
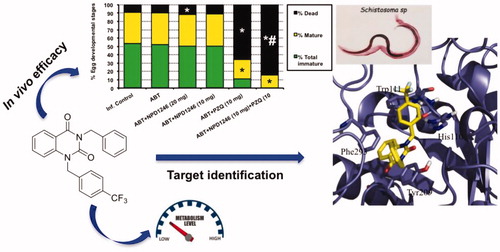
Abstract
A previous phenotypic screening campaign led to the identification of a quinazoline derivative with promising in vitro activity against Schistosoma mansoni. Follow-up studies of the antischistosomal potential of this candidate are presented here. The in vivo studies in a S. mansoni mouse model show a significant reduction of total worms and a complete disappearance of immature eggs when administered concomitantly with praziquantel in comparison with the administration of praziquantel alone. This fact is of utmost importance because eggs are responsible for the pathology and transmission of the disease. Subsequently, the chemical optimisation of the structure in order to improve the metabolic stability of the parent compound was carried out leading to derivatives with improved drug-like properties. Additionally, the putative target of this new class of antischistosomal compounds was envisaged by using computational tools and the binding mode to the target enzyme, aldose reductase, was proposed.
Graphical Abstract
Introduction
Schistosomiasis is a parasitic infectious disease caused by a trematode belonging to Schistosoma spp. Transmission occurs through contact with freshwater that is contaminated with larval forms (furcocercariae). Once in the human body, the larvae become adults in the blood vessels where the females release eggs. Part of the eggs is passed in the faeces or urine to continue the parasite’s life cycle by contaminating water while most become trapped in body tissues causing immune-inflammatory responses and progressive damage to organsCitation1. This neglected tropical disease is endemic in a number of tropical and subtropical countries representing a serious health problem especially in poor communities. The disease has recently also reached Europe, demonstrating the possibility to emerge in new geographical areas previously unknown related to migration movements and parasite genetic variantsCitation2.
Treatment and control of all forms of schistosomiasis fully rely on mass drug administration with the only available antischistosomal drug praziquantel (PZQ)Citation3. Even considering it is safe, effective, operationally convenient and low-cost, there is an increasing concern among the scientific community to anticipate PZQ therapeutic failureCitation4. The massive use for many years has clearly increased the risk of resistance development. This fact, together with the lack of efficacy against immatures makes the development of new drugs more urgentCitation5.
Historically, drug discovery for schistosomiasis has been based on phenotypic screening using whole-organism assays, however, new chemotherapeutics with known mechanism-of-action (MOA) are highly desirable to anticipate drug resistanceCitation6,Citation7. Particular advantages and disadvantages of phenotypic vs. target-based approaches are well known, and the combination of both strategies is logically the best way to move forward and optimise the drug discovery processCitation8,Citation9. Recognising the pivotal role of target identification and the challenging task of identifying the MOA for bioactive small molecules, significant progress has been made to develop a number of computational strategies to unveil the MOA of phenotypic hitsCitation10. In silico target identification offers chances for drug repurposing and for the detection of new links between disease and known targets. Large datasets such as ChEMBLCitation11 or PubChemCitation12 are now available and contain an impressive amount of biological data related to the activity of millions of ligands in multiple assays. As such, they provide an invaluable source of information for the development of knowledge-based approaches guided by computational techniquesCitation13.
In a previous in vitro phenotypic screening campaign using Schistosoma mansoni with worm killing as primary outcome, compounds from a selected library were successfully classified and prioritised based on potency and selectivityCitation14. The present work expands the evaluation of the antischistosomal activity for the quinazoline NPD-1246 (1) () which was one of the best in vitro “hits”, involving (i) in vivo evaluation in the S. mansoni-infected mouse model, (ii) a medicinal chemistry programme to obtain better drug-like compounds and (iii) exploration of the putative target using computational consensus methodology applying ligand-based approaches.
Figure 1. In vitro findings for NPD-1246 (1) previously reportedCitation14.

Materials and methods
Chemical procedures
Substrates were purchased from commercial sources and used without further purification. Melting points were determined with a Mettler Toledo MP70 apparatus. Flash column chromatography was carried out with automated silica gel column chromatography at medium pressure using silica gel (E. Merck, Grade 60, particle size 0.040–0.063 mm, 230–240 mesh ASTM) with the indicated solvent as eluent. Compounds were detected with UV light (254 nm). 1H NMR or 13C NMR experiments were obtained on the Bruker AVANCE-300 or Bruker AVANCE-500 spectrometers. 1H and 13C spectra were calibrated using residual chloroform or DMSO as an internal reference (CHCl3: δ 7.26 ppm and δ 77.3 ppm, respectively. DMSO: 2.54 ppm and 40.4 ppm, respectively). Chemical shifts for 1H NMR are reported as follows: chemical shift (δ ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, dd = double of doublets, ddd = doublet of double of doublets, td = triplet of doublets, m = multiplet), coupling constant (Hz), and integration. Chemical shifts for 13C NMR are reported in terms of chemical shift (δ ppm). Purity of assayed compounds was determined by elemental analysis recorded on Heraeus CHN-O-rapid analyser performed by the analytical department at CAI (UCM) and values were within ±0.4% of the theoretical values for all compounds.
Method A (R3=Me). General procedure for the synthesis of substituted 3-benzylquinazolin-2,4(1H,3H)-dione using anthranilate methyl ester derivatives as starting material. In a round‐bottomed flask with a magnetic stir bar, the corresponding anthranilate methyl ester is dissolved in toluene (5 mL/mmol) and cold at 0 °C. 1.1 equiv. of the corresponding isocyanate is added to the mixture, which is stirred overnight at rt. Then, 8 mL of toluene and 2 mL/mmol of NaOH (6 M) is added and the mixture is heated to 70 °C until complete conversion to the 3-substituted quinazolinedione (6 h). The reaction mixture is cooled in an ice bath and water (5 mL/mmol) is added, which causes a precipitate. This slurry is stirred for 30 min, filtered, washed with water (5 mL/mmol), and then heptanes (3 × 10 mL/mmol) before drying the solids under a stream of nitrogen. Products are generally obtained as white or off‐white solids and were used in the next step without further purification.
Method B (R3=H). General procedure for the synthesis of substituted 3-benzylquinazolin-2,4(1H,3H)-dione using anthranilic acid derivatives as starting material. In a round‐bottomed flask with a magnetic stir bar, the corresponding anthranilic acid derivative is dissolved in diethyl ether (8 mL/mmol) and 1.1 equiv. of the corresponding isocyanate is added drop by drop to the mixture, which is stirred overnight at rt. The solvent is rotary evaporated and replaced with EtOH (10 mL/mmol). Then, 2 mL/mmol of concentrated HCl (12.1 M) is added and the mixture is heated to 70 °C until complete conversion to the 3‐substituted quinazolinedione (3 h). The reaction mixture is cooled in an ice bath and water (5 mL/mmol) is added, which causes a precipitate. This slurry is stirred for 30 min, filtered, washed with water (5 mL/mmol), and then heptanes (3 × 10 mL/mmol) before drying the solids under a stream of nitrogen. Products are generally obtained as white or off‐white solids and were used in the next step without further purification.
General procedure for the synthesis of substituted 3-benzyl-1-(4-(trifluoromethyl)benzyl)quinazolin-2,4(1H,3H)-dione. In a round‐bottomed flask with a magnetic stir bar, substituted 3-benzylquinazolinedione previously obtained by method A or B is dissolved in DMF (3mL/mmol) and 1.5 equiv. of 4-(trifluoromethyl)benzyl chloride and 3 equiv. of NaHCO3 are added. The mixture is heated to 130 °C overnight. The reaction mixture is cooled and water (5mL/mmol) and EtOAc (15 mL/mmol) are added. Upon separation of the layers, the aqueous phase was extracted with EtOAc (3 × 10 mL). The organic layer was washed sequentially with sat. aq. NH4Cl, brine, then dried over Na2SO4 anhydrous. The desiccant was filtered and solvent removed under vacuum. The residue was purified by flash column chromatography using as eluents mixtures of solvents (EtOAc/hexane, 1:9–6:4) as indicated in each case to obtain the desired products.
3-Benzyl-1-(4-(trifluoromethyl)benzyl)quinazoline-2,4(1H,3H)-dione (1). Reagents: 3-benzylquinazolin-2,4(1H,3H)-dione (1.2 mmol) obtained by method A from methyl 2-aminobenzoate and 1-(isocyanatomethyl)benzene), 4-(trifluoromethyl)benzyl chloride (266 µL, 1.8 mmol), NaHCO3 (302 mg, 3.6 mmol) and DMF (6.0 mL). Purification: EtOAc/Hex (1:4). Yield: 207 mg, 42%. Whitish solid. mp: 160–162 °C. mp lit.Citation15: 160–161 °C.
3-(4-Fluorobenzyl)-1-(4-(trifluoromethyl)benzyl)quinazolin-2,4(1H,3H)-dione (2). Reagents: 3-(4-fluorobenzyl)quinazolin-2,4(1H,3H)-dione (1.2 mmol) obtained by method A from methyl 2-aminobenzoate and 1-fluoro-4-(isocyanatomethyl)benzene), 4-(trifluoromethyl)benzyl chloride (266 µL, 1.8 mmol), NaHCO3 (302 mg, 3.6 mmol) and DMF (6.0 mL). Purification: EtOAc/Hex (1:4). Yield: 102 mg, 20%. Whitish solid. mp: 142–144 °C. 1H NMR (300 MHz, CDCl3) δ 8.18 (dd, J = 7.9, 1.6 Hz, 1H), 7.57–7.39 (m, 5H), 7.26 (d, J = 8.0 Hz, 2H), 7.20–7.09 (m, 1H), 6.92 (t, J = 8.7 Hz, 3H), 5.34 (s, 2H), 5.21 (s, 2H). 13C NMR (75 MHz, CDCl3) δ 162.4 (d, JC-F=247 Hz), 161.5, 151.3, 139.7 (q, JC-F=2 Hz), 139.6, 135.3, 132.6 (d, JC-F = 3 Hz), 131.1 (d, JC-F= 8 Hz), 130.0 (q, JC-F = 32 Hz), 129.4, 126.7, 126.0 (q, JC-F = 4 Hz), 124.1 (q, JC-F = 272 Hz), 123.4, 115.7, 115.3 (d, JC-F = 22 Hz), 114.0, 47.0, 44.4. Anal. (C23H16F4N2O2) Calculated: C 64.49%, H 3.76%, N 6.54%. Found: C 64.19%, H 3.80%, N 6.49%.
1-(4-(Trifluoromethyl)benzyl)3-(4-methoxybenzyl)quinazolin-2,4(1H,3H)-dione (3). Reagents: 3-(4-methoxybenzyl)quinazolin-2,4(1H,3H)-dione (1.2 mmol) obtained by method A from methyl 2-aminobenzoate and 1-(isocyanatomethyl)-4-methoxybenzene), 4-(trifluoromethyl)benzyl chloride (266 µL, 1.8 mmol), NaHCO3 (302 mg, 3.6 mmol) and DMF (6.0 mL). Purification: EtOAc/Hex (1:4). Yield: 111 mg, 21%. Whitish solid. mp: 130–132 °C. 1H NMR (300 MHz, DMSO-d6) δ 8.21–8.11 (m, 1H), 7.80–7.66 (m, 3H), 7.62–7.52 (m, 2H), 7.42–7.25 (m, 4H), 6.98–6.87 (m, 2H), 5.53 (s, 2H), 5.18 (s, 2H), 3.77 (s, 3H). 13C NMR (75 MHz, DMSO-d6) δ 161.5, 158.9, 151.3, 141.7, 139.9, 135.9, 129.8, 129.5, 128.6, 128.3 (d, JC-F=32.1 Hz), 127.6, 126.0 (q, J = 3.8 Hz), 124.5 (q, J = 272 Hz), 123.6, 122.8, 115.4 (d, JC-F=21.4 Hz), 114.1, 55.5, 46.6, 44.4. Anal. (C24H19F3N2O3) Calculated: C 65.45%, H 4.35%, N 6.36%. Found: C 65.26%, H 4.47%, N 5.98%.
6-Bromo-3-(4-fluorobenzyl)-1-(4-(trifluoromethyl)benzyl)quinazolin-2,4(1H,3H)-dione (4). Reagents: 6-bromo-3-(4-fluorobenzyl)quinazolin-2,4(1H,3H)-dione (1.2 mmol) obtained by method B from 2-amino-5-bromobenzoic acid and 1-fluoro-4-(isocyanatomethyl)benzene), 4-(trifluoromethyl)benzyl chloride (191 µL, 1.5 mmol), NaHCO3 (302 mg, 3.6 mmol) and DMF (6.0 mL). Purification: EtOAc/Hex (1:4). Yield: 176 mg, 29%. Whitish solid. mp: 186–188 °C. 1H NMR (300 MHz, CDCl3) δ 8.29 (d, J = 2.4 Hz, 1H), 7.59–7.37 (m, 5H), 7.24 (d, J = 8.0 Hz, 2H), 6.92 (t, J = 8.7 Hz, 2H), 6.81 (d, J = 8.9 Hz, 1H), 5.31 (s, 2H), 5.19 (s, 2H). 13C NMR (75 MHz, CDCl3) δ 162.3 (d, JC-F = 247 Hz), 160.4, 150.9, 139.2 (q, JC-F = 2 Hz), 138.5, 138.1, 132.3 (d, JC-F = 2 Hz), 131.8, 131.2 (d, JC-F = 8 Hz), 130.3 (q, JC-F = 32 Hz), 126.6, 126.1 (q, JC-F = 4 Hz), 123.7 (q, JC-F = 272 Hz), 117.3, 116.4, 115.9, 115.3 (d, JC-F = 22 Hz), 47.2, 44.6. Anal. (C23H15BrF4N2O2) Calculated: C 54.46%, H 2.98%, N 5.52%. Found: C 54.50%, H 2.94%, N 5.51%.
6-Bromo-1-(4-(trifluoromethyl)benzyl)-3-(4-methoxybenzyl)quinazolin-2,4(1H,3H)-dione (5). Reagents: 6-bromo-3-(4-methoxybenzyl)quinazolin-2,4(1H,3H)-dione (1.2 mmol) obtained by method B from 2-amino-5-bromobenzoic acid and 1-(isocyanatomethyl)-4-methoxybenzene), 4-(trifluoromethyl)benzyl chloride (266 µL, 1.8 mmol), NaHCO3 (302 mg, 3.6 mmol) and DMF (6.0 mL). Purification: EtOAc/Hex (1:4). Yield: 405 mg, 65%. Whitish solid. mp: 147–149 °C. 1H NMR (300 MHz, CDCl3) δ 8.10 (d, J = 8.4 Hz, 1H), 7.61 (d, J = 8.1 Hz, 2H), 7.50 (d, J = 8.7 Hz, 2H), 7.42–7.29 (m, 3H), 7.18 (d, J = 1.6 Hz, 1H), 6.91–6.80 (m, 2H), 5.36 (s, 2H), 5.24 (s, 2H), 3.78 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 161.3, 159.6, 151.5, 140.8, 139.5 (q, JC-F = 2 Hz), 131.1, 131.1, 130.6 (q, JC-F = 32 Hz), 130.5, 129.1, 127.1, 127.1, 126.5 (q, JC-F = 4 Hz), 124.3 (q, JC-F = 272 Hz), 117.3, 115.0, 114.2, 55.6, 47.5, 45.1. Anal. (C24H18BrF3N2O3) Calculated: C 55.51%, H 3.49%, N 5.39%. Found: C 55.63%, H 3.55%, N 5.31%.
7-Bromo-3-(4-fluorobenzyl)-1-(4-(trifluoromethyl)benzyl)quinazolin-2,4(1H,3H)-dione (6). Reagents: 7-bromo-3-(4-fluorobenzyl)quinazolin-2,4(1H,3H)-dione (1.2 mmol) obtained by method B from 2-amino-4-bromobenzoic acid and 1-fluoro-4-(isocyanatomethyl)benzene), 4-(trifluoromethyl)benzyl chloride (266 µL, 1.8 mmol), NaHCO3 (302 mg, 3.6 mmol) and DMF (6.0 mL). Purification: EtOAc/Hex (1:4). Yield: 140 mg, 22%. Whitish solid. mp: 153–155 °C. 1H NMR (300 MHz, CDCl3) δ 8.13 (d, J = 8.4 Hz, 1H), 7.64 (d, J = 8.0 Hz, 2H), 7.50–7.41 (m, 2H), 7.44–7.32 (m, 3H), 7.22 (d, J = 1.6 Hz, 1H), 7.02 (t, J = 8.7 Hz, 2H), 5.39 (s, 2H), 5.28 (s, 2H). 13C NMR (75 MHz, CDCl3) δ 162.5 (d, JC-F = 247 Hz), 160.9, 151.0, 140.5, 139.0 (q, JC-F = 2 Hz), 132.3 (d, JC-F = 2 Hz), 131.1 (d, JC-F = 8 Hz), 130.7, 130.3 (q, JC-F = 32 Hz), 130.3, 126.9, 126.7, 126.2 (q, JC-F = 4 Hz), 123.8 (q, JC-F = 272 Hz), 117.0, 115.3 (d, JC-F = 22 Hz), 114.5, 47.14, 44.59. Anal. (C23H15BrF4N2O2) Calculated: C 54.46%, H 2.98%, N 5.52%. Found: C 54.43%, H 3.01%, N 5.50%.
7-Bromo-1-(4-(trifluoromethyl)benzyl)-3-(4-methoxybenzyl)quinazolin-2,4(1H,3H)-dione (7). Reagents: 7-bromo-3-(4-methoxybenzyl)quinazolin-2,4(1H,3H)-dione (1.2 mmol) obtained by method B from 2-amino-4-bromobenzoic acid and 1-(isocyanatomethyl)-4-methoxybenzene), 4-(trifluoromethyl)benzyl chloride (266 µL, 1.8 mmol), NaHCO3 (302 mg, 3.6 mmol) and DMF (6.0mL). Purification: EtOAc/Hex (1:4). Yield: 218 mg, 35%. Whitish solid. mp: 138–140 °C. 1H NMR (300 MHz, CDCl3) δ 8.39 (d, J = 2.4 Hz, 1H), 7.62 (dd, J = 8.2, 4.5 Hz, 3H), 7.56–7.47 (m, 2H), 7.34 (d, J = 8.0 Hz, 2H), 6.88 (m, 3H), 5.40 (s, 2H), 5.28 (s, 2H), 3.81 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 160.4, 159.2, 151.0, 139.3 (q, JC-F = 2 Hz), 138.5, 137.9, 131.8, 130.7, 130.2 (q, JC-F = 32 Hz), 128.7, 126.6, 126.1 (q, JC-F = 4 Hz), 123.8 (q, JC-F = 272 Hz), 117.4, 116.3, 115.8, 113.8, 55.2, 47.1, 44.8. Anal. (C24H18BrF3N2O3) Calculated: C 55.51%, H 3.49%, N 5.39%. Found: C 55.31%, H 3.54%, N 5.30.
6-Chloro-3-(4-fluorobenzyl)-1-(4-(trifluoromethyl)benzyl)quinazolin-2,4(1H,3H)-dione (8). Reagents: 6-chloro-3-(4-fluorobenzyl)quinazolin-2,4(1H,3H)-dione (1.2 mmol) obtained by method A from methyl 2-amino-5-chlorobenzoate and 1-fluoro-4-(isocyanatomethyl)benzene), 4-(trifluoromethyl)benzyl chloride (266 µL, 1.8 mmol), NaHCO3 (302 mg, 3.6 mmol) and DMF (6.0 mL). Purification: EtOAc/Hex (1:4). Yield: 55 mg, 10%. Whitish solid. mp: 160–162 °C. 1H NMR (300 MHz, CDCl3) δ 8.19 (d, J = 8.5 Hz, 1H), 7.62 (d, J = 8.1 Hz, 2H), 7.54 (dd, J = 8.6, 5.5 Hz, 2H), 7.34 (d, J = 8.0 Hz, 2H), 7.21 (dd, J = 8.5, 1.7 Hz, 1H), 7.05–6.95 (m, 3H), 5.37 (s, 2H), 5.26 (s, 2H). 13C NMR (75 MHz, CDCl3) δ 163.0 (d, JC-F = 247 Hz), 161.2, 151.5, 142.2, 140.9, 139.4 (q, JC-F = 2 Hz), 132.7 (d, JC-F = 3 Hz), 131.5 (d, JC-F = 8 Hz), 131.1, 130.7 (q, JC-F = 32 Hz), 127.1, 126.5 (q, JC-F = 4 Hz), 124.4, 124.1 (q, JC-F = 272 Hz), 115.7 (d, JC-F = 22 Hz), 114.5, 114.5, 47.5, 44.9. Anal. (C23H15ClF4N2O2) Calculated: C 59.69%, H 3.27%, N 6.05%. Found: C 59.62%, H 3.30%, N 6.01%.
6-Chloro-1-(4-(trifluoromethyl)benzyl)-3-(4-methoxybenzyl)quinazolin-2,4(1H,3H)-dione (9). Reagents: 6-chloro-3-(4-methoxybenzyl)quinazolin-2,4(1H,3H)-dione (1.2 mmol) obtained by method A from methyl 2-amino-5-chlorobenzoate and 1-(isocyanatomethyl)-4-methoxybenzene), 4-(trifluoromethyl)benzyl chloride (266 µL, 1.8 mmol), NaHCO3 (302 mg, 3.6 mmol) and DMF (6.0 mL). Purification: EtOAc/Hex (1:4). Yield: 387 mg, 71%. Whitish solid. mp: 145–147 °C. 1H NMR (300 MHz, CDCl3) δ 8.21 (d, J = 8.4 Hz, 1H), 7.64 (d, J = 8.1 Hz, 2H), 7.58–7.47 (m, 2H), 7.37 (d, J = 8.0 Hz, 2H), 7.22 (dd, J = 8.5, 1.7 Hz, 1H), 7.02 (d, J = 1.7 Hz, 1H), 6.91–6.83 (m, 2H), 5.39 (s, 2H), 5.27 (s, 2H), 3.81 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 160.8, 159.2, 151.2, 141.6, 140.5, 139.1 (q, JC-F = 2 Hz), 130.7, 130.2 (q, JC-F = 32 Hz), 128.8, 126.7, 126.1 (q, JC-F = 4 Hz), 124.0 (q, JC-F = 272 Hz), 123.9, 114.3, 114.0, 113.8, 55.2, 47.1, 44.7. Anal. (C24H18ClF3N2O3) Calculated: C 60.70%, H 3.82%, N 5.90%. Found: C 60.72%, H 3.80%, N 5.86%.
3-(4-Fluorobenzyl)-1-(4-(trifluoromethyl)benzyl)-6,7-dimethoxyquinazolin-2,4(1H,3H)-dione (10). Reagents: 3-(4-fluorobenzyl)-6,7-dimethoxyquinazolin-2,4(1H,3H)-dione (1.2 mmol) obtained by method A from methyl 2-amino-4,5-dimethoxibenzoate and 1-fluoro-4-(isocyanatomethyl)benzene), 4-(trifluoromethyl)benzyl chloride (266 µL, 1.8 mmol), NaHCO3 (302 mg, 3.6 mmol) and DMF (6.0 mL). Purification: EtOAc/Hex (1:2). Yield: 73 mg, 15%. Whitish solid. mp: 180–182 °C. 1H NMR (300 MHz, CDCl3) δ 7.62 (m, 3H), 7.57 (dd, J = 8.6, 5.5 Hz, 2H), 7.38 (d, J = 8.0 Hz, 2H), 7.02 (t, J = 8.7 Hz, 2H), 6.43 (s, 1H), 5.42 (s, 2H), 5.30 (s, 2H), 3.93 (s, 3H), 3.77 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 162.7 (d, JC-F = 246 Hz), 161.5, 155.6, 151.9, 146.2, 140.3 (q, JC-F = 2 Hz), 135.6, 133.2 (d, JC-F = 3 Hz), 131.4 (d, JC-F = 8 Hz), 130.6 (q, JC-F = 32 Hz), 127.1, 126.4 (q, JC-F = 4 Hz), 124.0 (q, JC-F = 272 Hz), 115.6 (d, JC-F = 22 Hz), 109.6, 108.5, 97.4, 56.7, 56.5, 47.7, 44.8. Anal. (C25H20F4N2O4) Calculated: C 61.48%, H 4.13%, N 5.74%. Found: C 60.67%, H 4.11%, N 5.68%.
1-(4-(Trifluoromethyl)benzyl)-6,7-dimethoxy-3-(4-methoxybenzyl)quinazolin-2,4(1H,3H)-dione (11). Reagents: 6-7-dimethoxy-3-(4-methoxybenzyl)quinazolin-2,4(1H,3H)-dione (1.2 mmol) obtained by method A from methyl 2-amino-4,5-dimethoxibenzoate and 1-(isocyanatomethyl)-4-methoxybenzene), 4-(trifluoromethyl)benzyl chloride (266 µL, 1.8 mmol), NaHCO3 (302 mg, 3.6 mmol) and DMF (6.0 mL). Purification: EtOAc/Hex (1:2). Yield: 72 mg, 12%. Whitish solid. mp: 171–173 °C. 1H NMR (300 MHz, CDCl3) δ 7.55 (s, 1H), 7.52 (d, J = 8.0 Hz, 2H), 7.44 (d, J = 8.6 Hz, 2H), 7.28 (d, J = 8.0 Hz, 2H), 6.78 (d, J = 8.7 Hz, 2H), 6.32 (s, 1H), 5.32 (s, 2H), 5.19 (s, 2H), 3.83 (s, 3H), 3.70 (s, 3H), 3.67 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 161.6, 159.5, 155.5, 151.9, 146.2, 140.5 (q, JC-F = 2 Hz), 135.6, 131.0, 130.5 (q, JC-F = 32 Hz), 129.7, 127.1, 126.4 (q, JC-F = 4 Hz), 124.3 (q, JC-F = 272 Hz), 114.1, 109.6, 108.6, 97.4, 56.6, 56.5, 55.6, 47.6, 45.0. Anal. (C26H23F3N2O5) Calculated: C 62.40%, H 4.63%, N: 5.60%. Found: C 62.09%, H 4.56%, N: 5.52%.
8-Bromo-3-(4-fluorobenzyl)-1-(4-(trifluoromethyl)benzyl)-6-methylquinazolin-2,4(1H,3H)-dione (12). Reagents: 8-bromo-3-(4-fluorobenzyl)-6-methylquinazolin-2,4(1H,3H)-dione (1.2 mmol) obtained by method B from 2-amino-3-bromo-6-methylbenzoic acid and 1-fluoro-4-(isocyanatomethyl)benzene), 4-(trifluoromethyl)benzyl chloride (266 µL, 1.8 mmol), NaHCO3 (302 mg, 3.6 mmol) and DMF (6.0 mL). Purification: EtOAc/Hex (1:4). Yield: 106 mg, 17%. Whitish solid. mp: 183–185 °C. 1H NMR (300 MHz, CDCl3) δ 8.00 (d, J = 2.2 Hz, 1H), 7.62 (d, J = 2.2 Hz, 1H), 7.47 (d, J = 8.1 Hz, 2H), 7.38 (dd, J = 8.6, 5.5 Hz, 2H), 7.17 (d, J = 8.0 Hz, 2H), 6.88 (t, J = 8.7 Hz, 2H), 5.66 (s, 2H), 5.11 (s, 2H), 2.29 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 162.3 (d, JC-F = 247 Hz), 160.7, 152.2, 142.9, 141.7 (q, JC-F = 2 Hz), 137.2, 135.2, 132.2 (d, JC-F = 2 Hz), 131.1 (d, JC-F = 8 Hz), 129.5 (q, JC-F = 32 Hz), 129.2, 126.5, 125.4 (q, JC-F = 4 Hz), 124.0 (q, JC-F = 272 Hz), 119.5, 115.2 (d, JC-F = 22 Hz), 107.5, 51.3, 44.6, 20.0. Anal. (C24H17BrF4N2O2) Calculated: C 55.35%, H 3.29%, N 5.37%. Found: C 55.35%, H 3.35%, N 5.35%.
8-Bromo-1-(4-(trifluoromethyl)benzyl)-3-(4-methoxybenzyl)-6-methylquinazolin-2,4(1H,3H)-dione (13). Reagents: 8-bromo-3-(4-methoxybenzyl)-6-methylquinazolin-2,4(1H,3H)-dione (1.2 mmol) obtained by this method: In round‐bottomed flask with a magnetic stir bar, 2-amino-3-bromo-5-methylbenzoic acid (1.0 equiv.) is dissolved in THF (5 mL/mmol) and 1.0 equiv. of 1-(isocyanatomethyl)-4-methoxybenzene is added to the mixture, which is stirred in an oil bath at 80 °C overnight. The solvent is rotary evaporated and replaced with EtOH (20 mL/mmol). Then, 2 mL/g of concentrated HCl (12.1 M) is added and the mixture is reheated to 70 °C until complete conversion to the 3‐substituted quinazolinedione (3 h). The reaction mixture is cooled in an ice bath and water (20 mL/g) is added, which causes a precipitate. This slurry is stirred for 30 min, filtered, washed with water (10 mL/mmol), and then heptanes (3 × 10 mL/mmol) before drying the solids under a stream of nitrogen); 4-(trifluoromethyl)benzyl chloride (266 µL, 1.8 mmol), NaHCO3 (302 mg, 3.6 mmol) and DMF (6.0 mL). Purification: EtOAc/Hex (1:4). Yield: 204 mg, 32%. Whitish solid. mp: 158–160 °C. 1H NMR (500 MHz, CDCl3) δ 8.10 (dd, J = 2.3, 0.9 Hz, 1H), 7.71 (dd, J = 2.3, 0.8 Hz, 1H), 7.57 (d, J = 8.1 Hz, 2H), 7.50–7.40 (m, 2H), 7.34–7.23 (m, 2H), 6.84 (d, J = 8.7 Hz, 2H), 5.76 (s, 2H), 5.19 (s, 2H), 3.80 (s, 3H), 2.39 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 161.1, 159.6, 152.6, 143.2, 142.2, 137.6, 135.5, 131.0, 129.6 (q, JC-F = 32 Hz), 129.6, 129.1, 126.9, 125.8 (q, JC-F = 4 Hz), 124.4 (q, JC-F = 272 Hz), 120.0, 114.1, 107.8, 55.6, 51.6, 45.2, 20.4. Anal. (C25H20BrF3N2O3) Calculated: C 56.30%, H 3.78%, N 5.25%. Found: C 56.20%, H 3.76%, N 5.18%.
Microsomal stability assays
Mouse or human liver microsomes (S9), reduced nicotinamide adenine dinucleotide phosphate (NADPH) generating system solutions and uridine glucuronosyltransferase (UGT) reaction mix (BD Biosciences) were kept at −80 °C. The test compounds and reference compound diclofenac were formulated in DMSO at 10 µM. The assay was carried out based on the BD Biosciences Guidelines for Use (TF000017 Rev1.0) with minor adaptations. The metabolic stability of the compounds was studied through the CYP450 superfamily (Phase-I metabolism) by fortification with NADPH and through UGT enzymes (Phase-II metabolism) by fortification with uridine diphosphate glucuronic acid (UDPGA). For CYP450 and other NADPH dependent enzymes, the compounds were incubated at 5 µM together with 0.5 mg/mL S9 in potassium phosphate buffer in a reaction started by the addition of 1 mM NADP. At defined time points, 20 µL was withdrawn from the reaction mixture and 80 µL cold acetonitrile (ACN) was added to inactivate the enzymes and precipitate the protein. The mixture was vortexed for 30 s and centrifuged at 4 °C for 5 min to collect the supernatant. For UGT enzymes, the compounds were incubated at 5 µM together with 0.5 mg/mL S9 in a reaction started by the addition of 2 mM UDPGA cofactor. The loss of parent compound was determined using liquid chromatography (UPLC) (Waters AquityTM) coupled with tandem quadrupole mass spectrometry (MS2) (Waters XevoTM), equipped with an electrospray ionisation (ESI) interface and operated in multiple reaction monitoring (MRM) mode.
In vivo antischistosomal efficacy
PZQ was obtained from Egyptian International Pharmaceutical Industries Company (EIPICO) and aminobenzotriazole (ABT) from Fluorochem Ltd.
Experimental animals
Male Swiss albino mice (CD-1) obtained from SBSC and weighing 18–20 g were housed under environmentally controlled room temperature of 20–22 °C, 12 h light/dark cycle and 50–60% humidity with food and water ad libitum throughout the acclimatisation and experimental periods. All the animal experiments were conducted in accordance with the Guide for Care and Use of Laboratory Animals and were approved by the Institutional Review Board of Theodor Bilharz Research Institute (TBRI).
Infection and experimental design
Mice were infected with S. mansoni cercariae [provided by Schistosome Biology Supply Center (SBSC)] using body immersionCitation16 by exposure to 80 ± 10 cercariae/mouse. Infected mice were divided into six groups: groups 1 and 2 were treated the vehicle and the CYP450 inhibitor ABT respectively; groups 3 and 4 were dosed for 5 days with NPD-1246 at 20 and 10 mg/kg, respectively, while group 5 was treated with PZQ at 10 mg/kg/day for 5 days. Group 6 was treated with NPD-1246 combined with PZQ, each at 10 mg/kg/day for 5 days starting from week 7 post-infection. To minimise first-pass elimination, ABT was administered at 100 mg/kg/day for 5 days 2 h prior to each compound administration. NPD-1246 and PZQ were freshly suspended in 2% Cremophore-EL (Sigma-Aldrich). All drug administrations were performed orally.
Parasitological criteria for cure
Ten days post-treatment, all mice were sacrificed and perfused, and the number of worms recovered (worm burden) was quantified and sexedCitation17. The number of eggs per gram of liver or intestinal tissue was countedCitation18. The percentage of egg developmental stages (oogram pattern) was studiedCitation19 in which eggs at different stages of maturity (from I to IV) were identified and counted. Mature eggs and dead eggs (granular, dark, and semi-transparent) were also counted in three fragments of intestine and the mean number of each stage was calculated.
Statistical analysis
The percentage reduction of worm or egg burden in each treated group was calculated. The 50% effective concentration (EC50) was calculated using Prism (GraphPad; Version 5.0) software using a variable slope for the sigmoidal curve with an upper limit of 100%. Results are expressed as mean ± SEM. A two-tailed, unpaired Student’s t-test was used to detect the significance of difference between the means of different groups. Results are considered significant if p value is <0.05.
In vitro S. mansoni worm killing
Stock solutions of 5 mM PZQ and the quinazoline derivatives 2–13 were prepared in DMSO. Concentrations of 100 µM, 50 µM, 25 µM, 10 µM and 5 µM were freshly prepared on the day of experiment in RPMI-1640 medium. All compounds were initially tested at 100 and 50 µM; those showing worm killing were further tested at 25, 10 and 5 µM.
Worms were obtained from SBSC of TBRI. Six to eight worms were placed in 12-well tissue culture plates and fresh RPMI-1640 medium (supplemented with glutamine, 20% newborn calf serum and streptomycin, penicillin, and gentamicin), containing the indicated concentration of the test compound was addedCitation20,Citation21. Worms were incubated overnight in a CO2 incubator at 37 °C. On the 2nd day, worms were examined by microscopy, washed three times with normal saline, fresh medium was added and the incubation was continued. On the 3rd day, worm motility was observed and on the 4th day, medium was changed again. On day 5 (end of the observation period), worms were microscopically examined for their motility and appearance. Each concentration was tested in duplicate, and the final recording of percent worm mortality was determined as the number of dead worms [contracted and opaque] divided by the total number of worms × 100. Negative controls used medium without additions or medium with 2% DMSO; positive control media containing identical concentrations of PZQ were tested in parallel.
S. mansoni ovipositing capacity
12-well tissue culture plates were used, each well containing 6–8 worms with at least one worm couple. Worms were incubated overnight in a CO2 incubator at 37 °C. Each concentration was tested in duplicate wells. On day 4, eggs were counted and discarded and the medium was changed. On day 5 (end of observation period) newly deposited eggs were counted. The final egg number is the total count of days 4 and 5 for each concentration tested and the final recording of the percentage of egg reduction was determined as [the total number of eggs on days 4 and 5 of control – the total number of eggs on days 4 and 5 of treated]/the total number of eggs on days 4 and 5 of control × 100.
Metabolic stability prediction
To determine the most likely sites of CYP450 mediated metabolism of NPD-1246, SMARTCypCitation22 was applied using the default settings. This tool determines the sites in a molecule that are susceptible to be metabolised using the 2D structure of the compound. It is based on a model that predicts the reactivity at C, S, N and P positions in a given ligand based on a series of over 40 rules derived from quantum chemical and calculations of energies required for oxidation using density functional theory (DFT). Finally, the atoms in the molecule are ranked according to these results.
Target prediction
With the objective of searching a potential MOA for this family of compounds, a target prediction study was performed. A consensus methodology was applied considering several approaches that involve ligand-based target prediction. Three different strategies were selected as representatives of the different tools available. Among them, polypharmacology browser (PPB)Citation23 was used using all the fingerprints combinations available in the tool and default parameters. This technique was selected because it applies an important number of fingerprints alone and in combination. Also, similarity ensemble approach (SEA) was selectedCitation24,Citation25 since both tools search in databases such as ChEMBL with an impressive amount of biological data to retrieve the most accurate results. These methods were used using default settings. From the list of potential targets, the first 35 were further considered. Finally, a search on the PDB using the main scaffold of the compounds was carried out to look for already crystallised structures.
Homology modelling
In order to produce the most accurate model for S. mansoni aldose reductase (G4LXS0), a template search was performed using the Swiss-Model serverCitation26. In the next step, the sequential alignment of the target protein was carried out with the different templates selected, the human aldose reductase (P15121) and S. japonicum Q5DD64 sequences. The templates and target sequences were retrieved from Uniprot databaseCitation27.
Due to the high identity with the S. japonicum protein, this template was selected to further build the target model using the Swiss-Model server. Once the model was built, the estimation of the protein model accuracy is required. For that purpose, model quality assessment was performed using different metrics. The local composite scoring function QMEAN (Qualitative Model Energy Analysis) allows discriminating good from bad models assessing geometrical aspects of the protein structure using several statistical descriptors. Energetic and geometric quality assessment was also performed using Ramachandran plotCitation28, ProSACitation29, ERRATCitation30 or VERIFY3DCitation31 tools.
Docking studies
Ligand preparation
The preparation of the test compounds and the 2D-to-3D conversion was carried out using the LigPrep toolCitation32, a module of the Schrödinger software package. This tool allows the preparation of molecules including different steps such as the calculation of the ionisation state of the molecules at a pH range, the addition of hydrogen atoms, and a final energy minimisation using the OPLS-2005 force fieldCitation33,Citation34. To perform the studies, physiological pH conditions were used to prepare the molecules, all of them were desalted and in the last step, the compounds were minimised as default.
Protein preparation
The structures of the proteins used in this study were preprocessed and refined using the Protein Preparation Wizard toolCitation35,Citation36 included on MaestroCitation37. H-bond assignment and calculation of the protonation state of the residues at physiological pH with a final restraint minimisation were carried out.
Docking studies
Automated docking protocol was used to assess the appropriate binding mode and suitable poses of the reference compound and the ligand. A Lamarckian genetic algorithmCitation38 method implemented in the programme AutoDock 4.2Citation39 was applied. For docking calculations, Gasteiger charges were added, rotatable bonds were set by AutoDock tools (ADT) and all torsions were allowed to rotate for the ligand. In all the cases, we used grid maps with a grid box size of 60 × 60 × 60 Å3 points and a grid-point spacing of 0.375 Å, using as centroid of the grid the key Trp111 presents in the catalytic site. The docking protocol consisted of 200 independent genetic algorithm runs, population size of 150 and maximum number of evaluation 250,000, while the remaining parameters were conserved as default. Final best-docked poses were grouped into clusters, within the default 2.0 Å RMSD. Best energetic and most representative docking clusters were analysed by visual inspection according to the binding energies and relative population provided by the software. Best poses in these clusters were considered as most reliable representatives of the ligand-binding mode and were further studied.
In the case of S. mansoni aldose reductase an additional strategy was carried out to optimise the structure of the model built. This strategy is called induced fit docking (IFD)Citation40,Citation41 and is based on fitting the ligand to the protein binding sites allowing changes in the residues geometry, mainly in their side chain orientations. First, PrimeCitation42 predicts the active site structure using the pose of compound NPD-1246 to rearrange nearby side chains of the protein and minimising the overall energy of the protein. Finally, each ligand is re-docked into its corresponding low energy protein structures and the resulting complexes are ranked according to docking score. Extra precision (XP) mode was used in a standard protocol and no constraints were set, residues were optimised to 5.0 Å of the ligand poses and the rest of the parameters were set as default.
Results
In vitro metabolic stability
NPD-1246 was exposed to mouse S9 microsomal fractions to investigate the in vitro metabolic stability through phase-I and phase-II metabolism (). The results indicate that the compound becomes extensively metabolised through phase-I but not through phase-II metabolism. After 30 min, only 27% of parent compound is left indicating poor metabolic stability. The reference drug diclofenac showed extensive phase-I and phase-II metabolism, validating the performance of the assay.
Table 1. In vitro metabolic stability of NPD-1246: percentage of parent compound remaining over time in the presence of mouse liver microsomes.
Activity in the S. mansoni-infected mouse model
NPD-1246 was previously shown to have relevant schistosomicidal activity potential in vitroCitation14. Since NPD-1246 proved to be metabolically unstable (), the compound was administered orally concomitantly with the CYP450 inhibitor ABT to counter metabolic degradationCitation43. Infected mice were treated either alone with NPD-1246 or in combination with PZQ. Control groups receiving ABT or ABT + PZQ were included as well.
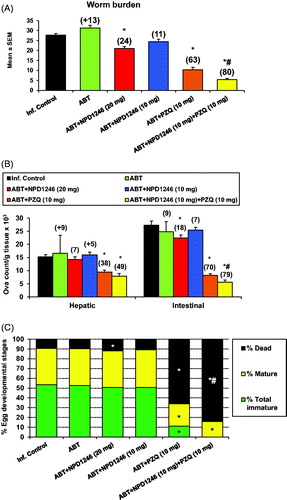
The pooled data of two experiments (number of mice ranged from 6 to 10/group in each experiment) upon a 5-day treatment with NPD-1246 at 20 mg/kg revealed a reduction in total worm burdens and intestinal tissue egg load by 24% and 18%, respectively (), accompanied with significant increase in the percentage of dead eggs when compared to the infected untreated group (). Administration of NPD-1246 at 10 mg/kg did not produce any significant change in these parameters with respect to the group dosed at 20 mg/kg (). Treatment with PZQ at 10 mg/kg significantly reduced total worms by 63% and hepatic and intestinal tissue egg loads by 38% and 70%. Total immature and mature eggs were also reduced with a significant increase in dead eggs. Finally, 5-day co-treatment of NPD-1246 and PZQ at 10 mg/kg/day revealed an enhanced reduction in total worms (80% vs. 63% for PZQ alone), intestinal tissue egg load (79% vs. 70% for PZQ alone) with complete disappearance of immature eggs and increase of dead eggs (84% vs. 66% for PZQ alone).
Figure 2. Effect of NPD-1246 alone (in a dose of 20 or 10 mg/kg/day) or in combination with PZQ (10 mg/kg/day each) for 5 days treatment on (A) worm burden, (B) tissue egg load and (C) oogram pattern in S. mansoni-infected mice sacrificed 10 days post end of treatment. *Significantly different from infected control at p < 0.05. #Significantly different from PZQ group at p < 0.05. Numbers above columns and between parentheses represent percentage change from infected control group.
Metabolic stability prediction
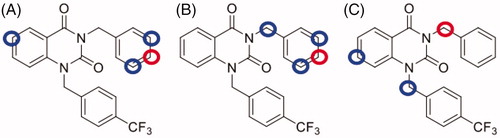
NPD-1246 shows promising antischistosomal activity potential in vivo, but was found metabolically unstable. A medicinal chemistry programme was therefore designed with the aim of improving its drug-like properties that would enable further development. Computational studies using SmartCypCitation22 were performed to identify the positions potentially susceptible for metabolic degradation. This software tool predicts CYP3A4, CYP2D6 and CYP2C9 effect on the target molecule and ranks atoms according to their probability to be modified by metabolism (Supplementary Figures S1–S3). Several potential sites were identified (). The most important of which are C6 in the quinazoline scaffold and the meta- and para-positions of the benzyl substituent in the N1.
Figure 3. Metabolic site prediction using SMARTCyp web server for NPD-1246. Top-ranked sites (red circles) and minor sites (blue circles) predicted to be metabolised by (A) CYP2C9, (B) CYP2D6 and (C) CYP3A4.
Design and synthesis of NPD-1246 derivatives
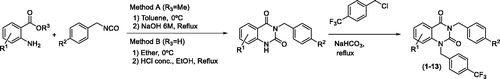
With the aim of increasing the metabolic stability of 3-benzyl-1-((4-trifluoromethyl)benzyl)quinazolin-2,4(1H,3H)-dione (NPD-1246, 1), different chemical modifications were prioritised to block the predicted positions. Methoxy and fluor were chosen as p-substituents for the benzyl tail attached to N3, while the benzene fused ring from the quinazoline remained without substituents or with halogen or alkyl groups attached to different positions. The synthesis was accomplished in two stages following previously described proceduresCitation15. In the first step, the reaction between the corresponding benzyl isocyanate and the anthranilic acid derivative yielded the substituted 3-benzylquinazolin-2,4(1H,3H)-dione used in the next step without further purification. In a second step, the mono-substituted quinazoline reacts with 4-(trifluoromethyl)benzyl chloride in basic media to obtain the desired di-substituted quinazolines 1–13 with moderate yields (Scheme 1). The compounds were characterised based on the analytical data detailed in the chemical procedures on the experimental part.
Scheme 1. Synthesis of 3-benzyl-1-(4-(trifluoromethyl)benzyl)quinazolin-2,4(1H,3H)-diones 1–13.
In vitro activity against S. mansoni
The new quinazolines 2–13 were initially tested at 100 and 50 µM against male and female worms and compared with the previously reported data for parent compound NPD-1246Citation14. The parameters for antischistosomal activity were worm mortality, motor activity alterations (sluggish worm movement or spastic contractions), unpairing and absence or reduction in egg numbers (). While almost all the new compounds are able to reduce the egg numbers and to separate or insult the coupling at both concentrations, 9 and 10 revealed 100% worm killing at 100 µM and 29% and 93% at 50 µM, respectively. Based on these data, both compounds were selected for further dose-titration. The EC50 for 9 was comparable to NPD-1246 (1) (47 µM vs. 50 µM) with 10 being slightly more active (EC50 25 µM) (). These values were obtained taking both males and females into account. When analysing each sex separately, the results remain similar pointing out that there are no obvious sex differences (). As for coupling and ova production, both compounds present a similar behaviour to NPD-1246 showing a significant reduction in egg numbers at 5 µM (i.e. the lowest concentration tested).
Table 2. Mature worm killing and ovipositing at 100 µM and 50 µM of new quinazolines (2–13) in comparison with previously reported data for NPD-1246 (1)Citation14.
Table 3. Mature (6 weeks old) worm killing and ovipositing under different concentrations of selected compounds in comparison with NPD-1246 (1) dataCitation14 previously reported.
Table 4. Mature (male & female) worm killing under different concentrations of selected compounds in comparison with NPD-1246 (1) dataCitation14 previously reported.
In vitro metabolic stability
The stability of 9 and 10 through phase-I and phase-II metabolism by mouse microsomes was investigated in a similar way than for NPD-1246 (). Both compounds presented a better profile than NPD-1246 with 51% and 39% of 9 and 10 remaining after 30 min, hence indicating acceptable stability. With human microsomes, the stability was even better with 61% and 83% of 9 and 10, respectively remaining after 30 min. None of the compounds was affected by phase-II metabolism.
Table 5. In vitro metabolic stability of 9 and 10: percentage of parent compound remaining over time in the presence of mouse and human liver microsomes.
Computational target deconvolution
Based on the confirmed in vitro and in vivo activity potential of the quinazoline family against S. mansoni, in silico studies were conducted to obtain additional information about their putative MOA. A consensus in silico methodology using tools based on ligand similarity was used, relying on the general assumption that related molecules will have similar activity and interaction patterns. Firstly, the PPBCitation23 was used as an exhaustive method in terms of fingerprint similarity. Secondly, the SEACitation24,Citation25 was selected to identify targets based on a group of known compounds rather than a single compound. As a third strategy, a structural comparison searching for similar structures in terms of chemical scaffold that had already been crystallised as protein inhibitors was performed using the protein data bank (PDB)Citation44. The three methodologies were sequentially applied to the reference compound NPD-1246.
The PPB search provides a list of potential targets, organisms and cell lines where a compound may have biological activity. In this study, the cell lines and organisms were discarded and only targets involving proteins and/or enzymes were conserved. The 35 top-ranked results are collected in Supplementary Table S1. The second approach using the SEA tool produced a ranking of targets, from which the top 35 were taken into account (Supplementary Table S2). Finally, the chemical scaffold search in the PDB retrieved five different results of crystallised quinazoline derivatives with different enzymes ().
Figure 4. Quinazoline-related structures crystallised with different target proteins according to the scaffold search in the PDB (access codes included).
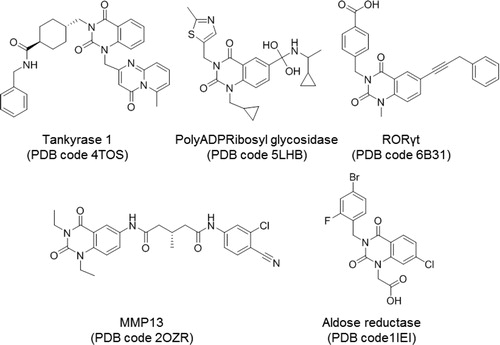
With the three methodologies combined one common target was revealed, namely the human aldose reductase. Moreover, one compound crystallised with this enzyme, zenarestat (PDB code 1IEI)Citation45 () retrieved in the search is a di-substituted quinazoline similar to NPD-1246.
Homology modelling
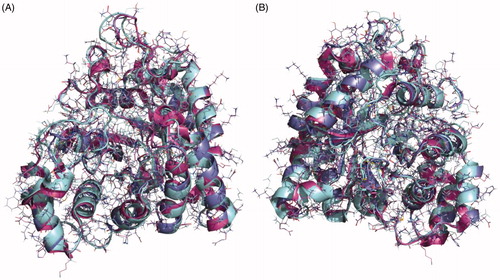
As the crystal structure of aldose reductase in S. mansoni was not available at the time of the study, a search for homologous structures was done to build an accurate model that would guide us in the computational studies on the potential binding mode of our compounds in the enzyme. The closest homologue with a crystal structure available is the aldose reductase of S. japonicum (PDB code 4HBK)Citation46 and a sequence alignment was carried out (Supplementary Figure S4). The sequences of the Schistosoma spp are almost identical showing an identity of 83.23% but the identity between the human and the S. mansoni proteins is 50.32%. The similarity between the Schistosoma enzymes is even higher considering that most of the mutations are related amino acids with similar properties. For this reason, the structure of aldose reductase from S. japonicum was chosen as the most accurate template to obtain a reliable model of our target protein using the Swiss-Model serverCitation26. The final structure was validated from a geometric and energetic point of view using several widely used metrics (Supplementary Table S3). More than 98% of the torsion angles are in allowed regions of the Ramachandran plotCitation28. It also presents an overall quality factor over 93.3% for ERRATCitation30 parameter and 92.9% according to the verify analysisCitation47. Superposition of the crystal structures of human aldose reductase (PDB code 1IEI), S. japonicum aldose reductase (PDB code 4HBK) and the homology model of S. mansoni aldose reductase is depicted in .
Figure 5. Superposition of the crystal structures of human aldose reductase 1IEI, depicted in cyan, S. japonicum aldose reductase 4HBK in magenta and the homology model of S. mansoni aldose reductase in purple, (A) front view and (B) back view.
Docking studies
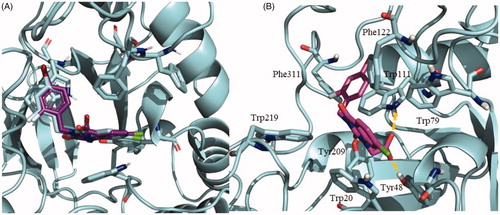
Docking studies were performed to assess the potential binding mode of NPD-1246 to S. mansoni aldose reductase. To validate our docking protocol, binding mode studies were first carried out with zenarestat, an inhibitor of the human aldose reductase crystallised with the enzyme (PDB code 1IEI)Citation45. It was observed that the majority of conformations for zenarestat were grouped in a single cluster with very good binding energy profile around −9.75 kcal/mol. This cluster was visually analysed and the best pose was compared to the crystal structure already available. The docking binding pose of zenarestat (depicted in purple) is almost identical to the one that was previously crystallised (shown in cyan) (). In a similar way to the crystal structure, the main interactions found are with aromatic residues from the catalytic site: hydrogen bonds with Tyr48 and Trp111, and several important aromatic interactions mainly critical with Trp20 and Trp111 ().
Figure 6. (A) Superimposition of zenarestat in the crystal structure 1IEI depicted in cyan and validation docking results depicted in purple show the high similarity between both poses in the human enzyme. (B) Detail of the zenarestat binding mode together with the main interactions found in the catalytic site of aldose reductase.
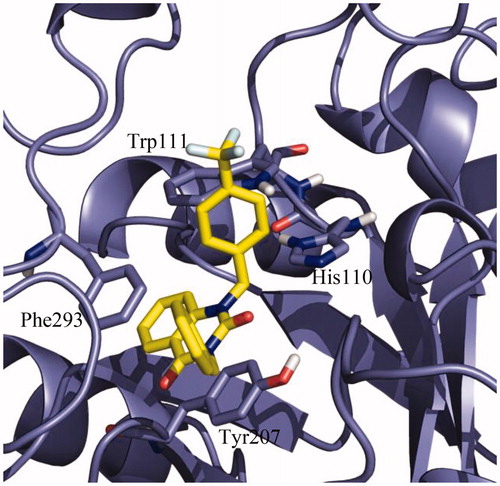
Once the protocol was validated, the potential binding mode of NPD-1246 in the aldose reductase of S. mansoni model was studied. Due to the fact that the model was based on 4HBK crystal, an apo structure of the S. japonicum aldose reductase, IFD studiesCitation40,Citation41 were performed. Once the model was optimised, a final docking with NPD-1246 was carried out using the previously validated protocol. A clear defined cluster was obtained in which the vast majority of the docking poses were grouped with very similar conformations. The best-ranked pose of this cluster, with a binding energy around −9.5 kcal/mol, was selected as representative of the binding mode of NPD-1246 in the protein. As shown in , the aromatic interactions are responsible for the stability of the ligand in the catalytic binding site, driving the ligand-binding process. Tyr207 and Phe293 are key to the stability, making face-to-face Π interactions with the quinazoline scaffold, while His110 and Trp111 are important making face-to-edge π interactions. As expected, due to the high similarity in terms of chemical structure between NPD-1246 and the novel derivatives, 9 and 10, the main interactions with the enzyme are maintained (Supplementary Figure S5).
Figure 7. Detail of the binding mode of NPD-01246 in the S. mansoni aldose reductase binding site.
Discussion
A previous phenotypic screening allowed us to successfully classify and identify promising compounds for further development based on antischistosomal in vitro potency. The selected quinazoline NPD-1246 showed an EC50 value of 50 µM in both males and females and a significant reduction in egg numbers at lower concentrations ()Citation14. Because metabolic stability is an important drug-like property to be considered for in vivo studies, the stability of NPD-1246 in the presence of mouse S9 microsomal fraction was checked. Unfortunately, the compound was extensively metabolised through phase-I metabolism (). Nevertheless, to check the in vivo activity potential, concomitant dosing with the CYP450 inhibitor ABT was carried out in the experimental S. mansoni-infected mouse model (control, NPD-1246, PZQ and combination of both).
NPD-1246 at 20 mg/kg orally showed a modest but significant reduction in total worm and intestinal egg loads together with an increase in the percentage of dead eggs. No significant differences were observed after administration at 10 mg/kg. More relevant to note is the synergy with PZQ when administered concomitantly with NPD-1246 at 10 mg/kg each (). Reduction of total worms was 80% vs. 63% for PZQ alone with a complete disappearance of immature eggs and significantly increased percentages of dead eggs (84% vs. 66% for PZQ). This fact is of pivotal importance because eggs are responsible for the pathology and transmission of the diseaseCitation1.
These results encouraged us to design NPD-1246 derivatives with improved metabolic stability. SMARTCypCitation22 software allowed us to identify position C6 in the quinazoline scaffold and meta- and para-positions of the benzyl substituents in the N1 as the most susceptible sites for metabolism by CYP450 (). Taking these predictions into account, a series of chemically related compounds with these positions blocked were synthesised following a two-step synthetic procedure (Scheme 1). The new quinazolines 2–13 were tested in vitro against S. mansoni and their activity compared with the parent compound NPD-1246 (). Two of them, 9 and 10 showed similar potency results as NPD-1246 but with an improved metabolic stability (), representing a significant improvement with respect to the original compounds.
The established antischistosomal potential of the quinazoline derivatives prompted us to investigate their MOA since target identification is becoming increasingly important to avoid/overcome drug resistance. The analysis of results obtained separately with three complementary computational methodologies (Supplementary Tables S1, S2 and ) led us to propose aldose reductase as the potential drug target for the quinazoline compounds.
S. japonicum aldose reductase was previously crystallised and antischistosomal activity of an inhibitor bearing two linked anthraquinone scaffolds was reportedCitation46. Although the role of S. japonicum aldose reductase is not fully understood, aldose reductase is believed to be an important antioxidant component in other organismsCitation48,Citation49. Moreover, antioxidant defense is an essential mechanism for schistosomes to face the damage from host immune and self-generated reactive oxygen species (ROS) and a number of redox-associated proteins have been already considered as key enzymes for drug developmentCitation50–53. Based on these evidences, and after the cloning and characterisation of S. japonicum aldose reductase, it was proposed as a potential drug target for schistosomiasis due to its possible role in the worm antioxidant mechanismCitation54.
All in all, these facts increased our interest in the quinazoline compounds as a new chemical class of inhibitors of this enzyme and a homology model of the S. mansoni aldose reductase was built based on the crystal structure of the S. japonicum counterpartCitation46 for checking the binding mode of NPD-1246. The catalytic site of aldose reductase in S. mansoni is highly hydrophobic with a high number of aromatic residues such as Trp20, Tyr48, Trp79, Trp111, Phe122 or Tyr207 among others, highlighted in the sequence alignment (Supplementary Figure S3). Docking calculations indicated that NPD-1246 and new quinazolines 9 and 10 are able to bind the catalytic site of the enzyme through important interactions with aromatic residues, such as Tyr207, His110, Trp111 and Phe293 ( and Supplementary Figure S5).
In conclusion, this study showed the antischistosomal potential of a new series of quinazoline derivatives through in vitro and in vivo studies and successful design of new derivatives to overcome the metabolic stability issues of the parent compound NPD-1246. The putative molecular target aldose reductase was identified by using complementary computational tools. This enzyme emerges as a new potential target to develop antischistosomal agents while the new quinazolines 9 and 10 represent improved candidates for further evaluation and development.
Supplemental Material
Download PDF (455 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Colley DG, Bustinduy AL, Secor WE, King CH. Human schistosomiasis. Lancet 2014;383:2253–64.
- Boissier J, Grech-Angelini S, Webster BL, et al. Outbreak of urogenital schistosomiasis in corsica (france): an epidemiological case study. Lancet Infect Dis 2016;16:971–9.
- Mutapi F, Maizels R, Fenwick A, Woolhouse M. Human schistosomiasis in the post mass drug administration era. Lancet Infect Dis 2017;17:e42–8.
- Aruleba RT, Adekiya TA, Oyinloye BE, et al. PZQ therapy: how close are we in the development of effective alternative anti-schistosomal drugs? Infect Disord Drug Targets 2019;19:337–49.
- Cioli D, Pica-Mattoccia L, Basso A, Guidi A. Schistosomiasis control: praziquantel forever? Mol Biochem Parasitol 2014;195:23–9.
- Thetiot-Laurent SA, Boissier J, Robert A, Meunier B. Schistosomiasis chemotherapy. Angew Chem Int Ed Engl 2013;52:7936–56.
- Ferreira LG, Oliva G, Andricopulo AD. Target-based molecular modeling strategies for schistosomiasis drug discovery. Future Med Chem 2015;7:753–64.
- Gilbert IH. Drug discovery for neglected diseases: molecular target-based and phenotypic approaches. J Med Chem 2013;56:7719–26.
- Heilker R, Lessel U, Bischoff D. The power of combining phenotypic and target-focused drug discovery. Drug Discov Today 2019;24:526–32.
- Sydow D, Burggraaff L, Szengel A, et al. Advances and challenges in computational target prediction. J Chem Inf Model 2019;59:1728–42.
- Gaulton A, Hersey A, Nowotka M, et al. The ChEMBL database in 2017. Nucleic Acids Res 2017;45:D945–4.
- Kim S, Chen J, Cheng T, et al. PubChem 2019 update: improved access to chemical data. Nucleic Acids Res 2019;47:D1102–D1109.
- Lounkine E, Keiser MJ, Whitebread S, et al. Large-scale prediction and testing of drug activity on side-effect targets. Nature 2012;486:361–7.
- Botros SS, William S, Sabra AA, et al. Screening of a PDE-focused library identifies imidazoles with in vitro and in vivo antischistosomal activity. Int J Parasitol Drugs Drug Resist 2019;9:35–43.
- Martinez A, Gil C, Castro A, et al. Benzothiadiazine dioxide human cytomegalovirus inhibitors: synthesis and antiviral evaluation of main heterocycle modified derivatives. Antivir Chem Chemother 2003;14:107–14.
- Liang YS, Bruce JI, Boyd DA. Laboratory cultivation of schistosome vector snails and maintenance of schistosome life cycles. Proc First Sino-Am Sym 1987;1:34–48.
- Duvall RH, DeWitt WB. An improved perfusion technique for recovering adult schistosomes from laboratory animals. Am J Trop Med Hyg 1967;16:483–6.
- Cheever AW. Conditions affecting the accuracy of potassium hydroxide digestion techniques for counting Schistosoma mansoni eggs in tissues. Bull World Health Organ 1968;39:328–31.
- Pellegrino J, Oliveira CA, Faria J, Cunha AS. New approach to the screening of drugs in experimental schistosomiasis mansoni in mice. Am J Trop Med Hyg 1962;11:201–15.
- Pica-Mattoccia L, Cioli D. Sex- and stage-related sensitivity of Schistosoma mansoni to in vivo and in vitro praziquantel treatment. Int J Parasitol 2004;34:527–33.
- Botros S, Pica-Mattoccia L, William S, et al. Effect of praziquantel on the immature stages of Schistosoma haematobium. Int J Parasitol 2005;35:1453–7.
- Rydberg P, Gloriam DE, Olsen L. The smartcyp cytochrome p450 metabolism prediction server. Bioinformatics 2010;26:2988–9.
- Awale M, Reymond JL. The polypharmacology browser: a web-based multi-fingerprint target prediction tool using chembl bioactivity data. J Cheminform 2017;9:11.
- Wang Z, Liang L, Yin Z, Lin J. Improving chemical similarity ensemble approach in target prediction. J Cheminform 2016;8:20.
- Keiser MJ, Roth BL, Armbruster BN, et al. Relating protein pharmacology by ligand chemistry. Nat Biotechnol 2007;25:197–206.
- Waterhouse A, Bertoni M, Bienert S, et al. Swiss-model: homology modelling of protein structures and complexes. Nucleic Acids Res 2018;46:W296–303.
- UniProt C. Uniprot: a worldwide hub of protein knowledge. Nucleic Acids Res 2019;47:D506–15.
- Lovell SC, Davis IW, Arendall WB, 3rd, et al. Structure validation by Cα geometry: ϕ,ψ and Cβ deviation. Proteins 2003;50:437–50.
- Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res 2007;35:W407–10.
- Colovos C, Yeates TO. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci 1993;2:1511–9.
- Eisenberg D, Luthy R, Bowie JU. VERIFY3D: assessment of protein models with three-dimensional profiles. Meth Enzymol 1997;277:396–404.
- Schrödinger Release 2015-4: Ligprep, Schrödinger, LLC, New York, NY, 2015.
- Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc 1996;118:11225–36.
- Banks JL, Beard HS, Cao Y, et al. Integrated modeling program, applied chemical theory (IMPACT). J Comput Chem 2005;26:1752–80.
- Sastry GM, Adzhigirey M, Day T, et al. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des 2013;27:221–34.
- Schrödinger Suite 2015-4 including Protein Preparation Wizard, Epik, Impact, and Prime, Schrödinger, LLC, New York, NY, 2015.
- Schrödinger Release 2015-4: Maestro, Schrödinger, LLC, New York, NY, 2015.
- Morris GM, Goodsell DS, Halliday RS, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comp Chem 1998;19:1639–62.
- Morris GM, Huey R, Lindstrom W, et al. Autodock4 and Autodocktools4: automated docking with selective receptor flexibility. J Comput Chem 2009;30:2785–91.
- Sherman W, Day T, Jacobson MP, et al. Novel procedure for modeling ligand/receptor induced fit effects. J Med Chem 2006;49:534–53.
- Friesner RA, Murphy RB, Repasky MP, et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem 2006;49:6177–96.
- Jacobson MP, Friesner RA, Xiang Z, Honig B. On the role of the crystal environment in determining protein side-chain conformations. J Mol Biol 2002;320:597–608.
- Watanabe A, Mayumi K, Nishimura K, Osaki H. In vivo use of the cyp inhibitor 1-aminobenzotriazole to increase long-term exposure in mice. Biopharm Drug Dispos 2016;37:373–8.
- Berman HM, Battistuz T, Bhat TN, et al. The protein data bank. Acta Crystallogr D Biol Crystallogr 2002;58:899–907.
- Kinoshita T, Miyake H, Fujii T, et al. The structure of human recombinant aldose reductase complexed with the potent inhibitor zenarestat. Acta Crystallogr D Biol Crystallogr 2002;58:622–6.
- Liu J, Dyer DH, Cheng J, et al. Aldose reductase from Schistosoma japonicum: crystallization and structure-based inhibitor screening for discovering antischistosomal lead compounds. Parasit Vectors 2013;6:162.
- Luthy R, Bowie JU, Eisenberg D. Assessment of protein models with three-dimensional profiles. Nature 1992;356:83–5.
- Spycher SE, Tabataba-Vakili S, O’Donnell VB, et al. Aldose reductase induction: a novel response to oxidative stress of smooth muscle cells. FASEB J 1997;11:181–8.
- Srivastava SK, Yadav UC, Reddy AB, et al. Aldose reductase inhibition suppresses oxidative stress-induced inflammatory disorders. Chem Biol Interact 2011;191:330–8.
- Alger HM, Williams DL. The disulfide redox system of Schistosoma mansoni and the importance of a multifunctional enzyme, thioredoxin glutathione reductase. Mol Biochem Parasitol 2002;121:129–39.
- Kuntz AN, Davioud-Charvet E, Sayed AA, et al. Thioredoxin glutathione reductase from Schistosoma mansoni: an essential parasite enzyme and a key drug target. PLoS Med 2007;4:e206.
- Sayed AA, Cook SK, Williams DL. Redox balance mechanisms in Schistosoma mansoni rely on peroxiredoxins and albumin and implicate peroxiredoxins as novel drug targets. J Biol Chem 2006;281:17001–10.
- Song L, Li J, Xie S, et al. Thioredoxin glutathione reductase as a novel drug target: evidence from Schistosoma japonicum. PLoS One 2012;7:e31456.
- Liu J, Wang J, Wang S, et al. Molecular cloning and characterization of Schistosoma japonicum aldose reductase. Parasitol Res 2013;112:549–58.