
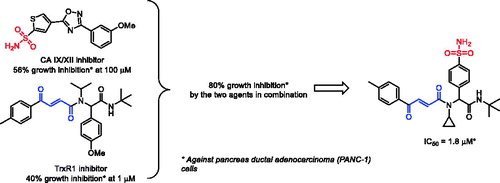
Abstract
A hypothesis that simultaneous targeting cancer-related carbonic anhydrase hCA IX and hCA XII isoforms (whose overexpression is a cancer cell’s defence mechanism against hypoxia) along with thioredoxin reductase (overexpressed in cancers as a defence against oxidative stress) may lead to synergistic antiproliferative effects was confirmed by testing combinations of the two inhibitor classes against pancreatic cancer cells (PANC-1). Combining both pharmacophoric motifs within one molecule led to a sharp increase of cytotoxicity. This preliminary observation sets the ground for a fundamentally new approach to anticancer agent design.
Graphical Abstract
1. Introduction
Growing malignant tumours possess numerous defence mechanisms which ensure survival of aberrantly proliferating cancer cells while normal cells in the surrounding tissues are being killed and replaced with the neoplasm.Citation1 Among these mechanisms, overexpression of membrane-bound isoforms of carbonic anhydrase (hCA IX and XII) protect tumours from hypoxia and provide defence against acidification of the environment surrounding the growing tumour.Citation2 Selective targeting of hCA IX and XII has been confirmed to lead to retardation of tumour growth and, ultimately, reduction of tumour size.Citation3 Recently, we reported on the discovery of potent 1,2,4-oxadiazole-based inhibitors 1–3 of these two cancer-related isoforms of carbonic anhydrase.Citation4,Citation5 These compounds possess subnanomolar to double-digit nanomolar potency against hCA IX and XII (), which makes them ideal tool compounds for further investigation of the role of these carbonic anhydrase isoforms in tumour growth.
Table 1. Chemical structures and inhibitory profile of carbonic anhydrase inhibitors 1–3 and thioredoxin reductase inhibitors 4–6 employed in this study (the pharmacophoric inhibitory motifs are highlighted red and blue, respectively).
Another important mechanism of tumour survival which can be considered a target for anticancer agent design,Citation6 is that providing tumour cell defence against oxidative stress (reactive oxygen species or ROS). In particular, tumour cells have been shown to overexpress thioredoxin reductase (TrxR, EC 1.8.1.9) which contributes to their resistant phenotype characterised by higher levels of ROS.Citation7 Thus, targeting TrxR1 (the most widespread cytosolic isoform of human TrxR) is an emerging approach to selective killing of cancer cells.Citation8 TrxR is a selenocysteine (Sec) enzyme which, along with NADPH and thioredoxin (Trx), constitutes the thioredoxin system responsible for maintaining Trx in the reduced bis-sulfhydryl state.Citation9 Among several classes of inhibitors possessing a variable degree of electrophilicity towards the catalytic Sec residue (reviewed by BellelliCitation10 and FangCitation11), we found covalent Michael acceptor inhibitors (such as Ugi-type adducts 4–6 which we dubbed “Ugi Michael Acceptors” or UMAs) to be particularly efficacious.Citation12 From the mechanistic standpoint, the inhibitory action of UMAs towards TrxR1 likely is exerted via the irreversible covalent trapping of the catalytic Sec residue (which exists in the ionised form at physiological pHCitation13) by the electrophilic β-benzoyl acrylamide (or acrylate) moiety present in 4–6. The inhibitory potency of these compounds was established at the submicromolar to low micromolar level () on a recombinant TrxR1 enzyme and compound 6 (DVD-445) was nominated a lead for further optimisation.
In this work, we aimed at verifying whether combinations of the said inhibitors from different classes (i.e. any of 1–3 with any of 4–6) would have stronger antiproliferative effects on tumour cells in comparison with a given single agent. With this idea in mind – along with the general understanding of the pharmacophoric moieties present in the carbonic anhydrase inhibitors (the zinc-binding primary sulphonamide group,Citation3 ZBG) and thioredoxin reductase inhibitor (the α,β-unsaturated Michael acceptor moietyCitation12) – we also aimed to combine these critical inhibitory motifs in the structure of a single agent and verify whether this will lead to potentiation of its cytotoxicity compared to inhibitors 1–6. Herein, we present the results of these studies.
2. Materials and methods
2.1. Chemical syntheses – general
All reagents and solvents were obtained from commercial sources and used without purification. All reactions implemented in an open flask without any protection from CO2 and H2O. Reactions were monitored by analytical thin-layer chromatography (TLC) Macherey-Nagel, TLC plates Polygram® Sil G/UV254. Visualisation of the developed chromatograms was performed by fluorescence quenching at 254 nm. 1H and 13C NMR spectra were measured on Bruker AVANCE DPX 400 (400 MHz for 1H and 100 MHz for 13C respectively). All chemical shifts (δ) are given in parts per million (ppm) with reference to solvent residues in DMSO-d6 (2.50 for proton and 39.52 for carbon) and coupling constant (J) are reported in hertz (Hz). Multiplicities are abbreviated as follows: s = singlet, d = doublet, t = triplet, q = quartette, m = multiplet, br = broad. Melting points were determined on Electrothermal IA 9300 series Digital Melting Point Apparatus. Mass spectra were recorded on microTOF spectrometers (ESI ionisation).
2.2. General Procedure 1: preparation of compounds 1–2 (GP1)
To a solution of carboxylic acid 7 (1.1 mmol) in dry DMSO (1.0 ml) CDI (195 mg, 1.2 mmol) was added. The reaction mixture was stirred at room temperature for 30 min, and then amidoxime 8 (1.0 mmol) was added. The reaction mixture was stirred at room temperature for another 18 h, then to the reaction mixture powdered NaOH (48 mg, 1.2 mmol) was added rapidly. The reaction mixture was stirred at room temperature for 2 h. Then the reaction mixture was diluted with cold water (20 ml). The resulting precipitate was filtered off, washed with cooled water (15 ml) and dried in air at 50 °C.
2.2.1. 4-(3-Cyclopropyl-1,2,4-oxadiazol-5-yl)thiophene-2-sulphonamide (1)
Yield 203 mg (75%). White solid, m.p. 311–313 °С. 1H NMR (400 MHz, DMSO) δ ppm 8.72 (d, J = 1.6 Hz, 1H), 7.97 (d, J = 1.6 Hz, 1H), 7.90 (s, 2H), 2.19 (m, 1H), 1.15 − 1.09 (m, 2H), 1.00 − 0.95 (m, 2H).). 13C NMR (101 MHz, DMSO) δ ppm 173.0 (C), 170.6 (C), 148.8 (C), 135.5 (CH), 128.5 (CH), 124.8 (C), 8.1 (CH2), 6.9 (C). HRMS (ESI, m/z): calculated for C9H9N3O3S2 [M + H]+ 272.0158; found 272.0152.
2.2.2. 4-(3-(3-Methoxyphenyl)-1,2,4-oxadiazol-5-yl)thiophene-2-sulphonamide (2)
Yield 222 mg (66%). Yellow solid, m.p. 167–169 °С. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.85 (d, J = 1.5 Hz, 1H), 8.10 (d, J = 1.5 Hz, 1H), 7.95 (s, 2H), 7.66 (d, J = 7.7 Hz, 1H), 7.57 − 7.55 (m, 1H), 7.52 (t, J = 8.0 Hz, 1H), 7.20 (dd, J = 8.3, 2.6 Hz, 1H), 3.86 (s, 3H).). 13C NMR (101 MHz, DMSO-d6) δ ppm 171.3 (C), 168.5 (C), 160.2 (C), 149.0 (C), 135.9 (CH), 131.0 (CH), 128.6 (CH), 127.7 (C), 124.7 (C), 119.9 (CH), 118.2 (CH), 112.5 (CH), 55.8 (CH3). HRMS (ESI, m/z): calculated for C13H11N3O4S2 [M + H]+ 338.0264; found 338.0266.
2.3. 3-(5-Cyclopropyl-1,2,4-oxadiazol-3-yl) benzenesulphonamide (3)
To a solution of amidoxime 9 (1.0 mmol) and methyl cyclopropanecarboxylate (1.5 mmol) in DMSO (1.0 ml) powdered NaOH (60 mg, 1.5 mmol) was rapidly added. The reaction mixture was stirred at room temperature for 2 h. The reaction mixture was diluted with cold water (20 ml). The resulting precipitate was filtered off, washed with water (15 ml) and dried in air at 50 °C.
Yield 151 mg (57%). Beige solid, m.p. 249–251 °С. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.40 (s, 1H), 8.17 (d, J = 6.7 Hz, 1H), 8.00 (d, J = 8.2 Hz, 1H), 7.76 (t, J = 7.7 Hz, 1H), 7.53 (s, 2H), 2.47 − 2.40 (m, 1H), 1.36 − 1.27 (m, 2H), 1.24 − 1.18 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ ppm 182.8 (C), 167.1 (C), 145.6 (C), 130.6 (CH), 130.4 (CH), 128.8 (CH), 127.4 (C), 124.6 (CH), 10.7 (CH), 7.7 (CH2). HRMS (ESI, m/z): calculated for C11H11N3O3S [M + H]+ 266.0594; found 266.0618.
2.4. General procedure for the preparation of compounds 4–6
Aldehyde (1 mmol) and amine (1 mmol) were combined in methanol (3 ml) and the resulting solution was stirred for 15 min at r. t. Isocyanide (1 mmol) and carboxylic acid (1 mmol) were added and the stirring continued for 24 h. The crystalline precipitate (formed in case of products 5a, 5i and 5j) was collected by filtration and washed with ether (3 ml); in all other cases, the solution was concentrated to dryness and the residue was purified by column chromatography on silica gel using dichloromethane-methanol (200:1) mixture as eluent.
2.4.1. (E)-N-(2-(tert-Butylamino)-1–(4-methoxyphenyl)-2-oxoethyl)-N-isopropyl-4-oxo-4-(p-tolyl)but-2-enamide (4)
Yield 77 mg (17%); White amorphous solid; 1H NMR (400 MHz, CDCl3) δ 8.01 − 7.85 (m, 3H), 7.55 (d, J = 15.1 Hz, 1H), 7.35 − 7.30 (m, 4H), 6.90 (d, J = 8.8 Hz, 2H), 6.33 (s, 1H), 4.95 (s, 1H), 4.42 − 4.28 (m, 1H), 3.83 (s, 3H), 2.45 (s, 3H), 1.47 (d, J = 6.7 Hz, 3H), 1.35 (s, 9H), 1.17 (d, J = 6.8 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 189.0, 169.4, 165.9, 159.1, 144.7, 134.4, 134.2, 133.8, 129.5, 129.2, 129.0, 128.7, 114.1, 63.0, 55.2, 51.3, 50.4, 28.6, 21.7, 21.6, 21.6; HRMS (ESI), m/z calcd for C27H34N2O4 [M + Na]+ 473.2411, found 473.2398.
2.4.2. (E)-N-(2-(tert-Butylamino)-1–(4-fluorophenyl)-2-oxoethyl)-N-cyclopropyl-4–(4-fluorophenyl)-4-oxobut-2-enamide (5)
Yield 97 mg (22%); Pale yellow amorphous solid; m.p.=115.5–117.1 °C; 1H NMR (400 MHz, CDCl3) δ 8.17 − 8.03 (m, 2H), 8.00 − 7.90 (m, 1H), 7.84 (d, J = 15.0 Hz, 1H), 7.48 − 7.38 (m, 2H), 7.24 − 7.14 (m, 2H), 7.13 − 7.05 (m, 2H), 5.79 (s, 1H), 5.64 (s, 1H), 2.74 − 2.52 (m, 1H), 1.40 − 1.29 (m, 9H), 1.21 − 1.05 (m, 1H), 1.02 − 0.91 (m, 1H), 0.85 − 0.65 (m, 2H); 13 C NMR (101 MHz, CDCl3) δ 188.0, 168.4, 167.6, 166.1 (d, J = 256.1 Hz), 162.5 (d, J = 248.5 Hz), 133.9, 133.8, 133.4 (d, J = 3.0 Hz), 131.6 (d, J = 9.5 Hz), 131.4 (d, J = 8.1 Hz), 131.2 (d, J = 3.4 Hz), 116.0 (d, J = 22.0 Hz), 115.6 (d, J = 21.5 Hz), 66.2, 51.6, 30.3, 28.6, 10.8, 9.2; HRMS (ESI), m/z calcd for C25H26F2N2O3 [M + Na]+ 463.1804, found 463.1794.
2.4.3. (E)-2-((2-(tert-Butylamino)-1–(4-fluorophenyl)-2-oxoethyl)(methyl)amino)ethyl 4–(4-fluorophenyl)-4-oxobut-2-enoate (6)
Yield 55 mg (12%); Yellowish amorphous solid; 1H NMR (400 MHz, CDCl3) δ 8.08 (dd, J = 8.7, 5.4 Hz, 2H), 7.95 (d, J = 15.5 Hz, 1H), 7.36 − 7.25 (m, 2H), 7.23 (t, J = 8.6 Hz, 2H), 7.05 (t, J = 8.6 Hz, 2H), 7.01 − 6.87 (m, 2H), 4.37 (td, J = 5.9, 2.3 Hz, 2H), 3.94 (s, 1H), 2.81–2.63 (m, 2H), 2.28 (s, 3H), 1.37 (s, 9H); 13 C NMR (101 MHz, CDCl3) δ 187.5, 170.1, 166.2 (d, J = 256.8 Hz), 165.3, 162.4 (d, J = 246.8 Hz), 136.6, 132.9 (d, J = 3.0 Hz), 132.1, 131.8 (d, J = 3.4 Hz), 131.6 (d, J = 9.5 Hz), 130.6 (d, J = 8.0 Hz), 116.2 (d, J = 22.0 Hz), 115.3 (d, J = 21.5 Hz), 75.1, 62.7, 53.3, 50.8, 40.1, 28.6; HRMS (ESI), m/z calcd for C25H28F2N2O4 [M + Na]+ 481.1915, found 481.1893.
2.5. N,N-Bis(2,4-dimethoxybenzyl)-4-formylbenzenesulphonamide (12)
A solution of 4-formylbenzenesulphonyl chlorideCitation14 (195 mg, 0.95 mmol), bis(2,4-dimethoxybenzyl)amine (302 mg, 0.95 mmol) and trimethylamine (193 mg, 1.91 mmol) in dichloromethane (10 ml) was stirred for 30 min at room temperature and then washed with 10% aq. HCl (10 ml), saturated NaHCO3 (2 × 10 ml) and brine (10 ml). The resulting solution was evaporated to dryness. Yield 430 mg, 93%; Yellow oil; 1H NMR (400 MHz, CDCl3) δ 10.08 (s, 1H), 7.99 − 7.84 (m, 2H), 7.79 (d, J = 8.3 Hz, 2H), 7.18 (d, J = 8.4 Hz, 2H), 6.41 (dd, J = 8.4, 2.4 Hz, 2H), 6.27 (d, J = 2.4 Hz, 2H), 4.45 (s, 4H), 3.79 (s, 6H), 3.62 (s, 6H); 13 C NMR (101 MHz, CDCl3) δ 191.0, 160.5, 158.2, 146.7, 138.0, 130.9, 129.6, 127.5, 116.6, 104.0, 98.0, 55.4, 54.9, 46.5.
2.6 (E)-N-(2-(tert-butylamino)-2-oxo-1–(4-sulphamoylphenyl) ethyl)-N-cyclopropyl-4-oxo-4-(p-tolyl)but-2-enamide (10)
A solution of N,N-bis(2,4-dimethoxybenzyl)−4-formylbenzenesulphonamide (97 mg, 0.2 mmol) and cyclopropylamine (11 mg, 0.2 mmol) in 2 ml of methanol was stirred for 15 min at room temperature, then tert-butyl isocyanide (17 mg, 0.2 mmol) and (E)−4-oxo-4-(p-tolyl)but-2-enoic acid (38 mg, 0.2 mmol) were added to the solution and the resulting mixture was stirred for 24 h at the same temperature. The solvent was removed under reduced pressure and the residue was purified by column chromatography using eluent ethyl acetate–hexane 1:1 (Rf=0.25). The Ugi adduct 13 thus obtained was dissolved in trifluoroacetic acid (3 ml) and the solution was stirred for 3 h. The solvent was evaporated and the residue was redissolved in 3 ml of methanol, filtered from insoluble impurities and evaporated. White semi-solid; Yield 12 mg, 12%; 1H NMR (400 MHz, Methanol-d4) δ 7.98 (d, J = 8.0 Hz, 2H), 7.96 − 7.91 (m, 2H), 7.87 (d, J = 15.3 Hz, 1H), 7.81 (d, J = 15.3 Hz, 1H), 7.56 (d, J = 8.2 Hz, 2H), 7.39 (d, J = 7.9 Hz, 2H), 5.90 (s, 1H), 2.78 − 2.55 (m, 1H), 2.45 (s, 3H), 1.38 (s, 9H), 1.16 − 1.04 (m, 1H), 0.83 (ddd, J = 9.6, 4.8, 2.2 Hz, 1H), 0.78 − 0.67 (m, 1H), 0.58 − 0.45 (m, 1H); 13 C NMR (101 MHz, Methanol-d4) δ 189.6, 169.0, 168.5, 145.0, 143.3, 140.1, 134.4, 133.8, 133.6, 130.0, 129.3, 128.7, 125.8, 65.3, 51.1, 29.3, 27.4, 20.3, 10.3, 9.0; HRMS (ESI/Q-TOF) m/z: [M + Na]+ Calcd for C26H31N3O5S 520.1877; Found 520.1874.
2.3. Cell viability assay
Human cell lines were maintained at 37 °C in humidified atmosphere containing air and 5% CO2 as previously described.Citation15 Pancreas ductal adenocarcinoma cells PANC-1 were obtained from Russian collection cell cultures at the RAS Institute of Cytology (Saint Petersburg, Russian Federation). Cell line were grown in Dulbecco’s Modified Eagle’s Medium-F12 (BioloT) containing 10% (v/v) heat-inactivated foetal calf serum (FCS, HyClone Laboratories, UT), 1% L-glutamine, 1% sodium pyruvate, 50 U/ml penicillin, and 50 μg/ml streptomycin (BioloT). Cytotoxicity of carbonic anhydrase inhibitors was evaluated using a routine colorimetric method with tetrazolium dye − 3–(4,5-dimethylthiazol-2-yl)−2,5-diphenyltetrazolium bromide (MTT). The cell lines were incubated for 48 h under normoxia and in the presence of the hypoxia-mimicking agent 50 μM CoCl2 with medium containing different concentrations of carbonic anhydrase inhibitors. Following treatment, Dulbecco’s Modified Eagle’s Medium-F12 (100 μl/well) and 20 μl of a 2.5 mg/ml MTT solution were added and cells were incubated for 1 h at 37 °C. The used cell density was 5 × 103 cells/200 μl/well in 96-well microtiter plates. After aspiration of the supernatants, the MTT-formazan crystals formed by metabolically active cells were dissolved in dimethyl sulphoxide (100 μl/well) and absorbance was measured at 540 nm and 690 nm in Varioskan LUX™ Multimode Microplate Reader (Thermo Scientific, Waltham, MA). Values measured at 540 nm were subtracted for background correction at 690 nm, and the data were plotted as a percent of control untreated samples.
3. Results and discussion
3.1. Chemistry
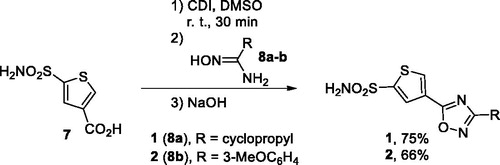
Compounds 1 and 2 were synthesised from common precursor − 5-sulphamoylthiophene-3-carboxylic acid (7) as described previously.Citation5 Carboxylic acid 7 was activated by treatment with CDI and the resulting imidazolide was reacted with amidoximes 8a-b. Addition of solid NaOH formed a superbase solution in DMSO which triggered cyclodehydration yielding compounds 1 and 2 in good yields (Scheme 1).
Scheme 1. Preparation of compounds 1 and 2.
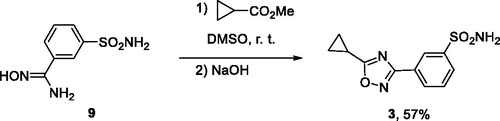
Isomeric 1,2,4-oxadiazole 3 was prepared also as described previously.Citation5 Amidoxime 9 was acylated with methyl cyclopropanecarboxylate in DMSO whereupon addition of solid NaOH, again, led to cyclodehydration to give compound 3 in 57% yield (Scheme 2).
Scheme 2. Preparation of compound 3.
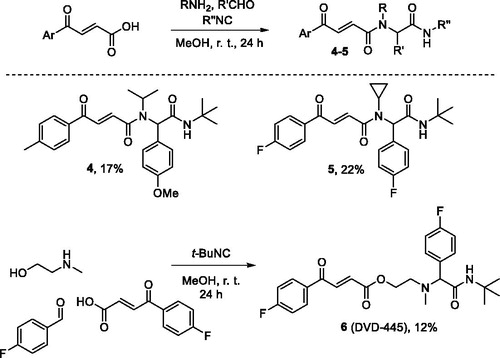
Diamides 4 and 5 were prepared via the traditional Ugi reaction. However, employing secondary b-(methylamino)ethanol as the amine component in the preparation of compound 6 (DVD-445) produced a different, amide ester scaffold (Scheme 3).Citation12
Scheme 3. Preparation of compounds 4–6.
For the synthesis of UMA/primary sulphonamide ZBG hybrid 10, the following synthetic strategy was adopted. KnownCitation14 sulphonyl chloride 11 was converted to bis-DMB-protected (DMB = 2,4-dimethoxybenzyl) aldehyde 12. The latter was involved in the Ugi reaction with cyclopropylamine, tert-butyl isocyanide, and p-toluoyl acrylic acid conducted at ambient temperature in methanol to give adduct 13 which was used in the next step without further purification. Removal of the DMB protecting groups with TFA gave the target UMA/ZBG hybrid 10 in modest yield (Scheme 4).
Scheme 4. Preparation of compound 10.
3.2. Biological evaluation
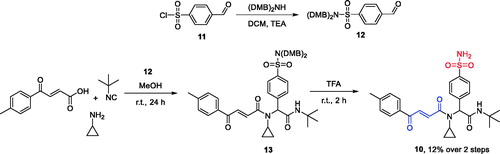
Initially, compounds 1–3 (100 μM) and 4–5 (1 μM) were tested as single agents against pancreas ductal adenocarcinoma cell line (PANC-1) to establish the basal cytotoxicity levels of the two groups of compounds exerting their cytotoxicity via entirely different mechanisms (hCA IX/XII inhibition and TrxR inhibition, respectively). As the result, only compounds 2 and 4 had a pronounced antiproliferative effect reducing the cell viability by >50% and >40%, respectively while the other four compounds (1, 3, 5–6) were only marginally cytotoxic at these concentrations ().
Figure 1. Cytotoxicity of compounds 1–3 (100 μM) and 4–6 (1 μM) as single agents against PANC-1 cell line.
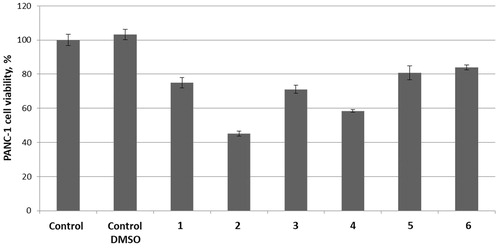
Next, we proceeded to text carbonic anhydrase inhibitors 1–3 (100 μM) in combination with thioredoxin reductase inhibitors 4–6 (1 μM). To our delight, in all cases, TrxR inhibitor 4 produced a strong potentiation of the carbonic anhydrase inhibitor’s antiproliferative activity. Addition of TrxR inhibitors 5 and 6 seemed to make little difference for the antiproliferative effect of carbonic anhydrase inhibitors 1 and 3. However, all three TrxR inhibitors 4–6 noticeably potentiated the cytotoxicity of carbonic anhydrase inhibitor 2 ().
Figure 2. Cytotoxicity towards PANC-1 cell line of carbonic anhydrase inhibitors 1 (A), 2 (B) and 3 (C) (100 μM) tested in combination with 1 μM concentrations of TrxR inhibitors 4, 5, or 6.
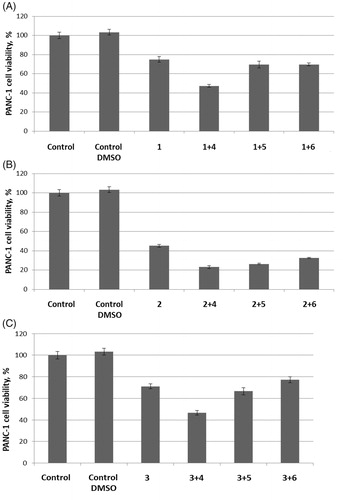
Encouraged by these findings, we also tested UMA/ZBG hybrid 10 against the same cell line. Pre-emptively expecting higher cytotoxicity of this compound compared to 1–3 and 4–5 taken as single agents or even in combination with each other, we tested compound 10 in the 1 − 100 μM concentration range. On increasing the concentration of compound 10 from 1 to 10 μM a sharp decrease in the number of viable cells was observed. To confirm this cytotoxic effect being dose-dependent, a full dose–response curve was obtained from which an IC50 value of 1.8 ± 0.4 μM was deduced ().
Figure 3. Cytotoxicity of compound 10 towards PANC-1 cell line.
4. Conclusions
We hypothesised that simultaneous targeting of two cancer cells’ defence mechanism – those against hypoxia and oxidative stress – by cooperative inhibiting carbonic anhydrase IX/XII and thioredoxin reductase, respectively, would have a synergistic effect. This was confirmed by testing several combinations of the previously reported CA IX/XII and TrxR inhibitors. These results led to an idea of combining CA and TrxR inhibitory pharmacophores (a primary sulphonamide zinc-binding groups and a Michael acceptor moiety, respectively) in the structure of a single agent. Testing a pilot compound conforming to this design principle demonstrated that the compound has a pronounced cytotoxic effect against pancreas ductal adenocarcinoma cells PANC-1 (IC50 = 1.8 ± 0.4 μM). This observation validated a new approach to the design of potential anticancer agents. Further studies are underway in our laboratories which are aimed at further investigating the structure-activity relationships of structurally diverse zinc-binding group-containing Michael acceptors as antiproliferative agents and to correlate the phenotypic assay data with the inhibition profile against the isolated recombinant enzymes. The results of these studies will be reported in due course.
Detailed experimental procedures for synthesis, complete characterization data and copies of 1H and 13 C NMR spectra are available online (Supplementary material).
Supplemental Material
Download PDF (399.1 KB)Acknowledgements
We thank the Research Centre for Magnetic Resonance, the Centre for Chemical Analysis and Materials Research of Saint Petersburg State University Research Park for obtaining the analytical data.
Disclosure statement
The authors declare no competing financial interest.
Additional information
Funding
Related Research Data
References
- Stewart TJ, Abrams SI. How tumours escape mass destruction. Oncogene 2008;27:5894–903.
- Schwartz L, Supuran CT, Alfarouk KO. The Warburg effect and the hallmarks of cancer. Anti-Cancer Agents Med Chem 2017;17:164–70.
- Supuran CT. Carbonic anhydrase inhibition and the management of hypoxic tumors. Metabolites 2017;7:48.
- Krasavin M, Shetnev A, Sharonova T, et al. Heterocyclic periphery in the design of carbonic anhydrase inhibitors: 1,2,4-oxadiazol-5-yl benzenesulfonamides as potent and selective inhibitors of cytosolic hCA II and membrane-bound hCA IX isoforms. Bioorg Chem 2018;76:88–97.
- Krasavin M, Shetnev A, Sharonova T, et al. Continued exploration of 1,2,4-oxadiazole periphery for carbonic anhydrase-targeting primary arene sulfonamides: discovery of subnanomolar inhibitors of membrane-bound hCA IX isoform that selectively kill cancer cells in hypoxic environment. Eur J Med Chem 2019;164:92–105.
- Bakulina O, Bannykh A, Jovanović M, et al. Design, synthesis, and biological evaluation of novel derivatives of dithiodiglycolic acid prepared via oxidative coupling of thiols. J Enzyme Inhib Med Chem 2019;34:665–71.
- Arner ES, Holmgren A. The thioredoxin system in cancer. Semin Cancer Biol 2006;16:420–6.
- Zhang J, Li X, Han X, et al. Targeting the thioredoxin system for cancer therapy. Trends Pharmacol Sci 2017;38:794–808.
- Bian M, Fan R, Zhao S, Liu W. Targeting the thioredoxin system as a strategy for cancer therapy. J Med Chem 2019;62:7309–21.
- Zhang B, Zhang J, Peng S, et al. Thioredoxin reductase inhibitors: a patent review. Exp Opin Ther Pat 2017;27:547–56.
- Saccoccia F, Angelucci F, Boumis G, et al. Thioredoxin reductase and its inhibitors. Curr Protein Pept Sci 2014;15:621–46.
- Jovanović M, Zhukovsky D, Podolski-Renić A, et al. Novel electrophilic amides amenable by the Ugi reaction perturb thioredoxin system via thioredoxin reductase 1 (TrxR1) inhibition: identification of DVD-445 as a new lead compound for anticancer therapy. Eur J Med Chem 2019;181:111580.
- Copeland PR. Making sense of nonsense: the evolution of selenocysteine usage in proteins. Genome Biol 2005;6:221.
- Kelly M, Kincaid J, Janagani S. 2-substituted and 4-substituted aryl nitrone compounds. US Patent Application No US20050182060A1, 2005.
- Hubina AV, Pogodaev AA, Sharoyko VV, et al. Self-assembled spin-labeled nanoparticles based on poly(amino acids). React Funct Polym 2016;100:173–80.