 4H), 7.07 (d, 1H, J = 5.1 Hz), 6.98 (d, 1H, J = 3.2 Hz), 6.79 (dd, 1H, J = 5.1, 3.2 Hz), 6.63 (s, 1H), 6.55 (d, 1H, J = 12.3 Hz), 6.23 (d, 1H, J = 12.3 Hz), 4.62 (s, 2H), 4.31 (s, 1H); MS m/z (%, fragment) (EI): 282 (M+, 100); HRMS (m/z): [M + H]+ calcd for 283.0827; found for 283.0848.
(Z)-4–(2-(oxazol-5-yl)vinyl)aniline (cis-22) was isolated (4.5 mg (7.2%)) as yellow oil; Rf (DCM) = 0.43; UV (EtOH) λmax/nm: 259 (13474), 265 (12817), 272 (13409); IR νmax/cm−1 (NaCl): 3412, 2917, 2844, 1737, 1632, 1462; UV (EtOH) λmax/nm: 259 (13474), 265 (12817), 272 (13409); 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.73 (s, 1H, H-Ox2), 7.40–7.33 (m, 4H), 7.32 (s, 2H), 6.91 (s, 1H), 6.67 (d, 1H, J = 12.5 Hz), 6.39 (d, 1H, J = 12.5 Hz); MS m/z (%, fragment) (EI): 186 (M+, 100); HRMS (m/z): [M + H]+ calcd for 186.0793; found for 186.0782.
Naphtho[1,2-d]oxazol-8-amine (23) was isolated (3.9 mg (6.4%)) as yellow oil; Rf (PE/DCM = 1:1) = 0.23; UV (EtOH) λmax/nm: 225 (18143), 353 (4881), 400 (4012); IR νmax/cm−1 (NaCl): 3414, 2922, 1637, 1601, 1533, 1508, 1255, 1222; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.37 (s, 1H, H-Ox2), 7.34 (d, 1H, J = 8.9 Hz), 6.93 (s, 1H), 6.86 (d, 1H, J = 10.0 Hz), 6.72 (d, 1H, J = 8.9 Hz), 6.65 (d, 1H, J = 10.0 Hz), 3.01 (s, 2H, NH2); MS m/z (%, fragment) (EI): 184 (M+, 100); HRMS (m/z): [M + H]+ calcd for 185.0637; found for 185.0644.
4.2. Reversible inhibition of cholinesterases by novel oxazole benzylamine compounds
Inhibition potency of novel compounds was evaluated for recombinant human AChE (prepared as described earlierCitation25 and kindly donated by Prof Palmer Taylor, Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California at San Diego, La Jolla, USA) and BChE isolated from human plasma (kindly donated by late Dr Douglas Cerasoli and Dr David Lenz, USAMRICD, Edgewood, MD). The inhibition mixture contained a 0.1 M phosphate buffer, pH 7.4, enzyme, tested compound, and reagent, 5,5'-dithiobis(2-nitrobenzoic acid) (DTNB, 0.3 mM; Sigma Chemical Co., St. Louis, MO, USA). Enzyme activity was measured upon addition of substrate, acetylthiocholine (ATCh, 0.2 or 0.1 mM; Sigma Chemical Co., St. Louis, MO, USA) by the Ellman methodCitation26 at 25 °C and 412 nm, on a Tecan Infinite M200PRO plate reader (Tecan Austria, GmbH, Salzburg, Austria). Due to the low solubility, a stock solution of the tested compounds was prepared in DMSO or methanol (Kemika, Zagreb, Croatia), and a corresponding solvent was in controls as well. The IC50 values were determined from at least three experiments by a nonlinear fit of the compound concentration logarithm values vs. % of enzyme activity using Prism6 software (GraphPad Prism 6 Software, San Diego, USA).
Supporting information
1H and 13 C NMR spectra of all the newly synthesised and isolated compounds, along with the 2 D NMR spectra of some compounds as well as AChE inhibition by trans-amino-5-arylethenyl-oxazole derivatives.
Supplemental Material
Download PDF (2.8 MB)Acknowledgement
The University of Zagreb short term scientific support under the title Synthesis and functionalization of novel (hetero)polycyclic photoproducts as potential cholinesterase inhibitors is gratefully acknowledged. The competent help by Željko Marinić in the NMR measurements is also appreciated.
Additional information
Funding
References
- Taylor P, Radić Z. The cholinesterases: from genes to proteins. Annu Rev Pharmacol Toxicol 1994;33:281–320.
- Sussman JL, Harel M, Frolow F, et al. Atomic structure of acetylcholinesterase from Torpedo Californica: a prototypic acetylcholine-binding protein. Science 1991;253:872–97.
- Kovarik Z. Amino acid residues conferring specificity of cholinesterases. Period Biol 1999;101:7–15.
- Radić Z, Pickering NA, Vellom DC, et al. Three distinct domains in the cholinesterase molecule confer selectivity for acetyl- and butyrylcholinesterase inhibitors. Biochemistry 1993;32:12074–84.
- Saxena A, Redman AMG, Jiang X, et al. Differences in active-site gorge dimensions of cholinesterase revealed by binding of inhibitors to human butyrylcholinesterase. Chem Biol Interact 1999;119 – 120:61–9.
- Taylor P, Radić Z, Hosea NA, et al. Structural bases for the specificity of cholinesterase catalysis and inhibition. Toxicol Lett 1995;82 – 83:453–8.
- Soreq H, Seidman S. Acetylcholinesterase – new roles for an old actor. Nat Rev Neurosci 2001;2:294–302.
- Silman I, Sussman JL. Acetylcholinesterase: 'classical' and 'non-classical' functions and pharmacology. Curr Opin Pharmacol 2005;5:293–302.
- Pienica C, Soreq H. MicroRNA regulators of cholinergic signaling link neuromuscular, cardiac and metabolic systems. Period Biol 2016;118:373–9.
- Chatonnet A, Lockridge O. Comparison of butyrylcholinesterase and acetylcholinesterase. Biochem J 1989;260:625–34.
- Çokugras AN. Butyrylcholinesterase: structure and physiological importance. Turk J Biochem 2003;28:54–61.
- Mesulam MM, Guillozet A, Shaw P, et al. Acetylcholinesterase knockouts establish central cholinergic pathways and can use butyrylcholinesterase to hydrolyze acetylcholine. Neuroscience 2002;110:627–39.
- Guillozet AL, Mesulam M-M, Smiley JF, Mash DC. Butyrylcholinesterase in the life cycle of amyloid plaques. Ann Neurol 1997;42:909–18.
- Mesulam MM, Geula C. Butyrylcholinesterase reactivity differentiates the amyloid plaques of aging from those of dementia. Ann Neurol 1994;36:722–7.
- Darvesh S, Walsh R, Kumar R, et al. Inhibition of human cholinesterases by drugs used to treat alzheimer disease. Alzheimer Dis Assoc Disord 2003;17:117–26.
- Bosak A, Ramić A, Šmidlehner T, et al. Design and evaluation of selective butyrylcholinesterase inhibitors based on Cinchona alkaloid scaffold. PLoS One 2018;13:e0205193.
- Bosak A, Opsenica DM, Šinko G, et al. Structural aspects of 4-aminoquinolines as reversible inhibitors of human acetylcholinesterase and butyrylcholinesterase. Chem Biol Interact 2019;308:101–9.
- Katalinić M, Bosak A, Kovarik Z. Flavonoids as inhibitors of human butyrylcholinesterase variants. Food Technol Biotechnol 2014;52:64–7.
- Šagud I, Škorić I, Burčul F. Naphthoxazoles and heterobenzoxazoles: cholinesterase inhibiting and antioxidant activity. Turk J Chem 2019;43:118–24.
- Šagud I, Šindler-Kulyk M, Škorić I, et al. Synthesis of naphthoxazoles by photocyclization of 4-/5-(phenylethenyl)oxazoles. Eur J Org Chem 2018;2018:3326–35.
- Fors BP, Krattiger P, Strieter E, Buchwald SL. Water-mediated catalyst preactivation: an efficient protocol for C-N cross-coupling reactions. Org Lett 2008;10:3505–8.
- Giacobini E. Cholinesterase inhibitors: new roles and therapeutic alternatives. Pharmacol Res 2004;50:433–40.
- Kumar A, Darreh-Shori T. DMSO: a mixed-competitive inhibitor of human acetylcholinesterase. ACS Chem Neurosci 2017;8:2618–25.
- Van Leusen AM, Hoogenboom BE, Siderius H. A novel and efficient synthesis of oxazoles from tosylmethylisocyanide and carbonyl compounds. Tetrahedron Letters 1972;13:2369–72.
- Cochran R, Kalisiak J, Küçükkilinç T, et al. Oxime-assisted acetylcholinesterase catalytic scavengers of organophosphates that resist aging. J Biol Chem 2011;286:29718–24.
- Ellman GL, Courtney KD, Andres V, Jr., Featherstone RM. New and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 1961;7:88–95.