
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Brain butyrylcholinesterase (BChE) is an attractive target for drugs designed for the treatment of Alzheimer’s disease (AD) in its advanced stages. It also potentially represents a biomarker for progression of this disease. Based on the crystal structure of previously described highly potent, reversible, and selective BChE inhibitors, we have developed the fluorescent probes that are selective towards human BChE. The most promising probes also maintain their inhibition of BChE in the low nanomolar range with high selectivity over acetylcholinesterase. Kinetic studies of probes reveal a reversible mixed inhibition mechanism, with binding of these fluorescent probes to both the free and acylated enzyme. Probes show environment-sensitive emission, and additionally, one of them also shows significant enhancement of fluorescence intensity upon binding to the active site of BChE. Finally, the crystal structures of probes in complex with human BChE are reported, which offer an excellent base for further development of this library of compounds.
Introduction
Alzheimer’s disease (AD) is the most common form of senile dementia, and its prevalence is expected to increase further in the coming decades due to ageing of the populations in the Western worldCitation1. AD is characterised by progressive impairment of cognition that is accompanied by behavioural decline, which results in loss of the ability to perform basic activities of daily lifeCitation2. As such, it causes distress for patients and caregivers, and it is becoming a large economic burden on societyCitation2.
The pathogenesis of AD is complex. It includes accumulation of the amyloid β protein (Aβ) and abnormal modifications and accumulation of the hyperphosphorylated protein tau, which is accompanied by oxidative stress. This leads to synaptic loss, selective neuronal death, and decreased brain concentrations of specific neurotransmitters, most notably of acetylcholine (ACh)Citation3. Following diagnosis of AD, it generally leads to death within 3 to 9 years; however the initial silent and asymptomatic stages start some 20 years before the onset of symptomsCitation4. Currently, the available treatments for AD include cholinesterase inhibitors and memantine, an N-methyl-D-aspartate (NMDA) receptor inhibitor. Unfortunately, these medications do not change the course of the disease nor the rate of patient decline, despite improving the quality of life for both patient and caregiverCitation5.
Cholinergic synapses are particularly affected by Aβ neurotoxicity, and the synaptic loss induces decreased brain ACh concentrations, which correlate in time with the cognitive impairment that is characteristic of AD. ACh is the neurotransmitter at cholinergic synapses, where its activity can be terminated by two cholinesterases: acetylcholinesterase (AChE) and butyrylcholinesterase (BChE). The activity of BChE is increased in certain regions of the brain of patients upon AD progressionCitation6. On the other hand, the expression of AChE decreases throughout the brain along with disease progression, thus making BChE an attractive target for the development of selective inhibitors for the treatment of AD, particularly in the later stages. Additionally, fewer side effects are expected for BChE inhibitors compared to the current AChE inhibitors due to the absence of BChE at cholinergic synapses outside of the brain; namely, at neuromuscular junctions and in the parasympathetic autonomous nervous systemCitation6. Furthermore, as BChE accumulates in Aβ plaques, it has the potential to serve as a biomarker of ADCitation6. The Aβ plaques are a central hallmark of AD, although a significant number of elderly people, and even young people, can have Aβ plaques without cognitive impairmentCitation7.
Future development of better treatments and more rapid and accurate diagnostic tools, and ultimately the prevention of AD, depends on a full understanding of the pathophysiology of AD, especially during the period before the onset of symptoms. The cholinesterases have important roles in the pathogenesis of AD, although their exact roles and their interplay in relation to the other hallmarks of AD remain poorly understood. New molecular tools to study cholinesterases might provide new insights into the pathophysiology of AD. Fluorescent probes are particularly useful here, due to the high sensitivity of fluorescence techniques and the potential for visualisation through various fluorescence microscopy techniques. Numerous AChE-specific fluorescent inhibitors have been reported, some of which are irreversible and some of which are competitiveCitation8–11. A tacrine-based fluorescent inhibitor with high affinity towards both AChE and BChE has been described, and this was shown to stain Aβ plaques when used as a probe for histochemical staining of brain tissueCitation12. Besides probes developed from inhibitors, two recently described BChE probes were developed as substrates for BChECitation13,Citation14. BChE converts the nonfluorescent substrate to highly fluorescent products. Furthermore, both probes are poor substrates for AChE or other types of esterases, thus providing high selectivity towards BChECitation13,Citation14. However, no BChE-selective probes derived from inhibitors were reported to date.
In the present study, we report on the two BChE-specific fluorescent probes that were derived from previously described highly potent and reversible BChE-selective inhibitorsCitation15–17. Although the incorporation of the fluorophore moieties had a negative impact on their BChE inhibition potency, the best three of these fluorescent inhibitors retained excellent activities in the low nanomolar range: probes 2A, 3A, and 3B. Moreover, significantly increased fluorescent intensity was observed upon binding of probe 2A to BChE. The crystal structures of these three best probes in complex with human BChE (huBChE) confirm the binding mode that was anticipated in the design phase.
Material and methods
Synthesis
Detailed schemes, synthetic procedures, and characterisation of each probe are given in the Supporting Information.
Inhibition of huAChE, mAChE, and huBChE
The inhibitory potencies of these probes against cholinesterases (i.e. huAChE, murine AChE [mAChE] and huBChE) were determined using the method of EllmanCitation18, as previously describedCitation19. The assays were carried out at room temperature in 100 mM phosphate buffer, pH 8.0, containing 333 µM Ellman reagent (5,5′-dithiobis(2-nitrobenzoic acid); DTNB), 500 µM substrate (huAChE/mAChE: acetylthiocholine; huBChE: butyrylthiocholine iodide [BSCh]) and huAChE/mAChE or huBChE (50 pM, 1 nM, respectively). The reactions were started by addition of the substrate, and the final dimethylsulphoxide (DMSO) carrier concentration did not exceed 1% (v/v). Formation of the yellow product was followed for 1 min as the change in the absorbance at 412 nm, using a microplate reader (Synerg H4; BioTek Instruments, Inc., USA). For screening for inhibitory activities, the compounds were assayed at 100 µM, in triplicate. The initial velocities in the absence (v0) and presence (vi) of the test compounds were calculated. The inhibitory potencies are expressed as the residual activities (RAs), according to Equation (1):
(1)
(1)
where b is the blank value.
For the IC50 measurements, serial dilutions were prepared for the compounds with RAs <50% at 100 µM. The IC50 values were calculated by plotting the RAs against the compound concentration according to a four-parameter logistic function, using the GraphPad Prism 8 software (GraphPad Software, USA). All of the measurements were repeated twice, each of which was in triplicate.
Kinetic studies
Chemicals
Purified huBChE was a generous gift from Drs. O. Lockridge and L. Schopfer. DTNB and BSCh were from Sigma. All other reagents used were of analytical grade. The experiments were carried out at 25 °C in 25 mM phosphate buffer, pH 7.0.
Inhibitory effects of 2C, 3A, and 3B on butyrylthiocholine iodide hydrolysis by purified huBChE
Three compounds (2C, 3A, 3B) were tested by measuring the progress curves for BSCh hydrolysis by huBChE in the absence and presence of each of these compounds at five to seven concentrations. Assays were carried out over 10 min, in a 0.6-ml cuvette, using the method of Ellman et alCitation18. The compound concentrations used were: 2C, 0–1 µM; 3A, 0–200 µM; and 3B, 0–50 nM.
The concentration of purified huBChE, which was always the final addition to these assays, was ∼0.5 nM. The hydrolysis of 50 µM BSCh was followed until completion (in most cases), in the presence of 1 mM DTNB. Reaching the plateau was important to get a precise estimate of the actual added BSCh concentration. For the calculation, the molar absorption coefficient of 13800 mol−1 cm−1 was used. All of the measurements were performed using a conventional UV/Vis spectrophotometer (Lambda45; Perkin-Elmer). Due to the experimental manipulation, a dead time of 10 s was added to each measurement, and all progress curves were extrapolated accordingly prior to the analysis.
Theoretical basis of the kinetics analysis
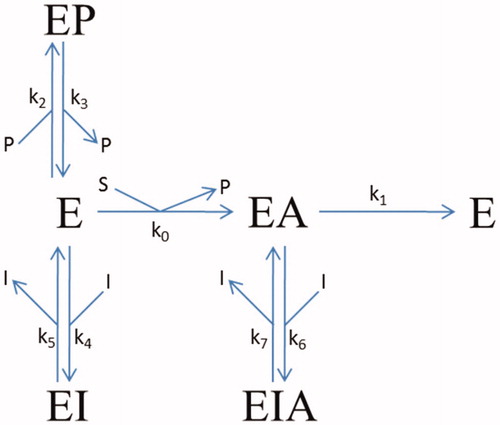
The progress-curve analysis was performed using the ENZO web application, implemented at www.enzo.cmm.ki.siCitation20–22. This programme is designed to generate differential equations from drawn reaction schemes and subsequently to fit the coefficients of these equations according to least squares methodology to reproduce the experimental data using a numerical integration algorithm. For determination of the kinetics mechanisms and parameters, the van Slyke-Cullen single intermediate reaction schemeCitation23 for substrate hydrolysis by huBChE was combined with a reversible mixed inhibition mechanism (). In all of the evaluations, the k1 of 814 s−1, the substrate specificity constant k1/Km of 1.0 × 108 M−1 −1s–t, and the inhibition constant of 54 µM for the binding of the thiocholine-TNB (k3/k2) in the hydrolysis of BSCh by purified huBChE were constrained, as these values had been determined previouslyCitation24.
Figure 1. Reaction scheme for the inhibition of BSCh hydrolysis by purified huBChE in the presence of DTNB by compounds 2C, 3A, and 3B. E, free enzyme; EA, acylated intermediate; S, substrate BSCh; P, all of the stoichiometrically released products (SCh-TNB, TNB–); and I, compounds (inhibitors) 2C, 3A, and 3B. The symbols for the constants are: kcat (k1), catalytic constant for BSCh turnover; Km (k0), Michaelis constant; Kp = k3/k2, inhibition constant for binding of the product thiocholine-TNB; K1 = k5/k4 and K2 = k7/k6, dissociation constants for binding of the compounds to the free and acylated enzyme, respectively.
X-ray crystallography of huBChE in complex with 2C, 3A, and 3B
Crystallization
Recombinant huBChE was produced in eukaryotic cells and insect cells as described previouslyCitation25,Citation26. The protein was purified by BChE-specific affinity chromatography (Hupresin; Chemforase, Rouen, France), followed by size exclusion chromatography (Superdex 200; GE Healthcare), as previously describedCitation26. Compounds 2C, 3A, and 3B were solubilised in 100% DMSO at 0.1 M, and the protein complexes were obtained by co-crystallization at 1 mM final ligand concentration (2C, 3B: 0.1 M MES, pH 6.5, 2.15 M (NH4)2SO4, 1% DMSO; 3A: 0.2 M ammonium acetate, 12% PEG 4000, pH 7.4) using the hanging drop method at 293 K. For 2C and 3B, the crystals were cryo-protected in a solution of 0.1 M MES, pH 6.5, 2.15 M (NH4)2SO4, 20% glycerol, 1 mM ligand, 1% DMSO before flash cooling in liquid nitrogen. For 3A, cryo-protection was achieved with the mother-liquor complemented with 17% glycerol and 1 mM 3A.
Structure determination
The X-ray diffraction data for 2C and 3B were collected on the ID23-1 beamline of the European Synchrotron Radiation Facility (Grenoble, France) at 100 K, with a Pilatus 6 M detector, and for 3A, on the ID23-2 beamline at 100 K with a Pilatus 2 M detector. For 2A and 3A, the detector frames were processed manually with XDSCitation27, and for 3B, automatically with autoPROCCitation28. Model refinement was performed using the Phoenix software suiteCitation29. The initial models were obtained by molecular replacement using Phaser-MR and the huBChE structure (PDB entry 1P0I), devoid of any ligands, glycans or water molecules. The electron densities in the active site gorge were examined, and allowed to undergo partial fitting with each compound. For 2C and 3B, ligand geometry restraints were processed using Phoenix eLBOWCitation30 and the semi-empirical quantum mechanical method (AM1). For 3A, the geometry restraints were generated using the PRODRG serverCitation31. Each model was refined by iterative cycles of Phoenix.refine and model building using CootCitation32. The huBChE structures in complex with 2C, 3A, and 3B have been deposited in the Protein Data Bank under accession numbers 6R6V, 6RUA, and 6R6W, respectively.
Results and discussion
Structure-activity relationships
Fluorescent probes that undergo emission changes upon binding to an enzyme active site can be used as “hot ligands”; i.e., in fluorescence-based competition binding assaysCitation33. The environment of the binding site is generally more stable and hydrophobic than the bulk, which can result in increased intensity of the fluorescence emission and a blue-shift of the emission maximumCitation34. For maximal sensitivity, a fluorophore should bind strongly and deeply into the active site. In the case of BChE, the active site is buried at the bottom of a deep aromatic gorge, at the entrance of which there is a peripheral binding site (main contributors, Asp70, Ala277). The active site features a choline-binding pocket (main contributor, Trp82) and an acyl binding pocket (main contributors, Trp231, Leu286, Phe329, Phe398), which accommodate the choline and butyrate moieties, respectively, of the substrate butyrylcholine (or its analogue, butyryl thiocholine). The catalytic activity features residues Ser198, His438, and Glu325.
The design of the initial fluorescent probe based on an inhibitor of BChE (, hit 1) began before its crystal structure in complex with huBChE was resolvedCitation13. In the first attempt, the naphthalene moiety of this amide-type inhibitor 1 was substituted with 7-(diethylamino)coumarin, to yield 1A (). 7-(Diethylamino)coumarin was selected because of its solvatochromic propertiesCitation35,Citation36. Additionally, to shorten the synthetic route, the indene moiety on the inhibitor 1 was substituted with a benzyl group. In retrospect, both of these modifications were far from optimal. The crystal structure of amide-type inhibitor 1 in complex with huBChE later showed that the naphthalene was tightly bound within the acyl binding pocket, and that substitution of naphthalene with a larger moiety was bound to, and accordingly resulted in, decreased activityCitation15. Replacement of the indene moiety by a benzene will also not have positively impacted on the affinity, as this eliminated the strong contribution to the binding of the cation-π interactions it established with Tyr332Citation13.
Figure 2. Structures of the parent non-fluorescent BChE inhibitors (compounds 1, 2, 3) and the synthesised fluorescent probes (1A, 2A–C, 3A–D). The IC50 values and residual activities (RAs) for inhibition of mAChE, huAChE, and huBChE are given.
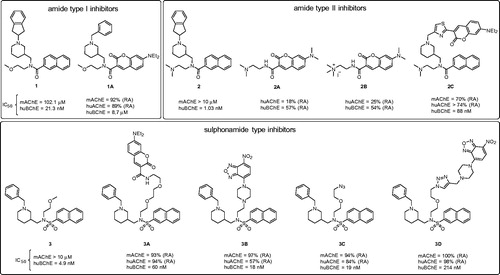
With the discovery of the picomolar selective BChE inhibitor 2Citation17, the design of an inhibitor with a fluorophore part positioned in the acyl binding pocket was revisited. We hypothesised that the loss of binding affinity that resulted from the substitution of naphthalene with coumarin might be compensated for by the strong cation-π interactions between its dimethylamino group and Trp82, as seen in the crystal structure of inhibitor 2 in complex with BChECitation17. Compounds 2A and 2B were synthesised without the piperidine moiety of the parent inhibitor 2 (). Additionally, the diethylamino group at position 7 of the coumarin scaffold was substituted with a less bulky dimethylamino group. To increase the interactions at the choline binding site, a compound with a permanent charge (2B) was designed, on the basis that compounds with a permanent ammonium cation can produce significantly stronger cation-π interactions compared with compounds with a tertiary amine moiety in the same positionCitation37. Indeed, the trimethylammonium fragment mimics the quarternary amine in substrate BSCh. Unfortunately, compounds 2A and 2B showed very low and unselective inhibition of BChE. Naphthalene was seen to be the optimal fit for the acyl binding pocket, and substitution with coumarin resulted in loss of inhibitory activity.
For the design of probe 2C (), a more conventional approach was therefore adopted, based on the crystal structure of inhibitor 2 in complex with huBChE. As the naphthalene and 2-(dimethylamino)ethyl moieties form crucial interactions with the active site, the indene group was replaced by a 7-(diethylamino)-3-(thiazol-2-yl)-coumarin moiety. Indeed, the indene only has non-specific interacts with the protein scaffold, and it is furthermore oriented out of the aromatic gorge, towards the bulk, which suggests more permissivity in terms of fluorescent group substitutions. Unsurprisingly, compound 2C showed lower huBChE potency when compared to parent inhibitor 2, which is known to be one of the best reversible inhibitors of huBChE. However, compound 2C maintained good affinity, with an IC50 of 88 nM.
The binding mode of inhibitor 3 is somewhat different compared to that of inhibitors 1 and 2Citation15–17. The common property of all of these three types of inhibitors is the position of the naphthalene moiety, which fits into the acyl binding pocket of huBChE. However, the piperidine groups of inhibitors 1 and 2 points towards the entrance of the aromatic active-site gorge, while for inhibitor 3, it fits into the choline-binding pocket and it is instead the methoxyethyl group that is oriented into the gorge. Therefore, elongation of the methoxymethyl group was the most straightforward approach to graft a fluorophore onto inhibitor 3, to yield 3A (). Compound 3A had its methoxyethyl group substituted with a triethylene glycol spacer, with 7-(diethylamino)coumarin at the distal end. This compound retained good BChE inhibitory activity, with an IC50 of 60 nM, but instead showed poor solubility in aqueous buffers. To avoid non-specific interactions, a more hydrophilic 1–(2-hydroxyethyl)piperazine spacer was introduced, with an environment-sensitive 7-nitro-2,1,3-benzoxadiazol-4-yl (NBD) fluorophore attached to the piperazine nitrogen. The resulting 3B () had the best in-vitro inhibitory activity of this series, with an IC50 of 18 nM, and showed improved solubility in aqueous solutions compared to 3A.
To save time in future studies by enabling easier substitution of the fluorophore moiety, we introduced a short linker with an azide into compound 3, which can react with any fluorophore that is functionalised by an alkyne group, according to “click chemistry” (copper(I)-catalyzed alkyne-azide cycloaddition)Citation38,Citation39. Compound 3C () had a short diethylene glycol linker with an azide, and it retained good inhibitory activity against huBChE. However, the introduction of a test fluorophore for compound 3D () resulted in substantial loss of inhibitory activity, making this approach less attractive here.
Kinetic studies of probes 2C, 3A, and 3B
Inhibition of BSCh hydrolysis by purified huBChE with 2C, 3A, and 3B
The actions of different concentrations of these three fluorescent inhibitors on huBChE were tested by measuring progress curves of product formation at approximately 50 µM BSCh (). The analysis of these progress curves revealed good agreement between the experimental curves and a theoretical model that defined a competitive reaction mechanism with binding of the inhibitor to both the free and the acylated enzyme. The dissociation constants are given in , and they show significantly lower affinities of all of the ligands for binding to the acylated enzyme (i.e. K2).
Figure 3. Time courses of product formation in the reactions between BSCh (54 μM) and purified huBChE (A, 0.47 nM; B, 0.49 nM; C, 0.43 nM) in the absence (red curves) and presence of 2C (5, 20, 50, 200 nM) (A), 3A (50, 100, 200, 500 nM, 1 μM) (B), and 3B (5, 10, 25, 33, 50 nM) (C).
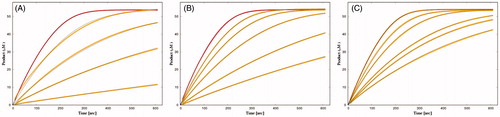
Table 1. Characteristic dissociation and rate constants for inhibition of huBChE by the fluorescent probes according to reaction scheme in .
Crystal structure of huBChE in complex with probes 2C, 3A, and 3B
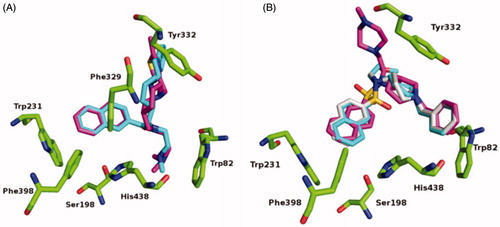
When compared to the previously determined structure of huBChE in complex with parent inhibitor 2 (PDB code 5NN0)Citation17, the fluorescent naphthalene–carboxamide derivative 2C adopts a similar binding mode, whereby the naphthalene moiety is positioned in the acyl-binding pocket through a T-stacking–like interaction with Trp231 (). The piperidine moiety interacts slightly deeper in the active-site gorge, stacking between Phe329 and Tyr332 (), while the dimethylamine moiety engages in cation-π interactions with Trp82. The remainder of the molecule is less defined. While the thiazole ring is positioned close to the parental indene moiety, no clear electron density is seen for the fluorescent coumarin-derivative moiety, which indicates that it does not interact with the protein scaffold and is likely to be free to adopt a variety of conformations, all of which will be characterised by the coumarin exposed to the bulk. The naphthalene–sulphonamide derivatives 3A and 3B have similar binding modes, where the naphthalene moiety points towards the acyl-binding pocket (). The piperidine moiety is inverted compared to the naphthalene carboxamide of 2C (5NN0), although it retains the same orientation as in the parent sulphonamide (5DYW)Citation16 (), where the benzyl ring protrudes into a highly hydrophobic pocket that is formed by residues Phe73, Trp82, Tyr332, Trp430, and Tyr440. As seen for the complex structure of parent inhibitor 3 with BChE, the sulphonamide oxygens of 3A and 3B form H-bonds with the hydroxyl group of Thr120. The piperazine ring of 3B points out of the aromatic gorge, towards the bulk; hence, as in the complex structure of BChE with 2C, no clear electron density is seen for the presumably highly flexible fluorescent NBD moiety. Similarly, no clear electron density is seen for the fluorescent coumarin and the triethyleneglycol linker of 3A, and only the first carbon attached to the sulphonamide nitrogen was modelled.
Figure 4. Crystal structures of 2C (A), 3A (B), and 3B (C) (purple stick models) bound to huBChE (green ribbon model; PDB codes 6R6V, 6RUA, 6R6W, respectively). Key residues in the active site are shown as green sticks. (B, C) The polar H-bond between Thr120 and the sulphonamide moiety of compounds 3A and 3B, respectively, is shown as yellow dashes (distance: B, 3.4 Å; C, 3.2 Å).
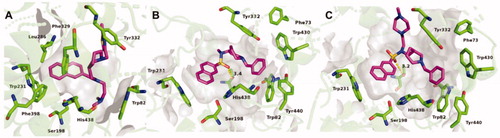
Figure 5. Alignments of crystal structures, with key residues in the active site, shown as green sticks. (A) Compound 2C (purple stick model; PDB code 6R6V) and parent naphthalene inhibitor 2 (cyan stick model; PDB code 5NN0) in their complexes with huBChE. (B) Compounds 3A (gray stick model; PDB code 6RUA), 3B (purple stick model; PDB code 6R6W) and parent sulphonamide inhibitor 3 (cyan stick model; PDB code 5DYW) in their complexes with huBChE.
Characterisation of fluorescence properties
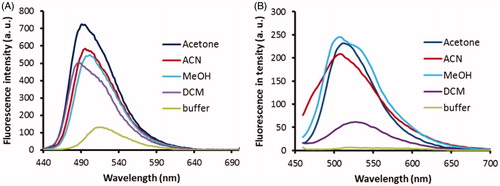
Only probes 2C and 3B had both good inhibitory potencies and sufficient aqueous solubility, and so these were further evaluated for their fluorescence properties. The low solubility of probe 3A might indeed lead to non-specific interactions with huBChE and limit is use, and it was therefore not included further. On the basis that the active site appears to be more hydrophobic than the bulk, the optimal probe should show strong emission in lipophilic solvents and no emission in polar solvents. In this respect, probe 3B proved ideal, as it showed strong emission in all of the lipophilic organic solvents, except dichloromethane, and very low fluorescence intensity in phosphate buffer at pH 8 (). Similarly, the emission intensities of probe 2C were also dependent on the solvent, although to a lesser extent than for probe 3B. However, probe 2C also showed substantial emission in phosphate buffer ().
Figure 6. Fluorescence spectra of probes 2C (A) and 3B (B) at 1 μM in the various solvents (as indicated). Excitation wavelengths: 440 nm, 380 nm, for probes 2C, 3B, respectively. a.u., arbitrary units.
The fluorescence properties of probes 2C and 3B were then examined when they were bound to huBChE. Probe 2C showed relatively strong emission in the buffer, and the addition of huBChE induced an approximately two-fold enhancement of its fluorescence intensity (). This mirrored the differences in the emission of probe 2C between the buffer solution and the organic solvents (). Improvements to the ratio of the fluorescence intensities between the bound and unbound probe might be obtained by the introduction of a more environment-sensitive fluorophore that can interact with the protein scaffold, in place of the 7-(diethylamino)-3-(thiazol-2-yl)-coumarin here, which does not. Addition of inhibitor 2 dislodged probe 2C form the active site and decreased the emission of 2C, nonetheless, the emission of 2C was still substantially higher compared to the emission of probe 2C in buffer. Most probably this is due to unspecific binding to protein surface and titration study (see Supplemental material) confirms this. Future development of probe 2C should thus be orientated towards lowering the lipophilicity, which should, in turn, reduce unspecific binding.
Figure 7. Fluorescence spectra of probes 2C (A) and 3B (B) at 1 μM in buffer without huBChE (blue), with huBChE at 500 nM (red) and with huBChE at 500 nM and inhibitor 2 at 1 μM (green). a.u. – arbitrary units.
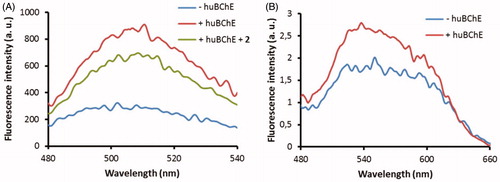
The weak fluorescence emission of probe 3B in the buffer was only marginally increased upon binding to huBChE, which indicated that the NBD fluorophore did not undergo significant changes in polarity. The crystal structure of probe 3B with huBChE confirmed this, as no full electron density was recorded for the NBD fluorophore and the linker of probe 3B. The NBD fluorophore would appear to lie in the polar environment at the entrance of the active site gorge, and surrounded by aqueous medium. For future development of probe 3B, it will be necessary to position the NBD fluorophore closer to the central scaffold, and to graft in additional functional groups that will position the fluorophore in a more hydrophobic environment. This will establish further interactions with the protein scaffold, thus providing the potential for a more pronounced increase in fluorescence emission upon binding to huBChE.
Rat brain slices
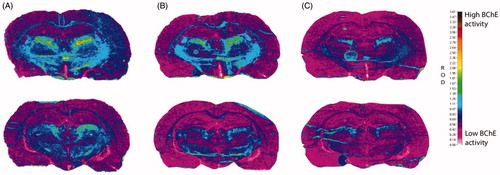
The inhibition of BChE by these highly specific fluorescent probes was pursued with a view to diagnosis and monitoring of progression of AD, by labelling the brain BChE that accumulates in disease-associated Aβ plaques. Inhibition of BChE in rat brain slices was therefore examined for probes 2C and 3B, to evaluate these two probes under more biologically relevant conditions. Sections of rat brain with the highest BChE activity were selected and the modified Koelle − Karnovsky histochemical method was used to detect BChE activityCitation16,Citation40–42. For these probes 2C and 3B, 1 mM was needed to achieve substantial inhibition of BChE, as measured by less formation of the dark-brown/black granular BChE reaction product (). The parent inhibitors 2 and 3 produced similar effects at 300 µM and 10 µM, respectively, which indicated the lower inhibition constants of the parent inhibitors.
Figure 8. Histochemical staining for BChE activity of coronal cryosections (10 μm) of a rat brain from two regions (top/bottom row) at the level of the thalamus. All sections were processed with 0.1 mM BW-284C51 in Koelle solution, to completely block AChE activity. The sections were incubated in the absence of the probes (A) and with probes 2C (1 mM; B) and 3B (1 mM; C). The relative optical density (ROD) pseudocolor scale for staining intensity of BChE activity is on the right.
Conclusions
We report here on the design, synthesis, kinetics and spectroscopic characterisation and binding modes of the two potent BChE-specific fluorescent probes. Design of these fluorescent probes was based on the crystal structures of previously described potent cholinesterase inhibitors in complex with their target, huBChE. Initially, a fluorophore was introduced in place of the naphthalene that binds to the acyl binding pocket of BChE, although this approach went no further. Instead, the scaffold was changed to ones that were already optimised to provide the crucial interactions with the active site gorge residues. The fluorophores were then introduced to be positioned to point outside the aromatic site gorge, towards the bulk. This approach was successful, as it yielded three promising probes, 2C, 3A, and 3B, which show good inhibition of BChE in the low nanomolar range while showing negligible inhibition of AChE. The fluorescence properties of the most promising probes, 2A and 3A, show that they both have environment-sensitive emission. Most notably, the emission of probe 2C is significantly enhanced upon binding to the active site, and thus can be used as a tool to study the activity of BChE. Additionally, inhibition of BChE activity of rat brain slices was investigated with probes 2C and 3B, as measured by the modified Koelle − Karnovsky histochemical method. Both probes successfully inhibited formation of the dark-brown/black granular BChE reaction product. Finally, the crystal structures of probes 2C, 3A, and 3B in complex with huBChE were obtained, which provide an excellent starting point for further optimisation of these fluorescent probes for BChE. In particular, the absence of electron density for the fluorophore moieties in these structures indicate that further improvement in inhibition potency and in environment sensitivity of the fluorescence emission can be provided by changes in the linker length and nature, as well as by grafting additional functional groups onto the fluorescent probes. This should provide tighter interactions with the protein scaffold, and thereby better fulfil the requirement for further evaluation as AD-specific fluorescent probes.
Supplemental Material
Download PDF (620.3 KB)Acknowledgements
The authors thank Dr. Chris Berrie for proof-reading of the manuscript.
Disclosure statement
The authors have no conflicts of interest to declare.
Additional information
Funding
Related Research Data
References
- Wang J, Gu BJ, Masters CL, et al. A systemic view of Alzheimer disease — insights from amyloid-β metabolism beyond the brain. Nat Rev Neurol 2017;13:612–23.
- Crous-Bou M, Minguillón C, Gramunt N, et al. Alzheimer’s disease prevention: from risk factors to early intervention. Alzheimers Res Ther 2017;9:71.
- Masters CL, Bateman R, Blennow K, et al. Alzheimer's disease. Nat Rev Dis Primers 2015;1:15056.
- Dubois B, Feldman HH, Jacova C, et al. Advancing research diagnostic criteria for Alzheimer's disease: the IWG-2 criteria. Lancet Neurol 2014;13:614–29.
- Weller J, Budson A. Current understanding of Alzheimer’s disease diagnosis and treatment. 1000Res 2018;7:1161.
- Darvesh S, Hopkins DA, Geula C. Neurobiology of butyrylcholinesterase. Nat Rev Neurol 2003;4:131–8.
- Macdonald IR, Maxwell SP, Reid GA, et al. Quantification of butyrylcholinesterase activity as a sensitive and specific biomarker of Alzheimer's disease. J Alzheimers Dis 2017;58:491–504.
- Saltmarsh JR, Boyd AE, Rodriguez OP, et al. Synthesis of fluorescent probes directed to the active site gorge of acetylcholinesterase. Bioorg Med Chem Lett 2000;10:1523–6.
- Huang X, Lee B, Johnson G, et al. Novel assay utilizing fluorochrome-tagged physostigmine (Ph-F) to in-situ detect active acetylcholinesterase (AChE) induced during apoptosis. Cell Cycle 2005;4:140–7.
- Amitai G, Ashani Y, Gafni A, et al. New fluorescent organophosphates as probes for studying aging-induced conformational changes in inhibited acetylcholinesterase. Neurochem Int 1980;2:199–204.
- Chao S, Krejci E, Bernard V, et al. A selective and sensitive near-infrared fluorescent probe for acetylcholinesterase imaging. ChemComm 2016;52:11599–602.
- Elsinghorst PW, Härtig W, Goldhammer S, et al. A gorge-spanning, high-affinity cholinesterase inhibitor to explore β-amyloid plaques. Org Biomol Chem 2009;7:3940–6.
- Yang S-H, Sun Q, Xiong H, et al. Discovery of a butyrylcholinesterase-specific probe via a structure-based design strategy. Chem Comm 2017;53:3952–5.
- Liu S-Y, Xiong H, Yang J-Q, et al. Discovery of butyrylcholinesterase-activated near-infrared fluorogenic probe for live-cell and in vivo imaging. ACS Sens 2018;3:2118–28.
- Brus B, Košak U, Turk S, et al. Discovery, biological evaluation, and crystal structure of a novel nanomolar selective butyrylcholinesterase inhibitor. J Med Chem 2014;57:8167–79.
- Košak U, Brus B, Knez D, et al. Development of an in-vivo active reversible butyrylcholinesterase inhibitor. Sci Rep 2016;6:39495.
- Košak U, Brus B, Knez D, et al. The magic of crystal-structure-based inhibitor optimization: development of a butyrylcholinesterase inhibitor with picomolar affinity and in-vivo activity. J Med Chem 2018;61:119–39.
- Ellman GL, Courtney KD, Andres V, et al. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 1961;7:88–95.
- Kosak U, Knez D, Coquelle N, et al. N-Propargylpiperidines with naphthalene-2-carboxamide or naphthalene-2-sulfonamide moieties: potential multifunctional anti-Alzheimer's agents. Bioorg Med Chem 2017;25:633–45.
- Bevc S, Konc J, Stojan J, et al. ENZO: a web tool for derivation and evaluation of kinetic models of enzyme catalyzed reactions. PLoS ONE 2011;6:e22265.
- Stojan J. The significance of low substrate concentration measurements for mechanistic interpretation in cholinesterases. Chem-Biol Interact 2013;203:44–50.
- Stojan J. Rapid mechanistic evaluation and parameter estimation of putative inhibitors in a single-step progress-curve analysis: the case of horse butyrylcholinesterase. Molecules 2017;22:1248.
- Kuzmič P. Application of the Van Slyke–Cullen irreversible mechanism in the analysis of enzymatic progress curves. Anal Biochem 2009;394:287–9.
- Stojan J. The mechanism and benefit of human butyrylcholinesterase activation by what would otherwise be inhibitors. Chem-Biol Interact 2019;308:350–6.
- Nachon F, Nicolet Y, Viguié N, et al. Engineering of a monomeric and low-glycosylated form of human butyrylcholinesterase. Eur J Biochem 2002;269:630–7.
- Brazzolotto X, Wandhammer M, Ronco C, et al. Human butyrylcholinesterase produced in insect cells: huprine-based affinity purification and crystal structure. Febs J 2012;279:2905–16.
- Kabsch W. Integration, scaling, space-group assignment and post-refinement. Acta Cryst D 2010;66:133–44.
- Vonrhein C, Flensburg C, Keller P, et al. Data processing and analysis with the autoPROC toolbox. Acta Cryst D 2011;67:293–302.
- Adams PD, Afonine PV, Bunkoczi G, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Cryst D 2010;66:213–21.
- Moriarty NW, Grosse-Kunstleve RW, Adams PD. Electronic ligand builder and optimization workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Cryst D 2009;65:1074–80.
- Schuttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr 2004;60:1355–63.
- Emsley P, Lohkamp B, Scott WG, et al. Features and development of Coot. Acta Cryst D 2010;66:486–501.
- Sakai H, Hirano T, Mori S, et al. 6-Arylcoumarins as novel nonsteroidal type progesterone antagonists: an example with receptor-binding-dependent fluorescence. J Med Chem. 2011;54(20):2055–7065.
- Eugenio Vázquez M, Rothman DM, Imperiali B. A new environment-sensitive fluorescent amino acid for Fmoc-based solid phase peptide synthesis. Org Biomol Chem 2004;2:1965–6.
- Goodey NM, Alapa MT, Hagmann DF, et al. Development of a fluorescently labeled thermostable DHFR for studying conformational changes associated with inhibitor binding. Biochem Biophys Res Commun 2011;413:442–7.
- Ikeda M, Yoshii T, Matsui T, et al. Montmorillonite − supramolecular hydrogel hybrid for fluorocolorimetric sensing of polyamines. J Am Chem Soc 2011;133:1670–3.
- Bissantz C, Kuhn B, Stahl M. A medicinal chemist’s guide to molecular interactions. J Med Chem 2010;53:5061–84.
- Rostovtsev VV, Green LG, Fokin VV, et al. A stepwise Huisgen cycloaddition process: copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew Chem Int 2002;41:2596–9.
- Tornøe CW, Christensen C, Meldal M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J Org Chem 2002;67:3057–64.
- Koelle GB, Friedenwald JS. A histochemical method for localizing cholinesterase activity. Proc Soc Exp Biol Med 1949;70:617–22.
- Darvesh S, Grantham DL, Hopkins DA. Distribution of butyrylcholinesterase in the human amygdala and hippocampal formation. J Comp Neurol 1998;393:374–90.
- Geula C, Nagykery N. Butyrylcholinesterase activity in the rat forebrain and upper brainstem: postnatal development and adult distribution. Exp Neurol 2007;204:640–57.