
Abstract
The hypothesis that sulfocoumarin acting as inhibitors of human carbonic anhydrase (CA, EC 4.2.1.1) cancer-associated isoforms hCA IX and – hCA XII is being able to also inhibit thioredoxin reductase was verified and confirmed. The dual targeting of two cancer cell defence mechanisms, i.e. hypoxia and oxidative stress, may both contribute to the observed antiproliferative profile of these compounds against many cancer cell lines. This unprecedented dual anticancer mechanism may lead to a new approach for designing innovative therapeutic agents.
Graphical Abstract
1. Introduction
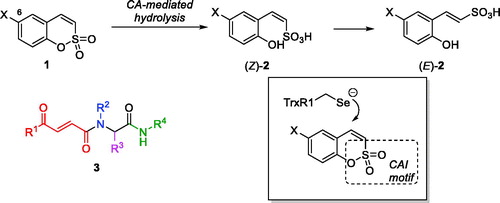
Earlier, we reported 6-substituted sulfocoumarins 1Citation1 (designed as isosteres of the structurally related coumarinsCitation2–6) as potent and remarkably isoform-selective inhibitors of the metallo-enzyme carbonic anhydrase (CA, EC 4.2.1.1)Citation7,Citation8. The ability of sulfocoumarins to selectively inhibit membrane-bound hCA IX and XII isoforms were attributed to the unique mechanism of action of these compounds whereby they act as prodrugs activated by CA-mediated hydrolysisCitation1–6. This makes these inhibitors fundamentally different from the classical carbonic anhydrase inhibitors (CAIs) – e.g. those of sulphonamide type which act by binding to the CA prosthetic zinc ion present in all isoforms, which makes designing isoform-selective sulphonamide CAIs particularly difficult. On the contrary, CA-mediated hydrolysis of sulfocoumarins 1 (as well as their progenitors coumarins) leads to the in situ formation of the Z-configured stiryl sulphonic acid (Z)-2 which is likely to isomerise to (E)-2, the active inhibitor form whose binding to CA was confirmed by X-ray crystallographyCitation1. This inhibitor activation and binding apparently occurs only in the protein environment of the two membrane-bound isoforms (hCA IX and XII) which makes these mechanistically distinct inhibitors ideal tools for targeting hypoxia survival mechanism in tumour cells providing which overexpression of precisely these two isoforms is considered responsible forCitation9. Indeed, selective targeting of hCA IX and XII has been confirmed to lead to retardation of tumour growth and, ultimately, reduction of tumour sizeCitation10.
Another principal mechanism of tumour survival which we have been recently tacklingCitation11,Citation12 as a target for anticancer agent design, is that providing tumour cell defence against oxidative stress (reactive oxygen species or ROS). In particular, tumour cells have been shown to overexpress thioredoxin reductase (TrxR, EC 1.8.1.9) which contributes to their resistant phenotype characterised by higher levels of ROSCitation13. Thus, targeting TrxR1 (the most widespread cytosolic isoform of human TrxR) has been investigated as an emerging approach to selective killing of cancer cellsCitation14. This selenocysteine (Sec) enzyme, along with NADPH and thioredoxin (Trx) is part of the Trx system and responsible for maintaining Trx in its reduced bis-sulfhydryl state. Among several classes of inhibitors of varying degree of electrophilicity towards the catalytic Sec residue (recently reviewed by BellelliCitation15 and FangCitation16), we found covalent Michael acceptor inhibitors (such as Ugi-type adducts 3 which we dubbed “Ugi Michael Acceptors” or UMAs) to be particularly efficaciousCitation12. The mechanism of inhibitory action of UMAs towards TrxR1 likely involves the irreversible covalent trapping of the selenide group of the catalytic Sec residue (which exists in the ionised form at physiological pHCitation17) by the electrophilic β-benzoylacrylamide moiety present in 3.
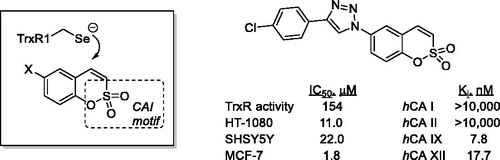
Considering the presence of a potential Michael acceptor moiety in sulfocoumarins 1, we hypothesised that in addition to their inhibitory activity towards hCAs, these compounds could potentially act as Michael acceptor-type TrxR inhibitors (), thus acting as dual inhibitors which target two cancer cell defence mechanisms at a time. Herein, we present our preliminary results obtained in the course of verifying this hypothesis.
Figure 1. Sulfocoumarins 1 and their CA inhibition mechanism, the previously reported Ugi Michael acceptor TrxR inhibitors (fragments originating from the four components of the Ugi reaction are colour-coded) and the hypothesis for dual CA/TrxR targeting verified in this work.
2. Materials and methods
2.1. Chemical syntheses – general
Reagents and starting materials were obtained from commercial sources (Sigma-Aldrich, St. Louis, MO) and used as received. The solvents were purified and dried by standard procedures prior to use; petroleum ether of boiling range 40–60° C was used. Flash chromatography was carried out using Merck silica gel (230–400 mesh). Thin-layer chromatography was performed on silica gel, spots were visualised with UV light (254 and 365 nM). Melting points were determined on an OptiMelt automated melting point system. IR spectra were measured on a Shimadzu FTIR IR Prestige-21 spectrometer. NMR spectra were recorded on Varian Mercury (400 MHz) spectrometer with chemical shifts values (d) in ppm relative to TMS using the residual DMSO-d6 signal as an internal standard. Elemental analyses were performed on a Carlo Erba CHNSeO EA-1108 apparatus. Starting material sulfocoumarins (4Citation18 and 5Citation19) were prepared as described previously. Alkynes employed in the synthesis of 1a–b are commercially available. Tetrazoles employed in the synthesis of 1c–d were prepared according to the literature protocolsCitation20,Citation21. All reagents for biological assays were purchased from Sigma (St. Louis, MO).
2.2. General procedure 1: preparation of sulfocoumarins 1a–b (GP1)
To a solution of 4 (1.0 equiv.) in dry THF (1 mL per mmol of 4) N,N-diisopropylethylamine (DIPEA) (50 equiv.), the appropriate alkyne (1.1, 2.0, or 5.0 equiv.), and CuI (2 equiv.) were added. The resulting mixture was stirred at room temperature under an argon atmosphere for 20 h. Saturated NH4Cl was added and extracted with EtOAc, washed with brine and dried over Na2SO4, and evaporated.
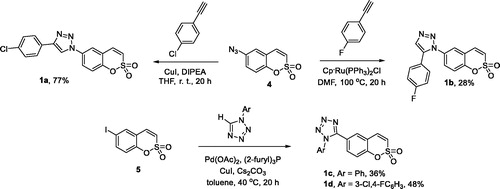
2.2.1. 4-(4-Chlorophenyl)-1-(2,2-dioxido-1,2-benzoxathiin-6-yl)-1H-1,2,3-triazole (1a)
Prepared from 4 (0.15 g, 0.67 mmol), 4-chlorophenylacetylene (0.18 g, 1.34 mmol), CuI (0.26 g, 1.34 mmol), and DIPEA (5.85 mL, 33.6 mmol) according to GP1. Crystallisation from ethanol afforded 1a as yellow crystalline solid (0.19 g, 77%). Mp 236–237 °C. IR (KBr, cm−1) νmax: 1369 (S–O), 1179 (S–O), and 1169 (S–O). 1H NMR (400 MHz, DMSO-d6) δ: 7.55–7.60 (m, 2H), 7.70 (d, J= 10.4 Hz, 1H), 7.75 (d, J= 8.9 Hz, 1H), 7.84 (d, J= 10.4 Hz, 1H), 7.92–7.97 (m, 2H), 8.12 (dd, J= 8.9, 2.7 Hz, 1H), 8.39 (d, J= 2.7 Hz, 1H), and 9.38 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ: 119.9, 120.2, 120.3, 121.4, 123.7, 124.0, 127.0, 128.9, 129.2, 132.9, 134.2, 135.8, 146.4, and 150.1. Anal. Calcd. for C16H10N3O3SCl (359.79): C, 53.41; H, 2.80; N, 11.68. Found: C, 53.22; H, 2.79; N, 11.32.
2.2.2. 1-(2,2-Dioxido-1,2-benzoxathiin-6-yl)-4-(4-fluorophenyl)-1H-1,2,3-triazole (1b)
Prepared from 4 (0.15 g, 0.67 mmol), 4-fluorophenylacetylene (0.16 g, 1.34 mmol), CuI (0.26 g, 1.34 mmol), and DIPEA (5.85 mL, 33.6 mmol) according to GP1. Yellow crystalline solid (0.19 g, 80%). Mp 224–225 °C. IR (KBr, cm−1) νmax: 1359 (S–O) and 1179 (S–O). 1H NMR (400 MHz, DMSO-d6) δ: 7.32–7.39 (m, 2H), 7.71 (d, J= 10.4 Hz, 1H), 7.75 (d, J= 8.9 Hz, 1H), 7.84 (d, J= 10.4 Hz, 1H), 7.94–8.00 (m, 2H), 8.12 (dd, J= 8.9, 2.6 Hz, 1H), 8.39 (d, J= 2.6 Hz, 1H), and 9.33 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ: 116.1 (d, J= 21.9 Hz), 119.8, 119.9, 120.2, 121.4, 123.7, 124.0, 126.6 (d, J= 3.2 Hz), 127.4 (d, J= 8.3 Hz), 134.2, 135.9, 146.6, 150.1, and 162.4 (d, J= 245.3 Hz). Anal. Calcd. for C16H10N3O3SF (343.33): C, 55.97; H, 2.94; N, 12.24. Found: C, 55.78; H, 2.94; N, 12.24.
2.2.3. 5-(2,2-Dioxido-1,2-benzoxathiin-6-yl)-1-phenyl-1H-tetrazole (1c)
Compound 5 (0.200 g, 0.649 mmol), 1-phenyl-1,2,3,4-tetrazoleCitation20 (0.190 g, 1.30 mmol), Cs2CO3 (0.233 g, 0.714 mmol), CuI (0.124 g, 0.649 mmol), Pd(OAc)2 (0.0146 g, 0.0649 mmol), and tris(2-furyl) phosphine (0.030 g, 0.130 mmol) were suspended in dry toluene (3 mL). The mixture was stirred at 40 °C under argon for 20 h, then EtOAc (20 mL) was added and the mixture was filtered through celite. Celite was washed with EtOAc (50 mL). The filtrate and washings were combined and concentrated under reduced pressure. The residue was purified by silica gel chromatography (petroleum ether/EtOAc 2:1) and additionally crystallised from EtOH to give 1c as yellow crystalline solid (0.076 g, 36%). Mp 189–190 °C. IR (KBr, cm−1) νmax: 1370 (S–O) and 1178 (S–O). 1H NMR (400 MHz, DMSO-d6) δ: 7.49–7.56 (m, 2H), 7.58–7.68 (m, 6H), 7.78 (d, 1H, J= 10.4 Hz), and 8.09–8.12 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ: 119.2, 119.3, 121.6, 123.6, 126.0, 130.0, 130.8, 131.0, 132.5, 133.8, 135.9, 152.2, and 152.4. Anal. Calcd. for C15H10N4O3S (326.33): C,55.21; H, 3.09; N, 17.17. Found: C, 55.25; H, 3.09; N, 17.08.
2.2.4. 1-(2,2-Dioxido-1,2-benzoxathiin-6-yl)-5-(4-fluorophenyl)-1H-1,2,3-triazole (1d)
To a solution of 5 (0.25 g, 1.12 mmol) and 4-fluorophenylacetylene (0.27 g, 2.24 mmol) in dry DMF (0.7 mL), Cp*Ru(PPh3)2Cl (0.01 mmol) was added and the resulting mixture was stirred at 100 °C under an argon atmosphere for 20 h. The solvent was removed under reduced pressure. The residue was purified by silica gel chromatography (petroleum ether/EtOAc 2:1) to give 1d as yellow crystalline solid (0.11 g, 28%). Mp 157–158 °C. IR (neat, cm−1) νmax: 1373 (S–O) and 1176 (S–O). 1H NMR (400 MHz, DMSO-d6) δ: 7.25–7.33 (m, 2H), 7.37–7.43 (m, 2H), 7.56 (dd, J= 8.8, 2.5 Hz, 1H), 7.61 (d, J= 8.8 Hz, 1H), 7.67 (d, J= 10.4 Hz, 1H), 7.75 (d, J= 10.4 Hz, 1H), 7.97 (d, J= 2.5 Hz, 1H), and 8.17 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ: 116.1 (d, J= 22.1 Hz), 119.6, 119.8, 122.4 (d, J= 3.2 Hz), 123.7, 127.1, 129.3, 131.1 (d, J= 8.8 Hz), 133.4, 133.6, 135.7, 137.0, 150.8, and 162.6 (d, J= 247.7 Hz). Anal. Calcd. for C16H10N3O3SF (343.33): C, 55.97; H, 2.94; N, 12.24. Found: C, 56.17; H, 2.93; N, 11.93.
2.3. Carbonic anhydrase inhibition assay
An Applied Photophysics stopped-flow instrument has been used for assaying the CA catalysed CO2 hydration activityCitation22. Phenol red (at a concentration of 0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 20 mM Tris (pH 8.3) as buffer, and 20 mM Na2SO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalysed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor, at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The uncatalysed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (0.1 mM) were prepared in distilled-deionised water and dilutions up to 0.005 nM were done thereafter with the assay buffer. Inhibitor and enzyme solutions were pre-incubated together for 15 min at room temperature prior to assay, in order to allow for the formation of the E–I complex. The inhibition constants were obtained by non-linear least-squares methods using PRISM 3 and the Cheng-Prusoff equation, as reported earlier, and represent the mean from at least three different determinations. All CA isoforms were recombinant ones obtained in-houseCitation23–26.
2.4. TrxR activity by DTNB reduction assay
Determination of TrxR activity in SHSY5Y cell lysate. TrxR activity in cell lysate was measured in 96-well plates using previously described methodsCitation27,Citation28. For TrxR activity measurement, compounds of different concentrations were incubated with 50 µg of cell lysate and 200 µM NADPH in a volume of 100 µL of 50 mM Tris–HCl and 1 mM EDTA, pH 7.5 (TE buffer), for different time points in 96-well plates at room temperature. Then, 100 µL of TE buffer containing DTNB and NADPH was added (final concentration: 2.5 mM and 200 µM, respectively), and the linear increase in absorbance at 412 nm during the initial 2 min was measured with a Tecan Infinite M1000 multifunctional microplate reader. TrxR activity was calculated as a percentage of enzyme activity of that of DMSO vehicle treated sample.
2.5. Cytotoxicity assay
Thus, monolayer tumour cell lines HT-1080 (human fibrosarcoma), SHSY5Y (human neuroblastoma), and MCF-7 (breast adenocarcinoma) were cultured in standard medium DMEM (Dulbecco’s modified Eagle’s medium) supplemented with 10% foetal bovine serum. About 2000–4000 cells per well (depending on line nature) were placed in 96-well plates and after 24 h compounds were added to the wells. Untreated cells were used as a control. The plates were incubated for 48 h, 37 °C, and 5% CO2. The number of surviving cells was determined using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolinium bromide (MTT). MTT-test: after incubating culture medium was removed and 200 µL fresh medium with 20 µL MTT (2 mg/mL in HBSS) was added in each well of the plate. After incubation (3 h, 37 °C, 5% CO2), the medium with MTT was removed and 200 µL DMSO were added at once to each sample. The samples were tested at 540 nm on Thermo Scientific Multiskan EX microplate photometer. The half-maximal inhibitory concentration (IC50) of each compound was calculated using Graph Pad Prism® 3.0 (GraphPad Software, La Jolla, CA).
3. Results and discussion
3.1. Chemistry
Compounds 1a and 1b were synthesised from azide 4 as described previouslyCitation18. CuI-catalysed Huisgen azide-alkyne cycloaddition gave 1,4-disubstituted 1,2,3-triazole 1a while employing RuII-catalysed protocol gave 1,5-disusbstituted 1,2,3-triazole 1b. For the synthesis of 1,5-disubstituted tetrazoles 1c–d, the previously describedCitation19 Pd-catalysed arylation of 1-aryl tetrazoles with aryl iodide 5 was employed (Scheme 1).
Scheme 1. Synthesis of compounds 1a–d.
3.2. Biological evaluation
To our utmost delight, when the previously establishedCitation18,Citation19 potent and selective inhibitory profile of compounds 1a–d towards cancer-related hCA IX and hCA XII isoforms was confirmed in reference to known CAI acetazolamide (AAZ), we have also found these compounds to display dose-dependent inhibition of TrxR activity in SHSY5Y cell lysate with IC50 values confidently residing in the 10−5…10−4 M range. Adding to the satisfaction over having our initial hypothesis regarding the dual CA/TrxR inhibitory effects of compounds 1, rather potent antiproliferative activity was established as evaluated against cultures of cancer cells such as HT-1080 (human fibrosarcoma), SHSY5Y (human neuroblastoma), and MCF-7 (breast adenocarcinoma). These findings are summarised in .
Table 1. Inhibitory profile towards four hCA isoforms, TrxR activity in SHSY5Y cell lysate and cytotoxicity towards cancer cell lines determined for compounds 1a–d (nd: not determined).
4. Conclusions
The previously described sulfocoumarins that were shown to potently and selectively inhibit cancer-related hCA IX and hCAXII isoforms (whose overexpression is a well-established mechanism of tumour cell defence against hypoxia) also display noticeable, dose-dependent inhibition of TrxR activity in cancer cell lysates. As overexpression of TrxR in cancer cells is a defence mechanism against oxidative stress, the established dual inhibition pattern constitutes a significant starting point for the design and discovery of new anticancer agents based on the dual targeting of the two defence mechanisms crucial for cancer cell survival. This communication opens a new line of research in our laboratories aimed at investigating the practical aspects of the new dual inhibitor design and establishing a well-understood link between inhibition of these two enzyme groups and the dual inhibitors’ antitumor activity. The results of this research will be reported in due course.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Tars K, Vullo D, Kazaks A, et al. Sulfocoumarins (1, 2-benzoxathiine-2, 2-dioxides): a class of potent and isoform-selective inhibitors of tumor-associated carbonic anhydrases. J Med Chem 2013;56:293–300.
- Maresca A, Temperini C, Vu H, et al. Non-zinc mediated inhibition of carbonic anhydrases: coumarins are a new class of suicide inhibitors. J Am Chem Soc 2009;131:3057–62.
- Touisni N, Maresca A, McDonald PC, et al. Glycosyl coumarin carbonic anhydrase IX and XII inhibitors strongly attenuate the growth of primary breast tumors. J Med Chem 2011;54:8271–7.
- Carta F, Maresca A, Scozzafava A, Supuran CT. 5- and 6-Membered (thio)lactones are prodrug type carbonic anhydrase inhibitors. Bioorg Med Chem Lett 2012;22:267–70.
- Maresca A, Temperini C, Pochet L, et al. Deciphering the mechanism of carbonic anhydrase inhibition with coumarins and thiocoumarins. J Med Chem 2010;53:335–44.
- Davis RA, Vullo D, Maresca A, et al. Natural product coumarins that inhibit human carbonic anhydrases. Bioorg Med Chem 2013;21:539–43.
- Alterio V, Di Fiore A, D’Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68.
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81.
- Schwartz L, Supuran CT, Alfarouk KO. The Warburg effect and the hallmarks of cancer. Anti-Cancer Agents Med Chem 2017;17:164–70.
- Supuran CT. Carbonic anhydrase inhibition and the management of hypoxic tumors. Metabolites 2017;7:48.
- Bakulina O, Bannykh A, Jovanović M, et al. Design, synthesis, and biological evaluation of novel derivatives of dithiodiglycolic acid prepared via oxidative coupling of thiols. J Enzyme Inhib Med Chem 2019;34:665–71.
- Jovanović M, Zhukovsky D, Podolski-Renić A, et al. Novel electrophilic amides amenable by the Ugi reaction perturb thioredoxin system via thioredoxin reductase 1 (TrxR1) inhibition: identification of DVD-445 as a new lead compound for anticancer therapy. Eur J Med Chem 2019;181:111580.
- Zhang J, Li X, Han X, et al. Targeting the thioredoxin system for cancer therapy. Trends Pharmacol Sci 2017;38:794–808.
- Arner ES, Holmgren A. The thioredoxin system in cancer. Semin Cancer Biol 2006;16:420–6.
- Saccoccia F, Angelucci F, Boumis G, et al. Thioredoxin reductase and its inhibitors. Curr Protein Pept Sci 2014;15:621–46.
- Zhang B, Zhang J, Peng S, et al. Thioredoxin reductase inhibitors: a patent review. Exp Opin Ther Pat 2017;27:547–56.
- Copeland PR. Making sense of nonsense: the evolution of selenocysteine usage in proteins. Genome Biol 2005;6:221.
- Grandane A, Tanc M, Zalubovskis R, Supuran CT. 6-Triazolyl-substituted sulfocoumarins are potent, selective inhibitors of the tumor-associated carbonic anhydrases IX and XII. Bioorg Med Chem Lett 2014;24:1256–60.
- Grandane A, Tanc M, Zalubovskis R, Supuran CT. Synthesis of 6-tetrazolyl-substituted sulfocoumarins acting as highly potent and selective inhibitors of the tumor-associated carbonic anhydrase isoforms IX and XII. Bioorg Med Chem Lett 2014;22:1522–8.
- Potewar TM, Siddiqui SA, Lahoti RJ, Srinivasan KV. Efficient and rapid synthesis of 1-substituted-1H-1,2,3,4-tetrazoles in the acidic ionic liquid 1-n-butylimidazolium tetrafluoroborate. Tetrahedron Lett 2007;48:1721–4.
- Voitekhovich SV, Vorob'ev AN, Gaponik PN, Ivashkevich OA. Synthesis of new functionally substituted 1-R-tetrazoles and their 5-amino derivatives. Chem Heterocycl Compd 2005;41:999–1004.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73.
- Maresca A, Carta F, Vullo D, Supuran CT. Dithiocarbamates strongly inhibit the β-class carbonic anhydrases from Mycobacterium tuberculosis. J Enzyme Inhib Med Chem 2013;28:407–11.
- Ekinci D, Kurbanoglu NI, Salamci E, et al. Carbonic anhydrase inhibitors: inhibition of human and bovine isoenzymes by benzenesulphonamides, cyclitols and phenolic compounds. J Enzyme Inhib Med Chem 2012;27:845–8.
- Ekinci D, Karagoz L, Ekinci D, et al. Carbonic anhydrase inhibitors: in vitro inhibition of α isoforms (hCA I, hCA II, hCA III, hCA IV) by flavonoids. J Enzyme Inhib Med Chem 2013;28:283–8.
- Alp C, Maresca A, Alp NA, et al. Secondary/tertiary benzenesulfonamides with inhibitory action against the cytosolic human carbonic anhydrase isoforms I and II. J Enzyme Inhib Med Chem 2013;28:294–8.
- Luthman M, Holmgren A. Rat liver thioredoxin and thioredoxin reductase: purification and characterization. Biochemistry 1982;21:6628–33.
- Arner ES, Zhong L, Holmgren A. Preparation and assay of mammalian thioredoxin and thioredoxin reductase. Meth Enzymol 1999;300:226–39.