
Abstract
A series of 2,4-disubstituted-5-(6-alkylpyridin-2-yl)-1H-imidazoles, 7a–c, 11a–h, and 16a–h has been synthesised and evaluated for their ALK5 inhibitory activity in an enzyme assay and in a cell-based luciferase reporter assay. Incorporation of a quinoxalin-6-yl moiety and a methylene linker at the 4- and 2-position of the imidazole ring, respectively, and a m-CONH2 substituent in the phenyl ring generated a highly potent and selective ALK5 inhibitor 11e. Docking model of ALK5 in complex with 11e showed that it fitted well in the ATP-binding pocket with favourable interactions.
1. Introduction
Transforming growth factor-β (TGF-β) is one of the most potent immunosuppressive cytokines in the tumour microenvironmentCitation1. Elevated serum levels of TGF-β commonly observed in patients with advanced colorectal cancerCitation2, breast cancerCitation3,Citation4, bladder carcinomaCitation5,Citation6, prostate cancerCitation7,Citation8, malignant melanomaCitation9, pancreatic ductal adenocarcinomaCitation10, and hepatocellular carcinomaCitation11 have been strongly associated with tumour progression and poor clinical outcome. Overexpression of TGF-β receptors has been implicated in cancerCitation12. In patients with advanced hepatocellular carcinoma treated with the first clinically available ALK5 inhibitor, galunisertib (1)Citation13, an approximately two-fold longer overall survival was observed in patients having a TGF-β1 response compared to patients who did not have a TGF-β1 responseCitation14. Therefore, TGF-β signalling pathway is an attractive target for development of cancer immunotherapeutic agents.
TGF-β signals through two distinct serine/threonine kinase receptors, the type I (activin receptor-like kinase 5 (ALK5)) and type II receptors. Small-molecule ALK5 inhibitors specifically inhibit the Smad pathway by competing with ATP at the hydrophobic ATP binding pocket of ALK5 kinase domain, which is essential for the phosphorylation of its substrates, Smad2/Smad3 proteins. Galunisertib progressed to phase 2/3 clinical trials against pancreatic carcinoma, glioblastoma, hepatocellular carcinoma, and myelodysplastic syndromeCitation13, however, Eli Lilly discontinued further clinical development of galunisertib in 2017. Recently, we developed an ALK5 inhibitor, vactosertib (2)Citation15, and it has progressed to phase 1b/2a clinical trials either alone or in combination with pembrolizumab, durvalumab, or pomalidomide against myelodysplastic syndrome, non-small cell lung cancer, gastric cancer, colon cancer, multiple myeloma, etc.Citation16. Another ALK5 inhibitor, LY3200882 (3) has entered a phase 1 clinical trialCitation16 (). Certain imidazo[2,1-b][1,3,4]thiadiazoles and 2,3,4-substituted 5,5-dimethyl-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazoles have been reported to possess ALK5 inhibitory activityCitation17,Citation18.
Figure 1. Small-molecule ATP-competitive ALK5 inhibitors in clinical trials.
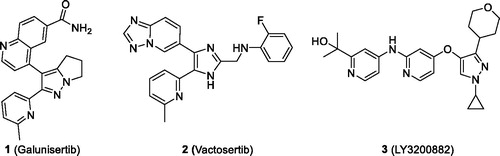
Vactosertib exhibited subnanomolar ALK5 inhibitory activity in a kinase assay and in a cell-based luciferase reporter assayCitation15, high selectivity against a panel of 320 protein kinases including p38αCitation15, moderate oral bioavailability in ratsCitation15, and high efficacy in animal models of cancerCitation19–21 and fibrosisCitation22–26. In this report, we examined whether structural modification of vactosertib could increase its subnanomolar ALK5 inhibitory activity, thus further increasing its selectivity. For this purpose, we replaced a [1,2,4]triazolo[1,5-a]pyridin-6-yl moiety of vactosertib with either a benzo[1,3]dioxol-5-yl or a quinoxalin-6-yl moiety and inserted either a methylene, an ethylene, or a propylene linker instead of a methyleneamino linker to optimise the distance between a central imidazole ring and a phenyl ring.
2. Materials and methods
2.1. Chemistry
1H NMR spectra were recorded on a Varian Unity-Inova 400 MHz instrument. The chemical shifts are reported in parts per million (ppm). For 1H NMR spectra, CDCl3 was used as solvent, and it served as the internal standard at δ 7.26. Infra-red spectra were recorded on a FT-infra-red spectrometer (Bio-Rad). Electrospray ionisation mass spectra (ESIMS) were obtained on a Q-Tof2 mass spectrometer (Micromass). Elemental analyses (C, H, and N) were used to determine the purity of all tested compounds, and the results were within ±0.4% of the calculated values (Carlo Erba 1106 elemental analyzer). Analytical thin-layer chromatography (TLC) was performed on Merck silica gel 60 F-254 glass plates. Medium-pressure liquid chromatography (MPLC) was performed using Merck silica gel 60 (230–400 mesh) with a YFLC-540 ceramic pump (Yamagen).
2.1.1. General procedure for the preparation of the 4-(benzo[1,3]dioxol-5-yl)-5–(6-methylpyridin-2-yl)-1H-imidazoles 6a–c
To a stirred solution of 1-(benzo[1,3]dioxol-5-yl)−2-(6-methylpyridin-2-yl)ethane-1,2-dione (4) (0.19 mmol) in AcOH (3 mL) were added NH4OAc (1.11 mmol) and 4-(2-oxoethyl)benzonitrile (5a), 4-(3-oxopropyl)benzonitrile (5b), or 4-(4-oxobutyl)benzonitrile (5c) (0.19 mmol), and the mixture was heated at 120 °C for 3 h. The pH of the cooled reaction mixture was adjusted to pH∼8 at 0 °C with 28% NH4OH solution in water, and the reaction mixture was extracted with CH2Cl2 (10 mL). The CH2Cl2 solution was washed with water (5 mL) and brine (5 mL), dried over anhydrous Na2SO4, filtered, and evaporated to dryness under reduced pressure. The residue was purified by MPLC on silica gel with CH2Cl2/MeOH as eluent to afford the 6a–c as a solid.
2.1.1.1. 4-((4-(Benzo[d][1,3]dioxol-5-yl)-5-(6-methylpyridin-2-yl)-1H-imidazol-2-yl)methyl)benzonitrile (6a)
Yield 30%; mp 176 − 179 °C; 1H NMR (CDCl3) δ 11.40 (br s, 1 H, NH), 7.45 (d, 2 H, 2 phenyl), 7.44 (overlapped, 1 H, pyridyl), 7.33 (d, 1 H, pyridyl), 7.25 (d, 2 H, 2 phenyl), 7.10 (d, 1 H, pyridyl), 7.07 (s, 1 H, piperonyl), 6.93 (d, 1 H, piperonyl), 6.84 (d, 1 H, piperonyl), 6.00 (s, 2 H, OCH2O), 4.09 (s, 2 H, CH2), 2.34 (s, 3 H, CH3); IR (CHCl3) 2229, 1574 cm−1; MS (EIS) m/z 395.13 (MH+).
2.1.1.2. 4-(2-(4-(Benzo[d][1,3]dioxol-5-yl)-5-(6-methylpyridin-2-yl)-1H-imidazol-2-yl)ethyl)benzonitrile (6b)
Yield 49%; mp 94 − 96 °C; 1H NMR (CDCl3) δ 10.53 (br s, 1 H, NH), 7.56 (d, 2 H, 2 phenyl), 7.43 (dd, 1 H, pyridyl), 7.31 (d, overlapped, 1 H, pyridyl) 7.29 (d, 2 H, 2 phenyl), 7.07 (d, 1 H, pyridyl), 7.04 (d, 1 H, piperonyl), 6.94 (d, 1 H, piperonyl), 6.84 (d, 1 H, piperonyl), 6.00 (s, 2 H, OCH2O), 3.13 (m, 2 H, CH2), 3.05 (m, 2 H, CH2), 2.48 (s, 3 H, CH3); IR (CHCl3) 3375, 2229, 1573 cm−1; MS (EIS) m/z 409.13 (MH+).
2.1.1.3. 4-(3-(4-(Benzo[d][1,3]dioxol-5-yl)-5-(6-methylpyridin-2-yl)-1H-imidazol-2-yl)propyl)benzonitrile (6c)
Yield 36%; mp 60 − 62 °C; 1H NMR (CDCl3) δ 10.63 (br s, 1 H, NH), 7.51 (d, 2 H, 2 phenyl), 7.42 (dd, 1 H, pyridyl), 7.30 (d, 1 H, pyridyl), 7.24 (d, 2 H, 2 phenyl), 7.10 − 7.04 (m, 2 H, 1 pyridyl and 1 piperonyl), 6.93 (d, 1 H, piperonyl), 6.82 (d, 1 H, piperonyl), 5.98 (s, 2 H, OCH2O), 2.74 (m, 4 H, 2 CH2), 2.47 (s, 3 H, CH3), 2.07 (m, 2 H, CH2); IR (CHCl3) 2228, 1573 cm−1; MS (EIS) m/z 423.14 (MH+).
2.1.2. General procedure for the preparation of the 4-(benzo[1,3]dioxol-5-yl)-5-(6-methylpyridin-2-yl)-1H-imidazoles 7a–c
A stirred solution of 6a–c (0.17 mmol), 6 N NaOH (0.04 mmol), and 28% H2O2 (0.59 mmol) in 95% EtOH (4 mL) was heated at 55 °C for 3 h. The reaction mixture was cooled to 0 °C and neutralised with 1 N HCl to pH∼8. The mixture was extracted with CH2Cl2 (30 mL), and the organic solution was washed with water (15 mL) and brine (15 mL), dried over anhydrous Na2SO4, filtered, and evaporated to dryness under reduced pressure. The residue was purified by MPLC on silica gel with CHCl3/MeOH or CH2Cl2/MeOH as eluent to afford the titled compounds 7a–c as a solid.
2.1.2.1. 4-((4-(Benzo[d][1,3]dioxol-5-yl)-5-(6-methylpyridin-2-yl)-1H-imidazol-2-yl)methyl)benzamide (7a)
Yield 55%; mp 248 − 250 °C; 1H NMR (CDCl3) δ 11.70 (br s, 1 H, NH), 7.45 (d, 2 H, 2 phenyl), 7.39 (dd, 1 H, pyridyl), 7.27 (d, 1 H, pyridyl), 7.10 (d, 2 H, 2 phenyl), 7.06 (overlapped, 1 H, pyridyl), 7.05 (s, 1 H, piperonyl), 6.87 (d, 1 H, piperonyl), 6.80 (d, 1 H, piperonyl), 6.39 (br s, 1 H, CONH), 5.95 (s, 2 H, OCH2O), 5.78 (br s, 1 H, CONH), 4.02 (s, 2 H, CH2), 2.27 (s, 3 H, CH3); IR (CHCl3) 3191, 1661 cm−1; MS (EIS) m/z 413.11 (MH+). Anal. Calcd for C24H20N4O3: C, 69.89; H, 4.89; N, 13.58. Found: C, 69.78; H, 4.95; N, 13.48.
2.1.2.2. 4-(2-(4-(Benzo[d][1,3]dioxol-5-yl)-5-(6-methylpyridin-2-yl)-1H-imidazol-2-yl)ethyl)benzamide (7b)
Yield 49%; mp 189−192 °C; 1H NMR (CDCl3) δ 10.85 (br s, 1 H, NH) 7.66 (d, 2 H, 2 phenyl), 7.42 (t, 1 H, pyridyl), 7.30 (d, 1 H, pyridyl), 7.19 (d, 2 H, 2 phenyl), 7.07 (d, overlapped, 1 H, pyridyl), 7.05 (s, 1 H, piperonyl), 6.93 (d, 1 H, piperonyl), 6.82 (d, 1 H, piperonyl), 6.37 (br s, 1 H, CONH), 5.99 (s, 2 H, OCH2O), 5.69 (br s, 1 H, CONH), 3.04 (m, 2 H, CH2), 3.01 (m, 2 H, CH2), 2.45 (s, 3 H, CH3); IR (CHCl3) 3441, 1653 cm−1; MS (EIS) m/z 427.10 (MH+). Anal. Calcd for C25H22N4O3: C, 70.41; H, 5.20; N, 13.14. Found: C, 70.23; H, 5.28; N, 12.98.
2.1.2.3. 4-(3-(4-(Benzo[d][1,3]dioxol-5-yl)-5-(6-methylpyridin-2-yl)-1H-imidazol-2-yl)propyl)benzamide (7c)
Yield 52%; mp 178−180 °C; 1H NMR (CDCl3) δ 10.60 (br s, 1 H, NH) 7.68 (d, 2 H, 2 phenyl), 7.41 (dd, 1 H, pyridyl), 7.30 (d, 1 H, pyridyl), 7.20 (d, 2 H, 2 phenyl), 7.10−7.05 (m, 2 H, 1 pyridyl and 1 piperonyl), 6.92 (d, 1 H, piperonyl), 6.82 (d, 1 H, piperonyl), 6.25 (br s, 1 H, CONH), 5.98 (s, 2 H, OCH2O), 5.75 (br s, 1 H, CONH), 2.73 (m, 4 H, 2 CH2), 2.49 (s, 3 H, CH3), 2.07 (m, 2 H, CH2); IR (CHCl3) 3183, 1660 cm−1; MS (EIS) m/z 441.12 (MH+). Anal. Calcd for C26H24N4O3: C, 70.89; H, 5.49; N, 12.72. Found: 70.63; H, 5.52; N, 12.66.
2.1.3. General procedure for the preparation of the 5-(6-alkylpyridin-2-yl)-1-hydroxy-4-(quinoxalin-6-yl)-1H-imidazoles 10a–h
To a stirred solution of 9a–d (0.23 mmol) in t-BuOMe (2.5 mL) were added either 5a or 3-(2-oxoethyl)benzonitrile (5d) (0.69 mmol) and NH4OAc (1.15 mmol) dissolved in MeOH (1.2 mL), and the mixture was stirred at room temperature overnight under argon atmosphere. The pH of the reaction mixture was adjusted to pH∼8 at 0 °C with saturated NaHCO3 solution. The reaction mixture was partitioned between CH2Cl2 (40 mL) and water (40 mL). The aqueous layer was extracted with CH2Cl2 (15 mL × 3). The combined organic solution was dried over anhydrous Na2SO4, filtered, and evaporated to dryness under reduced pressure. The residue was purified by MPLC on silica gel with CH2Cl2/MeOH as eluent to afford the titled compounds 10a–h as a solid.
2.1.3.1. 4-((1-Hydroxy-5-(6-methylpyridin-2-yl)-4-(quinoxalin-6-yl)-1H-imidazol-2-yl)methyl)benzonitrile (10a)
Yield 40%; mp 200–203 °C; 1H NMR (CDCl3) δ 8.86 (m, 2 H, 2 quinoxalinyl), 8.35 (d, 1 H, quinoxalinyl), 8.17 (d, 1 H, quinoxalinyl), 8.06 (dd, 1 H, quinoxalinyl), 7.62 (m, 2 H, 2 phenyl), 7.56 (m, 2 H, 2 phenyl), 7.52 (t, 1 H, pyridyl), 7.36 (d, 1 H, pyridyl), 7.06 (d, 1 H, pyridyl), 4.31 (s, 2 H, CH2), 2.61 (s, 3 H, CH3); IR (CHCl3) 3075, 2228, 1600, 1572 cm−1; MS (EIS) m/z 419.23 (MH+).
2.1.3.2. 4-((5-(6-Ethylpyridin-2-yl)-1-hydroxy-4-(quinoxalin-6-yl)-1H-imidazol-2-yl)methyl)benzonitrile (10b)
Yield 37%; mp 84–85 °C; 1H NMR (CDCl3) δ 8.86 (m, 2 H, 2 quinoxalinyl), 8.36 (d, 1 H, quinoxalinyl), 8.17 (d, 1 H, quinoxalinyl), 8.07 (dd, 1 H, quinoxalinyl), 7.62 (m, 2 H, 2 phenyl), 7.57 (m, 2 H, 2 phenyl), 7.54 (t, 1 H, pyridyl), 7.37 (d, 1 H, pyridyl), 7.08 (d, 1 H, pyridyl), 4.32 (s, 2 H, CH2), 2.90 (q, 2 H, CH2), 1.38 (t, 3 H, CH3); IR (CHCl3) 2229, 1599, 1572 cm−1; MS (EIS) m/z 433.18 (MH+).
2.1.3.3. 4-((1-Hydroxy-5-(6-isopropylpyridin-2-yl)-4-(quinoxalin-6-yl)-1H-imidazol-2-yl)methyl)benzonitrile (10c)
Yield 46%; mp 178–179 °C; 1H NMR (CDCl3) δ 8.86 (m, 2 H, 2 quinoxalinyl), 8.36 (d, 1 H, quinoxalinyl), 8.17 (d, 1 H, quinoxalinyl), 8.07 (dd, 1 H, quinoxalinyl), 7.62 (m, 2 H, 2 phenyl), 7.57 (m, 2 H, 2 phenyl), 7.55 (t, 1 H, pyridyl), 7.37 (d, 1 H, pyridyl), 7.09 (d, 1 H, pyridyl), 4.32 (s, 2 H, CH2), 3.13 (heptet, 1 H, CH), 1.38 (s, 3 H, CH3), 1.37 (s, 3 H, CH3); IR (CHCl3) 2229, 1598, 1571 cm−1; MS (EIS) m/z 447.22 (MH+).
2.1.3.4. 4-((5-(6-n-Butylpyridin-2-yl)-1-hydroxy-4-(quinoxalin-6-yl)-1H-imidazol-2-yl)methyl)benzonitrile (10d)
Yield 51%; mp 77–78 °C; 1H NMR (CDCl3) δ 8.86 (m, 2 H, 2 quinoxalinyl), 8.35 (d, 1 H, quinoxalinyl), 8.17 (d, 1 H, quinoxalinyl), 8.06 (dd, 1 H, quinoxalinyl), 7.62 (m, 2 H, 2 phenyl), 7.57 (m, 2 H, 2 phenyl), 7.53 (t, 1 H, pyridyl), 7.36 (dd, 1 H, pyridyl), 7.06 (d, 1 H, pyridyl), 4.31 (s, 2 H, CH2), 2.85 (t, 2 H, CH2), 1.76 (m, 2 H, CH2), 1.42 (m, 2 H CH2), 0.98 (t, 3 H, CH3); IR (CHCl3) 3402, 2228, 1608, 1572 cm−1; MS (EIS) m/z 461.20 (MH+).
2.1.3.5. 3-((1-Hydroxy-5-(6-methylpyridin-2-yl)-4-(quinoxalin-6-yl)-1H-imidazol-2-yl)methyl)benzonitrile (10e)
Yield 77%; mp 210–212 °C; 1H NMR (CDCl3) δ 8.86 (m, 2 H, 2 quinoxalinyl), 8.36 (d, 1 H, quinoxalinyl), 8.18 (d, 1 H, quinoxalinyl), 8.08 (dd, 1 H, quinoxalinyl), 7.76 (s, 1 H, phenyl), 7.70 (d, 1 H, phenyl), 7.53 (m, 1 H, phenyl), 7.51 (d, 1 H, phenyl), 7.43 (t, 1 H, pyridyl), 7.37 (d, 1 H, pyridyl), 7.07 (d, 1 H, pyridyl), 4.29 (s, 2 H, CH2), 2.63 (s, 3 H, CH3); IR (CHCl3) 3052, 2231, 1575 cm−1; MS (EIS) m/z 419.20 (MH+).
2.1.3.6. 3-((5-(6-Ethylpyridin-2-yl)-1-hydroxy-4-(quinoxalin-6-yl)-1H-imidazol-2-yl)methyl)benzonitrile (10f)
Yield 71%; mp 195–196 °C; 1H NMR (CDCl3) δ 8.85 (m, 2 H, 2 quinoxalinyl), 8.36 (d, 1 H, quinoxalinyl), 8.17 (d, 1 H, quinoxalinyl), 8.07 (dd, 1 H, quinoxalinyl), 7.75 (s, 1 H, phenyl), 7.70 (d, 1 H, phenyl), 7.54 (m, 2 H, 2 phenyl), 7.42 (t, 1 H, pyridyl), 7.38 (d, 1 H, pyridyl), 7.07 (d, 1 H, pyridyl), 4.29 (s, 2 H, CH2), 2.90 (q, 2 H, CH2), 1.38 (t, 3 H, CH3); IR (CHCl3) 2973, 2231, 1572 cm−1; MS (EIS) m/z 433.24 (MH+).
2.1.3.7. 3-((1-Hydroxy-5-(6-isopropylpyridin-2-yl)-4-(quinoxalin-6-yl)-1H-imidazol-2-yl)methyl)benzonitrile (10g)
Yield 59%; mp 146–147 °C; 1H NMR (CDCl3) δ 8.86 (m, 2 H, 2 quinoxalinyl), 8.36 (d, 1 H, quinoxalinyl), 8.18 (d, 1 H, quinoxalinyl), 8.08 (dd, 1 H, quinoxalinyl), 7.76 (m, 1 H, phenyl), 7.71 (m, 1 H, phenyl), 7.54 (m, 2 H, 2 phenyl), 7.43 (t, 1 H, pyridyl), 7.38 (dd, 1 H, pyridyl), 7.10 (dd, 1 H, pyridyl), 4.30 (s, 2 H, CH2), 3.14 (heptet, 1 H, CH), 1.39 (s, 3 H, CH3), 1.37 (s, 3 H, CH3); IR (CHCl3) 2967, 2231, 1597, 1572 cm−1; MS (EIS) m/z 447.22 (MH+).
2.1.3.8. 3-((5-(6-n-Butylpyridin-2-yl)-1-hydroxy-4-(quinoxalin-6-yl)-1H-imidazol-2-yl)methyl)benzonitrile (10h)
Yield 67%; mp 164–166 °C; 1H NMR (CDCl3) δ 8.85 (m, 2 H, 2 quinoxalinyl), 8.35 (d, 1 H, quinoxalinyl), 8.17 (d, 1 H, quinoxalinyl), 8.07 (dd, 1 H, quinoxalinyl), 7.75 (s, 1 H, phenyl), 7.70 (m, 1 H, phenyl), 7.53 (m, 2 H, 2 phenyl), 7.42 (t, 1 H, pyridyl), 7.37 (d, 1 H, pyridyl), 7.05 (d, 1 H, pyridyl), 4.29 (s, 2 H, CH2), 2.85 (t, 2 H, CH2), 1.76 (m, 2 H, CH2), 1.43 (m, 2 H, CH2), 0.98 (t, 3 H, CH3); IR (CHCl3) 2957, 2230, 1599, 1572 cm−1; MS (EIS) m/z 461.27 (MH+).
2.1.4. General procedure for the preparation of the 5-(6-alkylpyridin-2-yl)-4-(quinoxalin-6-yl)-1H-imidazoles 11a–h
To a stirred solution of 10a–h (0.63 mmol) in a mixture of EtOH (16 mL) and DMSO (4 mL) at room temperature were added 28% H2O2 (6.62 mmol) and 6 N NaOH (0.47 mmol). The mixture was warmed to 55 °C and stirred overnight, and to it, 1 N HCl solution was added to adjust to pH∼8 at 0 °C. The ethanol solvent was evaporated off under reduced pressure, and the residue was partitioned between CH2Cl2 (30 mL) and H2O (50 mL). The aqueous layer was saturated with NaCl and extracted with CH2Cl2 (30 mL × 3). The combined organic solution was washed with brine (30 mL), dried over anhydrous Na2SO4, filtered, and evaporated to dryness under reduced pressure. The residue was dissolved in anhydrous DMF (20 mL) and treated with triethyl phosphite (2.39 mmol). The mixture was heated at 110 °C for 3 days, cooled to room temperature, and evaporated to dryness under reduced pressure. The reaction mixture was partitioned between CH2Cl2 (30 mL) and water (50 mL), and the aqueous layer was extracted with CH2Cl2 (30 mL × 2). The combined organic solution was washed with saturated NaHCO3 solution (40 mL) and brine (50 mL), dried over anhydrous Na2SO4, filtered, and evaporated to dryness under reduced pressure. The residue was purified by MPLC on silica gel with CH2Cl2/MeOH as eluent to afford the titled compounds 11a–h as a solid.
2.1.4.1. 4-((5-(6-Methylpyridin-2-yl)-4-(quinoxalin-6-yl)-1H-imidazol-2-yl)methyl)benzamide (11a)
Yield 36%; mp 227–229 °C; 1H NMR (CDCl3) δ 12.01 (br s, 1 H, NH), 8.83 (m, 2 H, 2 quinoxalinyl), 8.38 (s, 1 H, quinoxalinyl), 8.15 (dd, 2 H, 2 quinoxalinyl), 7.55 (d, 2 H, 2 phenyl), 7.42 (dd, 1 H, pyridyl), 7.33 (d, 1 H, pyridyl), 7.21 (d, 2 H, 2 phenyl), 6.95 (d, 1 H, pyridyl), 6.62 (br s, 1 H, CONH), 5.83 (br s, 1 H, CONH), 4.13 (s, 2 H, CH2), 2.29 (s, 3 H, CH3); IR (CHCl3) 3185, 1665, 1616, 1572 cm−1; MS (EIS) m/z 421.14 (MH+). Anal. Calcd for C25H20N6O: C, 71.41; H, 4.79; N, 19.99. Found: C, 71.44; H, 4.65; N, 19.87.
2.1.4.2. 4-((5-(6-Ethylpyridin-2-yl)-4-(quinoxalin-6-yl)-1H-imidazol-2-yl)methyl)benzamide (11 b)
Yield 44%; mp 218–219 °C; 1H NMR (CDCl3) δ 11.62 (br s, 1 H, NH), 8.83 (s, 2 H, 2 quinoxalinyl), 8.39 (s, 1 H, quinoxalinyl), 8.15 (dd, 2 H, quinoxalinyl), 7.59 (d, 2 H, 2 phenyl), 7.45 (t, 1 H, pyridyl), 7.33 (d, 1 H, pyridyl), 7.26 (d, 2 H, 2 phenyl), 6.99 (d, 1 H, pyridyl), 6.45 (br s, 1 H, CONH), 5.83 (br s, 1 H, CONH), 4.17 (s, 2 H, CH2), 2.65 (q, 2 H, CH2), 1.09 (t, 3 H, CH3); IR (CHCl3) 3179, 1663, 1615, 1571 cm−1; MS (EIS) m/z 435.19 (MH+). Anal. Calcd for C26H22N6O: C, 71.87; H, 5.10; N, 19.34. Found: C, 71.57; H, 5.28; N, 19.12.
2.1.4.3. 4-((5-(6-Isopropylpyridin-2-yl)-4-(quinoxalin-6-yl)-1H-imidazol-2-yl)methyl)benzamide (11c)
Yield 35%; mp 151–152 °C; 1H NMR (CDCl3) δ 10.33 (br s, 1 H, NH), 8.84 (m, 2 H, 2 quinoxalinyl), 8.42 (s, 1 H, quinoxalinyl), 8.15 (s, 2 H, 2 quinoxalinyl), 7.76 (m, 2 H, 2 phenyl), 7.44 (m, 3 H, 2 phenyl and 1 pyridyl), 7.33 (d, 1 H pyridyl), 7.01 (d, 1 H, pyridyl), 6.10 (br s, 1 H, CONH), 5.60 (br s, 1 H, CONH), 4.28 (s, 2 H, CH2), 3.01 (heptet, 1 H, CH), 1.27 (s, 3 H, CH3), 1.25 (s, 3 H, CH3); IR (CHCl3) 3183, 1664, 1615, 1571 cm−1; MS (EIS) m/z 449.23 (MH+). Anal. Calcd for C27H24N6O: C, 72.30; H, 5.39; N, 18.74. Found: C, 72.25; H, 5.45; N, 18.61.
2.1.4.4. 4-((5-(6-n-Butylpyridin-2-yl)-4-(quinoxalin-6-yl)-1H-imidazol-2-yl)methyl)benzamide (11d)
Yield 42%; mp 170–171 °C; 1H NMR (CDCl3) δ 11.47 (br s, 1 H, NH), 8.84 (s, 2 H, 2 quinoxalinyl), 8.40 (s, 1 H, quinoxalinyl), 8.16 (m, 2 H, 2 quinoxalinyl), 7.61 (d, 2 H, 2 phenyl), 7.43 (t, 1 H, pyridyl), 7.33 (d, 1 H, pyridyl), 7.28 (d, 2 H, 2 phenyl), 6.97 (d, 1 H, pyridyl), 6.35 (br s, 1 H, CONH), 5.68 (br s, 1 H, CONH), 4.19 (s, 2 H, CH2), 2.64 (t, 2 H, CH2), 1.45 (m, 2 H, CH2), 1.24 (m, 2 H, CH2), 0.81 (t, 3 H, CH3); IR (CHCl3) 3182, 1661, 1616, 1570 cm−1; MS (EIS) m/z 463.24 (MH+). Anal. Calcd for C28H26N6O: C, 72.71; H, 5.67; N, 18.17. Found: C, 72.83; H, 5.56; N, 18.02.
2.1.4.5. 3-((5-(6-Methylpyridin-2-yl)-4-(quinoxalin-6-yl)-1H-imidazol-2-yl)methyl)benzamide (11e)
Yield 23%; mp 181 − 182 °C; 1H NMR (CDCl3) δ 8.79 (s, 2 H, 2 quinoxalinyl), 8.34 (s, 1 H, quinoxalinyl), 8.06 (s, 2 H, 2 quinoxalinyl), 7.71 (s, 1 H, phenyl), 7.55 (d, 1 H, phenyl), 7.41 (t, 1 H, pyridyl), 7.34 (d, 1 H, pyridyl), 7.29 (d, 1 H, phenyl), 7.19 (t, 1 H, phenyl), 6.96 (d, 1 H, pyridyl), 6.83 (br s, 1 H, CONH), 6.30 (br s, 1 H, CONH), 4.13 (s, 2 H, CH2), 2.36 (s, 3 H, CH3); IR (CHCl3) 3196, 1674, 1658 cm−1; MS (EIS) m/z 421.23 (MH+). Anal. Calcd for C25H20N6O: C, 71.41; H, 4.79; N, 19.99. Found: C, 71.26; H, 4.92; N, 19.85.
2.1.4.6. 3-((5-(6-Ethylpyridin-2-yl)-4-(quinoxalin-6-yl)-1H-imidazol-2-yl)methyl)benzamide (11f)
Yield 26%; mp 125 − 126 °C; 1H NMR (CDCl3) δ 8.84 (m, 2 H, 2 quinoxalinyl), 8.40 (s, 1 H, quinoxalinyl), 8.13 (d, 2 H, 2 quinoxalinyl), 7.85 (s, 1 H, phenyl), 7.72 (m, 1 H, phenyl), 7.57 (m, 1 H, phenyl), 7.44 (m, 2 H, phenyl and pyridyl), 7.34 (br d, 1 H, pyridyl), 7.00 (dd, 1 H, pyridyl), 6.20 (br s, 1 H, CONH), 5.60 (br s, 1 H, CONH), 4.28 (s, 2 H, CH2), 2.80 (q, 2 H, CH2), 1.28 (t, 3 H, CH3); IR (CHCl3) 3407, 1670, 1657 cm−1; MS (EIS) m/z 435.22 (MH+). Anal. Calcd for C26H22N6O: C, 71.87; H, 5.10; N, 19.34. Found: C, 71.89; H, 5.15; N, 19.24.
2.1.4.7. 3-((5-(6-Isopropylpyridin-2-yl)-4-(quinoxalin-6-yl)-1H-imidazol-2-yl)methyl)benzamide (11g)
Yield 29%; mp 114−115 °C; 1H NMR (CDCl3) δ 8.81 (m, 2 H, 2 quinoxalinyl), 8.37 (s, 1 H, quinoxalinyl), 8.11 (m, 2 H, 2 quinoxalinyl), 7.71 (s, 1 H, phenyl), 7.65 (m, 1 H, phenyl), 7.50 (d, 1 H, phenyl), 7.46 (t, 1 H, pyridyl), 7.36 (t, 1 H, phenyl), 7.35 (overlapped, 1 H, pyridyl), 7.02 (dd, 1 H, pyridyl), 6.37 (br s, 1 H, CONH), 5.70 (br s, 1 H, CONH), 4.22 (s, 2 H, CH2), 2.99 (heptet, 1 H, CH), 1.24 (s, 3 H, CH3), 1.22 (s, 3 H, CH3); IR (CHCl3) 3184, 1665, 1574 cm−1; MS (EIS) m/z 449.25 (MH+). Anal. Calcd for C27H24N6O: C, 72.30; H, 5.39; N, 18.74. Found: C, 72.03; H, 5.52; N, 18.67.
2.1.4.8. 3-((5-(6-n-Butylpyridin-2-yl)-4-(quinoxalin-6-yl)-1H-imidazol-2-yl)methyl)benzamide (11h)
Yield 27%; mp 110−111 °C; 1H NMR (CDCl3) δ 8.83 (m, 2 H, 2 quinoxalinyl), 8.41 (s, 1 H, quinoxalinyl), 8.13 (s, 2 H, 2 quinoxalinyl), 7.81 (s, 1 H, phenyl), 7.70 (d, 1 H, phenyl), 7.55 (d, 1 H, phenyl), 7.42 (m, 2 H, phenyl and pyridyl), 7.32 (br d, 1 H, pyridyl), 6.98 (d, 1 H, pyridyl), 6.20 (br s, 1 H, CONH), 5.60 (br s, 1 H, CONH), 4.27 (s, 2 H, CH2), 2.77 (t, 2 H), 1.63 (m, 2 H, CH2), 1.38 (m, 2 H, CH2), 0.93 (t, 3 H, CH3); IR (CHCl3) 3183, 1666, 1576 cm−1; MS (EIS) m/z 463.25 (MH+). Anal. Calcd for C28H26N6O: C, 72.71; H, 5.67; N, 18.17. Found: C, 72.89; H, 5.51; N, 18.03.
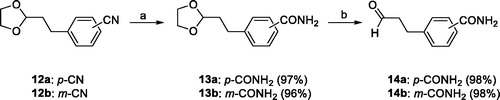
2.1.5. General procedure for the preparation of the 4-(3-oxopropyl)benzamide (14a) and 3-(3-oxopropyl)benzamide (14b)
To a stirred solution of 4-(2-(1,3-dioxolan-2-yl)ethyl)benzonitrile (12a) (1.50 g, 7.34 mmol) in MeOH (50 mL) at room temperature were added 28% H2O2 (25.70 mmol) and 6 N NaOH (7.34 mmol). The mixture was warmed to 55 °C and stirred for 2 h, and to it, 1 N HCl solution was added to adjust to pH∼8 at 0 °C. The MeOH was evaporated off under reduced pressure, and the residue was extracted with CH2Cl2 (30 mL × 3). The organic solution was washed with brine (30 mL), dried over anhydrous Na2SO4, filtered, and evaporated to dryness under reduced pressure. The residue was purified by MPLC on silica gel with MeOH/CH2Cl2 (1:19, then 1:9 (v/v)) as eluent to give 1.58 g (97%) of 4-(2-(1,3-dioxolan-2-yl)ethyl)benzamide (13a) as a solid. To a stirred solution of 13a (0.50 g, 2.26 mmol) in THF (22 mL) was added 1 N HCl solution (20 mL) at room temperature. The mixture was heated under reflux for 1 h and cooled to room temperature. After saturation with NaCl, the reaction mixture was extracted with CHCl3 (20 mL × 5). The combined organic solution was dried over anhydrous Na2SO4, filtered, and evaporated under reduced pressure to give 0.40 g (98%) of 4-(3-oxopropyl)benzamide (14a) as a solid which was used to the next step without further purification.
The 3-(3-oxopropyl)benzamide (14 b) was prepared by the same procedure as for 14a.
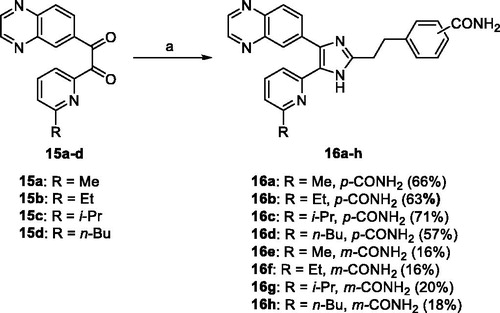
2.1.6. General procedure for the preparation of the 5-(6-alkylpyridin-2-yl)-4-(quinoxalin-6-yl)-1H-imidazoles 16a–h
To a stirred solution of 15a–d (3.79 mmol) in a mixture of t-BuOMe (35 mL) and MeOH (25 mL) were added either 14a or 14b (5.69 mmol) and NH4OAc (18.95 mmol), and the mixture was stirred at 30 °C overnight under argon atmosphere. The pH of the reaction mixture was adjusted to pH∼8 at 0 °C with saturated NaHCO3 solution. After removal of solvent, the reaction mixture was extracted with CH2Cl2 (25 mL × 3), and the organic solution was dried over anhydrous Na2SO4, filtered and evaporated to dryness under reduced pressure. The residue was purified by MPLC on silica gel with CH2Cl2/MeOH as eluent to afford the titled compounds 16a–h as a solid.
2.1.6.1. 4-(2-(5-(6-Methylpyridin-2-yl)-4-(quinoxalin-6-yl)-1H-imidazol-2-yl)ethyl)benzamide (16a)
Yield 66%; mp 145–147 °C; 1H NMR (CDCl3) δ 8.82 (m, 2 H, 2 quinoxalinyl), 8.36 (s, 1 H, quinoxalinyl), 8.09 (d, 2 H, 2 quinoxalinyl), 7.64 (d, 2 H, 2 phenyl), 7.43 (t, 1 H, pyridyl), 7.32 (d, 1 H, pyridyl), 7.17 (d, 2 H, 2 phenyl), 6.99 (d, 1 H, pyridyl), 6.58 (br s, 1 H, CONH), 6.09 (br s, 1 H, CONH), 3.06 (s, 4 H, 2 CH2), 2.45 (s, 3 H, CH3); IR (CHCl3) 3191, 1674, 1615, 1572 cm−1; MS (EIS) m/z 435.19 (MH+). Anal. Calcd for C26H22N6O: C, 71.87; H, 5.10; N, 19.34. Found: C, 71.65; H, 5.23; N, 19.30.
2.1.6.2. 4-(2-(5-(6-Ethylpyridin-2-yl)-4-(quinoxalin-6-yl)-1H-imidazol-2-yl)ethyl)benzamide (16b)
Yield 63%; mp 123–125 °C; 1H NMR (CDCl3) δ 11.40 (br s, 1 H, NH), 8.82 (m, 2 H, 2 quinoxalinyl), 8.37 (s, 1 H, quinoxalinyl), 8.11 (m, 2 H, 2 quinoxalinyl), 7.64 (d, 2 H, 2 phenyl), 7.47 (t, 1 H, pyridyl), 7.35 (br d, 1 H, pyridyl), 7.19 (d, 2 H, 2 phenyl), 7.01 (d, 1 H, pyridyl), 6.50 (br s, 1 H, CONH), 5.90 (br s, 1 H, CONH), 3.08 (s, 4 H, 2 CH2), 2.74 (q, 2 H, CH2), 1.19 (t, 3 H, CH3); IR (CHCl3) 3418, 1666, 1570 cm−1; MS (EIS) m/z 449.20 (MH+). Anal. Calcd for C27H24N6O: C, 72.30; H, 5.39; N, 18.74. Found: C, 72.55; H, 5.26; N, 18.61.
2.1.6.3. 4-(2-(5-(6-Isopropylpyridin-2-yl)-4-(quinoxalin-6-yl)-1H-imidazol-2-yl)ethyl)benzamide (16c)
Yield 71%; mp 113–115 °C; 1H NMR (CDCl3) δ 10.75 (br s, 1 H, NH), 8.83 (m, 2 H, 2 quinoxalinyl), 8.38 (s, 1 H, quinoxalinyl), 8.12 (m, 2 H, 2 quinoxalinyl), 7.70 (m, 2 H, 2 phenyl), 7.46 (t, 1 H, pyridyl), 7.34 (br s, 1 H, pyridyl), 7.27 (d, 2 H, 2 phenyl), 7.02 (d, 1 H, pyridyl), 6.33 (br s, 1 H, CONH), 5.85 (br s, 1 H, CONH), 3.14 (m, 4 H, 2 CH2), 3.00 (heptet, 1 H, CH), 1.26 (s, 3 H, CH3), 1.24 (s, 3 H, CH3); IR (CHCl3) 3446, 1652, 1626 cm−1; MS (EIS) m/z 463.21 (MH+). Anal. Calcd for C28H26N6O: C, 72.71; H, 5.67; N, 18.17. Found: C, 72.53; H, 5.82; N, 18.11.
2.1.6.4. 4-(2-(5-(6-n-Butylpyridin-2-yl)-4-(quinoxalin-6-yl)-1H-imidazol-2-yl)ethyl)benzamide (16d)
Yield 57%; mp 100–102 °C; 1H NMR (CDCl3) δ 11.55 (br s, 1 H, NH), 8.83 (m, 2 H, 2 quinoxalinyl), 8.36 (s, 1 H, quinoxalinyl), 8.11 (m, 2 H, 2 quinoxalinyl), 7.62 (d, 2 H, 2 phenyl), 7.46 (t, 1 H, pyridyl), 7.36 (br d, 1 H, pyridyl), 7.18 (d, 2 H, 2 phenyl), 7.00 (dd, 1 H, pyridyl), 6.50 (br s, 1 H, CONH), 5.80 (br s, 1 H, CONH), 3.07 (s, 4 H, 2 CH2), 2.69 (t, 2 H, CH2), 1.54 (m, 2 H, CH2), 1.27 (m, 2 H, CH2), 0.82 (t, 3 H, CH3); IR (CHCl3) 3411, 1657, 1621, 1569 cm−1; MS (EIS) m/z 477.23 (MH+). Anal. Calcd for C29H28N6O: C, 73.09; H, 5.92; N, 17.63. Found: C, 72.98; H, 5.85; N, 17.71.
2.1.6.5. 3-(2-(5-(6-Methylpyridin-2-yl)-4-(quinoxalin-6-yl)-1H-imidazol-2-yl)ethyl)benzamide (16e)
Yield 16%; mp 120−121 °C; 1H NMR (CDCl3) δ 8.82 (m, 2 H, 2 quinoxalinyl), 8.35 (s, 1 H, quinoxalinyl), 8.08 (s, 2 H, 2 quinoxalinyl), 7.62 (m, 2 H, 2 phenyl), 7.43 (t, 1 H, pyridyl), 7.32 (m, 2 H, phenyl and pyridyl), 7.29 (d, 1 H, phenyl), 6.99 (d, 1 H, pyridyl), 6.63 (br s, 1 H, CONH), 6.10 (br s, 1 H, CONH), 3.08 (m, 4 H, 2 CH2), 2.46 (s, 3 H, CH3); IR (CHCl3) 3192, 1658, 1581 cm−1; MS (EIS) m/z 435.20 (MH+). Anal. Calcd for C26H22N6O: C, 71.87; H, 5.10; N, 19.34. Found: C, 71.53; H, 5.35; N, 19.21.
2.1.6.6. 3-(2-(5-(6-Ethylpyridin-2-yl)-4-(quinoxalin-6-yl)-1H-imidazol-2-yl)ethyl)benzamide (16f)
Yield 16%; mp 119−121 °C; 1H NMR (CDCl3) δ 8.80 (s, 2 H, 2 quinoxalinyl), 8.34 (t, 1 H, quinoxalinyl), 8.06 (s, 2 H, 2 quinoxalinyl), 7.59 (m, 2 H, 2 phenyl), 7.47 (t, 1 H, pyridyl), 7.33 (d, 1 H, pyridyl), 7.23 (t, 1 H, phenyl), 7.01 (dd, 1 H, pyridyl), 6.80 (br s, 1 H, CONH), 6.25 (br s, 1 H, CONH), 3.06 (m, 2 H, CH2), 3.01 (m, 2 H, CH2), 2.71 (q, 2 H, CH2), 1.16 (t, 3 H, CH3); IR (CHCl3) 3317, 1657, 1581 cm−1; MS (EIS) m/z 449.26 (MH+). Anal. Calcd for C27H24N6O: C, 72.30; H, 5.39; N, 18.74. Found: C, 72.44; H, 5.25; N, 18.58.
2.1.6.7. 3-(2-(5-(6-Isopropylpyridin-2-yl)-4-(quinoxalin-6-yl)-1H-imidazol-2-yl)ethyl)benzamide (16g)
Yield 20%; mp 116−119 °C; 1H NMR (CDCl3) δ 8.82 (s, 2 H, 2 quinoxalinyl), 8.37 (s, 1 H, quinoxalinyl), 8.10 (m, 2 H, 2 quinoxalinyl), 7.69 (t, 1 H, phenyl), 7.62 (m, 1 H, phenyl), 7.46 (t, 1 H, pyridyl), 7.34 (m, 2 H, phenyl and pyridyl), 7.31 (t, 1 H, phenyl), 7.02 (dd, 1 H, pyridyl), 6.50 (br s, 1 H, CONH), 6.00 (br s, 1 H, CONH), 3.13 (s, 4 H, 2 CH2), 3.00 (heptet, 1 H, CH), 1.25 (s, 3 H, CH3), 1.24 (s, 3 H, CH3); IR (CHCl3) 3427, 1658 cm−1; MS (EIS) m/z 463.26 (MH+). Anal. Calcd for C28H26N6O: C, 72.71; H, 5.67; N, 18.17. Found: C, 72.86; H, 5.46; N, 18.23.
2.1.6.8. 3-(2-(5-(6-n-Butylpyridin-2-yl)-4-(quinoxalin-6-yl)-1H-imidazol-2-yl)ethyl)benzamide (16h)
Yield 18%; mp 115−118 °C; 1H NMR (CDCl3) δ 8.79 (s, 2 H, 2 quinoxalinyl), 8.32 (t, 1 H, quinoxalinyl), 8.05 (m, 2 H, 2 quinoxalinyl), 7.56 (m, 1 H, phenyl), 7.52 (s, 1 H, phenyl), 7.47 (t, 1 H, pyridyl), 7.34 (d, 1 H, pyridyl), 7.23 (overlapped, 1 H, phenyl), 7.19 (t, 1 H, phenyl), 7.00 (dd, 1 H, pyridyl), 6.90 (br s, 1 H, CONH), 6.23 (br s, 1 H, CONH), 3.04 (m, 2 H, CH2), 2.94 (m, 2 H, CH2), 2.66 (t, 2 H, CH2), 1.48 (m, 2 H, CH2), 1.23 (m, 2 H, CH2), 0.78 (t, 3 H, CH3); IR (CHCl3) 1657 cm−1; MS (EIS) m/z 477.30 (MH+). Anal. Calcd for C29H28N6O: C, 73.09; H, 5.92; N, 17.63. Found: C, 72.88; H, 6.15; N, 17.55.
2.2. Luciferase reporter assay
To establish HaCaT (3TP-luc) stable cells, cells were seeded on six-well plates. Cells were allowed to adhere overnight and then transfected with the p3TP-luc (neo) expression plasmid using PEI reagent (Sigma Aldrich). Transfected cells were cultured for four weeks in the presence of G418 (500 µg/mL). Several single clones were isolated and measured luciferase activity. The clone showing response to TGF-β1 treatment was used for reporter assay. HaCaT (3TP-luc) stable cells were seeded at 2.5 × 104 cells/well in 96-well plate and were allowed to adhere overnight. Cells were concomitantly treated with TGF-β1 (2 ng/mL) and indicated concentrations of ALK5 inhibitors in 0.2% FBS medium and incubated for 24 h at 37 °C in 5% CO2. Cell lysates were prepared using Luciferase Assay System (Promega) according to the manufacturer’s instruction, and luminescence was measured by a luminometer, Micro Lumat Plus (Berthold, Germany).
2.3. Cell permeability assay
Caco-2 cells were seeded in Transwell® polycarbonate filter at a density of 8 × 104 cells/filter and cultured for 21 days. Culture medium was removed from both apical (AP) and basolateral (BL) chambers of transwell, and the wells were rinsed three times with PBS. AP buffer (HBSS, pH 6.5) containing 10 mM MES and BL buffer (HBSS, pH 7.4) containing 10 mM HEPES were loaded in AP (500 µL) and BL (1500 µL) chambers, respectively, followed by incubation for 30 min at 37 °C. Then, test compound (100 µM) was added to the AP side and incubated for 2 h at 37 °C. After the incubation, BL buffer was collected and analysed using an UV spectrophotometer at a maximum wavelength (wavelength 225–357 nm).
2.4. Docking study
All computational works were performed on the Sybyl-X 2.1.1 (Tripos Inc., St Louis, MO, USA) molecular modelling package with CentOS Linux 5.4. operating system1Citation27.
2.4.1. Preparation of ligands and receptor
The 11e was prepared with sketch module embedded in Sybyl package and saved as mol2 format. After sketching the molecule, Gasteiger-Hückel charges were assigned to all atoms. To optimise the ligand, energy minimisation was conducted by the standard tripos force field with convergence to maximum derivatives of 0.001 kcal mol−1·Å−1. The X-ray structure of ALK5 complexed with 5,6-dihydro-4H-pyrrolo[1,2-b]pyrazole inhibitor was used as a receptor for docking (PDB id: 1RW8)Citation28. Receptor structure was retrieved from PDB (http://www.rcsb.org/) and optimised using structure preparation tool embedded in biopolymer module. Native ligand was extracted and all water molecules except key water molecule for water-mediated hydrogen bond network were deleted from the complex structure.
2.4.2. Molecular docking
To examine the binding poses of 11e, docking study was conducted using Surflex-Dock3 embedded in Tripos Sybyl X 2.1.1 software package. For docking performance, the active site was assigned as a protomol generated by using the native ligand in the X-ray structure. Flexible docking was carried out by using default parameter values (threshold = 0.5 and bloat = 0), producing 200 conformers as maximum number of poses per ligand. Binding affinity of each docking pose of ligand was calculated by Surflex-dock score and consensus scoring function (CScore). The total Surflex-Dock score was expressed as –logKd to represent binding affinities. To build the best docking model, key interactions between candidate compound and active site were investigated by visual inspection.
3. Results and discussion
3.1. Chemistry
To develop a potent, selective, and orally bioavailable ALK5 inhibitor, as our starting point, we designed three target molecules, 4-((4-(benzo[1,3]dioxol-5-yl)-5-(6-methylpyridin-2-yl)-1H-imidazol-2-yl)methyl)benzamide (7a) and its ethyl and n-propyl analogues, 7b and 7c to optimise the distance between the two pharmacophores, a central imidazole ring and a phenyl ring.
The 7a–c were prepared as shown in Scheme 1. The 1-(benzo[1,3]dioxol-5-yl)-2-(6-methylpyridin-2-yl)ethane-1,2-dione (4)Citation29 was condensed with 4-(2-oxoethyl)benzonitrile (5a)Citation30, 4-(3-oxopropyl)benzonitrile (5b)Citation31, or 4-(4-oxobutyl)benzonitrile (5c)Citation32 and NH4OAc in AcOH at 120 °C to produce the 2-((4-cyanophenyl)alkyl)imidazoles 6a–c in 30–49% yields. Transformation of the carbonitrile group of 6a–c to the corresponding carboxamide group was accomplished by treatment with 28% H2O2 and 6 N NaOH in EtOH at 55 °C to provide the 7a–c in 49–55% yields.
Scheme 1. Reagents and conditions: (a) NH4OAc, AcOH, 120 °C, 3 h; (b) 28% H2O2, 6 N NaOH, EtOH, 55 °C, 3 h.
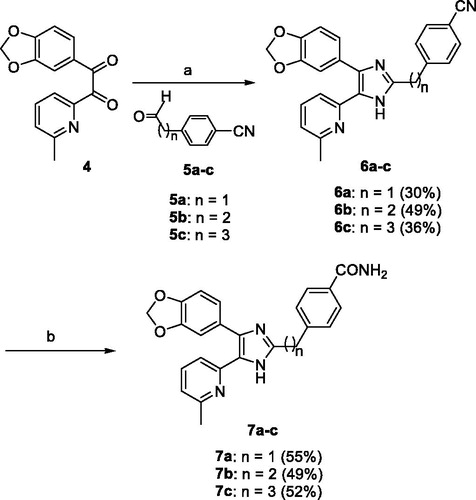
Since a methylene or an ethylene linker was proved to be more beneficial than a n-propylene linker in ALK5 inhibition in both a kinase assay and a cell-based luciferase reporter assay, we next synthesised a series of 5-(6-alkylpyridin-2-yl)-4-(quinoxalin-6-yl)-1H-imidazoles, 11a–h and 16a–h, to compare the impact of a benzo[1,3]dioxol-5-yl moiety and a quinoxalin-6-yl moiety in ALK5 inhibition.
The 11a–h were prepared as shown in Scheme 2. The 2-(6-alkylpyridin-2-yl)-1-(quinoxalin-6-yl)ethanones 8a–dCitation33 were treated with NaNO2 in 5 N HCl to give the 2-(6-alkylpyridin-2-yl)-2-(hydroxyimino)-1-(quinoxalin-6-yl)ethanones 9a–d in 88–99% yields. Condensation of 9a–d with either 5a or 3-(2-oxoethyl)benzonitrile (5d)Citation34 and NH4OAc in a mixture of t-BuOMe and MeOH at room temperature afforded the 5-(6-alkylpyridin-2-yl)-1-hydroxy-4-(quinoxalin-6-yl)-1H-imidazoles 10a–h in 37–77% yields. Conversion of the carbonitrile group of 10a–h to the carboxamide group and subsequent dehydroxylation with triethyl phosphite in anhydrous DMF at 110 °C for 3 days gave the target compounds 11a–h in 23–44% yields.
Scheme 2. Reagents and conditions: (a) NaNO2, 5 N HCl, rt, 1 h; (b) 5a or 3-(2-oxoethyl)benzonitrile (5d), NH4OAc, t-BuOMe/MeOH, rt, overnight, Ar atmosphere; (c) (i) 28% H2O2, 6 N NaOH, EtOH, DMSO, 55 °C, overnight; (ii) triethyl phosphite, anhydrous DMF, 110 °C, 72 h.
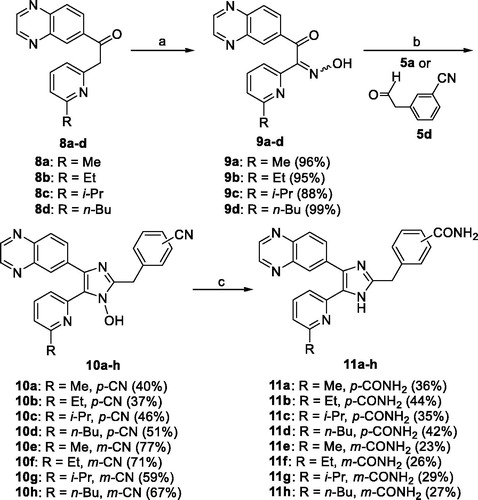
The requisite aldehydes, 4–(3-oxopropyl)benzamide (14a) and 3-(3-oxopropyl)benzamide (14b) for the synthesis of the target compounds 16a–h were prepared as shown in Scheme 3.
Scheme 3. Reagents and conditions: (a) 28% H2O2, 6 N NaOH, MeOH, 55 °C, 2 h; (b) 1 N HCl, THF, reflux, 1 h.
Selective hydrolysis of the carbonitrile group of 12aCitation31 and 12bCitation35 to the carboxamide group followed by acidic hydrolysis of 1,3-dioxolanyl protecting group of 13a and 13 b afforded 14a and 14 b in almost quantitative yield. The 2-(6-alkylpyridin-2-yl)-1-(quinoxalin-6-yl)ethane-1,2-diones 15a–dCitation33 was condensed with either 14a or 14 b and NH4OAc in a mixture of t-BuOMe and MeOH to obtain the 4-carboxamide analogues 16a–d in modest yields (57–71%) and the 3-carboxamide analogues 16e–h in lower yields (16–20%) (Scheme 4).
Scheme 4. Reagents and conditions: (a) 14a or 14 b, NH4OAc, t-BuOMe/MeOH, 30 °C, overnight, Ar atmosphere.
3.2. ALK5 inhibitory activity in an enzyme assay and in a cell-based luciferase reporter assay
To evaluate whether these potential inhibitors 7a–c, 11a–h, and 16a–h could inhibit ALK5, a kinase assay was performed using the purified human ALK5 kinase domain produced in Sf9 insect cells and casein as a substrate (). The ALK5 inhibitory activity of 7b (IC50 = 0.093 µM) was 2.4-fold and 3.6-fold higher than those of 7a (IC50 = 0.224 µM) and 7c (IC50 = 0.338 µM), respectively, in a kinase assay (). In a cell-based luciferase reporter assay using HaCaT (3TP-luc) stable cells, 7c (40% inhibition) was much less inhibitory than 7a (72% inhibition) and 7b (74% inhibition) at a concentration of 0.1 µM.
Table 1. ALK5 inhibitory activity and Caco-2 cell permeability of 2,4-disubstitubed-5–(6-alkylpyridin-2-yl)-1H-imidazoles 7a–c, 11a–h, and 16a–h.
Replacement of a benzo[1,3]dioxol-5-yl moiety in 7a and 7b with a quinoxalin-6-yl moiety increased ALK5 inhibitory activity, thus, the corresponding analogues, 11a (IC50 = 0.088 µM) and 16a (IC50 = 0.030 µM), were 2.5-fold and 3.1-fold more potent than 7a and 7b, respectively, in a kinase assay (). Similar to kinase assay, 11a (84% inhibition) and 16a (85% inhibition) were much more inhibiting than 7a and 7b in a luciferase reporter assay. It was previously demonstrated that a Me substituent at the six-position of the pyridine ring in the pyridyl-substituted ALK5 inhibitors significantly increased ALK5 inhibitory activityCitation36–38. Therefore, we examined the effect of bulkier alkyl groups such as Et, i-Pr, and n-Bu in ALK5 inhibition. The 6-ethylpyridyl analogues 11b, 11f, 16b, and 16f were 1.3-, 2.1-, 1.4-, and 1.4-fold less potent than the corresponding 6-methylpyridyl analogues 11a, 11e, 16a, and 16e, respectively. However, the 6-i-propylpyridyl analogues (11c, 11g, 16c, and 16g) and 6-n-butylpyridyl analogues (11d, 11h, 16d, and 16h) displayed 13–35-fold and 29–71-fold lower inhibitory activity compared to the respective 6-methylpyridyl analogues, indicating that a bulkier group than Et group cannot be accommodated favourably into the ATP binding pocket of ALK5. Regarding the length of a linker, an ethylene linker was generally more beneficial than a methylene linker in the ALK5 inhibition as shown in 7a and 7b, thus, 16a, 16b, 16c, and 16g were 2.9-, 2.7-, 3.4-, and 2.1-fold more inhibiting than the respective 11a, 11b, 11c, and 11g. Compounds 16d–f showed the similar level of potency to that of 11d–f, respectively. The position of a carboxamide group in the phenyl ring also influenced ALK5 inhibition. The m-CONH2 analogues 11e–h were 1.5–2.6-fold more inhibiting than the respective p-CONH2 analogues 11a–d in a series of compounds having a methylene linker, whereas the p-CONH2 analogues having an ethylene linker, 16a and 16 b, were 1.6-fold more inhibiting than the corresponding m-CONH2 analogues 16e and 16f. Compounds showing IC50 values of <0.05 µM in a kinase assay, 11e (IC50 = 0.036 µM), 16a, 16 b (IC50 = 0.041 µM), and 16e (IC50 = 0.047 µM) exhibited much higher inhibitory activity in a luciferase reporter assay, also (11e: 92%, 16a: 85%, 16b: 84%, 16e: 93%).
Among this series of compounds, 11e possessing the most potent ALK5 inhibitory activity and the highest permeability in Caco-2 cells was selected, and its ALK5 inhibitory activity was compared with those of potential competitors, galunisertib and vactosertib in a kinase assay and in a luciferase reporter assay. In a kinase assay, 11e (IC50 = 0.013 µM) showed the same level of potency to that of vactosertib (IC50 = 0.013 µM) and 6.6-fold higher potency compared to that of galunisertib (IC50 = 0.086 µM) (). Luciferase activity of HaCaT (3TP-luc) cells was increased by 65-fold after treatment of TGF-β1 (2 ng/mL), and 11e and vactosertib displayed the similar level of inhibition on the TGF-β1-induced luciferase reporter activity with IC50 values of 0.0196 µM and 0.0165 µM, respectively. Similar to a kinase assay, galunisertib displayed much lower inhibition (IC50 >0.1 µM) compared to 11e and vactosertib ().
Table 2. ALK5 and p38α inhibitory activity of 11e, 1, and 2.
The kinase domain of p38α is known to be one of the most homologous to that of ALK5Citation39, therefore, this enzyme was chosen to compare selectivity of 11e, galunisertib, and vactosertib. In a p38α kinase assay, IC50 values of 11e, galunisertib, and vactosertib were 0.288 µM, 0.320 µM, and 1.775 µM, respectively, thus, their selectivity indices (IC50 for p38α/IC50 for ALK5) were 22, 4, and 137, respectively. Although 11e was 5.5-fold more selective against p38α than galunisertib, it was 6.2-fold less selective than vactosertib.
3.3. Caco-2 cell permeability assay
To estimate oral absorption of target compounds, their permeability in a Caco-2 monolayer was evaluated at a concentration of 100 µM (). The 11e showed the highest permeability (56%), but 16f (6%), 16g (0%), and 16h (0%) showed very limited or no permeation in this assay, demonstrating that even simple structural modification markedly affected the permeability in this series of compounds.
3.4. Kinase profiling assay
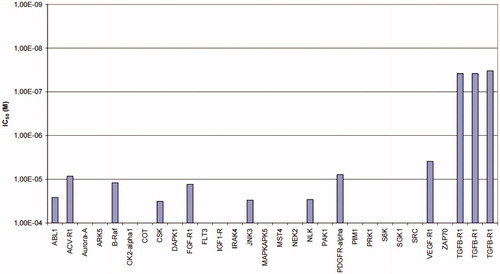
Considering the ALK5 inhibitory activity in both a kinase assay and a luciferase reporter assay and the Caco-2 cell permeability, 11e was selected as a candidate for preliminary kinase profiling. Because ALK5 is a serine/threonine kinase, we chose 19 serine/threonine kinases including ALK5 and an ALK family kinase, ACV-R1 (ALK5, ACV-R1, Aurora-A, ARK5, B-Raf, CK2α1, COT, DAPK1, IRAK4, JNK3, MAPKAPK5, MST4, NEK2, NLK, PAK1, PIM1, PRK1, S6K, SGK1) and 9 tyrosine kinases (ABL1, CSK, FGF-R1, FLT3, IGF1-R, PDGFR-α, SRC, VEGF-R1, ZAP70). Selectivity profiling of 11e using a panel of 28 protein kinases showed selectivity indices of >100 against all the kinases tested (ProQinase GmbH (Freiburg, Germany)) ().
Figure 2. Inhibitory profile of 11e in 28 protein kinase assays.
3.5. Docking model of ALK5 in complex with 11e
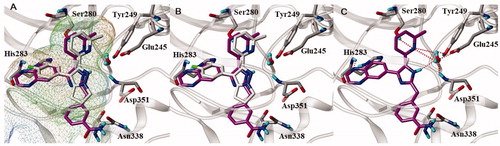
To determine the binding pose of 11e in the active site of ALK5, we executed docking modelling with flexible molecular docking programme Surflex-dockCitation40. We analysed the result of docking considering Surflex-dock docking score (–logKd) and consensus score (obtained from CScore moduleCitation27), and selected the poses of 11e with high scores (–logKd ≥7 and CScore ≥3). To select the best docked pose among them, we also identified key interactions between amino acid residues in the active site and 11e, in comparison with the X-ray pose of native ligand (3-(4-fluorophenyl)-2-(6-methylpyridin-2-yl)-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazole)Citation28. As shown in , 11e fits well into cavity of active site, with quinoxaline ring and methyl group of 6-methylpyridine ring occupying the hydrophobic pockets. Hydrogen bond interactions between 11e and ALK5 are exhibited in . The quinoxaline ring nitrogen acts as a hydrogen bond acceptor and interacts with NH of His283 in the backbone of hinge region of ALK5. The 6-methylpyridine ring nitrogen forms a water-mediated hydrogen bond network with the backbone NH of Asp351, and the side chain of both Tyr249 and Glu245. The imidazole ring NH of 11e is also involved in the water-mediated hydrogen bond network. The NH2 group of carboxamide in the meta position of phenyl ring interacts with the side chain of Asn338 by forming hydrogen bond, which cannot be formed for the compounds, 11a–d having the carboxamide group in the para position. Our docking model for the most active compound 11e well supports the key interactions for ALK5 inhibition which was previously reportedCitation28.
Figure 3. Docked pose of 11e in the active site of ALK5 (PDBid:1RW8). (A, B) 11e is (magenta carbon atoms) superimposed over the X-ray pose of native ligand (grey carbon atoms). The active site of ALK5 is shown as MOLCAD lipophilic potential surface map (A), and lipophilicity increases from blue (hydrophilic) to brown (liphophilic). Grey capped sticks represent key amino acid residues within the binding site, and the backbone of ALK5 is shown as ribbon. The bound water molecule in the X-ray structure is represented by ball and stick. (C) Intermolecular interaction between 11e and ALK5. Grey capped sticks represent key amino acid residues in the active site. Red dashed lines are hydrogen bonding interactions (<3.0 Å).
4. Conclusions
In this report, a series of 2,4-disubstituted-5-(6-alkylpyridin-2-yl)-1H-imidazoles, 7a–c, 11a–h, and 16a–h has been synthesised and evaluated for their ALK5 inhibitory activity in an enzyme assay and in a cell-based luciferase reporter assay. The structure–activity relationships in this series of compounds revealed that an ethylene linker at the two-position of the imidazole ring was the most beneficial in ALK5 inhibitory activity. Replacement of a benzo[1,3]dioxol-5-yl moiety at the four-position of the imidazole ring with a quinoxalin-6-yl moiety markedly increased ALK5 inhibitory activity. Regarding the alkyl substituent at the six-position of the pyridine ring, the compounds having a methyl or an ethyl substituent displayed much higher inhibitory activity than the compounds having an i-propyl or a n-butyl substituent. The m-CONH2 analogues were more inhibiting than the p-CONH2 analogues in compounds having a methylene linker, whereas the p-CONH2 analogues were more inhibiting than the m-CONH2 analogues in compounds having an ethylene linker. In a cell permeability assay using Caco-2 monolayer, 11e showed the highest permeability in this series of compounds. The 11e was equipotent to vactosertib, but much more potent than galunisertib in an ALK5 kinase assay and in a cell-based luciferase reporter assay. Although 11e was 5.5-fold more selective against p38α than galunisertib, it was 6.2-fold less selective than vactosertib. Therefore, it can be concluded that combination of replacement of a [1,2,4]triazolo[1,5-a]pyridin-6-yl moiety with a quinoxalin-6-yl moiety, insertion of a methylene linker instead of a methyleneamino linker, and a m-CONH2 substituent in the phenyl ring in vactosertib maintained its high ALK5 inhibitory activity, but decreased its selectivity against p38α. Selectivity profiling of 11e using a panel of 28 protein kinases showed that it is highly selective for ALK5. Our docking results demonstrate that 11e fits well in the ATP-binding pocket of ALK5 with favourable intermolecular interactions.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Pickup M, Novitskiy S, Moses HL. The roles of TGFβ in the tumour microenvironment. Nat Rev Cancer 2013;13:788–99.
- Tsushima H, Kawata S, Tamura S, et al. High levels of transforming growth factor β1 in patients with colorectal cancer: association with disease progression. Gastroenterology 1996;110:375–82.
- Ivanovic V, Todorovic-Rakovic N, Demajo M, et al. Elevated plasma levels of transforming growth factor- β1 (TGF- β1) in patients with advanced breast cancer: association with disease progression. Eur J Cancer 2003;39:454–61.
- Dave H, Shah M, Trivedi S, Shukla S. Prognostic utility of circulating transforming growth factor beta 1 in breast cancer patients. Int J Biol Markers 2012;27:53–9.
- Eder IE, Stenzl A, Hobisch A, et al. Transforming growth factors-β1 and β2 in serum and urine from patients with bladder carcinoma. J Urol 1996;156:953–7.
- Shariat SF, Kim JH, Andrews B, et al. Preoperative plasma levels of transforming growth factor β1 strongly predict clinical outcome in patients with bladder carcinoma. Cancer 2001;92:2985–92.
- Adler HL, McCurdy MA, Kattan MW, et al. Elevated levels of circulating interleukin-6 and transforming growth factor-β1 in patients with metastatic prostatic carcinoma. J Urol 1999;161:182–7.
- Shariat SF, Shalev M, Menesses-Diaz A, et al. Preoperative plasma levels of transforming growth factor beta1 (TGF-β1) strongly predict progression in patients undergoing radical prostatectomy. J Clin Oncol 2001;19:2856–64.
- Krasagakis K, Thölke D, Farthmann B, et al. Elevated plasma levels of transforming growth factor (TGF)-β1 and TGF-β2 in patients with disseminated malignant melanoma. Br J Cancer 1998;77:1492–4.
- Zhao J, Liang Y, Yin Q, et al. Clinical and prognostic significance of serum transforming growth factor-beta1 levels in patients with pancreatic ductal adenocarcinoma. Braz J Med Biol Res 2016;49:e5485.
- Lin TH, Shao YY, Chan SY, et al. High serum transforming growth factor-β1 levels predict outcome in hepatocellular carcinoma patients treated with sorafenib. Clin Cancer Res 2015;21:3678–84.
- Bierie B, Moses HL. Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer 2006;6:506–20.
- Herbertz S, Sawyer JS, Stauber AJ, et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des Devel Ther 2015;9:4479–99.
- Faivre S, Santoro A, Kelley RK, et al. Novel transforming growth factor beta receptor I kinase inhibitor galunisertib (LY2157299) in advanced hepatocellular carcinoma. Liver Int 2019;39:1468–77.
- Jin CH, Krishnaiah M, Sreenu D, et al. Discovery of N-((4-([1,2,4]triazolo[1,5-a]pyridin-6-yl)-5-(6-methylpyridin-2-yl)-1H-imidazol-2-yl)methyl)-2-fluoroaniline (EW-7197): a highly potent, selective, and orally bioavailable inhibitor of TGF-β type I receptor kinase as cancer immunotherapeutic/antifibrotic agent. J Med Chem 2014;57:4213–38.
- Batlle E, Massague J. Transforming growth factor-beta signaling in immunity and cancer. Immunity 2019;50:924–40.
- Patel HM, Sing B, Bhardwaj V, et al. Design, synthesis and evaluation of small molecule imidazo[2,1-b][1,3,4]thiadiazoles as inhibitors of transforming growth factor-β type-I receptor kinase (ALK5). Eur J Med Chem 2015;93:599–613.
- Reznickova E, Tenora L, Pospisilova P, et al. ALK5 kinase inhibitory activity and synthesis of 2,3,4-substituted 5,5-dimethyl-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazoles. Eur J Med Chem 2017;127:632–42.
- Yoon JH, Jung SM, Park SH, et al. Activin receptor-like kinase5 inhibition suppresses mouse melanoma by ubiquitin degradation of Smad4, thereby derepressing eomesodermin in cytotoxic T lymphocytes. EMBO Mol Med 2013;5:1720–39.
- Son JY, Park SY, Kim SJ, et al. EW-7197, a novel ALK-5 kinase inhibitor, potently inhibits breast to lung metastasis. Mol Cancer Ther 2014;13:1704–16.
- Naka K, Ishihara K, Jomen Y, et al. Novel oral transforming growth factor-β signaling inhibitor EW-7197 eradicates CML-initiating cells. Cancer Sci 2016;107:140–8.
- Park SA, Kim MJ, Park SY, et al. EW-7197 inhibits hepatic, renal, and pulmonary fibrosis by blocking TGF-β/Smad and ROS signaling. Cell Mol Life Sci 2015;72:2023–39.
- Kim MJ, Park SA, Kim CH, et al. TGF-β type I receptor kinase inhibitor EW-7197 suppresses cholestatic liver fibrosis by inhibiting HIF1α-induced epithelial mesenchymal transition. Cell Physiol Biochem 2016;38:571–88.
- Jun EJ, Park JH, Tsauo J, et al. EW-7197, an activin-like kinase 5 inhibitor, suppresses granulation tissue after stent placement in rat esophagus. Gastrointest Endosc 2017;86:219–28.
- Han K, Park JH, Yang SG, et al. EW-7197 eluting nano-fiber covered self-expandable metallic stent to prevent granulation tissue formation in a canine urethral model. PLoS One 2018;13:e0192430.
- Tsauo J, Song HY, Choi EY, et al. EW-7197, an oral transforming growth factor β type I receptor kinase inhibitor, for preventing peritoneal adhesion formation in a rat model. Surgery 2018;164:1100–8.
- Sybyl-X 2.1.1. SYBYL molecular modeling software. St. Louis (MO): Tripos Inc.; 2013.
- Sawyer JS, Beight DW, Britt KS, et al. Synthesis and activity of new aryl- and heteroaryl-substituted 5,6-dihydro-4H-pyrrolo[1,2-b]pyrazole inhibitors of the transforming growth factor-β type I receptor kinase domain. Bioorg Med Chem Lett 2004;14:3581–4.
- Gaster LM, Hadley MS, Harling JD, et al. Pyridinylimidazoles. Patent WO 01/62756 A1; 2001.
- Powers JC, Boduszek B, Oleksyszyn J, Basic alpha-aminoalkylphosphonate derivatives. Patent US 5,686,419 A; 1997.
- Kelly SM, Liquid crystals. Patent US 5,204,018 A; 1993.
- Diulgheroff N, Pirkes M, Pontiroli A, Villa M. Processes for preparing intermediates of pemetrexed. Patent WO 2008/021385 A2; 2008.
- Kim D-K, Jung SH, Lee HS, Dewang PM. Synthesis and biological evaluation of benzenesulfonamide-substituted 4-(6-alkylpyridin-2-yl)-5-(quinoxalin-6-yl)imidazoles as transforming growth factor-β type 1 receptor kinase inhibitors. Eur J Med Chem 2009;44:568–76.
- Pasternak A, Dejesus RK, Zhu Y, et al. Inhibitors of the renal outer medullary potassium channel. Patent WO 2012/058134 A1; 2012.
- Fleury-Brégeot N, Oehlrich D, Rombouts F, Molander GA. Suzuki-Miyaura cross-coupling of potassium dioxolanylethyltrifluoroborate and aryl/heteroaryl chlorides. Org Lett 2013;15:1536–9.
- Byfield SD, Major C, Laping NJ, Roberts AB. SB-505124 is a selective inhibitor of transforming growth factor-beta type I receptors ALK4, ALK5, and ALK7. Mol Pharmacol 2004;65:744–52.
- Gellibert F, Woolven J, Fouchet MH, et al. Identification of 1,5-naphthyridine derivatives as a novel series of potent and selective TGF-beta type I receptor inhibitors. J Med Chem 2004;47:4494–506.
- Kim D-K, Jang Y, Lee HS, et al. Synthesis and biological evaluation of 4(5)-(6-alkylpyridin-2-yl)imidazoles as transforming growth factor-β type 1 receptor kinase inhibitors. J Med Chem 2007;50:3143–7.
- Eyers PA, Craxton M, Morrice N, et al. Conversion of SB 203580-insensitive MAP kinase family members to drug-sensitive forms by a single amino-acid substitution. Chem Biol 1998;5:321–8.
- Spitzer R, Jain AN. Surflex-Dock: Docking benchmarks and real-world application. J Comput Aided Mol Des 2012;26:687–99.