
Abstract
Cyclic imides containing 3-benzenesulfonamide, oxime, and β-phenylalanine derivatives were synthesised and evaluated to elucidate their in vivo anti-inflammatory and ulcerogenic activity and in vitro cytotoxic effects. Most active anti-inflammatory agents were subjected to in vitro COX-1/2 inhibition assay. 3-Benzenesulfonamides (2–4, and 9), oximes (11–13), and β-phenylalanine derivative (18) showed potential anti-inflammatory activities with 71.2–82.9% oedema inhibition relative to celecoxib and diclofenac (85.6 and 83.4%, respectively). Most active cyclic imides 4, 9, 12, 13, and 18 possessed ED50 of 35.4–45.3 mg kg−1 relative to that of celecoxib (34.1 mg kg−1). For the cytotoxic evaluation, the selected derivatives 2–6 and 8 exhibited weak positive cytotoxic effects (PCE = 2/59–5/59) at 10 μM compared to the standard drug, imatinib (PCE = 20/59). Cyclic imides bearing 3-benzenesulfonamide (2–5, and 9), acetophenone oxime (11–14, 18, and 19) exhibited high selectivity against COX-2 with SI > 55.6–333.3 relative to that for celecoxib [SI > 387.6]. β-Phenylalanine derivatives 21–24 and 28 were non-selective towards COX-1/2 isozymes as indicated by their SI of 0.46–0.68.
1. Introduction
Nonsteroidal anti-inflammatory drugs (NSAIDs) are the drugs of choice for the treatment of inflammation and painCitation1,Citation2. NSAIDs are cyclooxygenase inhibitors and they include the COX-1 and COX-2 enzymesCitation1–3. The cyclooxygenase isozymes are responsible for the conversion of arachidonic acid to eicosanoids (prostaglandins)Citation1–3. In this process, the inhibition of the COX-2 isozyme elucidates the anti-inflammatory effects of NSAIDsCitation1–3 while the inhibition of the COX-1 isozyme is responsible for the major side effects displayed by NSAIDs, such as gastrointestinal implicationCitation1,Citation2. Alternatively, selective COX-2 inhibitors, including compounds that contain the pyrazole ring system, such as celecoxib (A) and SC-558 (B)Citation4 (), display anti-inflammatory activity with improved gastric profile protection compared to NSAIDsCitation4. Moreover, celecoxib, a COX-2 inhibitor () that displays an anti-inflammatory effect, has been investigated as an antitumor agentCitation5.
Figure 1. Some reported selective COX-2 inhibitors (A–H) and the designed compounds (I).
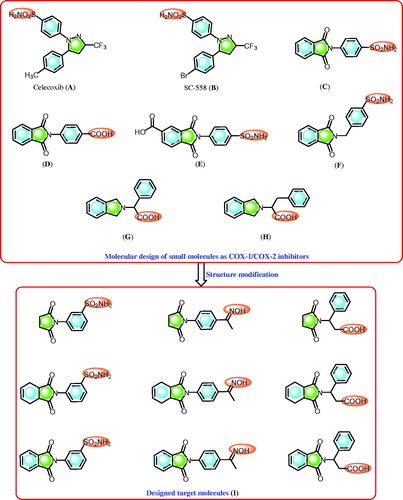
Compounds containing the sulphonamide moiety are interesting derivatives that possess versatile and potential biological activities, such as carbonic anhydrase inhibitorsCitation6–9, COX-1/2 inhibition ()Citation6a,Citation10,Citation11, and anti-inflammatory ()Citation6a,Citation10,Citation11 and antitumor activitiesCitation6a,Citation12. Recently, we reported the synthesis of phthalimide derivatives with potential anti-inflammatoryCitation6a,Citation10,Citation11, antitumorCitation6a,Citation13, hypoglycaemic, and antihyperlipidemic propertiesCitation14. In addition, some of these compounds were identified to display potent COX-2 and carbonic anhydrase inhibitory activitiesCitation6–11. Recently, phthalimide analogues such isoindolines bearing α-amino acids are reported as promising COX-1/COX-2 inhibitors ()Citation15.
Here, we report the design and synthesis of novel cyclic imides containing 3-benzenesulfonamide, acetophenone oxime, and β-phenylalanine scaffolds. The designed cyclic imides () were subjected to various analyses to: (i) investigate their in vivo anti-inflammatory and ulcerogenic activities, and their in vitro COX-1/2 inhibitory effects; (ii) study their in vitro cytotoxicity activity; (iii) explore their structure-activity relationships (SAR) based on their in vivo anti-inflammatory effects and in vitro COX-1/2 inhibitory activity with molecules containing substituted cyclic imides; (iv) compare the biological effects of 3-benzenesulfonamide to those of acetophenone oxime and β-phenylalanine based on in vivo anti-inflammatory and COX-1/2 inhibition; and (v) conduct a molecular docking study of the target derivatives to investigate their binding with the COX-2 isozyme.
2. Results and discussion
2.1. Chemistry
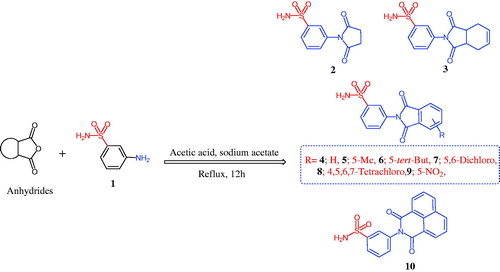
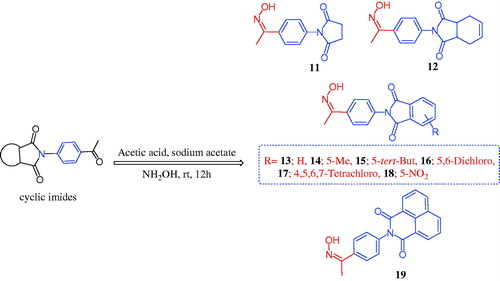
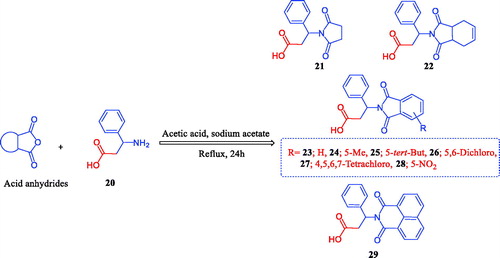
The chemistry for the synthesis of the target molecules 2–10, 11–19, and 21–29 is shown in Schemes 1–3. The rationale used for the synthesis of these compounds is based on two routes where a non-carboxylic moiety, such as 3-benzenesulfonamide and hydroxyiminoethyl (oxime), and a carboxylic moiety, such as an amino acid (β-phenylalanine), are inserted into a versatile cyclic imides to explore and compare the efficacy of the substituted cyclic imides for inhibiting COX-1/COX-2 and evaluating their anti-inflammatory activity.
Non-carboxylic cyclic imide-based 3-benzenesulfonamides 2–10 and oximes 11–19 were synthesised as indicated in Schemes 1 and 2. Different carboxylic acid anhydrides were refluxed with 3-aminobenzenesulfonamide (1) in glacial acetic acid in the presence of anhydrous sodium acetateCitation6–11 to obtain cyclic imides 2–10 of good yields (Scheme 1). Scheme 2 displays the process used to obtain the 2-[4-(1-(hydroxyimino)ethyl)phenyl] derivatives 11–19 via the stirring of 2-(4-acetylphenyl)-1,3-isoindolinediones and hydroxylamine hydrochloride in glacial acetic acid containing anhydrous sodium acetateCitation6b. The acid imides 21–29 were synthesised as indicated in Scheme 3. The 3-(1,3-dioxoisoindolin-2-yl)-3-phenylpropanoic acid derivatives 21–29 were obtained with 66–93% yield by refluxing the cyclic imides and β-phenylalanines in glacial acetic acid containing anhydrous sodium acetate (Scheme 3). The molecular structures of the newly synthesised compounds 2–10, 11–19, and 21–29 were verified by spectral analyses including mass, IR, 1H NMR, and 13C NMR spectra as indicated in the experimental section.
Scheme 1. Synthesis of 1,3-isoindolinediones incorporating 3-benzenesulfonamide 2–10.
Scheme 2. Synthesis of 1,3-isoindolinediones incorporating oxime 11–19.
Scheme 3. Synthesis of 1,3-isoindolinediones incorporating β-phenylalanine 21–29.
2.2. Biological activity
2.2.1. In vivo anti-inflammatory activity and SAR study
Twenty-seven compounds, 2–10, 11–19, and 21–29 and the reference drugs, diclofenac and celecoxib, were administered to the rat carrageenan-induced foot paw oedema model to explore the in vivo anti-inflammatory activities of such compounds. The percentage inhibition of paw oedema was calculated after 2 h of carrageenan treatment when maximal inhibition of carrageenan-induced paw oedema was observed ()Citation3,Citation16,Citation17.
Table 1. Results of anti-inflammatory activity of the tested compounds against carrageenan induced rat paw oedema in ratsa.
Data in show that most of the designed compounds decreased paw oedema by 21.2–82.9% (). The reference drugs, diclofenac and celecoxib, caused 83.4 and 85.6% oedema inhibition, respectively. The non-carboxylic derivatives 2–10 (sulphonamide) and 11–19 (oxime) were generally more potent than the corresponding carboxylic acid derivatives 21–29 (β-phenylalanine), as observed for the non-carboxylic cyclic imides 2–4, 9, 11–13, and 18. These derivatives displayed the highest anti-inflammatory activities with 71.2–82.9% paw oedema reduction compared to the acidic derivatives 21–23 and 28, with 42.5–64.4% paw oedema reduction. For sulphonamide derivatives 2–10, the succinimide derivative 2 displayed a greater anti-inflammatory activity than the naphthalimide derivative 10, with percentage paw oedema reduction of 71.2 and 49.2%, respectively. The conversion of succinimide 2 to the corresponding tetrahydrophthalimide 3 or phthalimide 4 led to an increase in the anti-inflammatory activity, with percentage paw oedema reduction of 77.0 and 77.8%, respectively. Electronic substitution on the phthalimide core has a direct effect on the anti-inflammatory effects. For example, the presence of the methyl or tert-butyl group at the 5-position led to a decrease in activity, as observed for compounds 5 and 6, which had a percentage paw oedema reduction of 57.7 and 40.9%, respectively. Replacing the alkyl group with halogen atoms resulted in derivatives with weak anti-inflammatory activity as observed for compounds 7 and 8 (29.7 and 26.9% paw oedema reduction, respectively). However, modify 5-methyl group with 5-nitro derivative resulted in a sharp increase in the anti-inflammatory effects, as observed for compounds 5 and 9, with 57.7 and 80.9% paw oedema reduction, respectively. Interestingly, converting the 3-benzenesulfonamides 2–10 (26.9–80.9% oedema inhibition) into the corresponding oxime derivatives 11–19 maintained the levels of the anti-inflammatory activity (25.1–82.9% oedema inhibition). In the latter compounds 11–19, it was evident that the derivatives containing succinimide 11, tetrahydrophthalimide 12, phthalimide 13, and 5-nitro phthalimide 18 had anti-inflammatory activity levels (75.6, 79.7, 80.4, and 82.9% oedema inhibition, respectively) higher than the corresponding analogues 14–17 and 19 (61.6, 48.1, 39.6, 25.1, and 68.8% oedema inhibition, respectively). Replacing 3-benzenesulfonamide with compounds that had a terminal carboxylic moiety at the cyclic imide core 21–29 caused a decrease in anti-inflammatory levels, with percentage oedema inhibition ranging from 21.2–64.4%. Similarly, the phthalimide derivative 23 had a higher activity than succinimide 21 or naphthalimide 29, with percentage paw oedema reduction of 64.4, 46.4, and 27.0%, respectively. In contrast to derivatives 3, 9, 13, and 18, introducing a nitro group, as observed for compound 23, on the phthalimide core resulted in derivative 28, with a 42.5% decrease in oedema inhibition. Finally, cyclic imides with 3-benzenesulfonamide 2–10 or oxime 11–19 fragments had the greatest levels of anti-inflammatory activity, which were almost similar to those of diclofenac and celecoxib, indicating that both derivatives exerted similar interactions at the COX receptor binding site.
shows the three graded doses of the anti-inflammatory activities of the most active cyclic imides 4, 9, 12, 13, 18, and celecoxib.
Table 2. Results of anti-inflammatory activity of compounds 4, 9, 12, 13, 18, and celecoxib against carrageenan induced rat paw oedema in rats at three graded doses.
2.2.2. Ulcerogenicity
The levels of ulcerogenic activity for the most active cyclic imides 4, 9, 12, 13, 18, along with the reference drugs, diclofenac and celecoxib, were determined according to the reported technique ()Citation6a,Citation17,Citation18. Evidently, the phthalimide derivatives 9 and 18 had very low levels of ulcerogenic activity relative to diclofenac and celecoxib, whereas phthalimides 4, 12, and 13 had an ulcerogenic level similar to celecoxib and less than that of diclofenac ().
Table 3. Ulcerogenic potential of the tested compounds 4, 9, 12, 13, 18, diclofenac and celecoxib in miceTable Footnote*.
2.2.3. Cytotoxicity
Six compounds were selected by the National Cancer Institute (Bethesda, MD) based on structural variations to evaluate the in vitro cytotoxicity of compounds 2–6 and 8. Thereafter, the results were compared to those of the reference drug, imatinib, as shown in . These compounds, which are derivatives of cyclic imides bearing 3-benzenesulfonamides 2–6 and 8, were administered in single doses of 10 µM in a full NCI 59 cell line panel assay. These cell lines were obtained from nine different organs including leukaemia, non-small cell lung, colon, CNS, melanoma, ovarian, renal, prostate, and breastCitation19. The results are displayed in and expressed as the percentage growth inhibition (GI %) caused by the test compounds. Based on the results, the positive cytotoxic effects (PCE) of the tested cyclic imides 2–6 and 8 at 10 μM were 2/59–5/59, while the reference drug, imatinib, had a PCE of 20/59 ().
Table 4. Antitumor activity of trimellitimides derivatives 2–6 and 8 presented as growth inhibition percentages (GI %) over 59 subpanel tumour cell lines.
2.2.4. COX-1/2 inhibition and SAR study
The derivatives of cyclic imides 2–6, 9–15, 18, 19, 21–24, and 28, which showed anti-inflammatory activity higher than the 30% oedema inhibition, were subjected to COX-1 and COX-2 inhibition assay, with the reference drug, celecoxib, using an ovine COX-1/COX-2 assay kit (Cayman Chemicals Inc., Ann Arbour, MI). The IC50 (μM) and the selectivity indices (SI; IC50 (COX-1)/IC50 (COX-2)) are listed in . Data in show the value >387.6 as a selectivity index (SI; IC50 (COX-1)/IC50 (COX-2)) of the reference drug, celecoxib, with IC50 values >50/0.129 μM for COX-1/COX-2. The COX-1/COX-2 assay indicated that cyclic imides bearing a 3-benzenesulfonamide or 2-[4-(1-(hydroxyimino)ethyl)phenyl] fragment, as observed for derivatives 2–5, 9, 11–14, 18, and 19, were considered to be potent COX-2 inhibitors with IC50 ≅ 0.15–0.90 μM and SI ≅ >333.3 to >55.6. The results of cyclic imides containing the non-carboxylic tails 2–4, 9, 11–13, 18, and 19, were comparable to that of the reference drug, celecoxib (IC50 = 0.129 μM; COX-2 (SI) > 387.6). In contrast, cyclic imides containing a carboxylic tail, as observed for derivatives 21–24 and 28, were non-selective COX-1/2 inhibitors with IC50 (COX-1) ≅ 10.9 − 24.8 μM and IC50 (COX-2) ≅ 22.3 − 36.3 μM and SI ≅ 0.46 − 0.68.
Table 5. In vitro cyclooxygenase (COX-1/COX-2) enzyme inhibition assay and calculated selectivity indices.
The inhibitors, cyclic imides 2–4, 9, 11–13, 18, and 19, were the most potent and active derivatives with IC50 values of 0.26 μM (SI > 192.3), 0.20 μM (SI > 250.0), 0.18 μM (SI > 277.8), 0.15 μM (SI > 333.3), 0.22 μM (SI > 227.3), 0.16 μM (SI > 312.5), 0.16 μM (SI > 312.5), 0.15 μM (SI > 333.3), and 0.28 μM (SI > 178.6), respectively, relative to celecoxib which had an IC50 value of 0.129 μM and SI value of >387.6. The cyclic imides incorporating oximes, as observed for compounds 11–14 and 19 (COX-2 (SI) >62.5 − 312.5), were more active than the corresponding cyclic imides bearing a 3-benzenesulfonamide tail, as observed for compounds 2–5 and 10 (COX-2 (SI) ≫14.3 − 277.8). Interestingly, compounds 9 and 18 containing the 3-benzenesulfonamide and oxime tails, respectively, had the same COX-2 (SI) value >333.3, which may be attributed to the same binding interaction with the COX-2 putative pocket. The presence of the carboxylic moieties on the cyclic imides, as observed for series 21–24 and 28, afforded non-selective COX-1/COX-2 inhibition with a relatively high COX-1 inhibition of IC50 ≅ 10.9–24.8 μM) and lower COX-2 inhibition of IC50 ≅ 22.3–36.3 μM). Compounds 22 and 23 had low COX-2 inhibition, with IC50 values of 25.1 and 22.3 μM, respectively, and high COX-1 inhibition, with IC50 values of 11.6 and 10.9 μM, respectively, compared to their 3-benzenesulfonamide and oxime analogues 3, 4, 12, and 13, with SI values of >250.0, 277.8, 312.5, and >312.5, respectively. Hence, the sulphonamide (–SO2NH2) and oxime (–C=NOH) fragments are important pharmacophores for the interaction with the COX-2 binding pocket.
The structure-activity relationships (SAR) of the test compounds 2–6, 9–15, 18, 19, 21–24, and 28 in the COX-1/2 inhibition assay revealed the following: (i) generally, the COX-2 isozyme was potentially affected by cyclic imides 2–5, 9, 11–14, 18, and 19, with IC50 values ranging from 0.15 − 0.90 μM and SI ≅ >333.3 to >55.6 compared to celecoxib (IC50 = 0.129 μM; COX-2 (SI) > 387.6); (ii) the non-carboxylic cyclic imides, including the 3-benzenesulfonamide and oxime tails 2–5, 9, 11–14, 18, and 19, were potent inhibitors of the COX-2 isozyme, while the carboxylic cyclic imide derivatives 21–24 and 28 were very weak or non-COX-2 inhibitors. Consequently, the sulphonamide (SO2NH2) and oxime (–C=NOH) fragments are an essential part of COX-2 inhibition and recognition; (iii) the cyclic imides with a nitro (NO2) group at the 5-position in compounds containing sulphonamide and oxime fragments, as observed for compounds 9 and 18, possessed the strongest COX-2 inhibitory activity among the tested compounds (IC50 = 0.15 μM and SI > 333.3); (iv) in contrast, the carboxylic cyclic imide substituted with a nitro (NO2) group at the 5-position, as observed for compound 28, had reduced COX-2 inhibitory activity (IC50 = 30.0 μM) and increased COX-1 inhibitory activity (IC50 = 18.2 μM and SI = 0.61); (v) the tetrahydrophthalimide and phthalimide core structures, as observed for compounds 3 (IC50 = 0.20 μM and SI > 250), 4 (IC50 = 0.18 μM and SI > 277.8), 12 (IC50 = 0.16 μM and SI > 312.5), and 13 (IC50 = 0.16 μM and SI > 312.5), had higher inhibitory activity than the succinimide derivatives, as observed for compounds 2 (IC50 = 0.26 μM and SI > 192.3) and 11 (IC50 = 0.22 μM and SI > 227.3). These aforementioned results indicate the importance of the double bonds for COX-2 inhibition; (vi) replacing the phthalimide fragments in compounds 4 and 13 (IC50 = 0.18 μM, and 0.16 μM, respectively), with the naphthalimide structure in compounds 10 and 19 decreasing the COX-2 inhibitory activity with IC50 of 3.5 μM and 0.28 μM, respectively; (vii) substituting the phthalimide core in compounds 4 (IC50 = 0.18 μM), 13 (IC50 = 0.16 μM) and 23 (IC50 = 19.3 μM) with the methyl moiety (CH3) at the 5-position in compounds 5, 14, and 24 resulted in a decrease in COX-2 inhibition (IC50 = 0.9 μM, 0.8 μM, and 36.30 μM, respectively) and an increase in COX-1 inhibition of compound 24 (IC50 = 24.8 μM); (viii) replacing the methyl fragment (CH3) in compounds 5 and 14 (IC50 = 0.9 μM, and 0.8 μM, respectively) with the bulky tert-butyl group (–C(CH3)3) in compounds 6 and 15 led to a decrease in COX-2 inhibition (IC50 = 4.0 μM, and 3.5 μM, respectively); (ix) converting the terminal sulphonamide or oxime fragments in compounds 3 (IC50 = 0.2 μM), 4 (IC50 = 0.18 μM), 12 (IC50 = 0.16 μM), and 13 (IC50 = 0.16 μM) into a carboxylic acid (–COOH) moiety containing the same cyclic imide scaffold found in compounds 22 and 23 resulted in a decrease in COX-2 inhibition (IC50 = 25.1 μM and 22.3 μM, respectively) and increase in COX-1 inhibition (IC50 = 11.6 μM and 10.9 μM, respectively). Briefly, the non-carboxylic cyclic imide scaffolds containing the 3-benzenesulfonamide or 2-[4-(1-(hydroxyimino)ethyl)phenyl] fragments, as observed for derivatives 2–5, 9, 11–14, 18, and 19, were the most active inhibitors of the COX-2 isozyme and some displayed higher levels of selective COX-2 inhibition (SI ≥ 250 − 333.3), which were comparable to levels for celecoxib (SI ≥ 387.6).
2.3. Molecular docking studies
The molecular modelling technique was used to establish and understand the binding mode of the most bioactive compoundsCitation21,Citation22. The selectivity of the cyclic imide analogues towards COX-2 was studied using a molecular docking protocol on the MOE 2008.10 programme obtained from Chemical Computing Group Inc. (Montreal, Canada)Citation23.
Molecular docking was performed to examine the best interaction between the most active compounds, 9 and 18, and the COX-2 pocket binding site (, lower panels). The crystallographic binding site on the COX-2 isozyme in complex with the SC-558 ligand, an analogue of celecoxib, was derived from Protein Data Bank (PDB code: 1CX2) (, upper panels)Citation4.
Figure 2. 3D interactions of compounds SC-558 ligand (upper panel), 9 (lower left panel), and 18 (lower right panel) with the active site of COX-2. Hydrogen bonds are depicted as red lines.
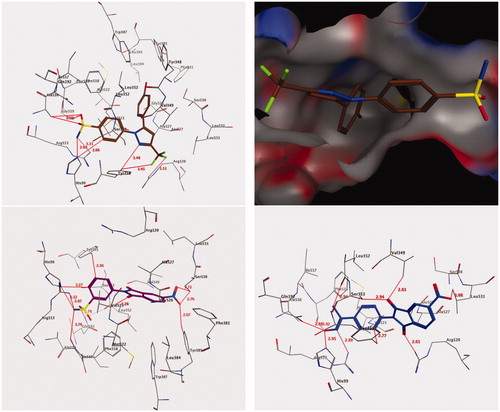
The scoring function and the hydrogen bond formed among these compounds and the surrounding amino acids were used to predict the interaction mode of compounds 9 and 18 in the putative active pocket of the COX-2 isozyme. shows the results of the docking studies for both compounds 9 and 18, which possess the common cyclic imide pharmacophore and a similar binding interaction, including H-bonding and hydrophobic interactions within the putative binding pocket. These compounds were inserted deep into the hydrophilic site of the COX-2 isozyme where the sulphonamide and oxime groups can interact with the hydrophilic pocket (His90, Gln192, and Arg513) through a network of hydrogen bonds; such binding interactions were similar to that of SC-558 co-crystallized in the COX-2 active siteCitation4. Additionally, the cyclic imide cores of compounds 9 and 18 were oriented at the top of the channel, which were involved in the hydrophobic interaction with amino acids, Ala527, Val349, Trp387, Val523, and Leu352. The terminal sulphonamide group (–SO2NH2) was responsible for the stability of the docked compound 9 through its conserved role in the binding pocket through the formation of suitable H-bonds with the key amino acids, Arg513 (2.87 Å), His90 (3.12 Å), Gln192 (2.78 Å), and Phe518 (3.76 Å) [, lower left panel]. Meanwhile, the phenyl ring of the 3-benzenesulfonamide formed two non-classical H-bonds by binding with amino acids, His90 (3.27 Å) and Tyr355 (2.96 Å), and an additional hydrophobic interaction with Ser353 (3.95 Å) through CH-pi interactions. The 5-nitro group of cyclic imide was also identified to form three H-bonds with the amino acids Tyr385 (2.67 Å) and Ser530 (2.76 Å and 2.72 Å), whereas one of the carbonyl group of cyclic imide interacted with the amino acid Val523 through a non-classical H-bond (3.20 Å) [, lower left panel].
In a similar manner, the complex generated by the docking of compound 18 [, lower right panel] showed that the terminal oxime moiety (C=N–OH) formed H-bonds with amino acids Arg513 (2.89 Å), His90 (2.95 Å), Gln192 (2.88 Å), and Phe518 (3.82 Å) similar to the sulphonamide moiety in compound 9 [, lower right panel]. Additionally, the methyl moiety of the oxime structure interacted with amino acid Leu352 through a non-classical H-bond (2.94 Å), while the N-phenyl moiety formed another non-classical H-bond with Tyr355 (2.77 Å) [, lower right panel]. Moreover, the two carbonyl groups of cyclic imide were held by one classical and two non-classical H-bonds with amino acids Arg120 (2.65 Å), Ser353 (2.94 Å), and Val349 (2.81 Å) [, lower right panel]. Finally, the 5-nitro group of cyclic imide formed a non-classical H-bond with Leu531 (3.08 Å). The binding interactions described herein are typical of the pyrazolic prototype (SC-558), which is a selective inhibitor of COX-2. Altogether, such findings confirm the molecular design of the reported class of anti-inflammatory 1,3-isoindoledione scaffoldsCitation10,Citation11.
3. Conclusions
The cyclic imide scaffolds bearing the non-carboxylic 3-benzenesulfonamide or 2-[4-(1-(hydroxyimino)ethyl)phenyl] fragments 2–10, and 11–19 and the carboxylic derivatives 21–29 were synthesised and evaluated for their in vivo anti-inflammatory and ulcerogenic activities and in vitro cytotoxicity. The derivatives revealed as the most active anti-inflammatory agents were also subjected to an inhibition assay with the COX-1/COX-2 enzymes. Among the tested compounds, cyclic imide derivatives bearing 3-benzenesulfonamides (2–4, 9), acetophenone oximes (11, 12, 13), and β-phenylalanine (18) exhibited remarkable anti-inflammatory activities (71.2–82.9% oedema inhibition) compared to the reference drugs, celecoxib and diclofenac (85.6 and 83.4% oedema inhibition, respectively). Cyclic imides attached to 3-benzenesulfonamide or oxime were stronger anti-inflammatory agents and selective COX-2 inhibitors than the carboxylic acid derivatives containing the same scaffolds. Compounds 2–4, 9, 11–13, 18, and 19 were the most potent COX-2 inhibitors (IC50 = 0.26, 0.20, 0.18, 0.15, 0.22, 0.16, 0.16, 0.15, and 0.28 μM, respectively) and demonstrated values comparable to that of celecoxib (IC50 = 0.129 μM). Some cyclic imide derivatives 2–6 and 8 were subjected to cytotoxic evaluation which showed weak positive cytotoxic effects (PCE = 2/59–5/59) compared to the standard drug, imatinib (PCE = 20/59). From the COX-2 inhibition assay and docking results, compounds 9 and 18 were recognised as the most active analogues, with the highest recognition at the COX-2 binding site and a correlation identified with the COX-2 selectivity indices.
4. Experimental
4.1. Chemistry
Melting points (uncorrected) were recorded on a Barnstead 9100 Electrothermal melting apparatus (APS Water Services Corporation, Van Nuys, CA) while the IR spectra were recorded on a FT-IR Perkin-Elmer spectrometer (PerkinElmer Inc., Waltham, MA). The 1H NMR and 13C NMR were measured in DMSO-d6 or CDCl3, on Bruker 700 or 500 and 176 or 125 MHz instruments, respectively (Bruker, Billerica, MA). Chemical shifts are reported in δ ppm. Mass spectra were recorded on an Agilent 6320 Ion Trap mass spectrometer (Agilent Technologies, Santa Clara, CA). C, H, and N were analysed at the Research Centre, College of Pharmacy, King Saud University, Saudi Arabia. The results were within ±0.4% of the theoretical values. Compounds 4, 8, 9, 12–17, and 22–24 were prepared according to a previously reported procedureCitation6b,Citation24.
4.1.1. General procedure for the synthesis of 1,3-isoindolediones 2–10
A mixture of an equimolar amount of 3-benzenesulfonamide and acid anhydrides (5.0 mmol) was heated under reflux for 12 h in glacial acetic acid (15 ml) containing anhydrous sodium acetate (0.69 g, 5.0 mmol) (Scheme 1). The reaction mixture was cooled, filtered, and the obtained solid was washed with water, dried, and re-crystallized.
4.1.1.1. 3-(2,5-Dioxopyrrolidin-1-yl)benzenesulfonamide (2)
M.P. 195–197°C, 90% yield (Ethanol); IR (KBr, cm−1) ν: 3337, 3246 (NH2), 1696 (C=O), 1339, 1157 (O = S=O); 1H NMR (500 MHz, DMSO-d6): δ 2.82 (s, 4H), 7.37 (s, 2H), 7.45 − 7.47 (t, 1H, J = 7.9 Hz), 7.60 − 7.63 (t, 1H, J = 7.9 Hz), 7.82 (s, 1H), 7.88 − 7.90 (d, 1H, J = 8.0 Hz); 13C NMR (125 MHz, DMSO-d6): δ 28.82, 124.78, 125.87, 129.69, 130.46, 133.19, 145.19, 176.68; C10H10N2O4S: m/z (254.3).
4.1.1.2. 3-(1,3-Dioxo-1,3,3a,4,7,7a-hexahydro-2H-isoindol-2-yl)benzenesulfonamide (3)
M.P. 159–161°C, 91% yield (Ethanol); IR (KBr, cm−1) ν: 3293, 3125 (NH2), 1773, 1699 (C=O), 1341, 1160 (O=S=O); 1H NMR (700 MHz, DMSO-d6): δ 2.30–2.32 (d, 2H, J = 12.6 Hz), 2.48–2.51 (d, 2H, J = 16.1 Hz), 3.35 (s, 2H), 5.98 (s, 2H), 7.46–7.47 (d, 1H, J = 7.0 Hz), 7.52 (s, 2H), 7.70 (s, 2H), 7.88–7.89 (d, 1H, J = 6.3 Hz); 13C NMR (176 MHz, DMSO-d6): δ 23.71, 40.35, 124.52, 126.03, 128.23, 130.26, 130.73, 133.22, 145.35, 179.55; C14H14N2O4S: m/z (306.3).
4.1.1.3. 3-(5-Methyl-1,3-dioxoisoindolin-2-yl)benzenesulfonamide (5)
M.P. 225–226°C, 86% yield (Methanol); IR (KBr, cm−1) ν: 3308, 3228 (NH2), 1779, 1717 (C=O), 1339, 1164 (O=S=O); 1H NMR (700 MHz, DMSO-d6): δ 2.54 (s, 3H), 7.55 (s, 2H), 7.69–7.71 (d, 1H, J = 7.8 Hz), 7.73–7.76 (q, 2H, J = 7.9 Hz), 7.82 (s, 1H), 8.87–7.91 (q, 2H, J = 7.8 Hz), 7.95 (s, 1H); 13C NMR (176 MHz, DMSO-d6): δ 21.89, 123.95, 124.40, 124.76, 125.57, 129.34, 130.12, 130.98, 132.31, 132.82, 135.69, 145.29, 146.35, 167.19, 167.29; C15H12N2O4S: m/z (316.4).
4.1.1.4. 3-(5-(tert-Butyl)-1,3-dioxoisoindolin-2-yl)benzenesulfonamide (6)
M.P. 151–153°C, 79% yield (Methanol); IR (KBr, cm−1) ν: 3354, 3264 (NH2), 1777, 1718 (C=O), 1375, 1164 (O=S=O); 1H NMR (700 MHz, DMSO-d6): δ 1.39 (s, 9H), 7.57 (s, 2H), 7.69–7.70 (d, 1H, J = 7.8 Hz), 7.74–7.77 (t, 1H, J = 7.8 Hz), 7.91–7.95 (q, 3H, J = 7.8 Hz), 7.96–7.97 (d, 2H, J = 6.4 Hz); 13C NMR (125 MHz, DMSO-d6): δ 31.26, 36.03, 120.81, 123.98, 124.83, 125.64, 129.46, 130.17, 131.09, 132.19, 132.27, 132.84, 145.28, 159.10, 167.09, 167.42; C18H18N2O4S: m/z (358.3).
4.1.1.5. 3-(5,6-Dichloro-1,3-dioxoisoindolin-2-yl)benzenesulfonamide (7)
M.P. 256–258°C, 71% yield (Methanol); IR (KBr, cm−1) ν: 3338, 3240 (NH2), 1779, 1718 (C=O), 1330, 1154 (O = S=O); 1H NMR (500 MHz, DMSO-d6): δ 7.50 (s, 2H), 7.65–7.66 (d, 1H, J = 8.3 Hz), 7.70–7.73 (t, 1H, J = 7.9 Hz), 7.92–7.93 (d, 1H, J = 7.9 Hz), 7.95–7.96 (t, 1H, J = 3.5 Hz), 8.21 (s, 2H); 13C NMR (125 MHz, DMSO-d6): δ 124.69, 125.93, 125.98, 130.01, 130.58, 131.76, 132.27, 138.37, 145.44, 165.27; C14H8Cl2N2O4S: m/z (371.2).
4.1.1.6. 3-(1,3-Dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)benzenesulfonamide (10)
M.P. 241–243°C, 76% yield (Methanol); IR (KBr, cm−1) ν: 3367, 3221 (NH2), 1776, 1711 (C=O), 1336, 1148 (O=S=O); 1H NMR (500 MHz, DMSO-d6): δ 7.39 (s, 2H), 7.52–7.53 (d, 1H, J = 7.8 Hz), 7.82–7.85 (m, 2H), 8.38–8.40 (d, 2H, J = 8.3 Hz), 8.43–7.45 (d, 2H, J = 8.3 Hz), 8.50–8.54 (m, 3H); 13C NMR (125 MHz, DMSO-d6): δ 118.94, 122.73, 125.96, 126.99, 127.32, 127.73, 129.76, 131.33, 133.15, 134.92, 135.85, 160.77, 163.97; C18H12N2O4S: m/z (352.3).
4.1.2. General procedure for the synthesis of 1,3-isoindoledione 11–19
A mixture of 1,3-isoindolinediones (5.0 mmol) and hydroxylamine hydrochloride (0.56 g, 8 mmol) was stirred at room temperature for 24 h in glacial acetic acid (20 ml) (Scheme 2). The reaction mixture was then filtered and the obtained solid was washed with water, dried, and re-crystallized.
4.1.2.1. 1-(4-(1-(Hydroxyimino)ethyl)phenyl)pyrrolidine-2,5-dione (11)
M.P. 184–186°C, 89% yield (Methanol); IR (KBr, cm−1) ν: 3358 (OH), 1774, 1690 (C=O), 1634 (C=N); 1H NMR (700 MHz, DMSO-d6): δ 2.12 (s, 3H), 2.52–2.54 (t, 2H, J = 6.7 Hz), 2.57–2.59 (t, 2H, J = 6.4 Hz), 7.58–7.61 (q, 4H, J = 9.5 Hz), 10.08 (s, 1H); 13C NMR (176 MHz, DMSO-d6): δ 11.83, 29.20, 31.50, 119.03, 131.94, 140.10, 152.95, 170.70, 174.32; C12H12N2O3: m/z (232.1).
4.1.2.2. 2-(4-(1-(Hydroxyimino)ethyl)phenyl)-5-nitroisoindoline-1,3-dione (18)
M.P. 269–271°C, 92% yield (Ethanol); IR (KBr, cm−1) ν: 3428 (OH), 1737, 1699 (C=O), 1645 (C=N); 1H NMR (700 MHz, DMSO-d6): δ 2.21 (s, 3H), 7.47–7.48 (d, 2H, J = 8.4 Hz), 7.81–7.82 (d, 2H, J = 8.3 Hz), 8.12–8.14 (t, 1H, J = 7.8 Hz), 8.26–8.27 (d, 1H, J = 7.4 Hz), 8.34–8.35 (d, 1H, J = 7.1 Hz), 11.39 (s, 1H); 13C NMR (176 MHz, DMSO-d6): δ 12.04, 123.37, 126.53, 127.55, 127.79, 128.88, 132.06, 134.01, 136.90, 137.45, 144.98, 152.95, 163.02, 165.60; C16H11N3O5: m/z (325.1).
4.1.2.3. 2-(4-(1-(Hydroxyimino)ethyl)phenyl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (19)
M.P. >300°C, 92% yield (Ethanol); Yield, 87%; IR (KBr, cm−1) ν: 3090 (OH), 1775, 1716 (C=O), 1649 (C=N); 1H NMR (500 MHz, DMSO-d6): δ 2.22 (s, 3H), 7.29–7.31 (t, 2H, J = 6.5 Hz), 7.76–7.77 (t, 2H, J = 6.5 Hz), 7.81–7.84 (t, 2H, J = 7.5 Hz), 8.38–8.40 (d, 2H, J = 8.5 Hz), 8.51–8.52 (d, 2H, J = 6.5 Hz), 11.14 (s, 1H); 13C NMR (125 MHz, DMSO-d6): δ 11.93, 122.82, 126.43, 127.29, 129.13, 129.53, 130.46, 131.27, 134.75, 136.01, 137.72, 146.22, 152.80, 162.82, 164.02; C20H14N2O3: m/z (330.1).
4.1.3. General procedure for the synthesis of 1,3-isoindoledione 21–29
A mixture of an equimolar amount of β-phenylalanine (0.83 gm, 5.0 mmol) and acid anhydrides (5.0 mmol) was heated under reflux for 24 h in glacial acetic acid (20 ml) containing anhydrous sodium acetate (0.69 g, 5.0 mmol) (Scheme 3). The reaction mixture was cooled and filtered, and the obtained solid was washed with water, dried, and re-crystallized.
4.1.3.1. 3-(2,5-Dioxopyrrolidin-1-yl)-3-phenylpropanoic acid (21)
M.P. 259–261°C, 93% yield (Ethanol); IR (KBr, cm−1) ν: 3111 (OH), 1743, 1702 (C=O); 1H NMR (700 MHz, DMSO-d6): δ 2.44–2.49 (m, 2H), 2.52–2.57 (m, 2H), 3.24–3.27 (dd, 1H, J = 11.9, 14.0 Hz), 3.37–3.39 (dd, 1H, J = 4.5, 14.1 Hz), 4.59–4.61 (dd, 1H, J = 4.5, 11.7 Hz), 7.09–7.10 (d, 2H, J = 7.3 Hz), 7.15–7.17 (t, 1H, J = 7.3 Hz), 7.22–7.24 (t, 2H, J = 7.5 Hz); 13C NMR (176 MHz, DMSO-d6): δ 28.02, 34.06, 55.53, 126.60, 128.68, 129.05, 139.32, 170.38, 177.55; C13H13NO4: m/z (247.2).
4.1.3.2. 3-(5-(tert-Butyl)-1,3-dioxoisoindolin-2-yl)-3-phenylpropanoic acid (25)
M.P.(0).119–121°C, 76% yield (Methanol); IR (KBr, cm−1) ν: 3219 (OH), 1739, 1703 (C=O); 1H NMR (700 MHz, DMSO-d6): δ 1.32 (s, 9H), 3.43–3.47 (t, 1H, J = 12.8 Hz), 3.52–3.55 (dd, 1H, J = 4.0, 14.4 Hz), 4.57–4.60 (dd, 1H, J = 4.0, 12.5 Hz), 7.06–7.08 (t, 1H, J = 7.3 Hz), 7.10–7.11 (d, 2H, J = 7.3 Hz), 7.15–7.18 (t, 2H, J = 7.6 Hz), 7.67–7.69 (d, 1H, J = 7.8 Hz), 7.72 (s, 1H), 7.79–7.81 (dd, 1H, J = 1.5, 7.9 Hz); 13C NMR (176 MHz, DMSO-d6): δ 31.22, 35.35, 35.83, 57.41, 119.93, 123.04, 126.28, 128.65, 128.80, 129.60, 131.50, 132.38, 140.75, 158.31, 168.60, 168.89, 174.40; C21H21NO4: m/z (351.2).
4.1.3.3. 3-(5,6-Dichloro-1,3-dioxoisoindolin-2-yl)-3-phenylpropanoic acid (26)
M.P. 205–207°C, 89% yield (Ethanol); IR (KBr, cm−1) ν: 2890 (OH), 1742, 1717 (C=O); 1H NMR (500 MHz, DMSO-d6): δ 3.28–3.33 (dd, 1H, J = 12.0, 14.0 Hz), 3.47–3.51 (dd, 1H, J = 4.7, 14.1 Hz), 5.12–5.15 (dd, 1H, J = 4.8, 11.7 Hz), 7.11–7.19 (m, 5H), 8.16 (s, 2H), 13.44 (s, 1H); 13C NMR (125 MHz, DMSO-d6): δ 34.33, 53.86, 126.19, 127.09, 128.82, 129.17, 130.81, 137.58, 138.56, 165.67, 170.17; C17H11Cl2NO4: m/z (364.2).
4.1.3.4. 3-Phenyl-3-(4,5,6,7-tetrachloro-1,3-dioxoisoindolin-2-yl)propanoic acid (27)
M.P. 280–282°C, 93% yield (Methanol); IR (KBr, cm−1) ν: 2870 (OH), 1741, 1715 (C=O); 1H NMR (500 MHz, DMSO-d6): δ 3.28–3.33 (t, 1H, J = 12.0 Hz), 3.49–3.53 (dd, 1H, J = 4.9, 14.3 Hz), 5.15–5.18 (dd, 1H, J = 4.7, 11.2 Hz), 7.13–7.21 (m, 5H), 13.47 (s, 1H); 13C NMR (125 MHz, CDCl3): δ 34.25, 54.18, 127.09, 127.48, 128.87, 129.11, 129.15, 137.53, 139.62, 163.03, 169.90; C17H9Cl4NO4: m/z (433.1).
4.1.3.5. 3-(5-Nitro-1,3-dioxoisoindolin-2-yl)-3-phenylpropanoic acid (28)
M.P.(0).191–193°C, 83% yield (Ethanol); IR (KBr, cm−1) ν: 2910 (OH), 1745, 1715 (C=O); 1H NMR (700 MHz, DMSO-d6): δ 3.29–3.34 (dd, 1H, J = 4.0, 16.0 Hz), 3.49–3.52 (dd, 1H, J = 6.9, 13.0 Hz), 5.15–5.18 (dd, 1H, J = 6.9, 16.0 Hz), 7.13–7.21 (m, 5H), 8.07–8.10 (t, 1H, J = 11.0 Hz), 8.16–8.18 (d, 1H, J = 10.2 Hz), 8.31–8.33 (d, 1H, J = 11.3 Hz); 13C NMR (176 MHz, DMSO-d6): δ 34.30, 54.03, 122.39, 127.12, 127.88, 128.63, 128.85, 129.20, 129.50, 129.66, 132.76, 137.50, 137.62, 144.88, 162.86, 165.51, 169.69, 170.07; C17H12N2O6: m/z (340.1).
4.1.3.6. 3-(1,3-Dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)-3-phenylpropanoic acid (29)
M.P. 151–153°C, 66% yield (Methanol); IR (KBr, cm−1) ν: 2930 (OH), 1708, 1667 (C=O); 1H NMR (700 MHz, DMSO-d6): δ 2.92–2.95 (t, 1H, J = 7.7 Hz), 4.24–4.26 (t, 1H, J = 7.6 Hz), 4.95–4.97 (dd, 1H, J = 7.6, 14.5 Hz), 7.28–7.31 (q, 3H, J = 7.4 Hz), 7.85–7.87 (t, 1H, J = 7.5 Hz), 7.90–7.93 (q, 2H, J = 7.6 Hz), 8.44 (s, 2H), 8.47-8.48 (s, 1H, J = 6.9 Hz), 8.51–8.54 (t, 2H, J = 7.5 Hz); 13C NMR (176 MHz, DMSO-d6): δ 33.96, 34.82, 54.73, 119.44, 122.42, 127.69, 128.02, 128.54, 128.95, 129.10, 129.40, 131.20, 132.95, 134.83, 135.19, 135.87, 139.21, 161.18, 163.51, 163.74; C21H15NO4: m/z (345.1).
4.2. Biological evaluation
4.2.1. Anti-inflammatory screening
Anti-inflammatory assessment of the newly synthesised compounds was carried out using an in vivo rat carrageenan-induced foot paw oedema model, as reported previouslyCitation16,Citation17. Compounds 6, 7, 8, 10, 11, 18, diclofenac, and celecoxib were tested at three different doses and their ED50 was determined.
4.2.2. Ulcerogenicity measurement
Ulcerogenicity was evaluated according to a previously reported methodCitation6a,Citation17,Citation18. The number and total length of the ulcers for each animal were measured and their averages were calculated and used as the ulcer indices.
4.2.3. In vitro cyclooxygenase (COX) inhibition assay
To determine the relative ability of the test compounds and reference drugs to inhibit COX-1/COX-2 isozymes, we used the colorimetric COX (ovine) inhibitor screening assay kit (Cayman Chemicals Inc., Ann Arbour, MI), according to the manufacturer’s instructionsCitation3,Citation10,Citation11,Citation20.
4.3. Docking methodology
Molecular modelling studies were performed using the 2007.09 software from Chemical Computing Group Inc. (Montreal, Canada). The docking protocol was similar to that mentioned in our previous reportCitation19.
Acknowledgements
The authors thank Ahmad Abbas at Department of Pharmacognosy, Faculty of Pharmacy, Mansoura University, Egypt for performing in vivo biological activity.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Related Research Data
References
- (a) Williams DA, Lemke TL, Non-steroidal anti-inflammatory drugs. In Lemke TL, Williams DA, ed. Foye’s principles of medicinal chemistry, 5th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2002:751–93. (b) Dubost JJ, Soubrier M, Sauvezie B. Le traitement de la polyarthrite rhumatoïde. Évolution des idées et des stratégies. Rev Med Interne 1999;20:171–8. (c) Dannhardt G, Kiefer W, Krämer G, et al. The pyrrole moiety as a template for COX-1/COX-2 inhibitors. Eur J Med Chem 2000;35:499–510. (d) Seibert K, Masferrer JL. Role of inducible cyclooxygenase (COX-2) in inflammation. Receptor 1994;4:17–23. (e) Fiorucci S, Meli R, Bucci M, Cirino G. Dual inhibitors of cyclooxygenase and 5-lipoxygenase. A new avenue in anti-inflammatory therapy? Biochem Pharmacol 2001;62:1433–8.
- (a) Balaji B, Hariharan S, Shah DB, Ramanathan M. Discovery of potential and selective COX-1 inhibitory leads using pharmacophore modelling, in silico screening and in vitro evaluation. Eur J Med Chem 2014;86:469–80. (b) Vane JR, Bakhle YS, Botting RM. Cyclooxygenases 1 and 2. Ann Rev Pharmacol Toxicol 1998;38:97–120. (c) Seibert K, Zhang Y, Leahy K, et al. Pharmacological and biochemical demonstration of the role of cyclooxygenase 2 in inflammation and pain. Proc Natl Acad Sci 1994;91:12013–7. (d) Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol 1971;231:232–5. (e) Kujubu DA, Fletcher BS, Varnum BC, et al. TIS10, a phorbol ester tumor promoter-inducible mRNA from Swiss 3T3 cells, encodes a novel prostaglandin synthase/cyclooxygenase homologue. J Biol Chem 1991;266:12866–72.
- (a) Crofford LJ. Crofford LJ. COX-1 and COX-2 tissue expression: Implications and predictions. J Rheumatol 1997;24:15–9. (b) El-Sayed MA-A, Abdel-Aziz NI, Abdel-Aziz AA-M, et al. Design, synthesis, and biological evaluation of substituted hydrazone and pyrazole derivatives as selective COX-2 inhibitors: molecular docking study. Bioorg Med Chem 2011;19:3416–24. (c) Abdel-Sayed MA, Bayomi SM, El-Sherbeny MA, et al. Synthesis, anti-inflammatory, analgesic, COX-1/2 inhibition activities and molecular docking study of pyrazoline derivatives. Bioorg Med Chem 2016;24:2032–42. (d) El-Sayed MA-A, Abdel-Aziz NI, Abdel-Aziz AA-M, et al. Synthesis, biological evaluation and molecular modeling study of pyrazole and pyrazoline derivatives as selective COX-2 inhibitors and anti-inflammatory agents. Part 2. Bioorg Med Chem 2012;20:3306–16.
- (a) Penning TD, Talley JJ, Bertenshaw SR, et al. Synthesis and biological evaluation of the 1,5-diarylpyrazole class of cyclooxygenase-2 inhibitors: identification of 4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide (SC-58635, Celecoxib). J Med Chem 1997;40:1347–65. (b) Kurumbail RG, Stevens AM, Gierse JK, et al. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature 1996;384:644–8. (c) Michaux C, Charlier C. Structural approach for COX-2 inhibition. Mini Rev Med Chem 2004;4:603–15.
- (a) Ghosh N, Chaki R, Mandal V, Mandal SC. COX-2 as a target for cancer chemotherapy. Pharmacol Rep 2010;62:233–44. (b) Dai ZJ, Ma XB, Kang HF, et al. Antitumor activity of the selective cyclooxygenase-2 inhibitor, celecoxib, on breast cancer in Vitro and in Vivo. Cancer Cell Int. 2012;12:53.
- (a) Abdel-Aziz AA-M, Angeli A, El-Azab AS, et al. Synthesis and anti-inflammatory activity of sulfonamides and carboxylates incorporating trimellitimides: dual cyclooxygenase/carbonic anhydrase inhibitory actions. Bioorg Chem 2019;84:260–8. (b) Abdel-Aziz AA-M, El-Azab AS, Abu El-Enin MA, et al. Synthesis of novel isoindoline-1,3-dione-based oximes and benzenesulfonamide hydrazones as selective inhibitors of the tumor-associated carbonic anhydrase IX. Bioorg Chem 2018;80:706–13. (c) Abdel-Aziz AA-M, El-Azab AS, Ekinci D, et al. Investigation of arenesulfonyl-2-imidazolidinones as potent carbonic anhydrase inhibitors. J Enzym Inhib Med Chem 2015;30:81–4. (d) El-Azab AS, Abdel-Aziz AA-M, Ayyad RR, et al. Inhibition of carbonic anhydrase isoforms I, II, IV, VII and XII with carboxylates and sulfonamides incorporating phthalimide/phthalic anhydride scaffolds. Bioorg Med Chem 2016;24:20–5. (e) Abdel-Aziz AA-M, El-Azab AS, Ceruso M, Supuran CT. Carbonic anhydrase inhibitory activity of sulfonamides and carboxylic acids incorporating cyclic imide scaffolds. Bioorg Med Chem Lett 2014;24:5185–9.
- (a) El-Azab AS, Abdel-Aziz AA-M, Bua S, et al. New anthranilic acid-incorporating N-benzenesulfonamidophthalimides as potent inhibitors of carbonic anhydrases I, II, IX, and XII: synthesis, in vitro testing, and in silico assessment. Eur J Med Chem 2019;181:111573. (b) El-Azab AS, Abdel-Aziz AA-M, Bua S, et al. Synthesis and comparative carbonic anhydrase inhibition of new Schiff’s bases incorporating benzenesulfonamide, methanesulfonamide, and methylsulfonylbenzene scaffolds. Bioorg Chem 2019;92:103225. (c) Abdel-Aziz AA-M, El-Azab AS, Bua S, et al. Design, synthesis, and carbonic anhydrase inhibition activity of benzenesulfonamide-linked novel pyrazoline derivatives. Bioorg Chem 2019;87:425–31.
- (a) El-Azab AS, Abdel-Aziz AA-M, Bua S, et al. Synthesis of benzensulfonamides linked to quinazoline scaffolds as novel carbonic anhydrase inhibitors. Bioorg Chem 2019;87:78–90. (b) Abdel-Aziz AA-M, El-Azab AS, Ghiaty AH, et al. 4-Substituted benzenesulfonamides featuring cyclic imides moieties exhibit potent and isoform-selective carbonic anhydrase II/IX inhibition. Bioorg Chem 2019;83:198–204.
- (a) Angeli A, Abdel-Aziz AA-M, Nocentini A, et al. Synthesis and carbonic anhydrase inhibition of polycyclic imides incorporating N-benzenesulfonamide moieties. Bioorg Med Chem 2017;25:5373–9. (b) Mohamed MA, Abdel-Aziz AA-M, Sakr HM, et al. Synthesis and human/bacterial carbonic anhydrase inhibition with a series of sulfonamides incorporating phthalimido moieties. Bioorg Med Chem 2017;25:2524–9. (c) Abdel-Aziz AA-M, Angeli A, El-Azab AS, et al. Synthesis and biological evaluation of cyclic imides incorporating benzenesulfonamide moieties as carbonic anhydrase I, II, IV and IX inhibitors. Bioorg Med Chem 2017;25:1666–71.
- (a) Abdel-Aziz AA-M, El-Azab AS, Abou-Zeid LA, et al. Synthesis, anti-inflammatory, analgesic and COX-1/2 inhibition activities of anilides based on 5,5-diphenylimidazolidine-2,4-dione scaffold: molecular docking studies. Eur J Med Chem 2016;115:121–31. (b) Al-Suwaidan IA, Alanazi AM, El-Azab AS, et al. Molecular design, synthesis and biological evaluation of cyclic imides bearing benzenesulfonamide fragment as potential COX-2 inhibitors. Part 2. Bioorg Med Chem Lett 2013;23:2601–5.
- (a) Alanazi AM, El-Azab AS, Al-Suwaidan IA, et al. Structure-based design of phthalimide derivatives as potential cyclooxygenase-2 (COX-2) inhibitors: anti-inflammatory and analgesic activities. Eur J Med Chem 2015;92:115–23. (b) Abdel-Aziz AA-M, El Tahir KEH, Asiri YA. Synthesis, anti-inflammatory activity and COX-1/COX-2 inhibition of novel substituted cyclic imides. Part 1: molecular docking study. Eur J Med Chem 2011;46:1648–55.
- (a) Abdel-Aziz AA-M, El-Azab AS, El-Subbagh HI, et al. Design, synthesis, single-crystal and preliminary antitumor activity of novel arenesulfonylimidazolidin-2-ones. Bioorg Med Chem Lett 2012;22:2008–14. (b) Abdel-Aziz AA-M, El-Azab AS, Alanazi AM, et al. Synthesis and potential antitumor activity of 7-(4-substituted piperazin-1-yl)-4-oxoquinolines based on ciprofloxacin and norfloxacin scaffolds: in silico studies. J Enzym Inhib Med Chem 2016;31:796–809. (c) El-Deeb IM, Bayoumi SM, El-Sherbeny MA, Abdel-Aziz AA-M. Synthesis and antitumor evaluation of novel cyclic arylsulfonylureas: ADME-T and pharmacophore prediction. Eur J Med Chem 2010;45:2516–30. (d) El-Sherbeny MA, Abdel-Aziz AA-M, Ahmed MA. Synthesis and antitumor evaluation of novel diarylsulfonylurea derivatives: molecular modeling applications. Eur J Med Chem 2010;45:689–97.
- (a) Abdel-Aziz AA-M. Novel and versatile methodology for synthesis of cyclic imides and evaluation of their cytotoxic, DNA binding, apoptotic inducing activities and molecular modeling study. Eur J Med Chem 2007;42:614–26. (b) El-Azab AS, Alanazi AM, Abdel-Aziz NI, et al. Synthesis, molecular modeling study, preliminary antibacterial, and antitumor evaluation of N-substituted naphthalimides and their structural analogues. Med Chem Res 2013;22:2360–75.
- Abdel-Aziz AA-M, El-Azab AS, Attia SM, et al. Synthesis and biological evaluation of some novel cyclic-imides as hypoglycaemic, anti-hyperlipidemic agents. Eur J Med Chem 2011;46:4324–9.
- Mancilla-Percino T, Trejo-Muñoz CR, Díaz-Gandarilla JA, et al. Isoindoline derivatives of α-amino acids as cyclooxygenase 1 and 2 inhibitors. Arch Pharm Chem Life Sci 2016;349:175–85.
- Winter CA, Risley EA, Nuss GW. Carrageenin-induced edema in hind paw of the rat as an assay for antiinflammatory drugs. Exp Biol Med 1962;111:544–7.
- El-Gamal MI, Bayomi SM, El-Ashry SM, et al. Synthesis and anti-inflammatory activity of novel (substituted)benzylidene acetone oxime ether derivatives: molecular modeling study. Eur J Med Chem 2010;45:1403–14.
- Adami E, Marazzi-Uberti E, Turba C. Pharmacological research on gefarnate, a new synthetic isoprenoid with an anti-ulcer action. Arch Int Pharmacodyn Ther 1964;147:113–45.
- (a) Al-Suwaidan IA, Alanazi AM, Abdel-Aziz AA-M, et al. Design, synthesis and biological evaluation of 2-mercapto-3-phenethylquinazoline bearing anilide fragments as potential antitumor agents: molecular docking study. Bioorg Med Chem Lett 2013;23:3935–41. (b) Mohamed MA, Ayyad RR, Shawer TZ, et al. Synthesis and antitumor evaluation of trimethoxyanilides based on 4(3H)-quinazolinone scaffolds. Eur J Med Chem 2016;112:106–13. (c) Al-Suwaidan IA, Abdel-Aziz AA-M, Shawer TZ, et al. Synthesis, antitumor activity and molecular docking study of some novel 3-benzyl-4(3H)quinazolinone analogues. J Enzym Inhib Med Chem 2016;31:78–89. (d) Alanazi AM, Al-Suwaidan IA, Abdel-Aziz AA-M, et al. Design, synthesis and biological evaluation of some novel substituted 2-mercapto-3-phenethylquinazolines as antitumor agents. Med Chem Res 2013;22:5566–77.
- (a) Uddin MJ, Rao PP, Knaus EE. Design and synthesis of acyclic triaryl (Z)-olefins: a novel class of cyclooxygenase-2 (COX-2) inhibitors. Bioorg Med Chem 2004;12:5929–40. (b) El-Husseiny WM, El-Sayed MA, Abdel-Aziz NI, et al. Structural alterations based on naproxen scaffold: synthesis, evaluation of antitumor activity and COX-2 inhibition, and molecular docking. Eur J Med Chem 2018;158:134–43. (c) El-Azab AS, Abdel-Aziz AA-M, Abou-Zeid LA, et al. Synthesis, antitumour activities and molecular docking of thiocarboxylic acid ester-based NSAID scaffolds: COX-2 inhibition and mechanistic studies. J Enzyme Inhib Med Chem 2018;33:989–98.
- (a) Goda FE, Abdel-Aziz AA-M, Ghoneim HA. Synthesis and biological evaluation of novel 6-nitro-5-substituted aminoquinolines as local anesthetic and anti-arrhythmic agents: molecular modeling study. Bioorg Med Chem 2005;13:3175–83. (b) Goda FE, Abdel-Aziz AA-M, Attef OA. Synthesis, antimicrobial activity and conformational analysis of novel substituted pyridines: BF3-promoted reaction of hydrazine with 2-alkoxy pyridines. Bioorg Med Chem 2004;12:1845–52. (c) El-Azab AS, Al-Omar MA, Abdel-Aziz AA-M, et al. Design, synthesis and biological evaluation of novel quinazoline derivatives as potential antitumor agents: molecular docking study. Eur J Med Chem 2010;45:4188–98.
- (a) Abdel-Aziz AA-M, Abou-Zeid LA, ElTahir KEH, et al. Synthesis, anti-inflammatory, analgesic, COX-1/2 inhibitory activities and molecular docking studies of substituted 2-mercapto-4(3H)-quinazolinones. Eur J Med Chem 2016;121:410–21. (b) Abdel-Aziz AA-M, Abou-Zeid LA, ElTahir KE, et al. Design, synthesis of 2,3-disubstitued 4(3H)-quinazolinone derivatives as anti-inflammatory and analgesic agents: COX-1/2 inhibitory activities and molecular docking studies. Bioorg Med Chem 2016;24:3818–28. (c) Alanazi AM, Abdel-Aziz AA-M, Shawer TZ, et al. Synthesis, antitumor and antimicrobial activity of some new 6-methyl-3-phenyl-4(3H)-quinazolinone analogues: in silico studies. J Enzyme Inhib Med Chem 2016;31:721–35. (d) El-Azab AS, Mary YS, Panicker CY, et al. DFT and experimental (FT-IR and FT-Raman) investigation of vibrational spectroscopy and molecular docking studies of 2-(4-oxo-3-phenethyl-3,4-dihydroquinazolin-2-ylthio)-N-(3,4,5-trimethoxyphenyl) acetamide. J Mol Struct 2016;1113:133–45.
- Molecular Operating Environment (MOE 2008.10) of Chemical Computing Group. Inc. Canada. Available from: http://www.chemcomp.com. [Last accessed 25 Oct 2019].
- (a) Sethi KK, Verma SM, Tanç M, et al. Carbonic anhydrase inhibitors: synthesis and inhibition of the cytosolic mammalian carbonic anhydrase isoforms I, II and VII with benzene sulfonamides incorporating 4,5,6,7-tetrachlorophthalimide moiety. Bioorg Med Chem 2013;21:5168–74. (b) Sethi KK, Verma SM, Tanç M, et al. Carbonic anhydrase inhibitors: synthesis and inhibition of the human carbonic anhydrase isoforms I, II, IX and XII with benzene sulfonamides incorporating 4- and 3-nitrophthalimide moieties. Bioorg Med Chem 2014;22:1586–95. (c) Scozzafava A, Mincione F, Menabuoni L, Supuran CT. Carbonic anhydrase inhibitors: topically acting antiglaucoma sulfonamides incorporating phthaloyl and phthalimido moieties. Drug Des Discov 2001;17:337–48. (d) Muller GW, U.S., Cyclic amides. 5698579, 16 Dec 1997. (e) Chowdhury S, Vaishnav R, Panwar N, Haq W. Regioselective β-csp 3-arylation of β-alanine: an approach for the exclusive synthesis of diverse β-aryl-β-amino acids. J Org Chem 2019;84:2512–22.