
Abstract
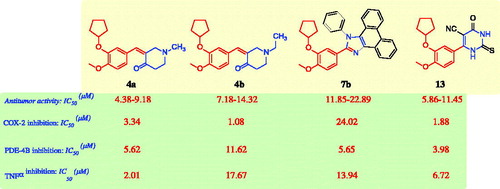
A series of 24 compounds was synthesised based on a 2-cyclopentyloxyanisole scaffold 3–14 and their in vitro antitumor activity was evaluated. Compounds 4a, 4b, 6b, 7b, 13, and 14 had the most potent antitumor activity (IC50 range: 5.13–17.95 μM), compared to those of the reference drugs celecoxib, afatinib, and doxorubicin. The most active derivatives 4a, 4b, 7b, and 13 were evaluated for their inhibitory activity against COX-2, PDE4B, and TNF-α. Compounds 4a and 13 potently inhibited TNF-α (IC50 values: 2.01 and 6.72 μM, respectively) compared with celecoxib (IC50=6.44 μM). Compounds 4b and 13 potently inhibited COX-2 (IC50 values: 1.08 and 1.88 μM, respectively) comparable to that of celecoxib (IC50=0.68 μM). Compounds 4a, 7b, and 13 inhibited PDE4B (IC50 values: 5.62, 5.65, and 3.98 μM, respectively) compared with the reference drug roflumilast (IC50=1.55 μM). The molecular docking of compounds 4b and 13 with the COX-2 and PDE4B binding pockets was studied.
Antitumor activity of new synthesized cyclopentyloxyanisole scaffold was evaluated.
The powerful antitumor 4a, 4b, 6b, 7b & 13 were assessed as COX-2, PDE4B & TNF-α inhibitors.
Compounds 4a, 7b, and 13 exhibited COX-2, PDE4B, and TNF-α inhibition.
Compounds 4b and 13 showed strong interactions at the COX-2 and PDE4B binding pockets.
Highlights
Graphical Abstract
Introduction
Cancer, the uncontrolled growth of cells that invade adjacent healthy tissues, is the most fatal disease in the worldCitation1. Therefore, the design and synthesis of new molecules with promising and potential antitumor activity is of great importanceCitation1–10. The clinical use of drug combinations has led to various side effects, whereas the use of single molecules that target multiple molecular mechanisms is the currently preferred therapeutic strategy and is under investigation by medicinal chemistsCitation11–13.
Cyclooxygenase-2 isoenzyme (COX-2) inhibitors, such as celecoxib (A; ), have been reported to have antitumor activitiesCitation8,Citation14,Citation15. The COX-2 isoenzyme is overexpressed in numerous human cancers, such as breast, lung, hepatocellular, gastric, ovarian, prostate, and colon cancersCitation8,Citation14–16. There are two anticancer mechanisms associated with COX-2 inhibition: the first, termed the COX-2-dependent anticancer mechanism, is selective inhibition with the restoration of normal apoptosis; the second is the COX-2-independent mechanism, which occurs through the induction of apoptosis or inhibition of cell proliferationCitation17. These results indicated that COX-2 enzyme inhibition was an interesting molecular target for the treatment of cancerCitation8,Citation14–17. In addition, phosphodiesterase isoenzyme 4 (PDE4) is responsible for inactivation and hydrolysis of 3′,5′-cyclic adenosine monophosphate (cAMP) and subdivided into four subtypes, PDE4A to PDE4DCitation18–20. The secondary messenger cAMP is important for various cellular processes such as proliferation, growth, migration, differentiation, and apoptosisCitation18–20. These isoenzymes of cAMP-PDE expressed in several cancer cells, such as colon cancer, melanoma, prostate cancer, myeloma, pancreatic cancer, B cell lymphoma, kidney cancer, and lung cancerCitation18–27. Recently, it was reported that PDE4 inhibitors possess antiproliferative effects, and inhibit the tumour cell growth of several types of cancers; thus, PDE4 inhibitors are a promising novel target for cancer therapyCitation18–27. Rolipram (B; )Citation18,Citation22,Citation23, roflumilast (C; )Citation18,Citation22,Citation23, Ro-20–1724 (D; )Citation23, and apremilast (E; )Citation23 are PDE4 inhibitors that reduced the growth of colon cancer cells through regulation of the level of intracellular cAMP, leading to the induction of apoptosis. Roflumilast (C; ) was approved by FDA as a PDE4 inhibitor and used for the treatment of chronic obstructive pulmonary diseaseCitation26 and was successfully tested in lung cancer and B-cell lymphomaCitation25. In contrast, an increase in the level of intracellular cAMP by the inhibition of PDE4 isoenzymes leads to inhibition of the production of tumour necrosis factor-alpha (TNF-α)Citation28. TNF-α is a central mediator of inflammation, and thus provides a molecular link between chronic inflammation and the development of malignanciesCitation29–32. In addition, TNF-α is overexpressed in various cancer cells such as liver cancer, kidney cancer, and gallbladder cancer and supports tumour growth and metastasisCitation29–32. The aforementioned results indicated that the inhibition of PDE4 enzyme activityCitation18–27 and the suppression of the production of TNF-αCitation28–32 are an interesting target for the treatment of cancer.
Figure 1. The structures of the reported antitumor agents (A–F) with COX-2 or PDE4 and the designed compounds 3–14.
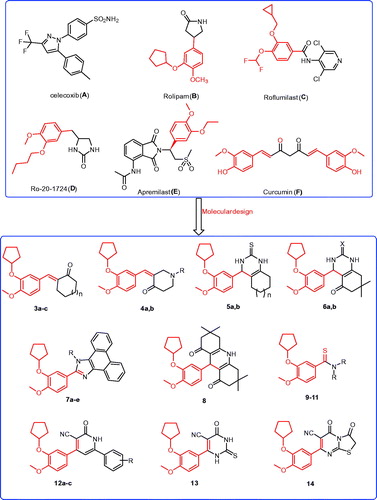
Compounds containing 2-cyclopentyloxyanisole analogues are reported to be PDE4 inhibitors with anticancer activities, such as rolipram (B; ), roflumilast (C; ), and apremilast (E; )Citation18,Citation22,Citation23. Meanwhile, compounds bearing chalcone structures constitute the main building block of several natural products with potential antitumor activity, such as curcumin (F; )Citation7,Citation9,Citation33. It was reported that curcumin exerts antitumor activity against colon cancer through inhibition of the COX-2 isoenzymeCitation34. Recently, curcumin was shown to have in vitro anti-angiogenic effects and in vivo anticancer activity through the inhibition of PDE isoenzymesCitation35. Indeed, several compounds possessing heterocyclic core structures, such as quinazolineCitation2–4, quinolineCitation9,Citation10, pyrimidineCitation36, pyridineCitation9, imidazoleCitation6, have potential antitumor activity.
Based on the aforementioned data, and to continue our efforts to develop new molecules as effective antitumor agents, we have reported (i) the synthesis of new derivatives incorporating chalcone derivatives based on the 2-cyclopentyloxyanisole core structure; (ii) the preparation of 2-cyclopentyloxyanisole bearing heterocyclic moieties such as quinazoline, quinoline, pyridine, pyrimidine, and imidazole ring systems; (iii) the synthesis of 2-cyclopentyloxyanisole bearing thioamide moieties; (iv) a comparison of the effectiveness of heterocyclic derivatives versus the chalcone and thioamide derivatives; and (v) an evaluation of the in vitro antitumor activity against different human cancers: liver cancer (HePG2 cell line), colon cancer (HCT-116 cell line), breast cancer (MCF-7 cell line), prostate cancer (PC3 cell line), and cervical cancer (HeLa cell line); (vi) a study of the structure–activity relationship (SAR) for the synthesised 2-cyclopentyloxyanisole structure with diverse substituent moieties regarding antitumor activities; (vii) an evaluation of the in vitro COX-2 and PDE4B, and TNF-α inhibitory abilities of the most promising compounds; and (viii) a molecular modelling study of the binding mode of the target molecules in the COX-2 and PDE 4 pockets.
Experimental methods
Chemistry
Melting points were recorded by using a Fisher-Johns melting point apparatus and were uncorrected. 1H NMR and 13C NMR spectra (500 MHz) were obtained in DMSO-d6 and CHCl3-d on a JOEL Nuclear Magnetic Resonance 500 spectrometer at Mansoura University, Faculty of Science, Egypt. Mass spectrometric analyses were performed by using a JEOL JMS-600H spectrometer at Mansoura University, Faculty of Science (Assiut, Egypt). The reaction times were determined by using a TLC technique on silica gel plates (60 F245, Merck, Kenilworth, NJ) and the spots were visualised by UV irradiation at 366 nm or 245 nm. The synthesis of 3-(cyclopentyloxy)-4-methoxybenzaldehyde (2) and 6-(3-(cyclopentyloxy)-4-methoxyphenyl)-4-oxo-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carbonitrile (13) are described elsewhereCitation18,Citation37,Citation38.
Synthesis of compounds 3a–c, 4a, and 4b
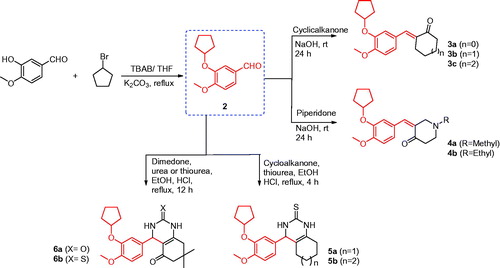
To a mixture of 3-(cyclopentyloxy)-4-methoxybenzaldehyde (2) (1.0 mmol, 0.22 g) and cyclic ketones (3.0 mmol) in ethanol (15 ml), NaOH (2.0 mmol, 0.08 g) was added whilst stirring at 0 °C. The reaction mixture was then stirred at room temperature for 24 h, poured on crushed ice, and the obtained solid was filtered, washed with water, and recrystallised from methanol (Scheme 1).
Scheme 1. Synthesis of the designed compounds 3–6.
2-(3-(Cyclopentyloxy)-4-methoxybenzylidene)cyclopentanone (3a)
Yield, 65%; melting point [MP] 252–254 °C. 1H NMR spectrum (DMSO-d6), δ, ppm: 1.53–1.56 (2H, m), 1.62–1.65 (4H, m), 1.70–1.74 (4H, m), 1.86–1.89 (2H, m), 2.89–2.91 (2H, m), 3.87 (3H, s), 4.74–4.77 (1H, m), 7.05–7.07 (1H, d, J = 8.0 Hz), 7.07–7.08 (1H, d, J = 8.0 Hz), 7.21 (1H, s), 7.74 (1H, s). IR spectrum, ν, cm−1: 2957, 2872, 1703, 1620, 954, 642. C18H22O3 MS: m/z 287 (M++1), 286 (M+).
2-(3-(Cyclopentyloxy)-4-methoxybenzylidene)cyclohexanone (3b)
Yield, 60%; MP 245–247 °C. 1H NMR spectrum (DMSO-d6), δ, ppm: 1.60–1.68 (6H, m), 1.81–1.93 (8H, m), 2.91–2.93 (2H, m), 3.85 (3H, s), 4.75–4.79 (1H, m), 7.03–7.04 (1H, d, J = 8.1 Hz), 7.06–7.07 (1H, d, J = 8.0 Hz), 7.25 (1H, s), 7.77 (1H, s). IR spectrum, ν, cm−1: 2953, 2870, 1705, 1621, 951, 638. C19H24O3 MS: m/z 301 (M++1), 300 (M+).
2-(3-(Cyclopentyloxy)-4-methoxybenzylidene)cycloheptanone (3c)
Yield, 63%; MP 250–252 °C. 1H NMR spectrum (DMSO-d6), δ, ppm: 1.50–1.60 (3H, m), 1.80–1.81 (2H, m), 1.82–1.85 (6H, m), 1.89–1.91 (5H, m), 2.68–2.71 (2H, m), 3.86 (3H, s), 4.73–4.75 (1H, m), 6.84 (1H, s), 6.86–6.88 (1H, d, J = 7.9 Hz), 6.89–6.90 (1H, d, J = 8.0 Hz), 7.44 (1H, s). IR spectrum, ν, cm−1: 2950, 2871, 1710, 1616, 954, 639. C20H26O3 MS: m/z 315 (M++1), 314 (M+).
3-(3-(Cyclopentyloxy)-4-methoxybenzylidene)-1-methylpiperidin-4-one (4a)
Yield, 70%; MP 253–255 °C. 1H NMR spectrum (DMSO-d6), δ, ppm: 1.55–1.58 (2H, m), 1.64–1.73 (4H, m), 1.79–1.86 (4H, m), 2.15 (2H, s), 2.42 (3H, s), 2.91–2.95 (2H, m), 3.71 (3H, s), 4.74–4.78 (1H, q, J = 5.5 Hz), 6.66–6.74 (2H, m), 6.89–6.94 (2H, m). IR spectrum, ν, cm−1: 2955, 2872, 1708, 1620, 956, 640. C19H25NO3 MS: m/z 317 (M++2), 316 (M++1), 315 (M+).
3-(3-(Cyclopentyloxy)-4-methoxybenzylidene)-1-ethylpiperidin-4-one (4b)
Yield, 68%; MP 249–251 °C. 1H NMR spectrum (DMSO-d6), δ, ppm: 1.29–1.32 (3H, t, J = 4.5 Hz), 1.52–1.54 (2H, m), 1.62–1.68 (4H, m), 1.81–1.85 (4H, m), 2.43–2.45 (2H, m), 2.88–2.92 (2H, m), 2.93–2.95 (2H, m), 3.73 (3H, s), 4.73–4.79 (1H, m), 6.66–6.77 (2H, m), 6.86–6.95 (2H, m). IR spectrum, ν, cm−1: 2954, 2870, 1708, 1624, 958, 644. C20H27NO3 MS: m/z 331 (M++2), 330 (M++1), 329 (M+).
Synthesis of compounds 5a and 5b
To a solution of 3-(cyclopentyloxy)-4-methoxybenzaldehyde (2) (5 mmol, 1.1 g), thiourea (5 mmol, 380 mg), and cyclic ketones (7.5 mmol) in ethanol (25 ml), four drops of concentrated hydrochloric acid were added. The reaction mixture was heated under reflux for 4 h, and the solvent was evaporated under vacuum. The obtained solid was dissolved in H2O and the solution was neutralised with ammonia solution. The precipitated solid was filtered, washed with water, and crystallised from ethanol (Scheme 1).
4-(3-(Cyclopentyloxy)-4-methoxyphenyl)-3,4,5,6,7,8-hexahydroquinazoline-2(1H)-thione (5a)
Yield, 55%; MP 199–201 °C. 1H NMR spectrum (CHCl3-d), δ, ppm: 0.80–0.86 (4H, m), 1.20–1.25 (4H, m), 1.83–1.89 (4H, m), 1.91–1.95 (4H, m), 3.83 (3H, s), 4.67 (1H, s), 4.78–4.93 (1H, m), 6.76 (1H, s), 6.80 (1H, s), 6.82 (1H, s), 6.83–6.86 (1H, d, J = 8.0 Hz), 7.13–7.16 (1H, d, J = 8.1 Hz). IR spectrum, ν, cm−1: 3422, 3240, 2960, 2871, 1630, 1260. C20H26N2O2S MS: m/z 360 (M++2), 359 (M++1), 358 (M+).
4-(3-(Cyclopentyloxy)-4-methoxyphenyl)-1,3,4,5,6,7,8,9-octahydro-2H-cyclohepta[d]pyrimidine-2-thione (5b)
Yield, 52%; MP 205–207 °C. 1H NMR spectrum (CHCl3-d), δ, ppm: 0.83–0.88 (6H, m), 1.19–1.24 (2H, m), 1.25–1.29 (2H, m), 1.61–1.66 (4H, m), 1.82–1.93 (4H, m), 3.84 (3H, s), 4.67 (1H, s), 4.78–4.92 (1H, m), 6.76 (1H, s), 6.81 (1H, s), 6.84 (1H, s), 6.81–6.85 (1H, d, J = 7.9 Hz), 7.10–7.12 (1H, d, J = 8.0 Hz). IR spectrum, ν, cm−1: 3426, 3243, 2963, 2873, 1632, 1262. C21H28N2O2S MS: m/z 374 (M++2), 373 (M++1), 372 (M+).
Synthesis of compounds 6a and 6b
To a solution of 3-(cyclopentyloxy)-4-methoxybenzaldehyde (2) (5 mmol, 1.1 g), urea or thiourea (5 mmol), and dimedone (7.5 mmol, 1.1 g) in ethanol (25 ml), four drops of concentrated hydrochloric acid were added. The reaction mixture was heated under reflux for 12 h and the solvent was evaporated under vacuum. The obtained solid was dissolved in H2O and the solution was neutralised by using ammonia solution. The precipitated solid was filtered, washed with water, and re-crystallised from DMF (Scheme 1).
4-(3-(Cyclopentyloxy)-4-methoxyphenyl)-7,7-dimethyl-4,6,7,8-tetrahydroquinazoline-2,5(1H,3H)-dione (6a)
Yield, 80%; MP 230–232 °C. 1H NMR spectrum (DMSO-d6), δ, ppm: 0.99 (3H, s), 1.02 (3H, s), 1.05 (1H, s), 1.54–1.58 (4H, m), 1.78–1.89 (4H, m), 2.40–2.43 (3H, t, J = 6.5 Hz), 3.74 (3H, s), 4.67 (1H, s), 4.72–4.76 (1H, q, J = 3.5 Hz), 6.67 (1H, s), 6.68 (1H, s), 6.70 (1H, s), 6.73–6.74 (1H, d, J = 6.5 Hz), 6.75–6.76 (1H, d, J = 6.5 H). IR spectrum, ν, cm−1: 3420, 3243, 2957, 2872, 1620, 1260. C22H28N2O4 MS: m/z 386 (M++2), 385 (M++1), 384 (M+).
4-(3-(Cyclopentyloxy)-4-methoxyphenyl)-7,7-dimethyl-2-thioxo-2,3,4,6,7,8-hexahydroquinazolin-5(1H)-one (6b)
Yield, 78%; MP 233–235 °C. 1H NMR spectrum (DMSO-d6), δ, ppm: 0.99 (3H, s), 1.01 (3H, s), 1.55 (2H, s), 1.77–1.82 (4H, m), 1.87–1.92 (4H, m), 2.44 (2H, s), 3.73 (3H, s), 4.68 (1H, s), 4.71–4.73 (1H, m), 6.66 (1H, s), 6.68 (1H, s), 6.69 (1H, s), 6.72–6.74 (1H, d, J = 7.5 Hz), 6.75–6.77 (1H, d, J = 6.5 Hz). IR spectrum, ν, cm−1: 3425, 3245, 2960, 2870, 1623, 1264. C22H28N2O3S MS: m/z 402 (M++2), 401 (M++1), 400 (M+).
Synthesis of compound 7a
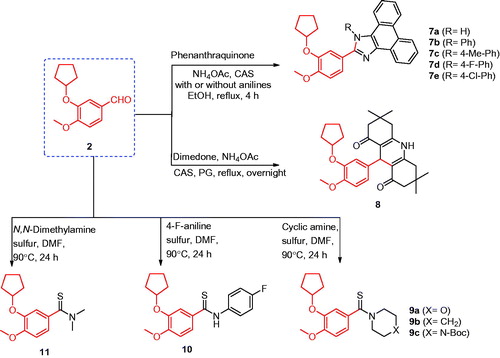
A mixture of 3-(cyclopentyloxy)-4-methoxybenzaldehyde (2) (5 mmol, 1.1 g), 9,10-phenanthraquinone (5 mmol, 1.04 g), ammonium acetate (15 mmol, 1.17 g), and CAS or iodine (5 mol%) in ethanol (25 ml) was heated under reflux for 4 h. The reaction mixture was cooled to room temperature, poured on crushed ice, and extracted with ethyl acetate. The extract was evaporated under vacuum to yield a precipitate, which was collected and re-crystallised from acetone (Scheme 2).
Scheme 2. Synthesis of the designed compounds 7–11.
2-(3-(Cyclopentyloxy)-4-methoxyphenyl)-1H-phenanthro[9,10-d]imidazole (7a)
Yield, 85%; MP 290–292 °C. 1H NMR spectrum (DMSO-d6), δ, ppm: 1.62 (2H, s), 1.79–1.82 (4H, m), 1.97 (2H, s), 3.84 (3H, s), 4.97 (1H, s), 7.17–7.18 (1H, d, J = 8.0 Hz), 7.62 (2H, s), 7.72 (2H, s), 7.87 (2H, s), 8.55–8.56 (2H, d, J = 6.5 Hz), 8.83–8.85 (2H, d, J = 7.5 Hz). 13C NMR spectrum (DMSO-d6), δ, ppm: 18.56, 23.68, 32.38, 55.67, 56.03, 79.95, 112.36, 112.85, 119.25, 121.88, 123.71, 125.02, 126.95, 127.07, 136.83, 147.14, 149.34, 150.92. IR spectrum, ν, cm−1: 3422, 2964, 2864, 930, 615. C27H24N2O2 MS: m/z 409 (M++1), 408 (M+).
Synthesis of compounds 7b–e
A mixture of 3-(cyclopentyloxy)-4-methoxybenzaldehyde (2) (5 mmol, 1.1 g), 9,10-phenanthraquinone (5 mmol, 1.04 g), ammonium acetate (15 mmol, 1.17 g), the appropriate aniline (5 mmol), and CAS or iodine (5 mol%) in ethanol (25 ml) was heated under reflux for 4 h. The formed precipitate was filtered, washed with ethanol, and crystallised from DMF (Scheme 2).
2-(3-(Cyclopentyloxy)-4-methoxyphenyl)-1-phenyl-1H-phenanthro[9,10-d]imidazole (7b)
Yield, 82%; MP 295–297 °C. 1H NMR spectrum (DMSO-d6), δ, ppm: 1.52 (2H, s), 1.60–1.65 (4H, m), 1.70–1.71 (2H, d, J = 6.5 Hz), 3.74 (3H, s), 4.49 (1H, s), 6.96–6.98 (2H, d, J = 8.0 Hz), 7.01–7.03 (1H, d, J = 8.0 Hz), 7.29–7.31 (2H, d, J = 7.5 Hz), 7.51–7.54 (1H, t, J = 7.5 Hz), 7.62–7.77 (7H, m), 8.67–8.68 (1H, d, J = 7.5 Hz), 8.85–8.87 (1H, d, J = 8.5 Hz), 8.90–8.92 (1H, d, J = 8.5 Hz). IR spectrum, ν, cm−1: 2960, 2869, 932, 618. C33H28N2O2 MS: m/z 485 (M++1), 484 (M+).
2-(3-(Cyclopentyloxy)-4-methoxyphenyl)-1-(4-methylphenyl)-1H-phenanthro[9,10-d]imidazole (7c)
Yield, 80%; MP 291–294 °C. 1H NMR spectrum (DMSO-d6), δ, ppm: 1.52 (4H, s), 1.64–1.68 (4H, m), 2.07 (3H, s), 3.75 (3H, s), 4.36 (1H, s), 6.89 (1H, s), 6.98–6.70 (1H, d, J = 8.5 Hz), 7.12–7.14 (1H, d, J = 8.0 Hz), 7.32–7.37 (2H, q, J = 9.0 Hz), 7.50–7.54 (3H, q, J = 7.5 Hz), 7.56–7.58 (2H, d, J = 8.0 Hz), 7.65–7.68 (1H, t, J = 7.5 Hz), 7.74–7.77 (1H, t, J = 7.5 Hz), 8.66–8.67 (1H, d, J = 7.5 Hz), 8.85–8.86 (1H, d, J = 8.5 Hz), 8.90–8.92 (1H, d, J = 9.0 Hz). IR spectrum, ν, cm−1: 2968, 2877, 942, 632. C34H30N2O2 MS: m/z 499 (M++1), 498 (M+).
2-(3-(Cyclopentyloxy)-4-methoxyphenyl)-1-(4-fluorophenyl)-1H-phenanthro[9,10-d]imidazole (7d)
Yield, 86%; MP 290–292 °C. 1H NMR spectrum (DMSO-d6), δ, ppm: 1.50–1.54 (2H, m), 1.60–1.64 (4H, m), 1.68–1.71 (2H, m), 3.86 (3H, s), 4.94–4.99 (1H, m), 6.85 (1H, s), 6.94–6.96 (1H, d, J = 7.5 Hz), 7.10–7.11 (1H, d, J = 7.5 Hz), 7.29–7.31 (2H, m), 7.40–7.49 (5H, m), 7.62–7.64 (1H, t, J = 8.0 Hz), 7.71–7.74 (1H, t, J = 8.0 Hz), 8.65–8.66 (1H, d, J = 8.5 Hz), 8.81–8.83 (1H, d, J = 9.0 Hz), 8.86–8.87 (1H, d, J = 8.5 Hz). IR spectrum, ν, cm−1: 2968, 2875, 940, 636. C33H27FN2O2 MS: m/z 505 (M++3), 503 (M++1), 502 (M+).
2-(3-(Cyclopentyloxy)-4-methoxyphenyl)-1-(4-chlorophenyl)-1H-phenanthro[9,10-d]imidazole (7e)
Yield, 84%; MP 294–296 °C. 1H NMR spectrum (DMSO-d6), δ, ppm: 1.52–1.56 (2H, m), 1.60–1.66 (4H, m), 1.69–1.73 (2H, m), 3.83 (3H, s), 4.91–4.95 (1H, m), 6.80 (1H, s), 6.94–6.96 (1H, d, J = 8.0 Hz), 7.12–7.14 (1H, d, J = 8.0 Hz), 7.25–7.29 (2H, m), 7.40–7.47 (5H, m), 7.61–7.63 (1H, t, J = 7.5 Hz), 7.72–7.73 (1H, t, J = 7.0 Hz), 8.65–8.67 (1H, d, J = 8.50 Hz), 8.79–8.81 (1H, d, J = 8.5 Hz), 8.84–8.86 (1H, d, J = 9.0 Hz). IR spectrum, ν, cm−1: 2965, 2873, 942, 635. C33H27ClN2O2 MS: m/z 520 (M++2), 519 (M++1), 518 (M+).
Synthesis of compound 8
To a solution of 3-(cyclopentyloxy)-4-methoxybenzaldehyde (2) (5 mmol, 1.1 g), dimedone (10 mmol, 1.47 g), and ammonium acetate (5 mmol, 0.39 g) in propylene glycol (20 ml), CAS or iodine (5 mol%) was added. The reaction mixture was heated under reflux overnight, cooled to room temperature, and poured on crushed ice. The obtained solid was filtered, washed with water, and re-crystallised from ethanol (Scheme 2).
9-(3-(Cyclopentyloxy)-4-methoxyphenyl)-3,3,6,6-tetramethyl-3,4,6,7,9,10-hexahydroacridine-1,8(2H,5H)-dione (8)
Yield, 77%; MP 286–287 °C. 1H NMR spectrum (DMSO-d6), δ, ppm: 0.86 (6H, s), 0.99 (6H, s), 1.53–1.55 (2H, m), 1.63–1.67 (4H, m), 1.69–1.71 (2H, m), 1.98–2.00 (2H, d, J = 6.5 Hz), 2.14–2.15 (2H, d, J = 5.5 Hz), 2.29–2.30 (2H, d, J = 5.5 Hz), 2.41–2.43 (2H, d, J = 6.5 Hz), 3.63 (3H, s), 4.55–4.58 (1H, q, J = 6.0 Hz), 4.72 (1H, s), 6.60–6.62 (1H, d, J = 8.0 Hz), 6.69–6.71 (2H, d, J = 6.0 Hz), 9.26 (1H, s). 13C NMR spectrum (DMSO-d6), δ, ppm: 23.52, 26.39, 29.16, 31.89, 32.07, 32.28, 50.27, 55.38, 79.38, 111.44, 111.59, 115.02, 119.52, 139.68, 146.14, 147.58, 148.95, 149.07, 194.39. IR spectrum, ν, cm−1: 3420, 2968, 2872, 1735, 1738. C29H37NO4 MS: m/z 464 (M++1), 463 (M+).
Synthesis of compounds 9a–c, 10, and 11
A solution of 3-(cyclopentyloxy)-4-methoxybenzaldehyde (2) (5 mmol, 1.1 g), appropriate amine derivatives (25 mmol), and precipitated sulphur (12.5 mmol, 0.40 g) in DMF (15 ml) was heated at 90 °C for 24 h. The reaction was monitored by TLC and, after completion, was cooled to room temperature and poured on crushed ice. The formed precipitate was filtered, washed with water, and re-crystallised from methanol (Scheme 2).
(3-(Cyclopentyloxy)-4-methoxyphenyl)(morpholino)methanethione (9a)
Yield, 70%; MP 190–192 °C. 1H NMR spectrum (DMSO-d6), δ, ppm: 1.55–1.56 (2H, d, J = 2.5 Hz), 1.67–1.70 (4H, m), 1.86–1.87 (2H, d, J = 4.0 Hz), 3.58–3.59 (4H, d, J = 3.0 Hz), 3.75 (3H, s), 4.27 (4H, s), 4.75–4.78 (1H, t, J = 5.5 Hz), 6.83–6.85 (2H, t, J = 8.0 Hz), 6.92–6.94 (1H, d, J = 8.0 Hz). IR spectrum, ν, cm−1: 2956, 2848, 1516, 1223, 1163, 925, 813, 631. C17H23NO3S MS: m/z 323 (M++2), 322 (M++1), 321 (M+).
(3-(Cyclopentyloxy)-4-methoxyphenyl)(piperidin-1-yl)methanethione (9b)
Yield, 72%; MP 193–195 °C. 1H NMR spectrum (DMSO-d6), δ, ppm: 1.50–1.57 (4H, q, J = 6.5 Hz), 1.66–1.69 (8H, t, J = 6.0 Hz), 1.85–1.86 (2H, d, J = 4.0 Hz), 3.52–3.53 (2H, d, J = 5.0 Hz), 3.75 (3H, s), 4.22–4.23 (2H, d, J = 5.5 Hz), 4.75–4.78 (1H, t, J = 6.0 Hz), 6.78–6.80 (2H, d, J = 7.5 Hz), 6.91–6.92 (1H, d, J = 8.5 Hz). IR spectrum, ν, cm−1: 2955, 2846, 1512, 1225, 1166, 920, 810, 630. C18H25NO2S MS: m/z 321 (M++2), 320 (M++1), 319 (M+).
tert-Butyl 4-(3-(cyclopentyloxy)-4-methoxyphenylcarbonothioyl)piperazine-1-carboxylate (9c)
Yield, 68%; MP 191–193 °C. 1H NMR spectrum (DMSO-d6), δ, ppm: 1.39 (9H, s), 1.53–1.55 (2H, m), 1.68–1.70 (4H, t, J = 4.5 Hz), 1.86–1.87 (2H, m, J = 4.5 Hz), 3.30–3.33 (4H, m), 3.56–3.59 (4H, m), 3.75 (3H, s), 4.75–4.77 (1H, t, J = 5.5 Hz), 6.84–6.86 (2H, t, J = 7.0 Hz), 6.92–6.95 (1H, t, J = 8.0 Hz). IR spectrum, ν, cm−1: 2958, 2848, 1514, 1224, 1160, 929, 812, 633. C22H32N2O4S MS: m/z 421 (M++1), 420 (M+).
3-(Cyclopentyloxy)-N-(4-fluorophenyl)-4-methoxybenzothioamide (10)
Yield, 75%; MP 194–196 °C. 1H NMR spectrum (DMSO-d6), δ, ppm: 1.55–1.58 (2H, d, J = 6.0 Hz) , 1.70–1.74 (4H, m), 1.91–1.94 (2H, d, J = 4.0 Hz), 3.81 (3H, s), 4.83–4.85 (1H, t, J = 5.5 Hz), 7.06–7.07 (1H, d, J = 7.5 Hz), 7.19–7.23 (2H, m), 7.25–7.28 (2H, m), 7.34 (1H, s), 7.49–7.50 (1H, d, J = 7.5 Hz), 8.48 (1H, s). IR spectrum, ν, cm−1: 2951, 2848, 1510, 1225, 1162, 921, 814, 633. C19H20FNO2S MS: m/z 347 (M++2), 346 (M++1), 345 (M+).
3-(Cyclopentyloxy)-4-methoxy-N,N-dimethylbenzothioamide (11)
Yield, 71%; MP 189–191 °C. 1H NMR spectrum (DMSO-d6), δ, ppm: 1.52–1.55 (2H, m), 1.68–1.70 (4H, t, J = 5.0 Hz), 1.71–1.73 (2H, m), 3.16 (3H, s), 3.46 (3H, s), 3.75 (3H, s), 4.75–4.77 (1H, t, J = 5.5 Hz), 6.84–6.87 (2H, m), 6.91–6.92 (1H, d, J = 8.5 Hz). 13C NMR spectrum (DMSO-d6), δ, ppm: 23.53, 32.15, 43.09, 43.98, 55.57, 79.47, 111.30, 113.04, 118.95, 135.54, 145.96, 149.78, 198.94. IR spectrum, ν, cm−1: 2956, 2851, 1514, 1229, 1159, 920, 814, 636. C15H21NO2S MS: m/z 280 (M++1), 279 (M+).
Synthesis of compounds 12a–c
A mixture of 3-(cyclopentyloxy)-4-methoxybenzaldehyde (2) (2 mmol, 0.44 g), the appropriate acetophenone derivatives (2 mmol), ethyl cyanoacetate (2 mmol, 0.23 g), and ammonium acetate (16 mmol, 1.24 g) in ethanol (10 ml) was heated under reflux for 16 h. The reaction mixture was cooled to room temperature, filtered, washed with ethanol, and re-crystallised from acetone (Scheme 3).
Scheme 3. Synthesis of the designed compounds 12–14.
4-(3-(Cyclopentyloxy)-4-methoxyphenyl)-2-oxo-6-phenyl-1,2-dihydropyridine-3-carbonitrile (12a)
Yield, 88%; MP > 300 °C; 1H NMR spectrum (DMSO-d6), δ, ppm: 1.57–1.58 (2H, d, J = 6.0 Hz), 1.71–1.76 (4H, m), 1.89–1.91 (2H, t, J = 11.5 Hz), 3.82 (3H, s), 4.88–4.90 (1H, m), 6.77 (1H, s), 7.10–7.12 (1H, d, J = 10.0 Hz), 7.30 (2H, s), 7.33 (1H, s), 7.51–7.56 (3H, m), 7.87–7.88 (2H, d, J = 5.0 Hz). 13C NMR spectrum (DMSO-d6), δ, ppm: 23.62, 32.27, 55.71, 79.71, 112.04, 114.40, 116.94, 121.41, 127.78, 128.08, 128.94, 131.13, 146.82, 151.58. IR spectrum, ν, cm−1: 3445, 2964, 2220, 1630, 1510, 1265, 810. C24H22N2O3 MS: m/z 387 (M++1), 386 (M+).
4-(3-(cyclopentyloxy)-4-methoxyphenyl)-2-oxo-6-(p-tolyl)-1,2-dihydropyridine-3-carbonitrile (12b)
Yield, 84%; MP > 300 °C. 1H NMR spectrum (DMSO-d6), δ, ppm: 1.56–1.57 (2H, t, J = 4.0 Hz), 1.70–1.76 (4H, m), 1.88–1.92 (2H, m), 2.36 (3H, s), 3.81 (3H, s), 4.86–4.89 (1H, m), 6.74 (1H, s), 7.10–7.12 (1H, d, J = 8.5 Hz), 7.28–7.30 (2H, q, J = 4.0 Hz), 7.31 (1H, s), 7.33–7.34 (2H, d, J = 8.0 Hz), 7.77–7.79 (2H, d, J = 7.0 Hz). 13C NMR spectrum (DMSO-d6), δ, ppm: 20.92, 23.59, 32.24, 55.69, 79.69, 112.02, 114.39, 116.98, 121.34, 127.63, 128.14, 129.49, 141.27, 146.77, 151.52. IR spectrum, ν, cm−1: 3447, 2959, 2216, 1629, 1514, 1263, 807; C25H24N2O3 MS: m/z 401 (M++1), 400 (M+).
4-(3-(Cyclopentyloxy)-4-methoxyphenyl)-6-(3,4-dichlorophenyl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (12c)
Yield, 81%; MP > 300 °C. 1H NMR spectrum (DMSO-d6), δ, ppm: 1.52–1.57 (2H, m), 1.69–1.76 (4H, m), 1.89–1.94 (2H, m), 3.82 (3H, s), 4.86–4.89 (1H, m), 7.11–7.13 (2H, d, J = 8.0 Hz), 7.30–7.31 (2H, d, J = 2.5 Hz), 7.31–7.32 (1H, d, J = 2.5 Hz), 7.33–7.34 (1H, d, J = 2.5 Hz), 7.79–7.81 (2H, d, J = 9.0 Hz). IR spectrum, ν, cm−1: 3443, 2964, 2222, 1635, 1508, 1268, 808. C24H20Cl2N2O3 MS: m/z 456 (M++2), 454 (M+).
Synthesis of compound 14
A mixture of compound 13 (1 mmol, 0.34 g), chloroacetic acid (1 mmol, 0.10 g), anhydrous sodium acetate (4 mmol, 0.33 g) in acetic anhydride (2 ml), and glacial acetic acid (10 ml) was heated under reflux for 24 h. The reaction mixture was cooled to room temperature and poured into crushed ice. The obtained solid was filtered, washed with water, and crystallised from methanol (Scheme 3).
7-(3-(Cyclopentyloxy)-4-methoxyphenyl)-3,5-dioxo-2,3-dihydro-5H-thiazolo[3,2-a]pyrimidine-6-carbonitrile (14)
Yield, 55%; MP 265–267 °C. 1H NMR spectrum (DMSO-d6), δ, ppm: 1.50–1.53 (2H, m), 1.69–1.72 (4H, m), 1.88–1.90 (2H, m), 3.79 (3H, s), 4.21 (2H, s), 4.76–4.79 (1H, m), 7.01 (1H, s), 7.35 (1H, s), 7.38 (1H, s). IR spectrum, ν, cm−1: 2962, 2229, 1655, 16,450, 1217, 986. C19H17N3O4S MS: m/z 385 (M++2), 383 (M+).
Biological evaluation
In vitro antitumor activity evaluation assay
The antitumor activity was performed by using the tetrazolium salt 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay in accordance with an established methodCitation39.
In vitro COX-2 inhibition assay
The colorimetric COX-2 inhibition assay was performed in accordance with the manufacturer’s instructions (Kit 560101, Cayman Chemical, Ann Arbour, MI)Citation40–42.
In vitro TNF-α inhibition assay
The concentration of TNF-α was measured by human-specific sandwich enzyme-linked immunosorbent assay (ELISA) in accordance with the manufacturer’s instructions (no. 589201, Cayman Chemical, Ann Arbour, MI)Citation43,Citation44.
Docking methodology
The molecular docking technique was performed by using MOE 2008.10, from the Chemical Computing Group Inc.Citation45 in accordance with previously established methodsCitation18,Citation40–42.
Results and discussion
Chemistry
The synthetic strategies used to obtain the target compounds are presented in Schemes 1–3. The O-alkylation of isovanillin (1) with bromocyclopentane was successively conducted in the presence of K2CO3 and a phase transfer catalyst tetrabutylammonium bromide (TBAB) in THF to obtain the key intermediate 3-cyclopentyloxy-4-methoxybenzaldehyde (2) that provided the core structure of phosphodiesterase-4 inhibitorsCitation37. Tetrabutylammonium bromide successively exhibited the character of phase transfer catalyst in an environmentally friendly procedure under mild conditionsCitation37.
Synthesis of compounds 3–6
First, the cyclocondensation of 3-cyclopentyloxy-4-methoxybenzaldehyde (2)Citation37 with cyclic ketones in the ethanolic solution of sodium hydroxide afforded chalcones 3a–c and 4a,b in good yields (Scheme 1). In addition, the one-pot cyclocondensation reaction of 2 with the cyclic ketone (cyclohexanone/cycloheptanone/dimedone) and urea or thiourea in ethanol containing few drops of concentrated hydrochloric acid yielded the quinazoline derivatives 5a,b and 6a,bCitation46, as shown in Scheme 1.
Synthesis of compounds 7–11
The synthesis of imidazole via multicomponent reactions (MCRs) was achieved through the cyclocondensation of 1,2-diketone, an aldehyde, and ammonium acetate using a catalytic amount of ceric ammonium sulphate (CAS) or molecular iodineCitation47,Citation48 (Scheme 2). Thus, a one-pot synthesis achieved phenanthroimidazole derivatives 7a–e in good yield via the cyclocondensation of 9,10-phenanthraquinone, 3-cyclopentyloxy-4-methoxybenzaldehyde (2), and ammonium acetate in the presence of 5% mole of iodine or CAS. Furthermore, acridinedione 8 was prepared by a one-pot, three-component cyclocondensation reaction of 3-cyclopentyloxy-4-methoxybenzaldehyde (2), 1,3-dicarbonyl compound (dimedone), and ammonium acetate in the presence of a catalytic amount of 5% CAS using polyethylene glycol (PEG) as a solventCitation49. Thioamides 9a–c, 10, and 11 were synthesisedCitation50 by the reaction of elemental sulphur (S8), 3-cyclopentyloxy-4-methoxybenzaldehyde (2), and secondary amines, such as piperidine, morpholine, N-Boc-piperazine, and dimethylamine, or primary amines, such as 4-fluoroaniline in dimethylformamide (DMF), under heating condition.
Synthesis of compounds 12–14
The MCRs of 3-cyclopentyloxy-4-methoxybenzaldehyde (2), ethyl cyanoacetate, an appropriate acetophenone, and ammonium acetate in EtOH at reflux temperature gave pyridine-3-carbonitrile derivatives 12a–c in good yield. In contrast, the reaction of 3-cyclopentyloxy-4-methoxybenzaldehyde (2) with ethyl cyanoacetate and thiourea in an ethanolic solution of K2CO3 afforded 6-(3-(cyclopentyloxy)-4-methoxyphenyl)-4-oxo-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carbonitrile (13)Citation18,Citation38. Compound 13 was cyclised with chloroacetic acid in the presence of acetic anhydride and anhydrous sodium acetate in glacial acetic acid to yield thiazolo[3,2-a]pyrimidine-3,5-dione derivative 14Citation51 (Scheme 3).
Biological evaluation
Antitumor evaluation using the MTT assay
Compounds 3a–c, 4a,b, 5a,b, 6a,b, 7a–e, 8, 9a–c, 10, 11, 12a–c, 13, and 14 were screened for their in vitro antitumor activity by using the standard 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay against five human cancers: HePG2, HCT-116, MCF-7, PC3, and HeLa cell linesCitation39. The antitumor activities of the synthesised compounds 3–14 and the reference drugs, celecoxib, afatinib, and doxorubicin, are shown in Citation8–10. Compounds 3a–c, incorporating the cycloalkanone core, possessed strong to weak antitumor activity against some of the investigated cell lines (IC50 ≅ 19.34–95.96 μM). Interestingly, the replacement of the cycloalkanone moieties, such as in compounds 3a–c, with a piperidin-4-one fragment, such as compound 4a,b, resulted in a sharp increase in antitumor activity (IC50 ≅ 4.38–14.32 μM) against all of the investigated five cell lines, compared with the reference drug, celecoxib (IC50 ≅ 25.6–36.08 μM), afatinib (IC50 values of 5.4–11.4 μM), and doxorubicin (IC50 ≅ 4.17–8.87 μM).
Table 1. In vitro antitumor activity of the designed compounds, celecoxib, afatinib, and doxorubicin against human tumour cells.
Moreover, the introduction of quinazoline-2-thione or pyrimidine-2-thione moieties, instead of a piperidin-4-one moiety, as in compounds 5a,b, resulted in a sharp decrease in antitumor activity against all the investigated five cancer cell lines, with IC50 values in the range 46.29–92.37 μM. In contrast, the replacement of the quinazoline-2-thione fragment, as in compound 5a, with quinazoline-2,5-dione and 2-thioxo-quinazolin-5-one fragments at the same position, such as compounds 6a and 6b, resulted in a sharp increase in antitumor activity against all the investigated cancer cell lines, for HePG2 (IC50 values of 18.53 and 16.05 μM, respectively), HCT-116 (IC50 values of 30.49 and 25.41 μM, respectively), MCF-7 (IC50 values of 28.62 and 10.27 μM, respectively), PC3 (IC50 values of 27.44 and 17.95 μM, respectively), and HeLa (IC50 values of 19.12 and 13.49 μM, respectively), compared with celecoxib (IC50 values of 25.6, 29.54, 31.28, 30.69, and 36.08 μM, respectively), afatinib (IC50 values of 5.4, 11.4, 7.1, 7.7, and 6.2 μM, respectively), and doxorubicin (IC50 values of 4.50, 5.23, 4.17, 8.87, and 5.57 μM, respectively).
Moreover, weak antitumor activity against some of the tested cancer cell lines was exhibited by some polycyclic derivatives incorporating imidazole and quinoline ring systems, such as compounds 7a and 7c (IC50 ≅ 53.18–90.34 μM), whereas compounds 7e and 8 showed moderate antitumor activity against some selected cancer cell lines (IC50 ≅ 29.8–46.97 μM). Unexpectedly, derivative 7b showed a sharp increase in antitumor activity compared with the structural analogues 7a, c, d, and 8, with IC50 values of 13.68, 19.67, 11.85, 22.89, and 17.18 μM against HeG2, HCT-116, MCF-7, PC3, and HeLa cancer cell lines, respectively.
In contrast, the introduction of thioamide fragments in the 2-cyclopentyloxyanisole scaffold resulted in variable antitumor activity against the tested cancer cell lines; for example, compounds 9a–c showed strong to moderate antitumor activity (IC50 ≅ 24.85–48.93 μM) in comparison with thioamide 10 (IC50 ≅ 47.32–79.12 μM) and 11 (IC50 ≅ 88.63–96.79 μM). Furthermore, replacement of the thioamide moiety with a pyridine fragment, such as in compounds 12a–c, retained the antitumor activity against all cancer cell lines, as indicated by their IC50 values in the range 38.14–83.42 μM. In contrast, the 2-cyclopentyloxyanisole scaffold bearing the pyrimidine ring system, such as compounds 13 and 14, exhibited strong antitumor activities against the cancer cell lines tested (IC50 ≅ 5.86–20.11 μM). In brief, the compounds 4a, 4b, 7b, and 13 exhibited the strongest antitumor activities among the designed compounds against the HeG2, HCT-116, MCF-7, PC3, and HeLa cancer cell lines (IC50 ≅ 4.38–22.89 μM).
Structure–activity relationship of antitumor activity
According to the aforementioned antitumor activity, the SARs for the designed compounds indicated the following. (i) N-Methylpiperidin-4-one derivative 4a and N-ethylpiperidin-4-one derivative 4b exhibited higher antitumor activity (IC50 ≅ 4.38–14.32 μM) than the corresponding cycloalkanones 3a–c (IC50 ≅ 19.34 to >100 μM). It was clear that the derivative with N-methylpiperidin-4-one 4a had greater antitumor activity against all tested cancer cell lines (IC50 ≅ 4.38–9.18 μM) than the N-ethylpiperidin-4-one derivative 4b (IC50 ≅ 7.18–14.32 μM). (ii) Similarly, cyclohexanone derivative 3b exhibited greater antitumor activity against MCF-7 (IC50=23.81 μM), PC3 (IC50=19.34 μM), and HeLa (IC50=26.11 μM) cancer cells than cyclopentanone derivative 3a (IC50 ≅ 51.43 to >100 μM), and cycloheptanone derivative 3c (IC50 ≅ 81.65 to >100 μM). (iii) Compounds incorporating a quinazoline fragment, such as quinazoline-2,5(1H,3H)-dione derivative 6a (IC50 ≅ 18.53–30.49 μM) and 2-thioxoquinazolin-5(1H)-one derivative 6b (IC50 ≅ 10.27–25.41 μM) showed higher antitumor activity than the corresponding derivatives quinazoline-2(1H)-thione 5a, and pyrimidine-2-thione 5b (IC50 ≅ 46.29–92.37 μM). (iv) The 2-cyclopentyloxyanisole scaffold bearing the bulky polycyclic 1H-phenanthro[9,10-d]imidazoles 7a,c,d,e (IC50 ≅ 29.89 to >100 μM), and acridine-1,8(2H,5H)-dione 8 (IC50 ≅ 41.82–70.52 μM) showed lower antitumor activity than the corresponding 2-cyclopentyloxyanisole scaffold bearing quinazoline moiety 6a,b (IC50 ≅ 10.27–30.49 μM). Interestingly, the derivative 7b with the phenyl ring at position 1 of 1H-phenanthro[9,10-d]imidazole core structure (IC50 ≅ 11.85–22.89 μM) showed a sharp increase in antitumor activity in comparison with derivatives 7a,c,d,e and had approximately similar activity with compound 6b (IC50 ≅ 10.27–25.41 μM). (v) The antitumor activities of the 2-cyclopentyloxyanisole scaffold bearing a methanethione fragment, such as N-(4-fluorophenyl)benzothioamide derivative 10 (IC50 ≅ 47.32–79.12 μM) and N,N-dimethylbenzothioamide derivative 11 (IC50 ≅ 88.63–96.79 μM), were less potent than derivatives that contained morpholinomethanethione derivative 9a (IC50 ≅ 29.23–48.13 μM), piperidin-1-ylmethanethione derivative 9b (IC50 ≅ 24.85–39.07 μM), and tert-butyl piperazine-1-carboxylate derivative 9c (IC50 ≅ 33.39–52.87 μM). (vi) The pyrimidine derivatives, 4-oxo-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carbonitrile derivative 13 and 3,5-dioxo-2,3-dihydro-5H-thiazolo[3,2-a]pyrimidine-6-carbonitrile derivative 14, had potent antitumor activities (IC50 ≅ 5.86–20.11 μM) compared with that of the pyridine derivatives, 6-aryl-2-oxo-1,2-dihydropyridine-3-carbonitriles 12a–c, which have moderate to weak antitumor activity (IC50 ≅ 38.14–83.42 μM) against all tested cancer cells. Briefly, the structure–activity correlation of antitumor activity revealed that compounds 4a, 4b, 6b, 7b, 13, and 14 were the most active compounds, whereas compound 7d was the only derivative that had no antitumor activity against any of the tested cancer cell lines.
COX-2 inhibition assay
Several compounds that possess COX-2 inhibition activity have shown potent antitumor activities that may be attributable to the role of the COX-2 enzyme in cell proliferationCitation8,Citation14–17. Accordingly, the four compounds (4a, 4b, 7b, and 13) that exhibited the greatest antitumor activity, as well as celecoxib (used as the reference drug) were subjected to colorimetric COX-2 inhibition assays by using a COX-2 assay kit (catalogue no. 560101, Cayman Chemicals Inc., Ann Arbour, MI). The measured IC50 (μM) values are shown in , and are expressed as the means of three acquired determinationsCitation40–42. The IC50 values of celecoxib for COX-2 inhibition are found to be 0.68 μM. It is clear that compounds 4b and 13 were found to be the most active inhibitors of COX-2, with IC50 values of 1.08 and 1.88 μM, respectively, whereas compound 4a exhibited lower COX-2 inhibitory effect with an IC50 value of 3.34 μM. In contrast, compound 7b showed a very low inhibitory effect, with an IC50 value for COX-2 inhibition of 24.02 μM. Briefly, a small heterocyclic substituent on the 2-cyclopentyloxyanisole core, such as the piperidine ring in compounds 4a and 4b and the pyrimidine ring in compound 13, exhibited higher COX-2 inhibition in comparison with the polycyclic 1H-phenanthro[9,10-d]imidazole in compound 7b. The reduced inhibitory effect of compound 7b on COX-2 may be attributed to the bulkiness of the polycyclic system, which interferes with the COX-2 binding interactions.
Table 2. In vitro inhibitory effects of COX-2, PDE-4B, and TNF-α of the antitumor compounds 4a, 4b, 7b, and 13.Table Footnotea
PDE-4B enzyme assay
Compounds that inhibit PDE4 were recently shown to possess effective antitumor activities owing to the overexpression of PDE4 in cancer and its role in cell proliferation and tumour cell growthCitation18–27. The compounds that were the most active antitumor agents, such as compounds 4a, 4b, 7b, and 13, were subjected to a PDE4B inhibition assay using roflumilast as a reference drug; the IC50 values are presented in . Compound 13 showed the highest inhibition against PDE4B, with an IC50 value of 3.98 μM comparable to that of the reference drug roflumilast (IC50=1.55 μM), whereas compounds 4a and 7b were found have moderate activity, with IC50 values of 5.62 and 5.65 μM, respectively. Compound 4b possessed the lowest activity against PDE4B, with an IC50 value of 11.62 μM. From the structural study of the tested derivatives, including 4a, 4b, 7b, and 13, we concluded that the 2-cyclopentyloxyanisole scaffold bearing a cyanopyrimidine fragment, such as compound 13, increased the PDE4B inhibitory activity in comparison with other heterocyclic derivatives.
TNF-α inhibition assay
TNF-α has been reported as a target for cancer treatment; presently, TNF antagonists are under clinical investigation in phase I and II trials as single agents for cancer therapyCitation29–32. Accordingly, compounds 4a, 4b, 7b, and 13, which are the most active antitumor agents, were subjected to the TNF-α inhibition assay using celecoxib as a reference drugCitation43; the IC50 values are presented in . Compound 4a possessed potent TNF-α inhibitory effect, with an IC50 value of 2.01 µM, comparable with the reference drug celecoxib (IC50=6.44 µM), whereas compound 13 was found to be an effective inhibitor, with an IC50 value of 6.72 µM, similar to the TNF-α inhibitory effect of the reference drug celecoxib (IC50=6.44 µM). In contrast, compounds 4b and 7b were the least active derivatives, with IC50 values of 17.67 and 13.94 µM, respectively.
Molecular modelling analysis
Molecular modelling and docking analysis is an important technique used to establish the theoretical interaction between the bioactive molecules and the target enzyme and receptor to understand their binding modeCitation52,Citation53. Therefore, a molecular docking analysis was performed by using MOE 2008.10 software and viewer utility (Chemical Computing Group Inc., Montreal, Canada) in accordance with the standard MOE procedureCitation45.
Docking with the COX-2 isoenzyme
The molecular interaction of the most active compounds, 4b and 13, with the COX-2 isoenzyme was studied by molecular docking. The crystal structure of the COX-2 isoenzyme interacting with its inhibitor SC-558 was obtained from the RSC Protein Data Bank (PDB code: 1CX2)Citation54. The putative binding site of the COX-2 isoenzyme (), which is responsible for the hydrogen bonds and hydrophobic interactions with its inhibitors, consists of key amino acid residues, such as Arg510, Gln192, Arg120, Tyr355, His90, Val523, Ser353, and Ile517. The docking procedure was validated by including the bound inhibitor SC-558 for a one-ligand run docking calculation.
Figure 2. Three-dimensional (3D) orientation of the docked ligand SC-558 (upper left panel); docked compounds 4b (lower left panel), and 13 (lower right panel) in the active pocket of the COX-2 enzyme (H bond interactions are shown as green lines). Upper right panel showed the alignment of SC-558, 4b, and 13 in the active pocket of the COX-2 enzyme.
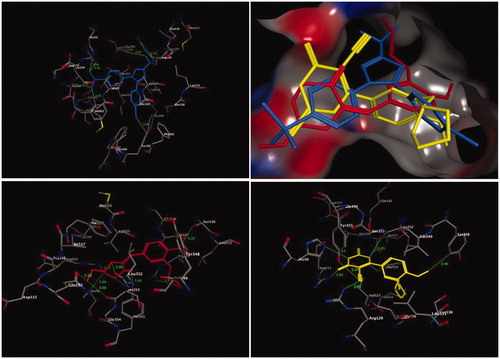
The bound ligand SC-558 exhibited two types of hydrogen bonds, classical and non-classical hydrogen bonds. Four classical hydrogen bonding interactions were observed with Arg513, His90, Arg120, and Tyr355. In addition, three non-classical hydrogen bonds connected the amino acids Tyr385, Phe518, and Ala516, and the benzenesulfonamide and 4-bromophenyl fragments of SC-558 through CH–O and CH–Br interactions (, upper panel).
Interestingly, compounds 4b and 13, which were the most active COX-2 inhibitors, were placed in the same binding site of the inhibitor SC-558 (). Compound 4b, which has nearly similar COX-2 inhibition activity as celecoxib, accommodated an orientation within the COX-2 binding site (, left lower panel), in which the N-ethylpiperdine-4-one fragment was located towards the secondary pocket of the COX-2 isoenzyme and interacted with the amino acid residues of Arg513, His90, Leu352, and Gln192. In general, when compound 4b was docked into the enzyme pocket, nine hydrogen bonds were formed with the surrounding amino acids lining the pocket. One of these interactions was a classical hydrogen bond between the carbonyl (C=O) group of the N-ethylpiperdine-4-one fragment and the OH group of the Tyr355 residue (3.06 Å). Moreover, eight non-bonding interactions, namely non-classical hydrogen bonds were formed, among the two bonds of the OH of the Tyr355 residue, and the C=O of the Leu352 residue with the CH2 of the piperdine-4-one moiety (3.44 Å, and 2.85 Å, respectively), and among two more bonds among the C=O fragments of the Gln192 and Ser353 residues and the CH3 moiety of N-ethylpiperdine-4-one (3.18 Å and 3.08 Å, respectively). The amino acid residues Arg513 and His90 formed additional two bonds between their HN groups and the CH2 of the piperdine-4-one ring (3.52 Å and 3.00 Å, respectively). Finally, the amino acid residues Arg120 and Ser530 formed two non-classical hydrogen bonds with the cyclopentyl and methoxyl moieties of the anisole core structure (NH–CH2, 2.87 Å; and CH2–OCH3, 3.22 Å, respectively). The overall outcome of the molecular docking of compound 4b, with respect to non-classical hydrogen bonds, showed that compound 4b had more hydrophobic interactions with the protein than the bound ligand SC-558.
The molecular docking analysis of compound 13 showed that the 4-oxo-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carbonitrile moiety was the main fragment responsible for COX-2 activity, which interacted with the surrounding amino acid residues of the active pocket of the COX-2 isoenzyme, such as Arg513, His90, Tyr348, Tyr355, and Arg120 (, right lower panel). Four classical and one non-classical hydrogen bonding interactions were formed between the abovementioned amino acid residues and compound 13. The nitrile group (CN) of compound 13 formed two classical hydrogen bonds with Arg120 (3.01 Å) and Tyr355 (3.24 Å), whereas the 4-oxo-tetrahydropyrimidine ring system interacted with amino acid residues Arg513 and His90 through two classical hydrogen bonds (2.81 Å and 3.11 Å, respectively). The final interaction was the hydrophobic interaction between Tyr348 and the methoxyl moiety of anisole through a CH2–π bond, with a non-bonding distance of 3.46 Å.
Docking with the PDE4B enzyme
The binding mode of the most active compound, 13, within the PDE4B enzyme was analysed by using molecular docking. The crystal structure of the PDE4B enzyme bound with its inhibitor roflumilast was obtained from the RSC Protein Data Bank (PDB code: 1XMU)Citation55. The binding site of the PDE4B enzyme (), which is responsible for the formation of coordination bonds, hydrogen bonds, and hydrophobic interactions with its inhibitor roflumilast, has three main sites for interaction: the solvent-filled metal coordination pocket, including both zinc and magnesium; the conserved residue Gln443; and the hydrophobic pocket. The amino acid residues Phe414, Ile410, Phe446, and Ile450 were the key residues that formed the tunnel, and were responsible for the accommodation of the hydrophobic interaction with the bound inhibitor, roflumilast. The molecular docking procedure was validated by performing a one-ligand run docking calculation for the bound inhibitor roflumilast. The results of the docking calculation of compound 13 are presented in (upper right panel). From the docking results, it was clear that the 2-cyclopentyloxyanisole scaffold and the pyrimidine ring adapted for hydrophobic recognition at the binding cavity lining with the amino acid residues Phe414, Ile410, Phe446, and Ile450 (, lower right panel), similar to the bound inhibitor roflumilast (, upper left panel). In contrast, the methoxyl group of the 2-cyclopentyloxyanisole scaffold formed a non-classical hydrogen bond with Ser442 (2.94 Å), whereas the conserved residue Gln443 interacted with the pyrimidine ring system through the nitrile moiety by the formation of hydrogen bond with a distance of 3.36 Å (, lower left panel). Moreover, the pyrimidine ring projected towards the metal-coordinating site filled with water molecules. Accordingly, the thione (C=S) moiety of the pyrimidine ring is coordinated with Zn and Mg ions, mediated by HOH2009, and formed a hydrogen bond with the amino acid residue His234. Meanwhile, the carbonyl oxygen (C=O) of the pyrimidine formed one hydrogen bond with Tyr233 (2.90 Å) and another two hydrogen bonds with the amino acid residues Asn395 and Asp392, mediated by HOH18. Finally, the internal NH group of pyrimidine ring was adapted to form a hydrogen bond with Asp392 mediated by HOH18.
Figure 3. Three-dimensional (3D) orientation of the docked roflumilast (upper left panel); docked compound 13 (upper right panel), in the active pocket of the PDE4B enzyme (H bond interactions are shown as green lines). Lower left panel showed near picture of compound 13 in the active pocket of the PDE4B enzyme. Lower right panel showed the hydrophobic interactions of compound 13 in the active pocket of the PDE4B enzyme.
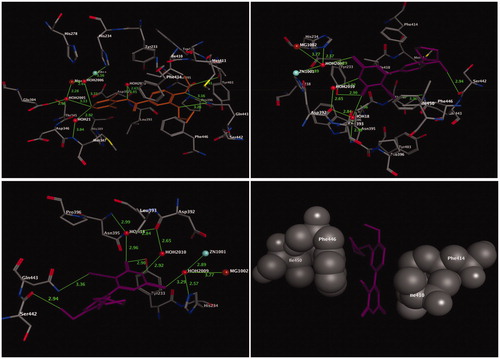
Briefly, in comparison of compound 13 with the bound inhibitor roflumilast, both compounds accommodated approximately similar interactions at the hydrophobic clamp site (Phe414, Ile410, Phe446, and Ile450) and the metal coordination site.
Conclusions
A series of compounds incorporating 2-cyclopentyloxyanisole scaffold bearing a variety of ring systems—cycloalkanones 3a–c and 4a–b, quinazolines 5a–b and 6a–b, fused imidazoles 7a–e, fused quinoline 8, thioamides 9a–c, 10, and 11, pyridines 12a–c, and pyrimidines 13 and 14 was synthesised. These compounds were evaluated for their in vitro antitumor activity in five human cancer cell lines: HePG2, HCT-116, MCF-7, PC3, and HeLa. The antitumor activity of compounds 4a, 4b, 6b, 7b, 13, and 14 indicated that these derivatives were the most potent antitumor agents among the tested compounds, with IC50 values of 5.13–17.95 μM in the tested cancer cell lines. The antitumor results of the synthesised compounds were comparable with the reference drug celecoxib (IC50 values of 25.6–36.08 μM), afatinib (IC50 values of 5.4–11.4 μM), and doxorubicin (IC50 values of 4.17–8.87 μM). In addition, the compounds that were most active as antitumor agents, 4a, 4b, 7b, and 13, were assayed for their ability to inhibit COX-2, PDE4B, and TNF-α. The results indicated that compounds 4b and 13 exhibited effective COX-2 inhibitory activity, with IC50 values of 1.08 and 1.88 μM, respectively, which were comparable with celecoxib (IC50=6.44 μM). In addition, compounds 4a and 13 inhibited the PDE4B enzyme, with an IC50 value of 5.62 and 3.98 μM, respectively, which was comparable with roflumilast (IC50=1.55 μM), whereas these compounds had potent TNF-α inhibitory effect, with IC50 values of 2.01 and 6.72 μM, respectively, which were comparable with the reference drug celecoxib (IC50=6.44 μM). Compounds 4b and 13 were docked into the COX-2 and PDE4B binding sites and exhibited similar binding characteristics to that of bound inhibitor SC-558 for the COX-2 enzyme and the bound inhibitor roflumilast for the PDE4B enzyme.
Acknowledgements
The authors thank the Deanship of Scientific Research and RSSU at the King Saud University for their technical support.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- (a) Avendańo C, Menéndez J. Medicinal chemistry of anticancer agents. Amsterdam: Elsevier; 2008. (b) Varmus H. The new era in cancer research. Science 2006;312:1162–5. (c) Eckhardt S. Recent progress in the development of anticancer agents. Curr Med Chem Anticancer Agents 2002;2:419–39.
- (a) El-Azab AS, Al-Omar MA, Abdel-Aziz AAM, et al. Design, synthesis and biological evaluation of novel quinazoline derivatives as potential antitumor agents: molecular docking study. Eur J Med Chem 2010;45:4188–98. (b) Al-Suwaidan IA, Alanazi AM, Abdel-Aziz AAM, et al. Design, synthesis and biological evaluation of 2-mercapto-3-phenethylquinazoline bearing anilide fragments as potential antitumor agents: molecular docking study. Bioorg Med Chem Lett 2013;23:3935–41. (c) Alanazi AM, Al-Suwaidan IA, Abdel-Aziz AAM, et al. Design, synthesis and biological evaluation of some novel substituted 2-mercapto-3-phenethylquinazolines as antitumor agents. Med Chem Res 2013;22:5566–77.
- (a) Alanazi AM, Abdel-Aziz AAM, Al-Suwaidan IA, et al. Design, synthesis and biological evaluation of some novel substituted quinazolines as antitumor agents. Eur J Med Chem 2014;79:446–54. (b) Alanazi AM, Abdel-Aziz AAM, Shawer TZ, et al. Synthesis, antitumor and antimicrobial activity of some new 6-methyl-3-phenyl-4(3H)-quinazolinone analogues: in silico studies. J Enzyme Inhib Med Chem 2016;31:721–35. (c) El-Azab AS, Al-Dhfyan A, Abdel-Aziz AAM, et al. Synthesis, anticancer and apoptosis-inducing activities of quinazoline–isatin conjugates: epidermal growth factor receptor-tyrosine kinase assay and molecular docking studies. J Enzyme Inhib Med Chem 2017;32:935–44.
- (a) Mohamed MA, Ayyad RR, Shawer TZ, et al. Synthesis and antitumor evaluation of trimethoxyanilides based on 4(3H)-quinazolinone scaffolds. Eur J Med Chem 2016;112:106–13. (b) Al-Suwaidan IA, Abdel-Aziz AAM, Shawer TZ, et al. Synthesis, antitumor activity and molecular docking study of some novel 3-benzyl-4(3H)quinazolinone analogues. J Enzyme Inhib Med Chem 2016;31:78–89. (c) El-Azab AS, Abdel-Aziz AAM, Ghabbour HA, et al. Synthesis, in vitro antitumour activity, and molecular docking study of novel 2-substituted mercapto-3-(3,4,5-trimethoxybenzyl)-4(3H)-quinazolinone analogues. J Enzyme Inhib Med Chem 2017;32:1229–39.
- (a) Abdel-Aziz AAM. Novel and versatile methodology for synthesis of cyclic imides and evaluation of their cytotoxic, DNA binding, apoptotic inducing activities and molecular modeling study. Eur J Med Chem 2007;42:614–26. (b) El-Azab AS, Alanazi AM, Abdel-Aziz NI, et al. Synthesis, molecular modeling study, preliminary antibacterial, and antitumor evaluation of N-substituted naphthalimides and their structural analogues. Med Chem Res 2013;22:2360–75.
- (a) El-Deeb IM, Bayoumi SM, El-Sherbeny MA, et al. Synthesis and antitumor evaluation of novel cyclic arylsulfonylureas: ADME-T and pharmacophore prediction. Eur J Med Chem 2010;45:2516–30. (b) Abdel-Aziz AAM, El-Azab AS, El-Subbagh HI, et al. Design, synthesis, single-crystal and preliminary antitumor activity of novel arenesulfonylimidazolidin-2-ones. Bioorg Med Chem Lett 2012;22:2008–14. (c) Alanazi AM, El-Azab AS, Al-Swaidan IA, et al. Synthesis, single-crystal, in vitro antitumor evaluation and molecular docking of 3-substitued 5,5-diphenylimidazolidine-2,4-dione derivatives. Med Chem Res 2013;22:6129–42.
- (a) Al-Suwaidan IA, Abdel-Aziz NI, El-Azab AS, et al. Antitumor evaluation and molecular docking study of substituted 2-benzylidenebutane-1,3-dione, 2-hydrazonobutane-1,3-dione and trifluoromethyl-1H-pyrazole analogues. J Enzyme Inhib Med Chem 2015;30:679–87. (b) El-Sherbeny MA, Abdel-Aziz AAM, Ahmed MA. Synthesis and antitumor evaluation of novel diarylsulfonylurea derivatives: molecular modeling applications. Eur J Med Chem 2010;45:689–97.
- (a) El-Husseiny WM, El-Sayed MAA, Abdel-Aziz NI, et al. Structural alterations based on naproxen scaffold: synthesis, evaluation of antitumor activity and COX-2 inhibition, and molecular docking. Eur J Med Chem 2018;158:134–43. (b) El-Azab AS, Abdel-Aziz AAM, Abou-Zeid LA, et al. Synthesis, antitumour activities and molecular docking of thiocarboxylic acid ester-based NSAID scaffolds: COX-2 inhibition and mechanistic studies. J Enzyme Inhib Med Chem 2018;33:989–98.
- El-Husseiny WM, El-Sayed MA, Abdel-Aziz NI, et al. Synthesis, antitumour and antioxidant activities of novel α,β-unsaturated ketones and related heterocyclic analogues: EGFR inhibition and molecular modelling study. J Enzyme Inhib Med Chem 2018;33:507–18.
- El-Sayed MA, El-Husseiny WM, Abdel-Aziz NI, et al. Synthesis and biological evaluation of 2-styrylquinolines as antitumour agents and EGFR kinase inhibitors: molecular docking study. J Enzyme Inhib Med Chem 2018;33:199–209.
- (a) Abdel-Aziz AAM, El-Azab AS, Alanazi AM, et al. Synthesis and potential antitumor activity of 7-(4-substituted piperazin-1-yl)-4-oxoquinolines based on ciprofloxacin and norfloxacin scaffolds: in silico studies. J Enzyme Inhib Med Chem 2016;31:796–809. (b) Abdel-Aziz AAM, Asiri YA, Al-Agamy MH. Design, synthesis and antibacterial activity of fluoroquinolones containing bulky arenesulfonyl fragment: 2D-QSAR and docking study. Eur J Med Chem 2011;46:5487–97.
- Bayomi SM, El-Kashef HA, El-Ashmawy MB, et al. Synthesis and biological evaluation of new curcumin analogues as antioxidant and antitumor agents: molecular modeling study. Eur J Med Chem 2015;101:584–94.
- (a) Stanković T, Dinic J, Podolski-Renić A, et al. Dual inhibitors as a new challenge for cancer multidrug resistance treatment. Curr Med Chem 2019;26:6074–106. (b) Raghavendra NM, Pingili D, Kadasi S, et al. Dual or multi-targeting inhibitors: the next generation anticancer agents. Eur J Med Chem 2018;143:1277–300.
- (a) Dai ZJ, Ma XB, Kang HF, et al. Antitumor activity of the selective cyclooxygenase-2 inhibitor, celecoxib, on breast cancer in vitro and in vivo. Cancer Cell Int 2012;12:53. (b) Ghosh N, Chaki R, Mandal V, et al. COX-2 as a target for cancer chemotherapy. Pharmacol Rep 2010;62:233–44.
- (a) Blanke C. Role of COX-2 inhibitors in cancer therapy. Cancer Invest 2004;22:271–82. (b) Basu GD, Pathangey LB, Tinder TL, et al. Mechanisms underlying the growth inhibitory effects of the cyclo-oxygenase-2 inhibitor celecoxib in human breast cancer cells. Breast Cancer Res 2005;7:422–35.
- (a) Vosooghi M, Amini M. The discovery and development of cyclooxygenase-2 inhibitors as potential anticancer therapies. Expert Opin Drug Discov 2014;9:255–67. (b) Claria J. Cyclooxygenase-2 biology. Curr Pharm Des 2003;9:2177–90.
- Gurpinar E, Grizzle WE, Piazza GA. COX-independent mechanisms of cancer chemoprevention by anti-inflammatory drugs. Front Oncol 2013;3:181–18.
- (a) Hirsh L, Dantes A, Suh BS, et al. Phosphodiesterase inhibitors as anti-cancer drugs. Biochem Pharmacol 2004;68:981–8. (b) Almatary AM, Elmorsy MA, El Husseiny WM, et al. Design, synthesis, and molecular modeling of heterocyclic bioisostere as potent PDE4 inhibitors. Arch Pharm (Weinheim) 2018;351:e1700403. (c) Sandeep G, Bhasker S, Ranganath YS. Phosphodiesterase as a novel target in cancer chemotherapy. Int J Pharmacol 2009;7:1.
- (a) Drees M, Zimmermann R, Eisenbrand G. 3′,5′-Cyclic nucleotide phosphodiesterase in tumor cells as potential target for tumor growth inhibition. Cancer Res 1993;53:3058–61. (b) Savai R, Pullamsetti SS, Banat GA, et al. Targeting cancer with phosphodiesterase inhibitors. Expert Opin Investig Drugs 2010;19:117–31.
- (a) Pullamsetti SS, Banat GA, Schmall A, et al. Phosphodiesterase-4 promotes proliferation and angiogenesis of lung cancer by crosstalk with HIF. Oncogene 2013;32:1121–34. (b) Selige J, Hatzelmann A, Dunkern TJ. The differential impact of PDE4 subtypes in human lung fibroblasts on cytokine-induced proliferation and myofibroblast conversion. Cell Physiol 2011;226:1970–80.
- (a) Domvri K, Zarogoulidis K, Ziogas N, et al. Potential synergistic effect of phosphodiesterase inhibitors with chemotherapy in lung cancer. J Cancer 2017;8:3648–56. (b) Yeo CD, Kim YA, Lee HY, et al. Roflumilast treatment inhibits lung carcinogenesis in benzo(a)pyrene-induced murine lung cancer model. Eur J Pharmacol 2017;812:189–95.
- (a) Kim DU, Kwak B, Kim SW. Phosphodiesterase 4B is an effective therapeutic target in colorectal cancer. Biochem Biophys Res Commun 2019;508:825–31. (b) Tsunoda T, Ota T, Fujimoto T, et al. Inhibition of phosphodiesterase-4 (PDE4) activity triggers luminal apoptosis and AKT dephosphorylation in a 3-D colonic-crypt model. Mol Cancer 2012;11:46.
- (a) Murata K, Sudo T, Kameyama M, et al. Cyclic AMP specific phosphodiesterase activity and colon cancer cell motility. Clin Exp Metastasis 2000;18:599–604. (b) Nishi K, Luo H, Ishikura S, et al. Apremilast induces apoptosis of human colorectal cancer cells with mutant KRAS. Anticancer Res 2017;37:3833–9.
- (a) Cooney JD, Aguiar RC. Phosphodiesterase 4 inhibitors have wide-ranging activity in B-cell malignancies. Blood 2016;128:2886–90. (b) Kelly K, Mejia A, Suhasini AN, et al. Safety and pharmacodynamics of the PDE4 inhibitor roflumilast in advanced B-cell malignancies. Clin Cancer Res 2017;23:1186–92.
- Kelly K, Mejia A, Suhasini AN, et al. Safety and pharmacodynamics of the PDE4 inhibitor roflumilast in advanced B-cell malignancies. Clin Cancer Res 2017;23:1186–92.
- (a) Parikh N, Chakraborti AK. Phosphodiesterase 4 (PDE4) inhibitors in the treatment of COPD: promising drug candidates and future directions. Curr Med Chem 2016;23:129–41. (b) Contreras S, Milara J, Morcillo E, et al. Selective inhibition of phosphodiesterases 4A, B, C and D isoforms in chronic respiratory diseases: current and future evidences. Curr Pharm Des 2017;23:2073–83.
- Mouratidis PX, Colston KW, Bartlett JB, et al. Antiproliferative effects of CC-8062 and CC-8075 in pancreatic cancer cells. Pancreas 2009;38:78–84.
- Balasubramanian G, Narayanan S, Andiappan L, et al. In vivo effective dibenzo[b,d]furan-1-yl-thiazoles as novel PDE-4 inhibitors. Bioorg Med Chem 2016;24:5702–16.
- (a) Brenner D, Blaser H, Mak TW. Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol 2015;15:362–74. (b) Lebrec H, Ponce R, Preston BD, et al. Tumor necrosis factor, tumor necrosis factor inhibition, and cancer risk. Curr Med Res Opin 2015;31:557–74.
- (a) Chuang MJ, Sun KH, Tang SJ, et al. Tumor-derived tumor necrosis factor-alpha promotes progression and epithelial–mesenchymal transition in renal cell carcinoma cells. Cancer Sci 2008;99:905–13. (b) Liu XL, Li FQ, Liu XL, et al. TNF-α, HGF and macrophage in peritumoural liver tissue relate to major risk factors of HCC recurrence. Hepatogastroenterology 2013;60:1121–6.
- (a) Mikami S, Mizuno R, Kosaka T, et al. Expression of TNF-α and CD44 is implicated in poor prognosis, cancer cell invasion, metastasis and resistance to the sunitinib treatment in clear cell renal cell carcinomas. Int J Cancer 2015;136:1504–14. (b) Zhu G, Du Q, Wang X, et al. TNF-α promotes gallbladder cancer cell growth and invasion through autocrine mechanisms. Int J Mol Med 2014;33:1431–40.
- (a) Yu M, Zhou X, Niu L, et al. Targeting transmembrane TNF-α suppresses breast cancer growth. Cancer Res 2013;73:4061–74. (b) Zidi I, Mestiri S, Bartegi A, et al. TNF-α and its inhibitors in cancer. Med Oncol 2010;27:185–98.
- (a) Katsori AM, Hadjipavlou-Litina D. Chalcones in cancer: understanding their role in terms of QSAR. Curr Med Chem 2009;16:1062–81. (b) Karthikeyan C, Moorthy N, Ramasamy S, et al. Advances in chalcones with anticancer activities. Recent Pat Anticancer Drug Discov 2014;10:97–115.
- (a) Goel A, Boland CR, Chauhan DP. Specific inhibition of cyclooxygenase-2 (COX-2) expression by dietary curcumin in HT-29 human colon cancer cells. Cancer Lett 2001;172:111–8. (b) Lev-Ari S, Strier D, Kazanov L, Madar-Shapiro L, et al. Celecoxib and curcumin synergistically inhibit the growth of colorectal cancer cells. Clin Cancer Res 2005;11:6738–44. (c) Lee SH, Lee GH, Park SY, et al. Apoptotic effects of curcumin via the regulation of COX-2/ VASP signaling molecules in MCF-7 breast cancer cells. Cancer Prev Res 2012;17:19–26.
- (a) Abusnina A, Keravis T, Zhou Q, et al. Tumour growth inhibition and anti-angiogenic effects using curcumin correspond to combined PDE2 and PDE4 inhibition. Thromb Haemost 2015;113:319–22. (b) Abusnina A, Keravis T, Yougbaré I, et al. Anti-proliferative effect of curcumin on melanoma cells is mediated by PDE1A inhibition that regulates the epigenetic integrator UHRF1. Mol Nutr Food Res 2011;55:1677–89. (c) Yi YX, Gaurav A, Akowuah GA. Docking studies of curcumin and analogues with various phosphodiesterase 4 subtypes. Curr Drug Discov Technol 2018.
- Ahmed NM, Youns M, Soltan MK, et al. Design, synthesis, molecular modelling, and biological evaluation of novel substituted pyrimidine derivatives as potential anticancer agents for hepatocellular carcinoma. J Enzyme Inhib Med Chem 2019;34:1110–20.
- Youssef KM, El-Sherbeny MA, El-Shafie FS, et al. Synthesis of curcumin analogues as potential antioxidant, cancer chemopreventive agents. Arch Pharm (Weinheim) 2004;337:42–54.
- Kambe S, Saito K, Kishi H, et al. A one-step synthesis of 4-Oxo-2-thioxopyrimidine derivatives by the ternary condensation of ethyl cyanoacetate, aldehydes, and thiourea. Synthesis 1979;4:287.
- (a) Denizot F, Lang R. Rapid colorimetric assay for cell growth and survival. J Immunol Methods 1986;89:271–7. (b) Vega-Avila E, Pugsley MK. An overview of colorimetric assay methods used to assess survival or proliferation of mammalian cells. West Pharmacol Soc 2011;54:10–4. (c) Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983;65:55–63.
- (a) Abdel-Aziz AAM, Angeli A, El-Azab AS, et al. Synthesis and anti-inflammatory activity of sulfonamides and carboxylates incorporating trimellitimides: dual cyclooxygenase/carbonic anhydrase inhibitory actions. Bioorg Chem 2019;84:260–8. (b) Uddin MJ, Rao PP, Knaus EE. Design and synthesis of acyclic triaryl (Z)-olefins: a novel class of cyclooxygenase-2 (COX-2) inhibitors. Bioorg Med Chem 2004;12:5929–40. (c) El-Sayed MA, Abdel-Aziz NI, Abdel-Aziz AAM, et al. Design, synthesis, and biological evaluation of substituted hydrazone and pyrazole derivatives as selective COX-2 inhibitors: molecular docking study. Bioorg Med Chem 2011;19:3416–24. (d) El-Sayed MA, Abdel-Aziz NI, Abdel-Aziz AAM, et al. Synthesis, biological evaluation and molecular modeling study of pyrazole and pyrazoline derivatives as selective COX-2 inhibitors and anti-inflammatory agents. Part 2. Bioorg Med Chem 2012;20:3306–16.
- (a) Abdel-Sayed MA, Bayomi SM, El-Sherbeny MA, et al. Synthesis, anti-inflammatory, analgesic, COX-1/2 inhibition activities and molecular docking study of pyrazoline derivatives. Bioorg Med Chem 2016;24:2032–42. (b) Abdel-Aziz AAM, El-Azab AS, Abou-Zeid LA, et al. Synthesis, anti-inflammatory, analgesic and COX-1/2 inhibition activities of anilides based on 5,5-diphenylimidazolidine-2,4-dione scaffold: molecular docking studies. Eur J Med Chem 2016;115:121–31.
- (a) Abdel-Aziz AAM, Abou-Zeid LA, ElTahir KEH, et al. Synthesis, anti-inflammatory, analgesic, COX-1/2 inhibitory activities and molecular docking studies of substituted 2-mercapto-4(3H)-quinazolinones. Eur J Med Chem 2016;121:410–21. (b) Abdel-Aziz AAM, Abou-Zeid LA, ElTahir KE, et al. Design, synthesis of 2,3-disubstitued 4(3H)-quinazolinone derivatives as anti-inflammatory and analgesic agents: COX-1/2 inhibitory activities and molecular docking studies. Bioorg Med Chem 2016;24:3818–28.
- Funakoshi-Tago M, Shimizu T, Tago K, et al. Celecoxib potently inhibits TNFα-induced nuclear translocation and activation of NF-κB. Biochem Pharmacol 2008;76:662–71.
- Gupta M, Kaur G. Aqueous extract from the Withania somnifera leaves as a potential anti-neuroinflammatory agent: a mechanistic study. J Neuroinflammation 2016;13:193–209.
- Molecular Operating Environment (MOE 2008.10) of Chemical Computing Group. Inc. Canada. Available from: http://www.chemcomp.com. 2008.
- El-Zahar MI, Abd El-Karim SS, Haiba ME. Synthesis and cytotoxic evaluation of some novel 6- (benzofuran-2-yl)-4-(4-fluorophenyl) pyridines. World J Chem 2008;4:182–194.
- (a) Davidson D, Weiss M, Jelling M. The action of ammonia on benzil. J Org Chem 1937;2:319–27. (b) Behmadi H, Roshani M, Saadati MS. Synthesis of phenanthrimidazole from 9,10-phenanthraquinone and aldehydes by molecular iodine as catalyst. Chin Chem Lett 2009;20:5–8.
- Arshia P, Azim A, Shaikh KA. Ceric ammonium nitrate catalyzed efficient one-pot synthesis of 2, 4, 5-triarylimidazoles. Res J Pharm Biol Chem Sci 2010;1:943–951.
- Neelam PP, Rajesh HV, Hitesh SP. Ceric Ammonium Nitrate (CAN)–Catalyzed Multicomponent Reactions: An efficient catalyst for green organic synthesis. Synthetic Commun 2015;45:2399–2425.
- (a) Daniel LP, Carsten B. Recent advances in the Willgerodt-Kindler reaction. Chem Soc Rev 2013;42:7870–7880. (b) Aghapoor K, Mohsenazadeh F, Khanalizadeh G, et al. The Willgerodt-Kindler reaction in water: high chemoselectivity of benzaldehydes over acetophenones. Monatshefte Chem Chem Month 2007;138:61.
- (a) El-Zahar MI, Abd El-Karim SS, Haiba ME, et al. Synthesis, antitumor activity and molecular docking study of novel benzofuran-2-yl pyrazole pyrimidine derivatives. Acta Pol Pharm Drug Res 2011;68:357–73. (b) Hussain SM, El‐Reedy AM, Hassan Rezk AM, et al. Reactions with 2-mercaptopyrimidines. Synthesis of some new thiazolo[3,2-a]- and triazolo[4,3-a]pyrimidines. J Heterocycl Chem 1987;24:1605–10.
- (a) Al-Suwaidan IA, Alanazi AM, El-Azab AS, et al. Molecular design, synthesis and biological evaluation of cyclic imides bearing benzenesulfonamide fragment as potential COX-2 inhibitors. Part 2. Bioorg Med Chem Lett 2013;23:2601–5. (b) Alanazi AM, El-Azab AS, Al-Suwaidan IA, et al. Structure-based design of phthalimide derivatives as potential cyclooxygenase-2 (COX-2) inhibitors: anti-inflammatory and analgesic activities. Eur J Med Chem 2015;92:115–23. (c) Abdel-Aziz AAM, El Tahir KEH, Asiri YA. Synthesis, anti-inflammatory activity and COX-1/COX-2 inhibition of novel substituted cyclic imides. Part 1: molecular docking study. Eur J Med Chem 2011;46:1648–55. (d) Abdel-Aziz AA, El-Azab AS, AlSaif NA, et al. Synthesis, anti-inflammatory, cytotoxic, and COX-1/2 inhibitory activities of cyclic imides bearing 3-benzenesulfonamide, oxime, and β-phenylalanine scaffolds: a molecular docking study. J Enzyme Inhib Med Chem 2020;35:610–21.
- (a) El-Gamal MI, Bayomi SM, El-Ashry SM, et al. Synthesis and anti-inflammatory activity of novel (substituted)benzylidene acetone oxime ether derivatives: molecular modeling study. Eur J Med Chem 2010;45:1403–14. (b) Goda FE, Abdel-Aziz AAM, Ghoneim HA. Synthesis and biological evaluation of novel 6-nitro-5-substituted aminoquinolines as local anesthetic and anti-arrhythmic agents: molecular modeling study. Bioorg Med Chem 2005;13:3175–83. (c) Goda FE, Abdel-Aziz AAM, Attef OA. Synthesis, antimicrobial activity and conformational analysis of novel substituted pyridines: BF3-promoted reaction of hydrazine with 2-alkoxy pyridines. Bioorg Med Chem 2004;12:1845–52. (d) El-Azab AS, Mary YS, Panicker CY, et al. DFT and experimental (FT-IR and FT-Raman) investigation of vibrational spectroscopy and molecular docking studies of 2-(4-oxo-3-phenethyl-3,4-dihydroquinazolin-2-ylthio)-N-(3,4,5-trimethoxyphenyl) acetamide. J Mol Struct 2016;1113:133–45. (e) El-Azab AS, Abdel-Aziz AA, Ahmed HEA, et al. Exploring structure–activity relationship of S-substituted 2-mercaptoquinazolin-4(3H)-one including 4-ethylbenzenesulfonamides as human carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2020;35:598–609.
- Kurumbail RG, Stevens AM, Gierse JK, et al. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature 1996;384:644–8.
- Card GL, England BP, Suzuki Y, et al. Structural basis for the activity of drugs that inhibit phosphodiesterases. Structure 2004;12:2233–47.