
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
A new series of thiobarbituric (thiopyrimidine trione) enamine derivatives and its analogues barbituric acid derivatives was synthesised, characterised, and screen for in vitro evaluation of α-glucosidase enzyme inhibition and anti-glycation activity. This series of compounds were found to inhibit α-glucosidase activity in a reversible mixed-type manner with IC50 between 264.07 ± 1.87 and 448.63 ± 2.46 µM. Molecular docking studies indicated that compounds of 3g, 3i, 3j, and 5 are located close to the active site of α-glucosidase, which may cover the active pocket, thereby inhibiting the binding of the substrate to the enzyme. Thiopyrimidine trione derivatives also inhibited the generation of advanced glycation end-products (AGEs), which cause long-term complications in diabetes. While, compounds 3a–k, 5, and 6 showed significant to moderate anti-glycation activity (IC50 = 31.5 ± 0.81 to 554.76 ± 9.1 µM).
1. Introduction
Diabetes mellitus (DM) is a disease which caused by a breakdown of carbohydrate metabolism, which plays a significant role in the development of long-term diabetic complications. According to the International Diabetes Federation, 693 million people will suffer from this condition by 2045Citation1,Citation2. DM can be categorised into three types: Type I (T1DM); Type II (T2DM); and gestational (GDM). About 80 − 90% of all DM patients are Type II (T2DM). Drug treatments of T2DM aim to decrease hepatic glucose production, enhance insulin action, and boost insulin secretion from β-pancreatic cells, or block α-glycosidase enzyme (carbohydrate digestive enzymes)Citation3–6. Therapeutic in individuals with this disease may lead to various complications, including kidney disease, disorders of the nervous system, leg amputation, heart disease and severe retinopathy up to blindnessCitation7.
Carbohydrate digestive enzymes are found in the brush border of the intestine. They catalyse the breaking down long-chain polysaccharides into absorbable monosaccharide units. Of these enzymes, α-glucosidases, which play a key role in the digestion and absorption of complex carbohydrates, and has emerged as target to maintain postprandial blood glucose control. α-Glucosidase inhibitors currently used to treat T2DM include acarbose (Precose), voglibose, and miglitolCitation8. However, these drugs are associated with several side effects, such as flatulence, stomach-ache, diarrhoea, and liver damageCitation9. Therefore, an increasing interest in exploring new drug candidates for glycosidase inhibition is neededCitation10–12.
Barbituric acid (BA) derivatives have been reported to have potential anti-hypertensiveCitation13, anti-cancerCitation14, anti-convulsantCitation15, anti-inflammatoryCitation16, anti-psychoticCitation17, and antitumor propertiesCitation18–21. Recently, these derivatives have also been reported as anti-diabetic agentsCitation22. On the other hand, thiobarbituric acid (TBA) analogues has been described to exert anti-inflammatoryCitation16,Citation23, immunotropicCitation24, anticonvulsantCitation25, and anti-hypnoticCitation25,Citation26, anti-neoplasticCitation27, and antitumor activitiesCitation28. De Belin et al.Citation29 reported a number of TBA derivatives as inhibitors of hypoxia-inducible factor 1 (HIF-1). Recently, Barakat et al.Citation30 described the synthesis of a new series of diethylammonium salts of aryl substituted TBA derivatives as α-glycosidase inhibitors. Therefore, given the relevance of TBA derivatives in medicinal chemistry, the design of new molecules containing the thiobarbituric moiety is an inspiring goal.
In continuation of our studies on the synthesis of biologically active compoundsCitation22,Citation30,Citation31, herein, we synthesised 1,3-diethylthiobarbiturate enamine derivatives and evaluated their in vitro α-glucosidase inhibitory and anti-glycation activities. In addition, molecular docking studies were performed to study the interactions of the compounds with the catalytic site of the enzyme using acarbose and evaluated their α-glucosidase inhibition capacity and the anti-glycation properties.
2. Results and discussion
2.1. Synthesis of the target compounds
Enamine derivatives 2aCitation32 and 2bCitation33 were prepared by reacting the commercially available compounds, 1,3-diethylthiobarbituric acid 1a or 1,3-dimethylbarbituric acid 1b with DMF in the presence of acetic anhydride as solvent for 2 h at 90 °C to afford 2a and 2b, respectively as a yellow crystalline solid in good yields. Compounds 2aCitation32 and 2bCitation33 were reacted with different amines in ethanol at RT to afford the target products 3a–k and 4a–d, respectively (Scheme 1) in excellent yields and purities, as observed from their spectral data. The reaction of 2a (2 equiv.) or its analogues 2b (2 equiv.) with the commercially available material 4-(aminomethyl)aniline (1 equiv.) under the same conditions described above gave the dimeric products 6 and 5, respectively as shown in Scheme 1. The structures of the products obtained were deduced by 1H- and 13C-NMR spectra (Supplementary material).
Scheme 1. Synthetic route for the synthesis of 3a–k, 4a–d, 5, and 6.
2.2. Biological activity
All the synthesised derivatives of TBA (3a–k), BA (4a–d), and the dimeric analogues 5 and 6 were evaluated for their capacity to inhibit α-glucosidase and protein glycation in vitro in comparison to acarbose (IC50 = 875.75 ± 2.08 µM) and rutin (IC50 = 54.59 ± 2.20 µM), as standard tested compounds ().
Table 1. Result of in vitro α-glucosidase enzyme inhibitor and anti-glycation activities.
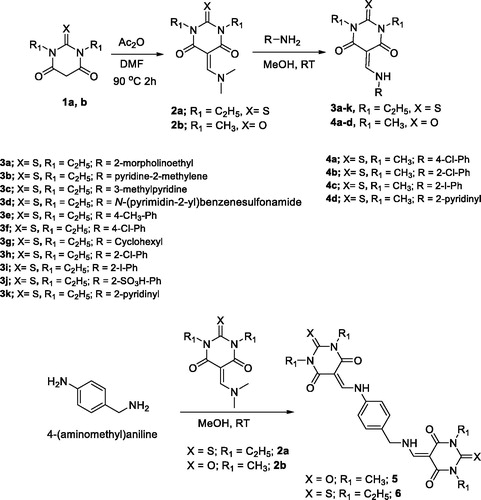
The results summarised in indicated that all the N,N′-dimethylbarbituric-based enamine acid derivatives 4a–d were completely inactive, while compounds 3a–k, 5, and 6 showed moderate to significant activity against protein glycation (IC50 = 554.76 ± 9.1 to 31.5 ± 0.81 µM). The dimeric moiety of TBA 6 via diaminobenezene linkage (IC50 = 31.5 ± 0.81 µM, ) was the most protein glycation inhibitor in this series of compounds, and showed more activity than the standard rutin (IC50 = 54.59 ± 2.20 µM). While, the dimeric analogues of BA 5 (IC50 = 554.76 ± 9.1 µM) was the least active.
On the other hand, substituted phenyl with an electron-withdrawing group such as a chlorine atom (a weak deactivating group) at the ortho position, showed a better anti-glycation activity than the same atom at the para position. Therefore, the change in the position had a remarkable effect on the anti-glycation activityCitation34 (3 h vs 3f, ). Halogen with a higher atomic weight and weaker electron-withdrawing effect, such as iodine at the ortho position, decreased the activity as compared to chlorine at the same position (3i vs 3 h). This observation could be attributed to the negative inductive effectCitation35,Citation36. In contrast, a strong electron-withdrawing group, such as sulphonic acid at ortho position, decreased the activity compared to chlorine and iodine in the ortho position (3j vs 3 h). Electron-donating group such as methyl (a weak donating group) at the para position yielded slightly better and a moderate activity as compared to the chlorine at the same position (3e vs 3f). On the other hand, replacing the 4-methylphenyl 3e by 2-pyridylmethylene 3b or 3-methylpyridyl 3c decreased the anti-glycation activity, and showed a comparable activity to compounds 3g and 3f as shown in . Compound with pyrimidine benzenesulfonamide 3d moiety decreased the activity, which is consistent with the result obtained for 3j with a strong withdrawing group. While, compounds with 2-morpholinoethyl 3a and cyclohexyl 3g moieties showed moderate activity against protein glycation.
The results summarised in indicated, once again, that none of the BA enamine derivatives showed any activity, while 3g, 3i, 3j, and 5 exerted a significant activity against α-glucosidase (IC50 = 264.07 ± 1.87 to 448.63 ± 2.46 µM). Of the series of compounds, thiopyrimidine trione derivative with higher atomic weight halogen, such as iodine at the ortho position, was the most active, exhibiting 3.3-fold higher activity than the standard acarbose. Compounds with a cyclohexyl ring 3g, sulphonic acid 3j, and the dimeric analogue of BA 5 showed twice the activity of the standard drug. The rest of the compounds did not show any activity.
Finally, the most two active compounds from the series are shown in . In conclusion, this work has demonstrated that the core of TBA-based enamine derivatives is a privileged structure for anti-glycation and α-glucosidase inhibition and thus deserves further investigation.
Figure 1. Lead compounds 3i and 6 with promising activities.
2.3. Molecular docking studies
Molecular docking provides significant insight into ligand-protein binding modes and mechanisms. Here, molecular docking studies were carried out to explore the binding modes of TBA derivatives with a notorious α-glucosidase, such as that of Baker’s yeast (Saccharomyces cerevisiae). We used our previously built homology model of α-glucosidase from the template (PDB ID: 3A4A)Citation30. Initially, the 3 D structures of all the ligands were built, protonated, and minimised by means of the MMFF94x force fieldCitation37, and using the molecular operating environment (MOE)Citation38 2018.04. All recently synthesised TBA derivatives and a reference inhibitor (acarbose) were docked into the active site of the receptor using the default parameters in MOE. Each complex was visually analysed for ligand–protein interactions, and their images were prepared using UCSF chimaera softwareCitation39.
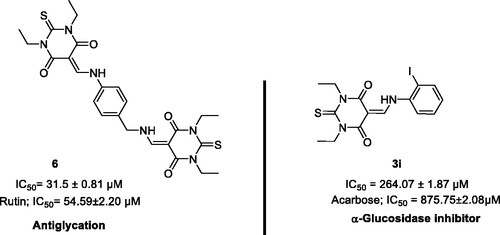
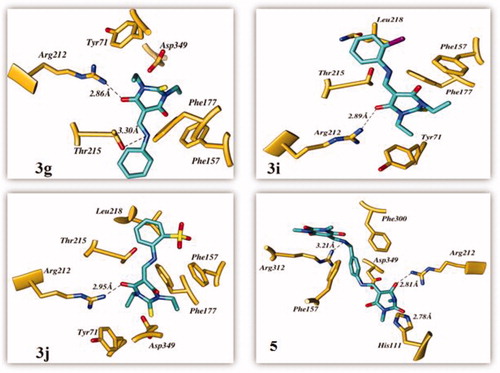
The top ranked conformer of TBA derivatives and standard (acarbose) were selected based on docking score. The docking score of the ligands 3g, 3i, 3j, and 5 and acarbose were −3.081, −4.909, −5.19, −5.642, and −4.382, respectively. The docking study revealed that the acarbose, and all the ligands accommodated into the binding pocket of the C-terminal domain of α-glucosidase. The clustering of standard and synthetic compounds at the allosteric site of the C-terminal domain is shown in .
Figure 2. Binding mode of thiobarbituric acid derivatives into the α-glucosidase binding cavity. For clarity, acarbose is shown in cyan. Compounds 3g, 3i, and 3j are indicated in pink, and 5 in green. The part of the enzyme in the background is shown as surface model.
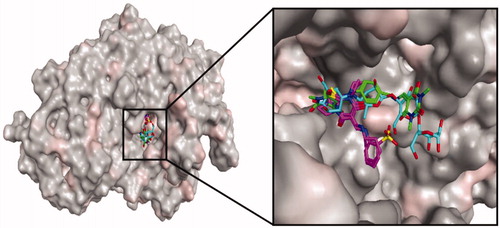
Acarbose occupied a large cavity in the binding sites of α-glucosidase due to its larger size, as compared to the synthetic compounds. The oxygen functionality of acarbose formed two hydrogen bonds with the active site residues, Arg212 and Arg439. Ring structures were involved in the π–π interactions with Phe177, His239, and Pro309. Moreover, residues Glu276, Glu304, and Asp349 interacted hydrophobically with the ligand ().
Figure 3. Interactions of acarbose with crucial residues of α-glucosidase.
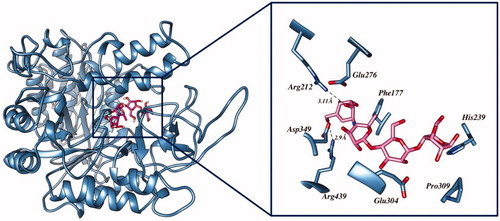
The carbonyl oxygen of the thiobarbituric ring of 3g, 3i, and 3j showed hydrogen bond interactions with crucial residue Arg212. Another hydrogen bond was observed between the nitrogen atoms of 3g with Thr215. These compounds were further stabilised through π–π interactions with the crucial active site residues Tyr71, Phe157, and Phe177. Additionally, π–π interactions were observed with the thiol ring of 3j through Phe177 and Tyr71. In the case of compound 3i, Phe157 was involved in forming halogen–π interactions. Moreover, hydrophobic interactions with the active site residues Phe157, Thr215, Leu218, and Arg349 stabilised these compounds. In the case of 5, the 2,4,6-trione ring-bearing oxygen atom formed hydrogen bonds with His111 and Arg212. Meanwhile, the amine functionality of the ligand also formed a hydrogen bond with residue Arg312. The benzene ring was involved in π–π interactions with Phe157 and Phe300. The hydrophobic interaction with crucial residue Arg349 also contributed to the binding of 5 with α-glucosidase. The interaction diagrams of all the ligands are shown in . The docking results of 5 were in good agreement with experimental results, thereby indicating that it could be a good candidate as α-glucosidase inhibitor.
Figure 4. The predicted binding interactions of compounds 3g, 3i, 3j, and 5 in the active site.
3. Conclusions
Several derivatives of barbitutic and thiobarbituric enamine derivatives were synthesised, characterised, and screened for in vitro evaluation of α-glucosidase enzyme inhibition and anti-glycation activity. The results reveal that the four monomeric compounds 4a–d derived from N,N′-dimethylbarbituric enamine derivatives showed no anti-glycation activity, while compounds derived from N,N′-diethylthiobarbituric enamine derivatives 3a–k exhibited moderate activity against protein glycation with IC50 in the range 70–550 µM. The most potent anti-glycation activity was showed by the dimeric product from N,N′-diethylthiobarbituric enamine 6 with an IC50 of 31.5 µM, while the dimeric analogue of N,N′-dimethylbarbituric enamine 5 showed less activity with an IC50 of 554.8 µM. The reported series of compounds were found to inhibit α-glucosidase activity in a reversible mixed-type manner with IC50 between 264 and 448 µM. The type and position of substituent on phenyl ring (enamine moiety) has great impact on the biological activity. In this regard, the moderate electron-withdrawing group, such as a chlorine atom at the ortho position 3 h showed greater activity compared to the same atom at the para position 3f. On the other hand, the presence of iodine at the ortho position decreased activity compared to chlorine in the same position (3i vs 3 h). The strong electron-withdrawing group, such as sulphonic acid showed decrease in activity compared to weak electron-donating group like methyl (3e).
The dimeric thiobarbituric derivative 6 showed better anti-glycation activity compared with the standard rutin, while thiobarbituric ortho-iodo-enamine derivative 3i showed a positive effect as α-glucosidase inhibitor compared to the standard acarbose.
Molecular docking studies indicated that compounds of 3g, 3i, 3j, and 5 are located close to the active site of α-glucosidase, which may cover the active pocket, thereby inhibiting the binding of the substrate to the enzyme.
This work has confirmed that the core of (thio)barbituric-based enamine derivatives are a privileged structure, because in addition of the previous described biological activity, they have shown activity for anti-glycation and α-glucosidase inhibition.
4. Experimental
4.1. General methods
All melting points were determined using Mel-Temp apparatus and are uncorrected. Thin layer chromatography (TLC) was performed on silica gel (Kiesel gel G, Merck) and spots were detected under UV light at 254 nm. FTIR Spectra were recorded in a KBr matrix on a Bruker Tensor 37 FTIR spectrophotometer. 1H-NMR spectra were recorded with a JEOL 400 MHz, 13C-NMR were recorded using the JEOL spectrophotometers, and the chemical shifts (δ) are given in ppm.
4.2. General procedure for the synthesis of 3a–k, 4a–d, 5, and 6
A solution of 2aCitation32 or 2bCitation33 (1 equiv.) was mixed with different amines (1 equiv.) in MeOH (10 ml) and stirred at room temperature for 10–120 min (TLC 20% EtOAc/n-hexane). The solvent was evaporated slowly, providing the corresponding solid products in excellent yields and purities.
4.2.1. 1,3-Diethyl-5-(((2-morpholinoethyl)amino)methylene)-2-thioxodihydropyrimidine-4,6(1H,5H)-dione (3a)
Compound 3a was synthesised from 2a and 4–(2-aminoethyl)morpholine following the general procedure, affording the product as a yellow powder in 81% yield; mp 135 °C; IR (KBr, cm−1): 3420, 2999, 2960, 2908, 2870, 1624, 1591, 1456; 1H-NMR (CDCl3, δ, ppm): 10.60 (brs, 1H, NH), 8.23 (d, 1H, J = 14.8 Hz, CH=), 4.50 (m, 4H, 2CH2), 3.72 (q, 2H, CH2), 3.54 (m, 4H, 2CH2), 2.60 (m, 2H, CH2), 2.50 (m, 4H, 2CH2), 1.25 (m, 6H, 2CH3); 13C-NMR (CDCl3 δ, ppm): 179.1, 163.0, 161.2, 160.6, 93.0, 66.9, 57.6, 53.6, 47.1, 43.0, 42.3, 12.5, 12.4; LC/MS (ESI): 341.44 [M + 1]+; Anal. Calcd for C15H24N4O3S: C, 52.92; H, 7.11; N, 16.46; Found: C, 53.01; H, 7.25; N, 16.59.
4.2.2. 1,3-Diethyl-5-(((pyridin-2-ylmethyl)amino)methylene)-2-thioxodihydropyrimidine-4,6(1H,5H)-dione (3b)
Compound 3b was synthesised from 2a and 2-picolylamine following the general procedure, affording the product as a pink powder in 83% yield; mp 154 °C; IR (KBr, cm−1): 3215, 3045, 2958, 2908, 2866, 1614, 1598, 1544,1463; 1H-NMR (CDCl3 δ, ppm): 11.01 (brs, 1H, NH), 8.63 (d, 1H, J = 5.2 Hz, Ar-H) , 8.39 (d, 1H, J = 14.0 Hz, CH=), 7.72 (t, 1H, J = 8.8 Hz, Ar-H), 7.27 (d, 1H, J = 8.8 Hz, Ar-H), 7.23 (m, 1H, Ar-H), 4.76 (d, 2H, J = 3.6 Hz, CH2), 4.56 (m, 4H, 2CH2), 1.28–124 (m, 6H, J = 16.4 Hz, 2CH3); 13C-NMR (CDCl3 δ, ppm): 179.1, 163.0, 161.2, 160.7, 154.1, 150.3, 137.3, 123.5, 121.7, 93.5, 55.1, 43.0, 42.3, 12.5, 12.4; LC/MS (ESI): 319.40 [M + 1]+; Anal. Calcd for C15H18N4O2S: C, 56.59; H, 5.70; N, 17.60; Found: C, 56.72; H, 5.81; N, 17.78.
4.2.3. 1,3-Diethyl-5-(((4-methylpyridin-2-yl)amino)methylene)-2-thioxodihydro pyrimidine-4,6(1H,5H)-dione (3c)
Compound 3c was synthesised from 2a and 2-amino-4-picoline following the general procedure, affording the product as a light yellow powder in 87% yield; mp 175 °C; IR (KBr, cm−1) 3215, 3157, 3045, 2958, 2908, 2866, 1614, 1598, 1544, 1463; 1H-NMR (CDCl3 δ, ppm):12.25 (d, 1H, J = 13.2 Hz, NH), 9.40 (d, 1H, J = 13.2 Hz, CH=), 8.27 (d, 1H, J = 5.2 Hz, Ar-H), 6.98 (d, 1H, J = 5.2 Hz, Ar-H), 6.86(s, 1H, Ar-H), 4.55 (m, 4H, 2CH2), 2.38 (s, 3H, CH3), 1.29 (m, 6H, 2CH3); 13C-NMR (CDCl3 δ, ppm):179.1, 163.3, 160.7, 152.5, 150.7, 149.6, 149.0, 122.9, 113.6, 95.8, 43.2, 42.5, 21.2, 12.5, 12.4; LC/MS (ESI): 319.40 [M + 1]+; Anal. Calcd for C15H18N4O2S: C, 56.59; H, 5.70; N, 17.60; Found: C, 56.81; H, 5.78; N, 17.79.
4.2.4. 4-(((1,3-Diethyl-4,6-dioxo-2-thioxotetrahydropyrimidin-5(2H)-ylidene)methyl)amino)-N-(pyrimidin-2-yl)benzenesulfonamide (3d)
Compound 3d was synthesised from 2a and sulphadiazine following the general procedure, affording the product as a yellow powder in 85% yield; mp 204 °C; IR (KBr, cm−1): 3421, 3116, 2958, 2860, 1618, 1591, 1508, 1440; 1H-NMR (DMSO-d6, δ, ppm): 12.20 (d, 1H, J = 14.0 Hz, NH), 8.72 (d, 1H, J = 14.0 Hz, NH), 8.52 (d, 1H, J = 4.4 Hz, CH=), 8.47 (d, 1H, J = 8.8 Hz, Ar-H), 8.0 (d, 1H, J = 8.8 Hz, Ar-H), 7.79 (d, 2H, J = 8.8 Hz, Ar-H), 7.10 (m, 1H, Ar-H), 6.57 (d, 2H, J = 8.8 Hz, Ar-H), 4.42 (m, 4H, 2CH2), 1.21 (m, 6H, 2CH3); 13C-NMR (DMSO-d6, δ, ppm): 178.9, 162.2, 160.5, 158.8, 157.8, 157.3, 154.3, 153.6, 142.4, 130.4, 129.8, 125.4,119.8, 116.1, 112.7, 95.7, 42.9, 42.4, 12.8, 12.7; LC/MS (ESI): 461.53 [M + 1]+; Anal. Calcd for C19H20N6O4S2: C, 49.55; H, 4.38; N, 18.25; Found: C, 49.66; H, 4.50; N, 18.41.
4.2.5. 1,3-Diethyl-2-thioxo-5-((p-tolylamino)methylene)dihydropyrimidine-4,6(1H,5H)-dione (3e)
Compound 3e was synthesised from 2a and 4-methylanline uracil following the general procedure, affording the product as a yellow powder in 89% yield; mp 139 °C; IR (KBr, cm−1): 3448, 3215, 3169, 2953, 2866, 1595, 1570, 1554, 1476, 1435, 1440; 1H-NMR (CDCl3, δ, ppm): 12.32 (d, 1H, J = 13.8 Hz, NH), 8.70 (d, 1H, J = 14.0 Hz, CH=), 7.27 (d, 2H, J = 8.0 Hz, Ar-H), 7.22 (dd, 2H, J = 8.0 Hz, Ar-H), 4.60(m, 4H, 2CH2), 2.36 (s, 3H, CH3), 1.30 (m, 6H, 2CH3); 13C-NMR (CDCl3, δ, ppm): 178.8, 163.2, 160.9, 152.9, 137.3, 135.5, 130.9, 118.2, 94.6, 43.2, 42.5, 12.4, 12.2; LC/MS (ESI): 317.41 [M + 1]+; Anal. Calcd for C16H19N3O2S: C, 60.55; H, 6.03; N, 13.24; Found: C, 60.32; H, 6.00; N, 13.43.
4.2.6. 5-(((4-Chlorophenyl)amino)methylene)-1,3-diethyl-2-thioxodihydropyrimidine-4,6(1H,5H)-dione (3f)
Compound 3f was synthesised from 2a and 4-chloroanline following the general procedure, affording the product as a yellow powder in 78% yield; mp 215 °C; IR (KBr, cm−1): 3302, 2958, 2908, 2866, 1614, 1587, 1545, 1504, 1438, 1409; 1H-NMR (CDCl3, δ, ppm): 12.32 (d, 1H, J = 14.0 Hz, NH), 8.65 (d, 1H, J = 14.0 Hz, CH=), 7.38 (d, 2H, J = 8.0 Hz, Ar-H), 7.25 (d, 2H, J = 8.8 Hz, Ar-H), 4.55 (m, 4H, 2CH2), 1.30 (t, 6H, J = 7.9 Hz, 2CH3); 13C-NMR (CDCl3, δ, ppm): 178.9, 163.2, 160.8, 152.8, 136.6, 132.6, 130.4, 119.4, 95.3, 43.2, 42.5, 12.5, 12.3; LC/MS (ESI): 338.82 [M + 1]+; Anal. Calcd for C15H16ClN3O2S: C, 53.33; H, 4.77; N, 12.44; Found: C, 53.54; H, 4.80; N, 12.63.
4.2.7. 5-((Cyclohexylamino)methylene)-1,3-diethyl-2-thioxodihydropyrimidine-4,6(1H,5H)-dione (3g)
Compound 3g was synthesised from 2a and cyclohexylamine following the general procedure, affording the product as a white powder in 84% yield; mp 105 °C; IR (KBr, cm−1): 3302, 2958, 2908, 2866, 1614, 1587, 1545, 1504, 1438, 1409; 1H-NMR (DMSO-d6, δ, ppm): 10.61 (brs, 1H, NH), 8.25 (d, 1H, J = 15.6 Hz, CH=), 4.52 (m, 4H, 2CH2), 3.40 (m, 1H, CH), 1.98 (m, 2H, CH2), 1.83 (m, 2H, CH2), 1.78 (m, 2H, CH2), 1.47 (m, 2H, 2CH2), 1.28 (m, 2H, 2CH3); 13C-NMR (DMSO-d6, δ, ppm): 179.0, 163.2, 161.3, 158.4, 92.6, 59.3, 42.9, 42.3, 33.5, 24.9, 24.3, 12.5, 12.4; LC/MS (ESI): 310.43 [M + 1]+; Anal. Calcd for C15H23N3O2S: C, 58.23; H, 7.49; N, 13.58; Found: C, 58.39; H, 7.53; N, 13.78.
4.2.8. 5-(((2-Chlorophenyl)amino)methylene)-1,3-diethyl-2-thioxodihydropyrimidine-4,6(1H,5H)-dione (3 h)
Compound 3h was synthesised from 2a and 2-chloroanline following the general procedure, affording the product as a yellow powder in 89% yield; mp 160 °C; IR (KBr, cm−1): 3302, 2958, 2908, 2866, 1614, 1587, 1545, 1504, 1438, 1409; 1H-NMR (CDCl3, δ, ppm): 12.62 (d, 1H, J = 13.2 Hz, NH), 8.73 (d, 1H, J = 14.0 Hz, CH=), 7.49 (m, 2H, Ar-H), 7.39 (t, 1H, J = 7.9 Hz, Ar-H), 7.21 (t, 1H, J = 8.0 Hz, Ar-H), 4.56 (m, 4H, 2CH2), 1.30(m, 6H, 2CH3); 13C-NMR (CDCl3, δ, ppm): 178.9, 163.0, 160.9, 152.1, 135.3, 130.7, 128.5, 127.3, 125.0, 116.9, 96.1, 43.2, 42.6, 12.5, 12.4; LC/MS (ESI): 338.82 [M + 1]+; Anal. Calcd for C15H16ClN3O2S: C, 53.33; H, 4.77; N, 12.44; Found: C, 53.53; H, 4.92; N, 12.60.
4.2.9. 1,3-Diethyl-5-(((2-iodophenyl)amino)methylene)-2-thioxodihydropyrimidine-4,6(1H,5H)-dione (3i)
Compound 3i was synthesised from 2a and 2-iodoanline following the general procedure, affording the product as a yellow powder in 83% yield; mp 175 °C; IR (KBr, cm−1): 3302, 2958, 2908, 2866, 1614, 1587, 1545, 1504, 1438, 1409; 1H-NMR (CDCl3, δ, ppm): 12.42 (d, 1H, J = 13.2 Hz, NH), 8.67 (d, 1H, J = 14.0 Hz, CH=), 791 (d, 1H, J = 7.2 Hz, Ar-H), 7.45 (t, 1H, J = 7.9 Hz, Ar-H), 7.36 (d, 1H, J = 8.0 Hz, Ar-H), 6.98 (t, 1H, J = 7.2 Hz, Ar-H), 4.53 (m, 4H, 2CH2), 1.30 (m, 6H, 2CH3); 13C-NMR (CDCl3, δ, ppm): 179.0, 162.8, 160.9, 153.0, 139.9, 130.0, 128.2,125.1, 117.7, 95.9, 89.9, 43.2, 42.5, 12.6, 12.4; LC/MS (ESI): 430.28 [M + 1]+; Anal. Calcd for C15H16IN3O2S: C, 41.97; H, 3.76; N, 9.79; Found: C, 41.88; H, 3.81; N, 10.01.
4.2.10. 2-(((1,3-Diethyl-4,6-dioxo-2-thioxotetrahydropyrimidin-5(2H)-ylidene)methyl) amino)benzenesulfonic acid (3j)
Compound 3j was synthesised from 2a and 2-aminobenzenesulfonic acid following the general procedure, affording the product as a yellow powder in 80% yield; mp 243 °C; IR (KBr, cm−1): 3302, 2958, 2908, 2866, 1614, 1587, 1545, 1504, 1438, 1409; 1H-NMR (DMSO-d6, δ, ppm): 13.01 (d, 1H, J = 14.8 Hz, NH), 8.62 (d, 1H, J = 14.4 Hz, CH=), 7.78 (d, 1H, J = 7.9 Hz, Ar-H), 7.64 (d, 1H, J = 8.0 Hz, Ar-H), 7.49 (t, 1H, J = 8.6 Hz, Ar-H), 7.30 (t, 1H, J = 8.4 Hz, Ar-H), 4.43 (m, 4H, 2CH2), 1.20 (m, 6H, 2CH3); 13C-NMR (DMSO-d6, δ, ppm): 178.9, 161.1, 160.9, 153.9, 138.6, 135.9, 130.7, 128.1, 124.6, 118.3, 95.3, 42.9, 42.2, 12.8. LC/MS (ESI): 384.44 [M + 1]+; Anal. Calcd for C15H17N3O5S2: C, 46.99; H, 4.47; N, 10.96; Found: C, 47.09; H, 4.53; N, 11.13.
4.2.11. 1,3-diethyl-5-((pyridin-2-ylamino)methylene)-2-thioxodihydropyrimidine-4,6(1H,5H)-dione (3k)
Compound 3k was synthesised from 2a and pyridin-2-amine following the general procedure, affording the product as a yellow powder in 83% yield; mp 243 °C; IR (KBr, cm−1): 3302, 2958, 2908, 2866, 1614, 1587, 1545, 1504, 1438, 1409; 1H-NMR (DMSO-d6, δ, ppm): 13.03 (d, 1H, J = 14.8 Hz, NH), 8.62 (d, 1H, J = 14.4 Hz, CH=), 7.76 (d, 1H, J = 7.6 Hz, Ar-H), 7.63 (d, 1H, J = 8.8 Hz, Ar-H), 7.49 (t, 1H, J = 9.6 Hz, Ar-H), 7.28 (t, 1H, J = 8.4 Hz, Ar-H), 4.46 (m, 4H, 2CH2), 1.22 (m, 6H, 2CH3); 13C-NMR (DMSO-d6, δ, ppm): 178.4, 160.5, 159.2, 138.1, 135.6, 130.7, 118.3, 94.6, 42.4, 41.7, 12.2. LC/MS (ESI): 305.35 [M + 1]+; Anal. Calcd for C14H16N4O2S: C, 55.25; H, 5.30; N, 18.41; Found: C, 55.38; H, 5.41; N, 18.59.
4.2.12. 5-(((4-Chlorophenyl)amino)methylene)-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione (4a)
Compound 4a was synthesised from 2b and 4-chloroanline following the general procedure, affording the product as a white powder in 87% yield; mp 197 °C; IR (KBr, cm−1): 3302, 2958, 2908, 2866, 1614, 1587, 1545, 1504, 1438, 1409;1H-NMR (CDCl3, δ, ppm):12.00 (d, 1H, J = 13.6 Hz, NH), 8.60 (d, 1H, J = 14.0 Hz, CH=), 7.36 (d, 2H, J = 8.8 Hz, Ar-H), 7.16 (d, 2H, J = 8.8 Hz, Ar-H), 3.32 (s, 6H, 2CH3); 13C-NMR (CDCl3, δ, ppm): 165.1, 162.6, 151.8, 136.8, 132.1, 130.3,119.2, 93.4, 28.1, 27.4; LC/MS (ESI): 294.71 [M + 1]+; Anal. for C13H12ClN3O3; Calcd: C, 53.16; H, 4.12; N, 14.31; Found: C, 53.15; H, 4.12; N, 14.33.
4.2.13. 5-(((2-Chlorophenyl)amino)methylene)-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione (4b)
Compound 4b was synthesised from 2b and 2-chloroanline following the general procedure, affording the product as a white powder in 87% yield; mp 202 °C; IR (KBr, cm−1): 3637, 3423, 3197, 2960, 2935, 1583, 1570, 1510, 1462; 1H-NMR (CDCl3, δ, ppm): 12.44 (d, 1H, J = 12.8 Hz, NH), 8.73 (d, 1H, J = 13.2 Hz, CH=), 7.48 (m, 2H, Ar-H), 7.36 (t, 1H, J = 7.2 Hz, Ar-H), 7.18 (m, 1H, Ar-H), 3.38 (s, 3H, CH3), 3.36 (s, 3H, CH3); 13C-NMR (CDCl3, δ, ppm): 164.9, 162.7, 151.9, 151.1, 135.4, 130.6, 128.5, 126.9, 124.7, 116.6, 94.4, 28.2, 27.5; LC/MS (ESI): 294.71 [M + 1]+; Anal. for C13H12ClN3O3; Calcd: C, 53.16; H, 4.12; N, 14.31; Found: C, 53.17; H, 4.11; N, 14.29.
4.2.14. 5-(((2-Iodophenyl)amino)methylene)-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione (4c)
Compound 4c was synthesised from 2b and 2-iodoanline following the general procedure, affording the product as a white powder in 89% yield; mp 285 °C; IR (KBr, cm−1): 3086, 3053, 2953, 2885,1598, 1560, 1516, 1463, 1371; 1H-NMR (CDCl3, δ, ppm): 12.77 (d, 1H, J = 14.8 Hz, NH), 8.55 (d, 1H, J = 14.0 Hz, CH=), 7.77 (d, 1H, J = 7.2 Hz, Ar-H), 7.57 (d, 1H, J = 8.0 Hz, Ar-H), 7.46 (t, 1H, J = 7.6 Hz, Ar-H), 7.26 (t, 1H, J = 7.2 Hz, Ar-H), 3.20 (s, 6H, 2CH3); 13C-NMR (CDCl3, δ, ppm):163.3, 162.9, 152.2, 151.9, 138.3, 136.4, 131.3, 128.1, 125.9, 118.5, 93.9, 28.2, 27.6; LC/MS (ESI): 386.16 [M + 1]+; Anal. for C13H12IN3O3; Calcd: C, 40.54; H, 3.14; N, 10.91; Found: C, 40.55; H, 3.15; N, 10.90.
4.2.15. 1,3-Dimethyl-5-((pyridin-2-ylamino)methylene)pyrimidine-2,4,6(1H,3H,5H)-trione (4d)
Compound 4d was synthesised from 2b and 2-aminopyridine following the general procedure, affording the product as a white powder in 85% yield; mp 287–290 °C; IR (KBr, cm−1): 3420, 2999, 2960, 2908, 2870, 1624, 1591, 1456; 1H-NMR (CDCl3, δ, ppm): 12.11 (brs, 1H, NH), 9.41 (d, 1H, J = 13.2 Hz, CH=), 8.43 (d, 1H, J = 4.4 Hz, Ar-H), 7.75 (dd, 1H, J = 8.0, 2.4 Hz, Ar-H), 7.16 (t, 1H, J = 7.6, Hz, Ar-H), 6.99 (d, 2H, J = 8.0 Hz, Ar-H), 3.36 (s, 6H, 2CH3); 13C-NMR (CDCl3, δ, ppm): 165.3, 162.6, 152.0, 151.3, 149.7, 149.3, 138.9, 121.4, 112.7, 94.3, 28.2, 27.5; LC/MS (ESI): 261.25 [M + 1]+; Anal. for C12H12N4O3; Calcd: C, 55.38; H, 4.65; N, 21.53; Found: C, 55.38; H, 4.64; N, 21.51.
4.2.16. 5-(((4-(((1,3-Dimethyl-2,4,6-trioxotetrahydropyrimidin-5(2H)-ylidene)methyl) amino)benzyl)amino)methylene)-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione (5)
Compound 5 was synthesised from 2b (2 equiv.) and 4-aminobenzylamine (1 equiv.) following the general procedure, affording the product as a white powder in 90% yield; mp 195 °C; IR (KBr, cm−1): 3302, 2958, 2908, 2866, 1614, 1587, 1545, 1504, 1438, 1409; 1H-NMR (DMSO-d6, δ, ppm): 10.48 (brs, 2H, NH), 8.25(d, 2H, J = 14.0 Hz, 2CH=), 7.01 (d, 2H, J = 8.0 Hz, Ar-H), 6.55 (d, 2H, J = 9.0 Hz, Ar-H), 4.50 (d, J = 6.4 Hz, 2H, CH2), 3.12 (s, 12H, 4CH3); 13C-NMR (DMSO-d6, δ, ppm): 168.1, 165.5, 160.6, 151.9, 134.4, 118.7, 145.2, 95.3, 28.1, 27.4; LC/MS (ESI): 455.44 [M + 1]+; Anal. Calcd for C21H22N6O6: C, 55.50; H, 4.88; N, 18.49; Found: C, 55.65; H, 4.93; N, 18.70.
4.2.17. 5-(((4-(((1,3-Diethyl-4,6-dioxo-2-thioxotetrahydropyrimidin-5(2H)-ylidene)methyl)amino)benzyl)amino)methylene)-1,3-diethyl-2-thioxodihydropyrimidine-4,6(1H,5H)-dione (6)
Compound 6 was synthesised from 2a (2 equiv.) and 4-aminobenzylamine (1 equiv.) following the general procedure, affording the product as a yellow powder in 86% yield; mp 247 °C; IR (KBr, cm−1): 3302, 2958, 2908, 2866, 1614, 1587, 1545, 1504, 1438, 1409; 1H-NMR (CDCl3, δ, ppm): 12.34 (d, 1H, J = 13.6 Hz, NH), 10.77 (m, 1H, NH), 8.68 (d, 1H, J = 14.0 Hz, CH=), 8.29 (d, 1H, J = 13.6 Hz, CH=), 7.39 (m, 4H, Ar-H), 4.65 (d, 2H, J = 6.4 Hz, CH2), 4.46 (m, 8H, 4CH2), 1.35 (m, 12H, 4CH3); 13C-NMR (CDCl3, δ, ppm): 179.0, 178.8, 163.3, 163.2, 161.0, 160.7, 160.4, 152.7, 138.4, 133.6, 129.7, 129.6, 118.9, 118.8, 95.4, 93.6, 53.6, 43.2, 43.0, 42.5, 42.4, 12.5, 12.4, 12.3, 12.2; LC/MS (ESI): 543.67 [M + 1]+; Anal. Calcd for C25H30N6O4S2: C, 55.33; H, 5.57; N, 15.49; Found: C, 55.54; H, 5.69; N, 15.66.
4.3. Protocol for in vitro α-glucosidase inhibition assay
The assay protocol for was performed spectrophotometrically following the reported methodCitation22, where α-glucosidase from S. cerevisiae (G0660-750UN, Sigma Aldrich) was dissolved in phosphate buffer (pH 6.8, 50 mM). Test compounds were dissolved in 70% DMSO. 20 μL of test sample, 20 μL of enzyme and 135 μL of buffer were added to 96-well plates and incubated for 15 min at 37 °C. After incubation, 25 μL of p-nitrophenyl-α-d-glucopyranoside (0.7 mM, Sigma Aldrich) was added and changes in absorbance were monitored for 30 min at 400 nm. The test compound was replaced by DMSO (7.5% final) as control. Acarbose (Acarbose, Sigma Aldrich) was used as a standard inhibitor.
4.4. Protocol for anti-glycation assayCitation40,Citation41
The assay was performed following Gutierrez. R. M. P, with slight modifications. In brief, Bovine Serum Albumin solution (10 mg/mL) was prepared in 100 mM of phosphate buffer pH 7.4 containing 3 mM sodium azide as antimicrobial agent. A methylglyoxal solution of 14 mM was also prepared in the same buffer. 1-mM concentrations of the test compounds and standard inhibitor were prepared in dimethyl sulfoxide (DMSO). Each well of a 96-well plate contained 20 µL of inhibitor, 50 µL of BSA, 50 µL of methylglyoxal and 80 µL of phosphate buffer, while the control contained 20 µl of DMSO instead of test compound. The total reaction volume was 200 µL. The reaction mixture was then incubated for 9 days at 37 ° C. After incubation, each sample was examined for the development of specific fluorescence (excitation 330 nm; emission 420 nm) against a blank on a microplate reader (Spectramax M2 Devices, CA, USA).
4.5. Calculation of inhibitory activity
The percentage inhibition of advanced glycation end (AGEs) products formation by the test sample versus control was calculated using the following formula:
Supplemental Material
Download PDF (2.5 MB)Acknowledgements
The authors would like to extend their sincere appreciation to the Deanship of Scientific Research at King Saud University for providing funding to this research group (RGP-234, Saudi Arabia).
Disclosure statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Related Research Data
References
- NCD Risk Factor Collaboration (NCD-RisC). Worldwide trends in diabetes since 1980: a pooled analysis of 751 population-based studies with 4*4 million participants. Lancet 2016;387:1513–30.
- Cho NH, Shaw JE, Karuranga S, et al. IDF diabetes Atlas: global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res Clin Pract 2018;138:271–81.
- Chiarelli F, Marzio DD. Peroxisome-proliferator-activated receptor-g agonists and diabetes: current evidence and future perspectives. Vasc. Health Risk Manage 2008;4:297–304.
- Fuhlendorff J, Rorsman P, Kofod H, et al. Stimulation of insulin release by repaglinide and glibenclamide involves both common and distinct processes. Diabetes 1998;47:345–51.
- Gloster TM, Davies GJ. Glycosidase inhibition: assessing mimicry of the transition state. Org Biomol Chem 2010;8:305–20.
- He Z-X, Zhou Z-W, Yang Y, et al. Overview of clinically approved oral antidiabetic agents for the treatment of type 2 diabetes mellitus. Clin Exp Pharmacol Physiol 2015;42:125–38.
- Deshpande AD, Harris-Hayes M, Schootman M. Epidemiology of diabetes and diabetes-related complications. Phys Ther 2008;88:1254–64.
- Sonama V, Kakkar R. Isatin and its derivatives: a survey of recent syntheses, reactions, and applications. Med Chem Commun 2019;10:351–36.
- Chai TT, Kwek MT, Ong HC, Wong FC. Water fraction of edible medicinal fern Stenochlaena palustrisis a potenta-glucosidase inhibitor with concurrent antioxidant activity. Food Chem 2015;186:26–31.
- de Melo EB, Gomes A, da S, Carvalho I. α- and β Glucosidase inhibitors: chemical structure and biological activity. Tetrahedron 2006;62:10277–302.
- Ghani U. Re-exploring promising a-glucosidase inhibitors for the potential development into oral anti-diabetic drugs: finding needle in the haystack. Eur J Med Chem 2015;103:133–62.
- Saeedi M, Hadjiakhondi A, Nabavi SM, Manayi A. Heterocyclic compounds: effective a-amylase and a-glucosidase inhibitors. Curr Top Med Chem 2016;17:428–40.
- Min-Kyu P, Yun-Hee R, Hyo-Jung L, et al. Antiplatelet and antithrombotic activity of indole-3-carbinol in vitro and in vivo. Phytother Res 2008;22:58–64.
- Penthala NR, Yerramreddy TR, Madadi NR, Crooks PA. Synthesis and in vitro evaluation of N-alkyl-3-hydroxy-3-(2-imino-3-methyl-5-oxoimidazolidin-4-yl)indolin-2-one analogs as potential anticancer agents. Bioorg Med Chem Lett 2010;20:4468–71.
- Goodman LS, Gilman A. The pharmacological basis of therapeutics. New Delhi: McGraw-Hill; 1991:358–360.
- Radwan MAA, Ragab EA, Sabry NM, El-Shenawy SM. Synthesis and biological evaluation of new 3-substituted indole derivatives as potential anti-inflammatory and analgesic agents. Bioorg Med Chem 2007;15:3832–41.
- Madadi NR, Penthala NR, Brents LK, et al. Evaluation of (Z)-2-((1-benzyl-1H-indol-3-yl)methylene)quinuclidin-3-one analogues as novel, high affinity ligands for CB1 and CB2cannabinoid receptors. Bioorg Med Chem Lett 2013;23:2019–21.
- Maquoi E, Sounni NE, Devy L, et al. Anti-invasive, antitumoral, and antiangiogenic efficacy of a pyrimidine-2,4,6-trione derivative, an orally active and selective matrix metalloproteinases inhibitor. Clin Cancer Res 2004;10:4038–47.
- Milanesi E, Costantini P, Gambalunga A, et al. The mitochondrial effects of small organic ligands of BCL-2 sensitization of DCL-2-overexpressing cells to apoptosis by a pyrimidine-2,4,6-trione derivatives. J Biol Chem 2006;28:10066–72.
- Duan JJ-W, Lu ZH, Wasserman ZR, et al. Non-hydroxamate 5-phenylpyrimidine-2,4,6-trione derivatives as selective inhibitors of tumor necrosis factor alpha converting enzyme. Bioorg Med Chem Lett 2005;15:2970–3.
- Hong TJ, Park H, Kim YJ, et al. Identification of new Hsp90 inhibitors by structure-based virtual screening. Bioorg Med Chem Lett 2009;19:4839–53.
- Barakat A, Soliman SM, Al-Majid AM, et al. Synthesis and structure investigation of novel pyrimidine-2,4,6-trione derivatives of highly potential biological activity as anti-diabetic agent. J Mol Struct 2015;1098:365–76.
- Badawey E, El-Ashmawey I. Nonsteroidal antiinflammatory agents-part 1: antiinflammatory, analgesic and antipyretic activity of some new 1-(pyrimidin-2-yl)-3-pyrazolin-5-ones and 2-(pyrimidin-2-yl)-l,2,4,5,6,7-hexahydro-3H-indazol-3-ones. Eur J Med Chem 1998;33:349–61.
- Goodman LS, Gilman A. The pharmacological basis of therapeutics. New Delhi: McGraw-Hill; 1991.
- Andrews G. Medical pharmacology. Saint Louis: The CV Mosby Company; 1976.
- Foye WO. Principles of medicinal chemistry. London: Lea & Febiger; 1989.
- Guerin DJ, Mazeas D, Musale MS, et al. Uridine phosphorylase inhibitors: chemical modification of benzyloxybenzyl-barbituric acid and its effects on urdpase inhibition. Bioorg Med Chem Lett 1999;9:1477–80.
- Rayburn ER, Ezell SJ, Zhang R. Anti-inflammatory agents for cancer therapy. Mol Cell Pharmacol 2009;1:29–43.
- De Belin JY, Martin MR, Fin PW, et al. Barbituric acid analogs as therapeutic agents. WO 01/93841 A2; 2001.
- Barakat A, Ali M, Al-Majid AM, Yousuf S, et al. Synthesis of thiobarbituric acid derivatives: in vitro α-glucosidase inhibition and molecular docking studies. Bioorg Chem 2017;75:99–105.
- Barakat A, Islam MS, Al-Majid AM, et al. Synthesis, in vitro biological activities and in silico study of dihydropyrimidines derivatives. Bioorg Med Chem 2015;23:6740–8.
- Kulinich AV, Derevyanko NA, Ishchenko AA. Synthesis and spectral properties of cyanine dyes-derivatives of 10,10-dimethyl-7,8,9. 10-tetrahydro-6H-Pyrido[1,2-a]Indolium. J Photochem Photobiol 2008;198:119–25.
- Gorobets NY, Yousefi BH, Belaj F, Kappe CO. Rapid microwave-assisted solution phase synthesis of substituted 2-pyridone libraries. Tetrahedron 2004;60:8633–44.
- Gong Z, Xie Z, Qiu J, Wang G. Synthesis, biological evaluation and molecular docking study of 2-substituted-4,6-diarylpyrimidines as α-glucosidase inhibitors. Molecules 2017;22:1865.
- Khan KM, Khan M, Karim A, et al. Xanthine oxidase inhibition by 5-aryledene N,N’-dimethylbarbituric acid derivatives. J Chem Soc Pak 2013;35:495–8.
- AlSharif AM. Synthesis of new pyrimidinone derivatives and their respective biological activity assessment. Oriental J Chem 2019;35:658–67.
- Halgren TA. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J Comput Chem 1996;17:490–519.
- Molecular Operating Environment (MOE), 2013.08; Chemical Computing Group ULC, 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2018.” 2018. 2018.
- Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput Chem 2004;25:1605–12.
- Daroux M, Prevost G, Maillard-Lefebvre H, et al. Advance glycation end-products: Implications for diabetic and non-diabetic nephropathies. Diabetes Metab 2010;36:1–10.
- Gutierrez R. Inhibition of advanced glycation end-product formation by Origanum majorana L. In vitro and in streptozotocin-induced diabetic rats. Evid Based Compl Alter Med 2012;2012:1–8.