
Abstract
As one of the world’s five terminally ills, tumours can cause important genetic dysfunction. However, some current medicines for tumours usually have strong toxic side effects and are prone to drug resistance. Studies have found that farnesyltransferase inhibitors (FTIs) extracted from natural materials have a good inhibiting ability on tumours with fewer side effects. This article describes several FTIs extracted from natural materials and clarifies the current research progress, which provides a new choice for the treatment of tumours.
Executive summary
Farnesyltransferase (FTase)
When Ras gene is activated, it becomes an oncogene with oncogenic activity. Ras protein plays a crucial role in cancer cells. FTase is the first step to activate Ras protein.
FTIs not only have anti-tumour effects, but also makes great contribution to the treatment of plasmodium falciparum, parasitic diseases and progeria.
There are still some problems with FTIs, but natural products FTIs are worthy of further research as a new class of low-toxic, safe and effective anticancer drugs. Summarised natural product-derived FTase inhibitors
This review summarised several FTIs extracted from natural materials and clarifies their anti-tumour activity (IC50 value) and structure, providing a reference for further research on tumour therapy.
1. Introduction
Tumour refers to the local mass formed by abnormal proliferation of local tissue under the action of various tumorigenic factors. It is one of the world’s five terminally ill diseases including motor neuron disease, AIDS, leukaemia, and rheumatoid arthritis. Cancer cells have unique properties of invasion and metastasis. Tumours disrupt cell junctions and cell signalling, leading to important gene dysfunctionCitation1. At present, the main methods for treating tumours are still traditional treatments such as surgical treatment, chemotherapy, and radiation therapy, which usually have strong toxic side effects and are prone to drug resistance. With the development of related disciplines such as molecular biology, research on anti-tumour drugs has also shifted to new anti-tumour drugs targeting multiple links in the mechanism of tumour development. These drugs, for example, targeting cell signalling molecules: including protein tyrosine kinase inhibitors, farnesyltransferase inhibitors (FTIs), mitogen-activated protein kinase (MAPK) signalling pathway inhibitors, cell cycle regulators, etc., are localised to target cell-specific bio-macromolecules, thereby inhibiting the growth and metastasis of tumour cells, rather than killing cells directly. Among them, FTIs are one of the hotspots in recent years, which have attracted the attention of many famous research institutions at home and abroad.
2. Brief introduction to Ras protein and FTIs
The Ras protein mainly regulates the differentiation and proliferation of cells, and is called the “molecular switch” in the transmission of cellular signalling networks. Ras protein is a low molecular weight protein that is distributed inside the cell membrane and has the function of binding to guanine nucleotides, which plays an significant role in cell growth, proliferation, development, differentiation, and cancer cell productionCitation2. However, if Ras protein is always in an activated state, it will have a continuous stimulating effect on the growth and proliferation of cells, which makes the cells in a state of continuous proliferation or even canceration. Therefore, Ras protein has a very close relationship with tumour generation and development.
Table 1. Anticancer active ingredients of natural materials.
As a precursor protein of cytoplasm, Ras protein needs some modifications including prenylation, proteolysis, carboxymethylation, and palmification to exert all biological activitiesCitation9. The prenylation of the Ras protein requires three enzymes in turn: farnesyltransferase (FTase), geranylgeranyl transferase I (GGTaseI), and geranylgeranyl transferase II (GGTaseII). Farnesyltran is the first step in post-translational modification of Ras. Therefore, the search for suitable FTIs is an important research direction to inhibit Ras protein and thus inhibit the occurrence of cancer.
Table 2. Anticancer active ingredients of natural materials.
FTIs not only have anti-tumour effects, but also have a great contribution to the treatments of Plasmodium falciparum and parasitic diseases. In recent years, the study of FTIs has a certain breakthrough in the treatment of premature aging and antiviral fieldCitation16,Citation17. According to the structural analysis and catalytic mechanism of FTase, FTIs can be divided into four typesCitation18: (1) CAAX (C is cysteine, A is an aliphatic amino acid, and X is serine or methionine) tetrapeptides and their analogues; (2) farnesyl phosphonate (FPP) mimetic; (3) double substrate mimetic; (4) natural product. Natural products have been reported as a major source of lead compounds, and a variety of natural products that could inhibit the activity of FTase have been reportedCitation19. This article describes several FTIs extracted from natural materials and clarifies the current research progress ().
Table 3. Anticancer active ingredients of natural materials.
3. Natural products FTIs
3.1. Quinones
Quinones are a class of chemical constituents with a quinoid structure, which are mainly classified into four types: benzoquinone, naphthoquinone, phenanthrenequinone, and anthraquinone. This article summarises the inhibition of FTase by natural products and their derivatives isolated from Tectona grandis L., sponges, A. camphorata, Streptomyces sp., and Dichrostachys cinerea, with IC50 values ranging from 0.031 μM to 100 μM. It is found that phenanthraquinone compounds extracted from Adocia sp. Sponge have a better inhibitory effect on FTase, especially compounds 9 and 10, for their similarity FPP. There are many types and a large number of anthraquinones, including compounds 1 to 6, 11 to 14 ( and ).
Figure 1. Chemical structures of quinones extracts from (a) Tectona grandis L. and their derivatives (1–6); (b) sponges and their derivatives (7–14).
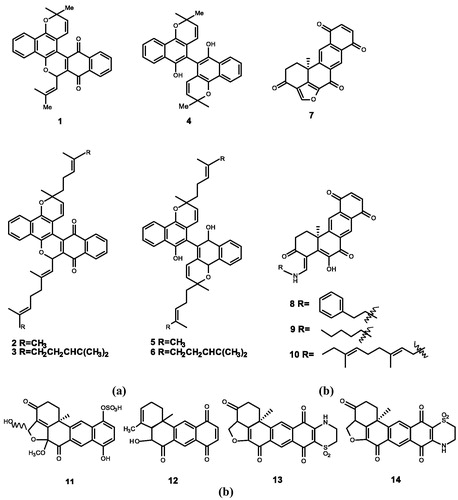
Figure 2. Chemical structures of quinones extracts from (a) A. camphorata (15); (b) Streptomyces sp. (16–18); (c) Dichrostachys cinerea (19–30).
3.1.1. Tectona grandis L
Tectona grandis L. belongs to the Lamiaceae family, which is native to Myanmar, Thailand, India, Indonesia, Laos, etc. Currently, it is widely introduced into Yunnan, Guangdong, Guangxi, Fujian, and TaiwanCitation31. It plays an important role in pharmacological effects such as antibacterial, anti-arthritic, anti-oxidant, and wound healingCitation32,Citation33 ().
Table 4. Anticancer active ingredients of natural materials.
Tecomaquinone I was originally extracted from Tectona grandis L. by RomanisCitation43. It is not only inhibited human and T. brucei FTase (IC50 = 0.065 and 0.112 μM), but also exhibited moderate activity inhibition against Plasmodium falciparum (IC50 = 3.44 ± 0.20 μM). Cadelis et al.Citation3 synthesised and analysed a series of derivatives of tecomaquinone I. They found that derivative 2 showed good inhibitory activity against human and T. brucei FTase (IC50 = 1.1 and 2.7 μM), but the inhibitory activity of derivative 3 with the longer side chain added at the same position was significantly reduced.
Sandermann and DietrichsCitation44 found a new natural product from Tectona grandis L., tectol (4). Tectol showed moderate inhibition against FTase (IC50 = 2.09 μM)Citation3. Cadelis et al.Citation3 synthesised and analysed the derivatives of tectol. They found, as opposed to tecomaquinone I, the derivative with longer side chain (6) has stronger inhibitory activity (IC50 = 1.8 μM). They believed that tecomaquinone I can be used as a novel scaffold to develop more effective FTIsCitation3.
3.1.2. Sponge
Sponge belongs to Porifera and is distributed in the ocean, lakes and streams. Terpenoids and terpenoids extracted from sponges play an important role in combating malaria, anti-inflammatory, antibacterial, antiviralCitation45.
Schmitz and BloorCitation46 isolated halenaquinone (7) from Adocia sp. sponge, synthesised the corresponding derivatives on the basis of them, and detected that these inhibitors have certain cytotoxicity. Cao et al.Citation5 isolated many halenaquinone-type polyketides from marine sponges of the genus Xestospongia by bioassay directed fractionation techniques. Wang et al.Citation4 synthesised some derivatives of halenaquinone, evaluated a series of biological targets including FTase, and determined that halenaquinone and derivative 8, 9, and 10 have FTase, phospholipase A2 and Plasmodium falciparum inhibition. Among them, compound 10 has the strongest inhibitory effect on FTase (IC50 = 0.031 μM).
Cao et al.Citation5,Citation47,Citation48 further isolated the sponges and found two compounds that inhibited FTase, xestosaprol C methylacetal (11), and orhalquinone (12). Compound 11 possess significant inhibitory activity against human FTase in µM range (IC50 = 4.34 µM). Compound 12 showed inhibitory activity against human and yeast FTase (IC50 = 0.40 μM), with a stronger inhibitory effect than xestosaprol C methylacetal. At the same time, Orhalquinone is a moderate growth inhibitor of P. falciparumCitation48. Besides, Cao et al.Citation5 also found that 3-ketoadociaquinone A (13) and 3-ketoadociaquinone B (14) have inhibitory effects on FTase (IC50 = 4.19 and 9.27 µM) and GGTase (IC50 = 1.08 and 3.89 µM).
3.1.3. Antrodia camphorata
Antrodia camphorata belongs to Taiwanofungus and grows on the inner wall of the decaying heartwood of Taiwan’s Cinnamomum kanehirae, with the effect of lowering cholesterol, anti-cancer, anti-Alzheimer’s diseaseCitation49–51.
Wu et al.Citation52 extracted antroquinonol (15), a novel compound with anti-inflammatory activities, from the mycelium of Antrodia camphorataCitation53,Citation54. Ho et al.Citation6 measured the inhibitory effect of Antroquinonol on various of tumours such as human lung cancer, liver cancer, and leukaemia. Antroquinonol plays a major role in interrupting the function of Ras and Ras-related GTP-binding proteins by inhibiting the activity of FTase and GGTase in cancer cells, ultimately resulting in cell death. At present, antroquinonol is in the phase 2 clinical trials in patients with non-small cell lung cancerCitation6,Citation55,Citation56.
3.1.4. Streptomyces sp
Streptomyces sp. belongs to Actinomyces and is widely found in soil. It exhibits a wide range of anti-microbial, anti-parasitic, and anti-oxidant activities and it is an important species for the study of antibioticsCitation57,Citation58.
Hara et al.Citation7 isolated UCF 116 compounds from Streptomyces sp. Among these compounds, UCF 116A (16) and UCF 116B (17) have a strong inhibitory effect against bovine brain FTase (IC50 = 1.2 and 0.6 μM), whereas UCF 116C (18) has a weaker inhibitory effect (100 μM)Citation59. Compounds 16 and 17 selectively inhibit rabbit reticulocyte lysate FTase (IC50 = 4 μM) and have a weak inhibitory effect on GTaseCitation60,Citation61. The kinetic analysis indicated that UCF 116B is competitive inhibitor of Ras protein, and UCF 116 compounds are all Ras-competitive and non-CAAX mimetic type inhibitors of FTase. It is further concluded that the substituent at the terminal amide linkage of the three compounds, the cyclohexenecarboxylalanine moiety of the compound 17 and the modification of the substituents at the C-11 site all have a non-negligible inhibitory activity against the FTase. More effective FTIs can be found based on these findings.
3.1.5. Dichrostachys cinerea
Dichrostachys cinerea belongs to Leguminosae and is native to Africa and India. The trunk can be used for myogenic, analgesic, hemostasis, dysentery, diarrhoea, etc. The bark can be used to treat intestinal parasites, syphilis, and leprosyCitation62,Citation63.
Long et al.Citation8 extracted a novel compound from Dichrostachys cinerea, which was named dichrostachine A (19). To fully study compound 19, they extracted other secondary metabolites from the roots, stems, and barks of Dichrostachys cinerea, a total of 18 compounds (dichrostachine A-R), all of which belong to the meroterpene derivatives. After determining their structures and synthesising the corresponding derivatives, using sch-66336 as the reference compound, they determined that dichrostachine A(19), B(20), C(21), D(22), E(23), G(24), H(25), L(26), M(27), O(28), P(29), and R(30) have the activity of inhibiting FtaseCitation64. Compound 25 has the highest inhibitory activity (IC50 = 1.8 μM). The second is compound 27 (IC50 = 3 μM), while compound 27 has high cytotoxicity.
3.2. Terpenoids
Terpenes take isoprene unit as the basic structural unit, and are mainly classified into monoterpenes, sesquiterpenes, and diterpenes. This article summarises the inhibition of FTase by natural products and their derivatives isolated from Aplidium conicum, Penicillium sp., Stachybotrys kampalensis, and plant essential oils. Most of the natural products and their derivatives extracted from Aplidium conicum have strong FTase inhibitory activity, and the activity varies with the side chain, whereas some monoterpenoids extracted from plant essential oils are weak. Derivatives with side chains of farnesyl group generally have good inhibitory activity. Triterpenoids 51–57 are all derived from mildew, and their activities vary greatly with the groups. Some of the monoterpenoids extracted from plant essential oils contain the least amount of isoprene unit and possess the weakest activity. Although terpenes take isoprene unit as the basic structural unit, they are mostly closed-loop structures, and only groups with chain isoprene groups or similar structures can help to inhibit FTase ( and ).
Figure 3. Chemical structures of terpenoids extracts from Aplidium conicum and their derivatives (31–42).
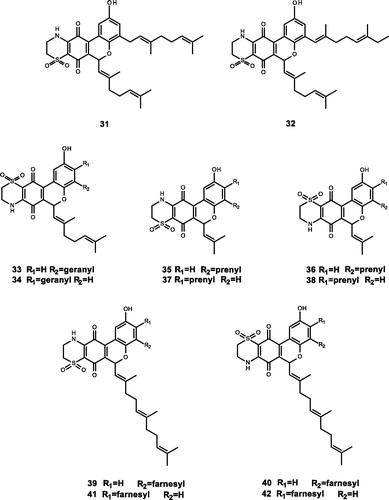
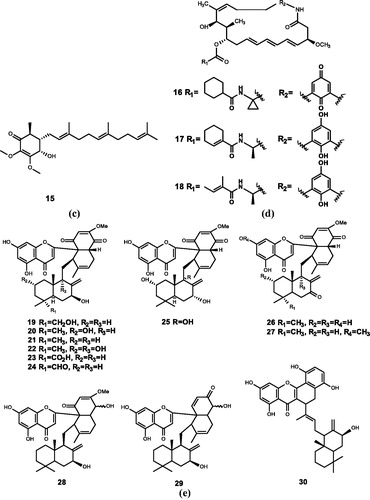
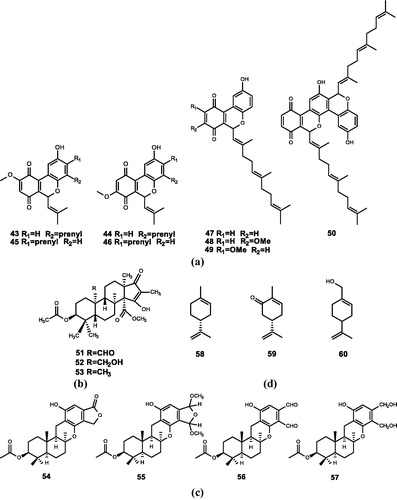
Figure 4. Chemical structures of terpenoids extracts from (a) Aplidium conicum and their derivatives (43–50); (b) Penicillium sp. (51–53); (c) Stachybotrys kampalensis and their derivatives (54–57); (d) plant essential oils (58–60).
3.2.1. Aplidium conicum
Aplidium conicum belongs to Ascidiacea, which is widely distributed in the ocean and has good medical value and edible value. Its extracts can promote apoptosisCitation65,Citation66.
Aiello et al.Citation67 extracted two marine meroterpenoids, thiaplidiaquinones A (31) and thiaplidiaquinones B (32), which could induce the apoptosis of Jurkat cells, from the ascidian Aplidium conicum. Compound 31 (IC50 = 0.78 and 0.74 μM) and Compound 32 (IC50 = 1.22 and 3.04 μM) have inhibitory effects against both human and T. brucei FTase. Harper et al.Citation10 obtained the corresponding dioxothiazine isomers (33) and (34). Compounds 31, 33, and 34 were identified to be potent inhibitors of human and T. brucei FTase, and the IC50 values are in the range of nM. Compounds 33 and 34 have moderate inhibitory activity against Plasmodium falciparum, and compound 33 also has moderate antiproliferative activity against melanocyte cell lines. To further investigate the structure–activity relationship of thiaplidiaquinones (31–34), Cadelis et al. synthesised a series of derivatives (35–50), among which 17 derivatives have inhibitory activity against FTase. The test results show that the derivatives with the farnesyl side chain (39–42) have stronger FTase inhibitory activity than the derivatives with the isoprene side chain (35–38)Citation11.
Grayfer et al.Citation68 found that the modification of the prenylated side chain of the natural product can regulate biological activity. For example, an increase in the number of prenyl units on the side chain of mallotojaponin B may enhance anti-malarial and FTase inhibitory activity. Whereas studies on tecomaquinone I indicated that this increase leads to a decrease in FTase activity. After synthesis and determination of derivatives of compounds 31 and 32, it was found that this series of compounds have strong or weak inhibitory effects on FTase, and most of the compounds also have moderate antimalarial activity.
3.2.2. Penicillium sp
Penicillium sp. belongs to Trichocomaceae and grows on decaying fruits, vegetables, meat, and various moist organic substances. Its metabolites show excellent antibacterial and anti-inflammatory activities, moreover, good killing effect on larvae such as Culex quinquefasciatusCitation69,Citation70.
Shiomi et al.Citation71 extracted andrastin A (51), andrastin B (52), and andrastin C (53) from Penicillium sp. FO-3929. These three compounds have inhibitory effects on FTase in a dose-dependent manner (IC50 = 24.9, 47.1, and 13.3 μM)Citation12.
3.2.3. Stachybotrys kampalensis
Stachybotrys kampalensis belongs to chartrum, which is commonly found in soil and can use plants to produce celluloseCitation72. Stachybotrys can help the treatment of cancerCitation73.
Singh et al.Citation13 isolated kampanol A-C (54–56) from Stachybotrys kampalensis and synthesised a derivative of 56. Compounds 54, 55 are found to have inhibitory activity against human recombinant FPTase by biological activity assay (IC50 = 7 and 13 μM). Nevertheless, 56 and its derivative (57) have weak inhibitory activities (IC50 = 560 and 460 μM). Besides, none of the four compounds have inhibitory activity against GGTase.
3.2.4. Plant essential oils
Plant essential oils are extracted from a variety of plants. Major of them are monoterpenoids. These compounds have antibacterial and insecticidal effectsCitation74. Several monoterpenoids described here have a certain inhibitory effect on FTase.
Crowell et al.Citation75 extracted the monoterpenoid d-limonene (58), one of the end products of the metabolism of mevalonate in plant cells, which is widely found in orange peel and some plant essential oils. Compound 58 is one of the few natural anti-tumour products known to have both chemopreventive and chemotherapeutic effects. Besides, d-limonene is also a very effective chemotherapeutic agent, which can cause 80% complete regression of rat breast cancer induced by chemical factorsCitation76–78. Currently, d-limonene has entered phase I clinical studies in the UKCitation79.
Crowell et al.Citation80 demonstrated that d-carvone (59), which is a d-limonene analogue, can inhibit cell growth. Hardcastle et al.Citation15 suggested that compound 59 may be a metabolite of limonene in vivo, a potential FTI (IC50 = 5700 µM).
Perillyl alcohol (60), similar to d-limonene, is a monoterpenoid derived from the essential oils of cherries and other plants. It also has the effect of inhibiting cell growth and weak FTase and GGTase inhibition (IC50 = 1000 and 700 µM)Citation14,Citation81.
3.3. Lactones
Lactone refers to the organic matter produced by dehydration of both carboxyl group and hydroxyl group in the same molecule, which is mainly classified according to the size of the ring. We summarise the inhibition of FTase by natural products and their derivatives isolated from Xanthium strumarium L., unidentified fungus, and Artemisia. In such compounds, the IC50 value of the natural product as a function of side chain groups varies widely. When the groups are the same, the activity of the compounds which are isomers will also vary greatly. For example, the compounds 71, 72 and the compounds 74, 75 are positional isomers, but their activities differ by more than 100-fold ().
Figure 5. Chemical structures of lactones extracts from (a) Xanthium strumarium L. (61–62); (b) unidentified fungus (63–64); (c) Artemisia (65–75).
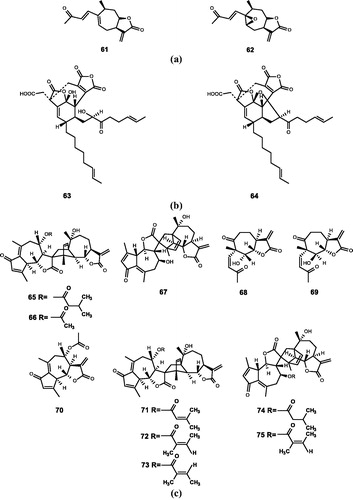
3.3.1. Xanthium strumarium L
Xanthium strumarium L. belongs to Compositae and is widely distributed in China. It can be used as raw material for paint, soap, and linoleum. Its fruit extracts have the ability of anti-bacterial, anti-oxidation, anti-diabetes, and preventing arthritisCitation82,Citation83.
Kim et al.Citation20 extracted two xanthanolide sesquiterpene lactones, 8-epi-xanthatin (61) and 8-epi-xanthatin epoxide (62) from the leaves of Xanthium strumarium L. The concentration of compound 61 required to obtain FTase inhibition is higher than that of compound 62 (IC50 = 64 and 58 μM). The results of in-depth study of compound 61 show that compound 61 can inhibit the growth of T. brucei rhodesiense, T. cruzi, L. donovani, and P. falciparum with IC50 values of 0.33, 11.3, 0.6, and 6.5 μM, respectively. However, the cytotoxicity of compound 61 reached 22.1 μM for rat myoblast cells. In other words, its side effects are greatCitation84.
Matsuo et al.Citation85 separated two milder compounds from Xanthium strumarium L. leaves. Although the inhibitory activities of the two compounds were weaker than that of the compound 61, their side effects were smaller and saferCitation86,Citation87.
3.3.2. Unidentified fungus
Dabrah et al.Citation21,Citation88,Citation89 found two compounds CP-225917 (63) and CP-263114 (64) from unidentified fungi, which have some characteristics of Phoma sp. The structures of the two compounds were analysed by means of extensive NMR measurements, UV spectra, and FT-IR spectraCitation89. It was verified by experiments that both compounds inhibited FTase in the brain of mice (IC50 = 6 and 20 μM).
3.3.3. Artemisia
As Compositae, Artemisia is extremely strong and widely distributed, which is distributed in temperate regions of the northern hemisphere. Artemisia has good medicinal value, can be used to treat malaria and cancer. In addition, Artemisia also has antibacterial and anti-oxidant effectsCitation90,Citation91.
Lee et al.Citation22 isolated a compound from the leaves of Artemisia sylvatica Maxim in 1998. It was determined to be arteminolide (65) by NMR, IR, HREIMS, NOESY spectrum, and mass spectral data. Compound 65 has broad biological activity and selectively inhibits FTase (IC50 = 0.36 μM)Citation92–94.
Later, Lee et al.Citation23 isolated five compounds from the methanol extract of Artemisia sylvatica in 2000. After testing and comparing the spectral data with the reported spectral dataCitation95, it was confirmed that the five compounds were artanomaloide (66), 8-acetylarteminolide (67), arteminone (68 and 69), and dehydromatricarin (70). All the five belong to sesquiterpene lactone. Among them, compounds 68 and 69 are identified as stereoisomers. The inhibitory effects of five compounds on FTase were examined by scintillation proximity assay (SPA). The results show that compound 67 selectively inhibited recombinant rat FTase (IC50 = 1.8 μM) and weaker inhibitory activity against recombinant rat GGTase (IC50 ≫ 100 μM). Compounds 66, 67, 68, and 69 also had some inhibitory effects on FTase (IC50 = 22, 85, 82, and 300 μM). This result is important for studying the pharmacological activities of sesquiterpene lactones. Due to the special structure of 8-acetylarteminolide, Lee et al. speculated that it might be a useful lead compound for the development of anticancer drugs. Subsequent studies show that 8-acetylarteminolide does have an inhibitory effect against tumour cellsCitation96,Citation97.
Lee et al.Citation24 isolated some compounds from Artemisia argyi and obtained the corresponding chemical structure through experiments in 1998. They renamed the arteminolide (65) as arteminolide A, and the other compounds were named arteminolide B (71), arteminolide C (72), arteminolide D (73), artanomaloide A (74), and artanomaloide C (75). Among them, compounds 74, 75 are regioisomers. In vitro enzyme assays and kinetic experiments revealed that compounds 71–73 have strong inhibitory effect on recombinant human FTase with IC50 values ranging from 1.7 to 1.1 μM. However, the IC50 values of artanomaloides are all over 100 μM. Arteminolides and artanomaloides have almost no inhibitory activity against rat squalene synthase and recombinant rat GGTase.
3.4. Polycarboxylic acids
Polycarboxylic acids are a class of carboxylic acids containing a plurality of carboxyl groups. We summarises the inhibition of FTase by polycarboxylic acids isolated from Chaetomella sp., Amaumarcus niger, Mollisia sp., and Phoma sp. The inhibitions of these natural products are significant. After analysing the activity and structure of each compound, it was found that several polycarboxylic acid compounds in this review had groups similar to FPP and could compete with FPP to inhibit FTase, so almost all these polycarboxylic acids compounds had good inhibitory activity. The side chain groups of polycarboxylic acids have a great influence on the activity. For example, the cis-orientation of dicarboxylic acid and the CH on the alkyl side can make the inhibitory effect more significant, while the cis-diacid composed of shorter alkyl chain can make the compound lose its inhibitory activity ( and ).
Figure 6. Chemical structures of polycarboxylic acids extracts from (a) Chaetomella acutiseta and their derivatives (76–82); (b) Amaumarcus niger and Mollisia sp. (87–93).
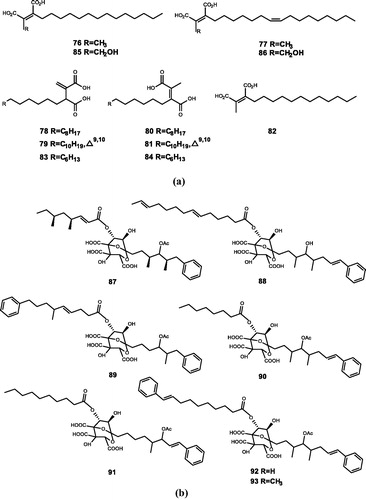
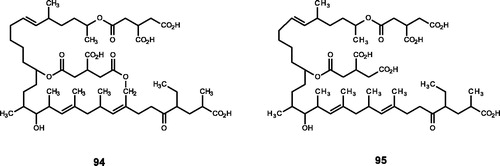
Figure 7. Chemical structures of polycarboxylic acids extracts from Actinoplanes sp. and their derivatives (94–95).
3.4.1. Chaetomella sp
Chaetomella sp. belongs to Sphaeropsidaceae, which uses many plants as its host to survive and has good adaptability to various environment, thus it is distributed all over the worldCitation98. Chaetomella sp. is an important material for the production of TaxolCitation99, which has a certain contribution to the treatment of cancer.
Singh et al.Citation100 extracted chaetomellic acids A (76) and B (77) from Chaetomella acutiseta and confirm that they can inhibit recombinant human FTase. Subsequently, compounds 76 and 77 were confirmed to have reversible inhibitory abilities against FTase (IC50 = 55 and 185 nM), and selective inhibitory abilities against bovine brain GGTase (IC50 = 92 and 54 μM)Citation25. Lingham et al.Citation101 further verified that compounds 76 and 77, similar to FPP, are reversible inhibitors of FTase. Singh et al.Citation26 synthesised the isomeric diacids (78, 79, 80, 81) of chaetomellic acids A and B, together with C-12 chain compound chaetomellic acid C (82) and its isomers (83, 84). They also found chaetomellic acids D (85) and E (86) from a mycelial broth. These compounds all have strong or weak FTase inhibitory abilities, but no inhibitory activity against GGTase. The results of their work show that the cisoid orientation of the dicarboxylic acid and the alkyl chain of the appropriate length play an important role in the inhibitory activity against FTase. The trans- and vinyl-diacids reduce the inhibitory activity of the compound, and the cis-diacids consisting of a shorter alkyl chain were inactive against FTase.
3.4.2. Amaumarcus niger, Mollisia sp., and Phoma sp
Amaumarcus niger is a keratinophilic fungus which is mostly distributed in the soil. Most of the extracts from Amaumarcus niger have strong activity. Mollisia sp. belongs to the Mollisiaceae and is mainly distributed in warm and humid areas. Although Mollisia sp. causes some diseases, some of its extracts can be used as new antibiotics and have contributed to the field of medicineCitation102. Phoma sp. is an endophytic fungus widely distributed in the ocean. It has considerable application value. Its isolated products can achieve the effect of treating cancer by inducing apoptosis and autophagy of cancer cellsCitation103. For example, terpenoids extracted from its culture medium show good antiviral activityCitation104, and its metabolites can be used to prepare herbicidesCitation105.
Zaragozic acid A (87), B (88), C (89), D (90), and D2 (91) extracted from keratinophilic fungus Amaumarcus niger belong to tricarboxylic acid. The structure was identified by 2D NMR and mass spectral experimentsCitation106–109. The five compounds all have FTase inhibitory activity, by competing with FPP. Among them, compounds 89, 90, and 91 have the strongest activity (IC50 = 150, 100, and 100 nM), whereas compound 87 and 88 have relatively weak activity (IC50 = 250 and 1000 nM)Citation25,Citation27. Tanimoto et al.Citation110 extracted zaragozic acid D3 (92) and zaragozic acid D3 8-methylester (93) from the fungus Mollisia sp. Both of the compounds have strong inhibitory activities on FTase and GGTaseCitation28. Pedretti et al.Citation111 performed a detailed docking analysis of zaragozic acid extracted from Phoma sp. by molecular docking and found that zaragozic acid have strong FTase inhibitory ability by interacting with residues at two FPP binding sites.
3.4.3. Actinoplanes sp
Actinoplanes sp. belongs to Micromonosporaceae, which grows in soil, fresh water, and plant remnants. Its hyphae produce a variety of antibiotics against bacteria and tumoursCitation112,Citation113.
Singh et al.Citation29 originally extracted actinoplanic acid A (94) from Actinoplanes sp. and determined its structure. Compound 94 was found to compete with FPP and was also a selective inhibitor of FTase (IC50 = 230 nM). Subsequently, actinoplanic acid B (95) was extracted from Actinoplanes sp., which also exerts selective and competitive inhibition of FTase and has stronger inhibitory activity (IC50 = 50 nM)Citation30,Citation114. When analysing the structure, we found that compounds 94 and 95 carried more groups similar to FPP, just as Singh et al. found that they could compete with FPP to inhibit FTase, with strong inhibitory activity.
3.5. Phenolics
The phenolic compound refers to a compound having a hydroxyl group on an aromatic hydrocarbon and is classified into monohydric phenol and polyhydric phenol. This review summarises phenolic compounds isolated from several fungi with a small IC50 difference and similar inhibitory abilities against FTase. After observing of the structure and activity of the compounds, it was found that compounds 96 and 97 had more complicated structures than other phenolic compounds and were less effective in inhibiting FTase ( and ).
Figure 8. Chemical structures of phenolics extracts from (a) Streptomyces sp. (96–97); (b) Fusidium griseum (98); (c) Phoma sp. (99–100); (d) Preussia isomera and Harmonema dematioides (101–106).
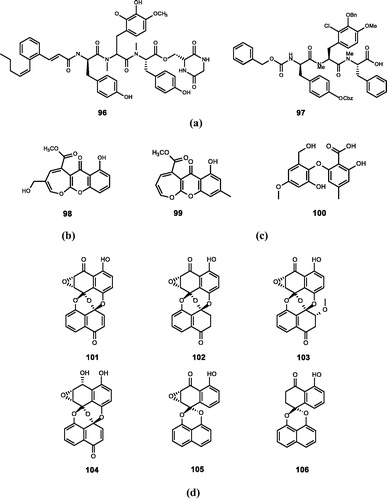
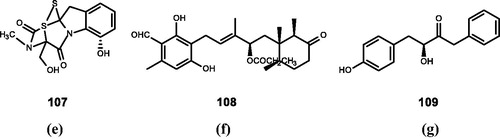
Figure 9. Chemical structures of phenolics extracts from (e) Aspergillus, Trichoderma, and Penicillium (107); (f) Cylidrocarpon lucidum (108); (g) Paecilomyces sp. (109).
3.5.1. Streptomyces sp
Streptomyces sp. is the highest Actinomyces and has a good adaptability to the environment. It is of great value to the development of antibiotics and cancer treatment.
Hinterding et al.Citation115 isolated pepticinnamin E (96) from secondary metabolites of Streptomyces sp. It is the first Ras protein substrate competition inhibitor derived from natural products. What is more, subsequent studies show that it is a dual substrate (Ras protein and FPP) competition inhibitor (IC50 = 42 μM)Citation34,Citation115,Citation116. Structurally, compound 96 mimics the two substrates of FTase, CAAX, and FPP.
Thutewohl and WaldmannCitation35 synthesised a library of compounds including 51 analogues based on the structure of compound 96 by a change in eight structural parameters. Among them, the seventh derivative (97) has an inhibitory activity similar to that of compound 96 (IC50 = 67 μM).
3.5.2. Fusidium sp
Fusidium sp. belongs to Moniliaceae and has important medical value. The antibiotic fusidic acid extracted from Fusidium sp. has been used for disease treatment since 1962Citation117,Citation118. Fusidic acid not only has good in vitro antibacterial effect, but also has strong anti-inflammatory effects in vivoCitation119.
Singh et al.Citation36 extracted fusidienol (98) from Fusidium griseum, which inhibited bovine brain FTase and human FTase (IC50 = 0.3 and 2.7 μM).
Fusidienol A (99) is an FTase inhibitor extracted from Phoma sp., which is similar in structure and function to compound 98. Compound 99 has a strong inhibitory activity on recombinant human FTase (IC50 = 1.8 μM) but does not inhibit GGTase wellCitation37.
3.5.3. Phuma sp
As an endophytic fungus, Phuma sp. has the effect of treating cancer and antiviral activity.
Jayasuriya et al.Citation38 extracted three natural products from Phuma sp. The structure and activity of these compounds were determined experimentally. Only barceloneic acid A (100) was found to have FTase inhibitory activity (IC50 = 40 μM).
3.5.4. Preussia sp. and Harmonema dematioides
Preussia sp. is a new Chinese strain discovered in 2013 and belongs to endophytic fungi. Its isolates have strong antiplasmodial, antibacterial, and antioxidant activitiesCitation120,Citation121. Harmonema dematioides is also an endophytic fungus. It produces secondary metabolites that inhibit the biosynthesis of sterols and thus have a good therapeutic effect on heart diseaseCitation122.
Zink et al.Citation39 studied the structure–activity relationships of preussomerin G (101), H (102), I (103), D (104), deoxypreussomerin A (105) and B (106) extracted from the coprophilous fungus Preussia isomera, and the endophytic fungus Harmonema dematioidesCitation123–125 which are fungal metabolites. They have a certain inhibitory activity on FTase. Among them, compound 101 has the strongest activity, not only against FTase, but also GGTase (IC50 = 1.2 and 20 μM).
3.5.5. Aspergillus, Trichoderma, and Penicillium
Aspergillus belongs to Aspergillaceae and is widely distributed in nature. Although it causes food spoilage and is extremely toxic, it has a key role in food fermentation. Aspergillus also has a positive contribution to antibacterial effectsCitation126. Trichoderma belongs to Hypocreaceae and is mainly distributed in rot, seeds, plant residues, organic fertilisers, soil, and air. Trichoderma has good medicinal value. For example, it can be used to produce cellulaseCitation127, producing antibiotics, and so onCitation128. Living on saprophytic life, Penicillium sp. can cause food to rot, but it also has certain antibacterial and anti-inflammatory effects.
Shah et al.Citation40,Citation129 extracted gliotoxin (107) from Aspergillus, Trichoderma, and Penicillium, which is a sulphur-containing mycotoxin. Compound 107 has modest FTase inhibitory activity (IC50 = 1.1 μM).
3.5.6. Cylidrocarpon lucidum
Cylidrocarpon lucidum is a fungus with excellent medicinal value. Its metabolites can form cyclosporin for treating rheumatoid arthritisCitation130.
Singh et al.Citation41 extracted cylindrol A (108) from Cylidrocarpon lucidum, which is a bicyclic resorcinaldehyde cyclohexanone propionate derivative. It could inhibit bovine FTase (IC50 = 2.2 μM).
3.5.7. Paecilomyces sp
Paecilomyces sp. belongs to Discellaceae, and its research is still not very thorough. It is a group of fungi that are only found in asexual or sexual stages. Its extracts have a considerable effect on antileishmanial and enhanced immunityCitation131,Citation132.
Uchida et al.Citation42 extracted kurasoin A (109) from a cultured broth of Paecilomyces sp. FO-3684, and determined the structure of this compound for the first time. It was found to have a dose-dependent inhibition against FTase (IC50 = 59 μM).
4. Future perspective
So far, many compounds with FTase inhibitory activity have been discovered and synthesised. What is more, many of them are now in the clinical trials. Oral FTIs developed in recent years have high bioavailability and selective antitumor activity in vivo, and their potential effects may eventually be used to treat malignant tumours in humans. FTIs have also made significant achievements in the treatment of diseases such as premature aging and leukaemiaCitation133–135.
It is worth noting that half of the compounds listed here with IC50 values less than 1 μM are from belong to polycarboxylic acid compounds. The importance of polycarboxylic acid compounds in the inhibition of FTase activity was demonstrated. The structural similarity of these polycarboxylic acid compounds to FPP may be the main reason for their effective FTase inhibitory activity. In this review, compounds containing chain groups are similar to FPP, which can compete with FPP to inhibit FTase. However, FPP is also substrate of other enzymes, FPP-competitive inhibitors may inhibit the physiological function of them, causing some harm to the human body. As far as the current results concerned, natural products FTIs are worthy of further research as a new class of low toxicity, safe, and effective anticancer drugs. Apart from further structural modifications based on the existing structural types to enhance the selectivity and in vivo activity of FTIs, a rich library of natural compounds is also worthy of attention. In recent years, many FTIs of novel structures have been found in natural products. It is expected that FTIs will be a promising field of research for new anticancer drugs, and will provide more new options for the clinical treatment of malignant tumours.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Wittekind C, Neid M. Cancer invasion and metastasis. Oncology 2005;69:14–6.
- Rodenhuis S. Ras and human tumors. Semin Cancer Biol 1992;3:241–7.
- Cadelis MM, Bourguet-Kondracki ML, Dubois J, et al. Discovery and preliminary structure–activity relationship studies on tecomaquinone I and tectol as novel farnesyltransferase and plasmodial inhibitors. Bioorg Med Chem 2016;24:3102–7.
- Wang J, Bourguet-Kondracki ML, Longeon A, et al. Chemical and biological explorations of the electrophilic reactivity of the bioactive marine natural product halenaquinone with biomimetic nucleophiles. Bioorg Med Chem Lett 2011;21:1261–4.
- Cao S, Foster C, Brisson M, et al. Halenaquinone and xestoquinone derivatives, inhibitors of Cdc25B phosphatase from a Xestospongia sp. Bioorg Med Chem 2005;13:999–1003.
- Ho C-L, Wang J-L, Lee C-C, et al. Antroquinonol blocks Ras and Rho signaling via the inhibition of protein isoprenyltransferase activity in cancer cells. Biomed Pharmacother 2014;68:1007–14.
- Hara M, Soga S, Itoh M, et al. UCF116, new inhibitors of farnesyltransferase produced by Streptomyces. J Antibiot 2000;53:720–3.
- Long C, Marcourt L, Raux R, et al. Meroterpenes from Dichrostachys cinerea inhibit protein farnesyl transferase activity. J Nat Prod 2009;72:1804–15.
- Yu C, Han W, Shi T, et al. PTPIP51, a novel 14-3-3 binding protein, regulates cell morphology and motility via Raf-ERK pathway. Cell Signal 2008;20:2208–20.
- Harper JL, Khalil IM, Shaw L, et al. Structure–activity relationships of the bioactive thiazinoquinone marine natural products thiaplidiaquinones A and B. Mar Drugs 2015;13:5102–10.
- Cadelis MM, Bourguet-Kondracki ML, Dubois J, et al. Structure–activity relationship studies on thiaplidiaquinones A and B as novel inhibitors of Plasmodium falciparum and farnesyltransferase. Bioorg Med Chem 2017;25:4433–43.
- Omura S, Inokoshi J, Uchida R, et al. Andrastins A–C, new protein farnesyltransferase inhibitors produced by Penicillium sp. FO-3929. I. Producing strain, fermentation, isolation, and biological activities. J Antibiot (Tokyo). 1996;49:414–7.
- Singh SB, Zink DL, Williams M, et al. Kampanols: novel ras farnesyl-protein transferase inhibitors from Stachybotrys kampalensis. Bioorg Med Chem Lett 1998;8:2071–6.
- Gelb MH, Tamanoi F, Yokoyama K, et al. The inhibition of protein prenyltransferases by oxygenated metabolites of limonene and perillyl alcohol. Cancer Lett 1995;91:169–75.
- Hardcastle I, Rowlands M, Moreno Barber A, et al. Inhibition of protein prenylation by metabolites of limonene. Biochem Pharmacol 1999;57:801–9.
- Chakrabarti D, Da Silva T, Barger J, et al. Protein farnesyltransferase and protein prenylation in Plasmodium falciparum. J Biol Chem 2002;277:42066–73.
- Wiesner J, Kettler K, Sakowski J, et al. Farnesyltransferase inhibitors inhibit the growth of malaria parasites in vitro and in vivo. Angew Chem (Int Ed. in English) 2004;43:251–4.
- Tamanoi F. Inhibitors of ras farnesyltransferases. Trends Biochem Sci 1993;18:349–53.
- Bhide RS, Patel DV, Patel MM, et al. Rational design of potent carboxylic acid based bisubstrate inhibitors of ras farnesyl protein transferase. Bioorg Med Chem Lett 1994;4:2107–12.
- Kim YS, Kim JS, Park SH, et al. Two cytotoxic sesquiterpene lactones from the leaves of Xanthium strumarium and their in vitro inhibitory activity on farnesyltransferase. Planta Med 2003;69:375–7.
- Moorthy NSH, Sousa SJ, Ramos MA, Fernandes P. Farnesyltransferase inhibitors: a comprehensive review based on quantitative structural analysis. Curr Med Chem. 2013;20:4888–923.
- Lee SH, Kim MJ, Bok SH, et al. Arteminolide, an inhibitor of farnesyl transferase from Artemisia sylvatica. J Org Chem 1998;63:7111–3.
- Lee S-H, Kang H-M, Song H-C, et al. Sesquiterpene lactones, inhibitors of farnesyl protein transferase, isolated from the flower of Artemisia sylvatica. Tetrahedron 2000;56:4711–5.
- Lee SH, Kim HK, Seo JM, et al. Arteminolides B, C, and D, new inhibitors of farnesyl protein transferase from Artemisia argyi. J Org Chem 2002;67:7670–5.
- Gibbs JB, Pompliano DL, Mosser SD, et al. Selective inhibition of farnesyl-protein transferase blocks ras processing in vivo. J Biol Chem 1993;268:7617–20.
- Singh SB, Jayasuriya H, Silverman KC, et al. Efficient syntheses, human and yeast farnesyl-protein transferase inhibitory activities of chaetomellic acids and analogues. Bioorg Med Chem 2000;8:571–80.
- Dufresne C, Wilson K, Singh S, et al. Zaragozic acids D and D 2: potent inhibitors of squalene synthase and of Ras farnesyl-protein transferase. J Nat Prod 1993;56:1923–9.
- Tanimoto T, Ohya S, Tsujita Y. Inhibitory activity to protein prenylation and antifungal activity of zaragozic acid D3, a potent inhibitor of squalene synthase produced by the fungus, Mollisia sp. SANK 10294. J Antibiot (Tokyo). 1998;51:428–31.
- Singh SB, Liesch JM, Lingham RB, et al. Actinoplanic acid A: a macrocyclic polycarboxylic acid which is a potent inhibitor of Ras farnesyl-protein transferase. J Am Chem Soc 1994;116:11606–7.
- Singh SM, Liesch JB, Lingham R, et al. Structure, chemistry, and biology of actinoplanic acids: potent inhibitors of ras farnesyl-protein transferase. J Org Chem 1995;62:7896–910.
- Hansen OK, Changtragoon S, Ponoy B, et al. Genetic resources of teak (Tectona grandis Linn. f.)—strong genetic structure among natural populations. Tree Genet Genomes 2014;11:802–18.
- Vyas P, Yadav DK, Khandelwal P. Tectona grandis (teak) – a review on its phytochemical and therapeutic potential. Nat Prod Res 2019;33:2338–54.
- Senthilkumar N, Nandhakumar E, Priya P, et al. Synthesis of ZnO nanoparticles using leaf extract of Tectona grandis (L.) and their anti-bacterial, anti-arthritic, anti-oxidant and in vitro cytotoxicity activities. New J Chem 2017;41:10347–56.
- Hinterding K, Hagenbuch P, Rétey J, Waldmann H. Synthesis and in vitro evaluation of the Ras farnesyltransferase inhibitor pepticinnamin E. Angew Chem Int Ed 1998;37:1236–9.
- Thutewohl M, Waldmann H. Solid-phase synthesis of a pepticinnamin E Library. Bioorg Med Chem 2003;11:2591–615.
- Singh S. Fusidienol: a novel inhibitor of Ras farnesyl-protein transferase from Fusidium griseum. Tetrahedron Lett 1994;35:4693–6.
- Singh SB, Ball RG, Zink DL, et al. Fusidienol A: a novel Ras farnesyl-protein transferase inhibitor from Phoma sp. J Org Chem 1997;62:7485–8.
- Jayasuriya H, Ball RG, Zink DL, et al. Barceloneic acid A, a new farnesyl-protein transferase inhibitor from a Phoma species. J Nat Prod 1995;58:986–91.
- Singh SB, Zink DL, Liesch JM, et al. Preussomerins and deoxypreussomerins: novel inhibitors of Ras farnesyl-protein transferase. J Org Chem 1994;59:6296–302.
- Van der Pyl D, Inokoshi J, Shiomi K, et al. Inhibition of farnesyl-protein transferase by gliotoxin and acetylgliotoxin. J Antibiot 1992;45:1802–5.
- Singh SB, Zink DL, Bills GF, et al. Cylindrol A: a novel inhibitor of Ras farnesyl-protein transferase from Cylindrocarpon lucidum. Tetrahedron Lett 1995;36:4935–8.
- Uchida R, Shiomi K, Inokoshi J, et al. ChemInform abstract: kurasoins A and B, new protein farnesyltransferase inhibitors produced by Paecilomyces sp. FO-3684. Part 1. Producing strain, fermentation, isolation, and biological activities. ChemInform 1997;28:932–34.
- Romanis R. LXXXVIII—certain products from teak. Preliminary notice. J Chem Soc Trans 1887;51:868–71.
- Sandermann W, Dietrichs H-H. Chemische Untersuchungen an Teakholz. Holzforschung 1959;13:137–48.
- Liang Y-Q, Liao X-J, Lin J-L, et al. Spongiains A–C: three new spongian diterpenes with ring A rearrangement from the marine sponge Spongia sp. Tetrahedron 2019;75:3802–8.
- Schmitz FJ, Bloor SJ. Xesto- and halenaquinone derivatives from a sponge, Adocia sp., from Truk lagoon. J Org Chem 1988;53:3922–5.
- Desoubzdanne D, Marcourt L, Raux R, et al. Alisiaquinones and alisiaquinol, dual inhibitors of Plasmodium falciparum enzyme targets from a new caledonian deep water sponge. J Nat Prod 2008;71:1189–92.
- He F, Mai HL, Longeon A, et al. Novel adociaquinone derivatives from the Indonesian sponge Xestospongia sp. Marine Drugs 2015;13:2617–28.
- Takeda R, Chen M, Chen P-N, et al. Effects of Antrodia camphorata mycelia extract containing antroquinonol on lowering low-density lipoprotein cholesterol: a randomized double-blind study. J Pharm Nutr Sci 2017;7:73–80.
- Shi Y, Yang S, Lee DY, Lee C. Increasing anti-Aβ-induced neurotoxicity ability of Antrodia camphorata-fermented product with deep ocean water supplementary. J Sci Food Agric 2016;96:4690–701.
- Yen IC, Yao C-W, Kuo M-T, et al. Anti-cancer agents derived from solid-state fermented Antrodia camphorata mycelium. Fitoterapia 2015;102:115–9.
- Wu S-H, Ryvarden L, Chang T-T. Antrodia camphorata (“niu-chang-chih”), new combination of a medicinal fungus in Taiwan. Bot Bull Acad Sin (Taipei). 1996;38:273–5.
- Tsai P-Y, Ka S-M, Chao T-K, et al. Antroquinonol reduces oxidative stress by enhancing the Nrf2 signaling pathway and inhibits inflammation and sclerosis in focal segmental glomerulosclerosis mice. Free Radic Biol Med 2011;50:1503–16.
- Kumar KJS, Chu F-H, Hsieh H-W, et al. Antroquinonol from ethanolic extract of mycelium of Antrodia cinnamomea protects hepatic cells from ethanol-induced oxidative stress through Nrf-2 activation. J Ethnopharmacol 2011;136:168–77.
- Angamuthu V, Shanmugavadivu M, Nagarajan G, Velmurugan BK. Pharmacological activities of antroquinonol – mini review. Chem-Biol Interact 2019;297:8–15.
- Lee W-T, Lee T-H, Cheng C-H, et al. Antroquinonol from Antrodia camphorata suppresses breast tumor migration/invasion through inhibiting ERK-AP-1- and AKT-NF-κB-dependent MMP-9 and epithelial–mesenchymal transition expressions. Food Chem Toxicol 2015;78:33–41.
- Saravana Kumar P, Duraipandiyan V, Ignacimuthu S. Isolation, screening and partial purification of antimicrobial antibiotics from soil Streptomyces sp. SCA 7. Kaohsiung J Med Sci 2014;30:435–46.
- Karthik L, Kumar G, Kirthi AV, et al. Streptomyces sp. LK3 mediated synthesis of silver nanoparticles and its biomedical application. Bioprocess Biosyst Eng 2014;37:261–7.
- Hara M, Akasaka K, Akinaga S, et al. Identification of Ras farnesyltransferase inhibitors by microbial screening. Proc Natl Acad Sci USA 1993;90:2281–5.
- Del Villar K, Urano J, Guo L, Tamanoi F. A mutant form of human protein farnesyltransferase exhibits increased resistance to farnesyltransferase inhibitors. J Biol Chem 1999;274:27010–7.
- Khosravi-Far R, Clark GJ, Abe K, et al. Ras (CXXX) and Rab (CC/CXC) prenylation signal sequences are unique and functionally distinct. J Biol Chem 1992;267:24363–8.
- Shandukani PD, Tshidino SC, Masoko P, Moganedi KM. Antibacterial activity and in situ efficacy of Bidens pilosa Linn and Dichrostachys cinerea Wight et Arn extracts against common diarrhoea-causing waterborne bacteria. BMC Complement Altern Med 2018;18:171.
- Abou Zeid AH, Hifnawy MS, Mohammed RS, Sleem AA. Lipoidal contents, analgesic and antipyretic activities of the aerial parts of Dichrostachys cinerea L. J Herbs Spices Med Plants 2015;21:118–28.
- Liu M, Bryant MS, Chen J, et al. Antitumor activity of SCH 66336, an orally bioavailable tricyclic inhibitor of farnesyl protein transferase, in human tumor xenograft models and Wap-ras transgenic mice. Cancer Res 1998;58:4947.
- Pereira DM, Valentão P, Andrade PB. Meroterpenes from marine invertebrates: chemistry and application in cancer. In: Handbook of anticancer drugs from marine origin. In: Kim S-K, editor. Cham: Springer International Publishing; 2015. p. 423–37.
- Menna M, Aiello A, D’Aniello F, et al. Conithiaquinones A and B. Tetracyclic cytotoxic meroterpenes from the Mediterranean Ascidian Aplidium Conicum. Eur J Org Chem 2013;2013:3241–6.
- Aiello A, Fattorusso E, Luciano P, et al. Antitumor effects of two novel naturally occurring terpene quinones isolated from the Mediterranean Ascidian Aplidium conicum. J Med Chem 2005;48:3410–6.
- Grayfer TD, Grellier P, Mouray E, et al. Mallotojaponins B and C: total synthesis, antiparasitic evaluation, and preliminary SAR studies. Org Lett 2016;18:708–11.
- Ragavendran C, Manigandan V, Kamaraj C, et al. Larvicidal, histopathological, antibacterial activity of indigenous fungus Penicillium sp. against Aedes aegypti L and Culex quinquefasciatus (Say) (Diptera: Culicidae) and its acetylcholinesterase inhibition and toxicity assessment of zebrafish (Danio rerio). Front Microbiol 2019;10:427–44.
- Li F, Sun W, Zhang S, et al. New cyclopiane diterpenes with anti-inflammatory activity from the sea sediment-derived fungus Penicillium sp. TJ403-2. Chin Chem Lett 2019.
- Shiomi K, Uchida R, Inokoshi J, et al. Andrastins A ∼ C, new protein farnesyltransferase inhibitors, produced by Penicillium sp. FO-3929. Tetrhedron Lett 1996;37:1265–8.
- Jong SC, Davis EE. Contribution to the knowledge of Stachybotrys and Memnoniella in culture. Mycotaxon 1976;3:409–85.
- Dearborn DG, Yike I, Sorenson WG, et al. Overview of investigations into pulmonary hemorrhage among infants in Cleveland, Ohio. Environ Health Perspect 1999;107:495–9.
- Hummelbrunner LA, Isman MB. Acute, sublethal, antifeedant, and synergistic effects of monoterpenoid essential oil compounds on the tobacco cutworm, Spodoptera litura (Lep., Noctuidae). J Agric Food Chem 2001;49:715–20.
- Crowell PL, Ren Z, Lin S, et al. Structure–activity relationships among monoterpene inhibitors of protein isoprenylation and cell proliferation. Biochem Pharmacol 1994;47:1405–15.
- Maltzman TH, Hurt LM, Elson CE, et al. The prevention of nitrosomethylurea-induced mammary tumors by d-limonene and orange oil. Carcinogenesis 1989;10:781–3.
- Wattenberg LW, Sparnins VL, Barany G. Inhibition of N-nitrosodiethylamine carcinogenesis in mice by naturally occurring organosulfur compounds and monoterpenes. Cancer Res 1989;49:2689–92.
- Wattenberg LW, Coccia JB. Inhibition of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone carcinogenesis in mice by d-limonene and citrus fruit oils. Carcinogenesis 1991;12:115–7.
- Bailey HH, Levy D, Harris LS, et al. A phase II trial of daily perillyl alcohol in patients with advanced ovarian cancer: Eastern Cooperative Oncology Group Study E2E96. Gynecol Oncol 2002;85:464–8.
- Crowell PL, Lin S, Vedejs E, Gould MN. Identification of metabolites of the antitumor agent d-limonene capable of inhibiting protein isoprenylation and cell growth. Cancer Chemother Pharmacol 1992;31:205–12.
- Crowell PL, Ayoubi AS, Burke YD. Antitumorigenic effects of limonene and perillyl alcohol against pancreatic and breast cancer. In: American Institute for Cancer Research, ed. Dietary phytochemicals in cancer prevention and treatment. Boston (MA): Springer; 1996. p. 131–6.
- Ingawale AS, Sadiq MB, Nguyen LT, Ngan TB. Optimization of extraction conditions and assessment of antioxidant, α-glucosidase inhibitory and antimicrobial activities of Xanthium strumarium L. fruits. Biocatal Agric Biotechnol 2018;14:40–7.
- Lin B, Zhao Y, Han P, et al. Anti-arthritic activity of Xanthium strumarium L. extract on complete Freund’s adjuvant induced arthritis in rats. J Ethnopharmacol 2014;155:248–55.
- Nour AM, Khalid SA, Kaiser M, et al. The antiprotozoal activity of sixteen asteraceae species native to Sudan and bioactivity-guided isolation of xanthanolides from Xanthium brasilicum. Planta Med 2009;75:1363–8.
- Matsuo K, Yokoe H, Shishido K, Shindo M. Synthesis of diversifolide and structure revision. Tetrahedron Lett 2008;49:4279–81.
- Vasas A, Hohmann J. Xanthane sesquiterpenoids: structure, synthesis and biological activity. Nat Prod Rep 2011;28:824–42.
- Feng J, Lei X, Bao R, et al. Enantioselective and collective total syntheses of xanthanolides. Angew Chem 2017;129:16541–5.
- Dabrah TT, Harwood HJ, Huang LH, et al. ChemInform Abstract: CP-225,917 and CP-263,114, novel Ras farnesylation inhibitors from an unidentified fungus. Part 1. Taxonomy, fermentation, isolation, and biochemical properties. J Antibiot 2010;50:1–7.
- Dabrah TT, Kaneko T, Massefski WB, Whipple E. CP225,917 and CP263,114: novel Ras farnesylation inhibitors from an unidentified fungus. 2. Structure elucidation. J Am Chem Soc 1997;119:1594–8.
- Efferth T. From ancient herb to modern drug: artemisia annua and artemisinin for cancer therapy. Semin Cancer Biol 2017;46:65–83.
- Megdiche-Ksouri W, Trabelsi N, Mkadmini K, et al. Artemisia campestris phenolic compounds have antioxidant and antimicrobial activity. Ind Crops Prod 2015;63:104–13.
- Robles M, Aregullin M, West J, Rodriguez E. Recent studies on the zoopharmacognosy, pharmacology and neurotoxicology of sesquiterpene lactones. Planta Med 1995;61:199–203.
- Picman AK. Biological activities of sesquiterpene lactones. Biochem Syst Ecol 1986;14:255–81.
- Chen WJ, Moomaw JF, Overton L, et al. High level expression of mammalian protein farnesyltransferase in a baculovirus system. The purified protein contains zinc. J Biol Chem 1993;268:9675–80.
- Jakupovic J, Tan RX, Bohlmann F, et al. Sesquiterpene lactones from Artemisia ludoviciana. Phytochemistry 1991;30:1573–7.
- Li L, Liu H, Tang C, et al. Cytotoxic sesquiterpene lactones from Artemisia anomala. Phytochem Lett 2017;20:177–80.
- Sadhu SK, Hirata K, Li X, et al. Flavonoids and sesquiterpenoids, constituents from Eupatorium capillifolium, found in a screening study guided by cell growth inhibitory activity. J Nat Med 2006;60:325–8.
- Li Y, Zhou Z, Lou J, et al. Biological characteristics of Chaetomella sp. (Sphaeropsidaceae), a novel causal agent of sansevieria leaf spot disease. Sci Silvae Sin 2015;51:121–6.
- Gangadevi V, Muthumary J. A novel endophytic taxol-producing fungus Chaetomella raphigera isolated from a medicinal plant, Terminalia arjuna. Appl Biochem Biotechnol 2009;158:675–84.
- Singh S, Zink DM, Liesch J, et al. ChemInform abstract: isolation and structure of chaetomellic acids A and B from Chaetomella acutiseta: farnesyl pyrophosphate mimic inhibitors of Ras farnesyl-protein transferase. ChemInform 2010;24(43).
- Lingham RB, Silverman KC, Bills GF, et al. Chaetomella acutiseta produces chaetomellic acids A and B which are reversible inhibitors of farnesyl-protein transferase. Appl Microbiol Biotechnol 1993;40:370–4.
- Weber D, Sterner O, Anke T. Mollisianitrile, a new antibiotic from Mollisia sp. A59-96. Zeitschrift Für Naturforschung C 2007;62:567–70.
- Wu HY, Yang FL, Li LH, et al. Ergosterol peroxide from marine fungus Phoma sp. induces ROS-dependent apoptosis and autophagy in human lung adenocarcinoma cells. Sci Rep 2018;8:17956.
- Liu S-S, Jiang J-X, Huang R, et al. A new antiviral 14-nordrimane sesquiterpenoid from an endophytic fungus Phoma sp. Phytochem Lett 2019;29:75–8.
- Izelmar T, Tássia CC, Luciana L, et al. Formulation of a bioherbicide with metabolites from Phoma sp. Sci Hortic 2018;241:285–92.
- Hensens OD, Dufresne C, Liesch JM, et al. The zaragozic acids: structure elucidation of a new class of squalene synthase inhibitors. Tetrahedron Lett 1993;34:399–402.
- Bergstrom JD, Kurtz MM, Rew DJ, et al. Zaragozic acids: a family of fungal metabolites that are picomolar competitive inhibitors of squalene synthase. Proc Natl Acad Sci USA 1993;90:80–4.
- Wilson K, Burk R, Biftu T, et al. Zaragozic acid A, a potent inhibitor of squalene synthase: initial chemistry and absolute stereochemistry. J Org Chem. 1992;57:7151–8.
- Dufresne C, Wilson KE, Zink D, et al. The isolation and structure elucidation of zaragozic acid C, a novel potent squalene synthase inhibitor. Tetrahedron 1992;48:10221–6.
- Tanimoto T, Hamano K, Onodera K, et al. Biological activities of novel zaragozic acids, the potent inhibitors of squalene synthase, produced by the fungus, Mollisia sp. SANK 10294. J Antibiot (Tokyo). 1997;50:390–4.
- Pedretti A, Villa L, Vistoli G. Modeling of binding modes and inhibition mechanism of some natural ligands of farnesyl transferase using molecular docking. J Med Chem 2002;45:1460–5.
- Huang H, Gao P, Zhao Q, Hu H-F. Construction of a mutant of Actinoplanes sp. N902-109 that produces a new rapamycin analog. Chin J Nat Med 2018;16:210–8.
- Liliya H, Bohdan O, Andriy L, et al. A gene cluster for the biosynthesis of moenomycin family antibiotics in the genome of teicoplanin producer Actinoplanes teichomyceticus. Appl Microbiol Biotechnol 2016;100:7629–38.
- Silverman KC, Cascales C, Genilloud O, et al. Actinoplanic acids A and B as novel inhibitors of farnesyl-protein transferase. Appl Microbiol Biotechnol 1995;43:610–6.
- Hinterding K, Hagenbuch P, Rétey J, Waldmann H. Synthesis and in vitro evaluation of the farnesyltransferase inhibitor pepticinnamin E. Chem Eur J 1999;5:227–36.
- Shiomi K, Yang H, Inokoshi J, et al. Pepticinnamins, new farnesyl-protein transferase inhibitors produced by an actinomycete. II. Structural elucidation of pepticinnamin E. J Antibiot 1993;46:229–34.
- Whitby M. Fusidic acid in the treatment of methicillin-resistant Staphylococcus aureus. Int J Antimicrob Agents 1999;12:S67–S71.
- Godtfredsen WO, Jahnsen S, Lorck H, et al. Fusidic acid: a new antibiotic. Nature 1962;193:987.
- Wu P-P, He H, Hong WD, et al. The biological evaluation of fusidic acid and its hydrogenation derivative as antimicrobial and anti-inflammatory agents. Infect Drug Resist 2018;11:1945–57.
- Talontsi FM, Lamshöft M, Douanla-Meli C, et al. Antiplasmodial and cytotoxic dibenzofurans from Preussia sp. harboured in Enantia chlorantha Oliv. Fitoterapia 2014;93:233–8.
- Babita P, Keshab B, Datta BH. Antimicrobial and antioxidant activities of two polyketides from lichen-endophytic fungus Preussia sp. Zeitschrift fur Naturforschung C J Biosci 2018;73:161–3.
- Trenin A. Fungal secondary metabolites that inhibit sterol biosynthesis. Appl Biochem Microbiol 1998;34:117–23.
- Weber HA, Baenziger NC, Gloer JB. Structure of preussomerin A: an unusual new antifungal metabolite from the coprophilous fungus Preussia isomera. J Am Chem Soc 1990;112:6718–9.
- Weber HA, Gloer J. The preussomerins: novel antifungal metabolites from the coprophilous fungus Preussia isomera Cain. J Org Chem 1991;56:4355–60.
- Polishook JD, Dombrowski AW, Tsou NN, et al. Preussomerin D from the endophyte hormonema dematioides. Mycologia 1993;85:62–4.
- Gonçalves SS, Souza ACR, Chowdhary A, Meis JF, et al. Epidemiology and molecular mechanisms of antifungal resistance in Candida and Aspergillus. Mycoses 2016;59:198–219.
- Schuster A, Schmoll M. Biology and biotechnology of Trichoderma. Appl Microbiol Biotechnol 2010;87:787–99.
- Dennis C, Webster J. Antagonistic properties of species-groups of Trichoderma: I. Production of non-volatile antibiotics. Trans Br Mycol Soc 1971;57:25–39.
- Shah DT, Larsen B. Clinical isolates of yeast produce a gliotoxin-like substance. Mycopathologia 1991;116:203–8.
- Richardson C, Emery P. Clinical use of cyclosporin in rheumatoid arthritis. Drugs 1995;50:26–36.
- Su-Jin J, Eun-Soo J, Eun-Kyung C, et al. Immunomodulatory effects of a mycelium extract of Cordyceps (Paecilomyces hepiali; CBG-CS-2): a randomized and double-blind clinical trial. BMC Complement Altern Med 2019;19:77–85.
- Braun GH, Ramos HP, Candido ACBB, et al. Evaluation of antileishmanial activity of harzialactone a isolated from the marine-derived fungus Paecilomyces sp. Nat Prod Res 2019;33:1–4.
- Gabriel D, Shafry DD, Gordon LB, Djabali K. Intermittent treatment with farnesyltransferase inhibitor and sulforaphane improves cellular homeostasis in Hutchinson-Gilford progeria fibroblasts. Oncotarget 2017;8:64809–26.
- Tanaka T, Ikegami Y, Nakazawa H, et al. Low-dose farnesyltransferase inhibitor suppresses HIF-1α and snail expression in triple-negative breast cancer MDA-MB-231 cells in vitro. J Cell Physiol 2017;232:192–201.
- Ding H, McDonald JS, Yun S, et al. Farnesyltransferase inhibitor tipifarnib inhibits Rheb prenylation and stabilizes Bax in acute myelogenous leukemia cells. Haematologica 2014;99:60–9.