
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Aiming to deepen the structure–activity relationships of the two P-glycoprotein (P-gp) modulators elacridar and tariquidar, a new series of amide and ester derivatives carrying a 6,7-dimethoxy-2-phenethyl-1,2,3,4-tetrahydroisoquinoline scaffold linked to different methoxy-substituted aryl moieties were synthesised. The obtained compounds were evaluated for their P-gp interaction profile and selectivity towards the two other ABC transporters, multidrug-resistance-associated protein-1 and breast cancer resistance protein, showing to be very active and selective versus P-gp. Two amide derivatives, displaying the best P-gp activity, were tested in co-administration with the antineoplastic drug doxorubicin in different cancer cell lines, showing a significant sensitising activity towards doxorubicin. The investigation on the chemical stability of the derivatives towards spontaneous or enzymatic hydrolysis, showed that amides are stable in both models while some ester compounds were hydrolysed in human plasma. This study allowed us to identify two chemosensitizers that behave as non-transported substrates and are characterised by different selectivity profiles.
Graphical Abstract
Introduction
The chemotherapy against cancer is often undermined by resistance that tumour cells develop to cytotoxic compounds after an exposure period. Multidrug resistance (MDR) was described as acquired resistance developed towards a multiplicity of structurally unrelated chemotherapeutic drugsCitation1, to which cells could also have never been exposedCitation2. The MDR phenotype is often related to an enhanced energy-dependent drug efflux, elicited by the overexpression of transmembrane proteins that extrude substrates in a unidirectional way and reduce the concentration of antitumor drugs below the active dose. In human MDR cells, these proteins are part of the ATP-binding cassette (ABC) transporter familyCitation3,Citation4, and use the energy deriving from ATP hydrolysis for this active transport.
In addition to be overexpressed in cancer cells, these proteins are also present in many healthy tissues where they arouse different physiological and pharmacological actionsCitation5, by regulating the permeability of biological membranes. In fact, they are often involved in the protection of these tissues from xenobiotics and can influence ADME properties and bioavailability of many drugs. The human genome encodes 49 ABC transmembrane proteins, divided into seven subfamilies (ABC-A to ABC-G), based on the similarity of their amino acid sequencesCitation6. Three transport proteins: P-glycoprotein (P-gp, ABCB1)Citation7,Citation8, multidrug-resistance-associated protein-1 (MRP1, ABCC1)Citation9,Citation10, and breast cancer resistance protein (BCRP, ABCG2)Citation11,Citation12 have mainly been associated with MDRCitation13–15.
Studies on P-gp showed the first evidences of a relationship between its overexpression and tumour responses to cytotoxic drugs; then also MRP1 and BCRP overexpressionCitation16,Citation17 was highlighted in different kind of tumours. Evidences also suggest that these three pumps can be co-expressedCitation18.
Since the discovery of P-gp, the identification of compounds that act as inhibitors of P-gp and then of the sister proteins has been considered a possible way to fight against MDR. These compounds can be defined as chemo-sensitizers or MDR reversers; they should restore the efficacy of cytotoxic agents in resistant cancer cells if co-administered with the drugs which are substrates of the transportersCitation19.
A great number of compounds showing P-gp modulating activity has been synthesised and studied, and were classified as first, second- or third-generationCitation20,Citation21 but none of these compounds has overcome the clinical trials, because no substantial benefits have been established. In fact, they often showed a low potency; moreover, they interacted with CYP4503A4 and altered the pharmacokinetic profiles of the co-administered antitumor agents, leading therefore also to increased toxicityCitation22,Citation23, although some of the latest MDR-reversing compounds show a safer profile. Therefore, aiming to overcome the obstacles described above, the search for safer ABC-transporter dependent MDR modulators is still pursued.
Two of the most interesting third-generation chemo-sensitizers are the tetrahydroisoquinoline derivatives elacridar (GF120918 or GW120918)Citation24 and tariquidar (XR9576)Citation25. These compounds displayed a high affinity towards the ABC proteins and a reduced effect on cytochromes, and few pharmacokinetic interactions with the cytotoxic drugsCitation26. Disappointingly, clinical trialsCitation27 failed to show an improvement of the efficacy of antitumor agents after co-administration of these compoundsCitation28,Citation29, and they have not been approved for therapy.
Elacridar and tariquidar share a common structural characteristic, the presence of a 6,7-dimethoxy-2-phenethyl-1,2,3,4-tetrahydroisoquinoline moiety connected to an aryl substituted amide function (). Early studies indicated that both derivatives are not specific for P-gp because they are also able to bind the BCRP transporterCitation30. Subsequent structure-activity relationship studies suggested that the 6,7-dimethoxy-2-phenethyl-1,2,3,4-tetrahydroisoquinoline nucleus is essential for inhibiting the two transporter proteins P-gp and BCRP while changes at the aryl-substituted amide moiety elicit variations in the selectivityCitation31. In the case of tariquidar, recent evidences indicated that this molecule is able to bind also the MRP1 transporterCitation32; the same compound was shown to potentiate the sensitivity to paclitaxel in resistant cells transfected by another member of the ABCC family, the MRP7 proteinCitation33.
Chart 1. Tariquidar and elacridar.
Therefore, despite the failure in clinical trials, tariquidar and elacridar have been considered lead compounds in the search for new MDR modulators, and a large number of analogues of these two derivatives have been synthesised.
Our research is part of this field of interest and aims to achieve information about the structural characteristics that confer potency and/or selectivity towards this family of transporters. In a previous studyCitation34, we reported a new series of derivatives bearing the 6,7-dimethoxy-2-phenethyl-1,2,3,4-tetrahydroisoquinoline moiety linked, as elacridar and tariquidar, to an aryl substituted amide, on which different aryl nucleus have been inserted (); we also synthesised the corresponding isosteric ester derivatives. The aryl residues were selected based on their presence in other compounds bearing different skeletons, acting as very potent MDR reversersCitation35–40. The obtained compounds were studied to evaluate their P-gp interaction profile and selectivity towards MPR1 and BCRP, using Madin-Darby Canine Kidney (MDCK) transfected cells.
Figure 1. (A) Compounds described in the previous studyCitation34; (B) compounds synthesised in the present work.
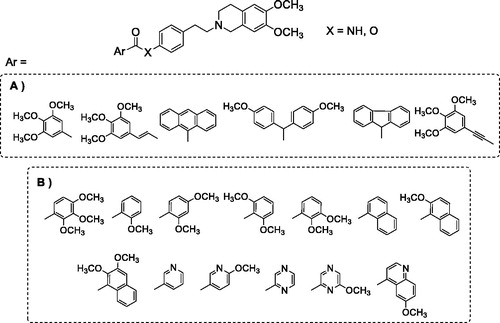
Both amide and ester derivatives were active, although less potent with respect to lead derivatives and, in general, more selective towards P-gp. Interestingly, biological data indicated that in these series of tetrahydroisoquinoline derivatives the presence of an amide function was not essential for modulating the transporter proteins; and the presence of an ester function in place of an amide seemed to influence P-gp selectivity. As a continuation of this study, aiming to better evaluate the features for selectivity towards the three transporters and to deepen the structure–activity relationships of the series, we designed and synthesised a new series of amide and ester derivatives characterised by the presence of a 6,7-dimethoxy-2-phenethyl-1,2,3,4-tetrahydroisoquinoline scaffold linked to additional different aryl moieties (). The new aryl moieties are methoxy-substituted phenyl or naphthyl nuclei, or nitrogen-containing hetero-aromatic residues.
The synthesised compounds were evaluated for their P-gp interaction profile and selectivity towards the two other ABC transporters, MPR1 and BCRP. The P-gp interacting-mechanism was investigated combining three assays: (i) apparent permeability (Papp) determination (BA/AB) in Caco-2 cell monolayer; (ii) ATP cell depletion in cells overexpressing the transporter (MDCK-MDR1); (iii) the inhibition of Calcein-AM transport in MDCK-MDR1 cells. The activity on MRP1 and BCRP was evaluated on cancer cell lines overexpressing each transporter (MDCK-MRP1 and MDCK-BCRP cells), by measuring the inhibition of the efflux of the pro-fluorescent probe Calcein-AM in MDCK-MRP1 cells or the fluorescent probe Hoechst 33342 in MDCK-BCRP cells.
Furthermore two selected compounds were further tested alone and in co-administration with the antineoplastic drug doxorubicin in different cancer cell lines with various levels of P-gp.
Finally, the stability of amide and ester derivatives was investigated in phosphate buffer solution (PBS) and human plasma, and the degradation profiles of the molecules were evaluated.
Material and methods
Chemistry
All melting points were taken on a Büchi apparatus and are uncorrected. NMR spectra were recorded on a Bruker Avance 400 spectrometer (400 MHz for 1H-NMR, 100 MHz for 13 C-NMR). 1H and 13 C NMR spectra were measured at room temperature (25 °C) in an appropriate solvent. 1H and 13 C chemical shifts are expressed in ppm (δ) referenced to TMS. Spectral data are reported using the following abbreviations: s = singlet, d = doublet, dd = doublet of doublets, t = triplet, td = triplet of doublets, m = multiplet, and coupling constants are reported in Hz, followed by integration.
Chromatographic separations were performed on a silica gel column by gravity chromatography (Kieselgel 40, 0.063–0.200 mm; Merck) or flash chromatography (Kieselgel 40, 0.040–0.063 mm; Merck). Yields are given after purification, unless otherwise stated.
The high resolution mass spectrometry (HRMS) analysis was performed with a Thermo Finnigan LTQ Orbitrap mass spectrometer equipped with an electrospray ionisation source (ESI). The accurate mass measure was carried out by introducing, via syringe pump at 10 μL min−1, the sample solution (1.0 μg mL−1 in mQ water: acetonitrile 50:50), and the signal of the positive ions was acquired. The proposed experimental conditions allowed to monitoring the protonated molecules of studied compounds ([M + H]+ species), that they were measured with a proper dwell time to achieve 60,000 units of resolution at Full Width at Half Maximum (FWHM). The elemental composition of compounds was calculated on the basis of their measured accurate masses, accepting only results with an attribution error less than 2.5 ppm and a not integer RDB (double bond/ring equivalents) value, in order to consider only the protonated speciesCitation41.
Compounds were named following IUPAC rules as applied by ChemBioDraw Ultra 14.0 software. When reactions were performed in anhydrous conditions, the mixtures were maintained under nitrogen. Free bases 1–26 were transformed into the hydrochloride by treatment with a solution of acetyl chloride (1.1 eq) in anhydrous CH3OH. The salts were crystallised from abs. ethanol/petroleum ether.
General procedures for the synthesis of the amide derivatives 1–13
Compounds were synthesised using two different general procedures.
General procedure A. To a solution of the aniline 27Citation34 (0.25 mmol) in 5 ml of an. CH2Cl2 or an. CH3CN at 0 °C, the proper carboxylic acid (0.30 mmol), DMAP (0.20 mmol) and EDC hydrochloride (0.45 mmol) were added. The reaction mixture was stirred at 0 °C for 1 h, then at room temperature for 48 h. The mixture was treated with CH2Cl2 and the organic layer was washed with water and with a saturated solution of NaHCO3. After drying with Na2SO4, the solvent was removed under reduced pressure. The residue was then purified by flash chromatography using the appropriate eluting system, obtaining the desired compound as an oil.
General procedure B. The proper carboxylic acid (0.30 mmol) was transformed into the acyl chloride by reaction with SOCl2 (4 eq) in 5 ml of CHCl3 (free of ethanol) or an. CH3CN at 60 °C for 6–8 h. The reaction mixture was cooled to rt, and the solvent was removed under reduced pressure. The mixture was then treated twice with cyclohexane and the solvent removed under reduced pressure. The acyl chloride obtained was dissolved in CHCl3 (free of ethanol), and the aniline 27Citation34 (0.25 mmol) was added. The mixture was kept at rt for 24 h, then the organic layer was washed twice with a saturated solution of NaHCO3. After drying with Na2SO4, the solvent was removed under reduced pressure. The residue obtained was purified by flash chromatography using the appropriate eluting system, yielding the desired compound as an oil.
In both procedures A and B, all the compounds were transformed into the corresponding hydrochloride as solid. The salts were crystallised from abs. ethanol/petroleum ether.
N-(4–(2-(6,7-dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl)-2,3,4-trimethoxybenzamide (1)
Procedure B in CHCl3 (free of ethanol), starting from aniline 27 and 2,3,4-trimethoxybenzoic acid.
Free base: chromatographic eluent: CH2Cl2/MeOH/NH4OH 97:3:0.3. Yield: 15.1%.
1H NMR (400 MHz, CDCl3) δ: 9.92 (s, 1H, NH); 7.98 (d, J = 8.8 Hz, 1H, CH arom.); 7.60 (d, J = 8.4 Hz, 2H, CH arom.); 7.23 (d, J = 8.4 Hz, 2H, CH arom.); 6.82 (d, J = 8.8 Hz, 1H, CH arom.); 6.60 (s, 1H, CH arom.); 6.53 (s, 1H, CH arom.); 4.05 (s, 3H, OCH3); 3.92 (s, 3H, OCH3); 3.90 (s, 3H, OCH3); 3.84 (s, 3H, OCH3); 3.83 (s, 3H, OCH3); 3.69 (s, 2H, NCH2Ar); 2.96–2.89 (m, 2H, CH2); 2.88–2.75 (m, 6H, CH2) ppm. 13 C NMR (100 MHz, CDCl3) δ: 162.71 (C=O); 156.78 (C); 152.14 (C); 147.66 (C); 147.32 (C); 136.69 (C); 135.84 (C); 129.26 (CH arom.); 126.96 (CH arom.); 125.97 (C); 125.91 (C); 120.33 (CH arom.); 119.00 (C); 111.38 (CH arom.); 109.49 (CH arom.); 107.89 (CH arom.); 61.97 (OCH3); 61.05 (OCH3); 59.93 (CH2); 56.10 (OCH3); 55.95 (OCH3); 55.91 (OCH3); 55.46 (CH2); 50.91 (CH2); 33.24 (CH2); 28.35 (CH2) ppm. ESI-HRMS (m/z) calculated for [M + H]+ ion species C29H35N2O6= 507.2490, found 507.2492.
Hydrochloride: mp 208–211 °C.
N-(4–(2-(6,7-dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl)-2-methoxybenzamide (2)
Procedure A in an. CH2Cl2, starting from aniline 27 and 2-methoxybenzoic acid.
Free base: chromatographic eluent: CH2Cl2/MeOH/NH4OH 97:3:0.3. Yield: 88.9%.
1H NMR (400 MHz, CDCl3) δ: 9.71 (s, 1H, NH); 8.22 (d, J = 8.0 Hz, 1H, CH arom.); 7.56 (d, J = 8.4 Hz, 2H, CH arom.); 7.41 (t, J = 8.0 Hz, 1H, CH arom.); 7.18 (d, J = 8.4 Hz, 2H, CH arom.); 7.06 (t, J = 8.0 Hz, 1H, CH arom.); 6.95 (d, J = 8.0 Hz, 1H, CH arom.); 6.55 (s, 1H, CH arom.); 6.49 (s, 1H, CH arom.); 3.96 (s, 3H, OCH3); 3.79 (s, 3H, OCH3); 3.78 (s, 3H, OCH3); 3.60 (s, 2H, NCH2Ar); 2.90–2.67 (m, 8H, CH2) ppm. 13C NMR (100 MHz, CDCl3) δ: 163.13 (C=O); 157.19 (C); 147.59 (C); 147.27 (C); 136.50 (C); 136.09 (C); 133.16 (CH arom.); 132.39 (CH arom.); 129.17 (CH arom.); 126.29 (C); 126.06 (C); 121.83 (C); 121.59 (CH arom.); 120.58 (CH arom.); 111.57 (CH arom.); 111.44 (CH arom.); 109.56 (CH arom.); 60.03 (CH2); 56.21 (OCH3); 55.95 (OCH3); 55.91 (OCH3); 55.56 (CH2); 50.96 (CH2); 33.33 (CH2); 28.53 (CH2) ppm. ESI-HRMS (m/z) calculated for [M + H]+ ion species C27H31N2O4= 447.2278, found 447.2279.
Hydrochloride: mp 226–228 °C.
N-(4–(2-(6,7-dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl)-2,4-dimethoxybenzamide (3)
Procedure B in CHCl3 (free of ethanol), starting from aniline 27 and 2,4-dimethoxybenzoic acid.
Free base: chromatographic eluent: CH2Cl2/MeOH/NH4OH 97:3:0.3. Yield: 54.7%.
1H NMR (400 MHz, CDCl3) δ: 9.69 (s, 1H, NH); 8.26 (d, J = 8.8 Hz, 1H, CH arom.); 7.60 (d, J = 8.4 Hz, 2H, CH arom.); 7.23 (d, J = 8.4 Hz, 2H, CH arom.); 6.66 (dd, J = 8.8 Hz, 2.2 Hz, 1H, CH arom.); 6.62 (s, 1H, CH arom.); 6.55 (s, 1H, CH arom.); 6.54 (d, J = 2.2 Hz, 1H, CH arom.); 4.03 (s, 3H, OCH3); 3.88 (s, 3H, OCH3); 3.86 (s, 3H, OCH3); 3.85 (s, 3H, OCH3); 3.73 (s, 2H, NCH2Ar); 3.00–2.94 (m, 2H, CH2); 2.93–2.77 (m, 6H, CH2) ppm. 13C NMR (100 MHz, CDCl3) δ: 163.70 (C=O); 163.04 (C); 158.51 (C); 147.66 (C); 147.31 (C); 136.78 (C); 135.43 (C); 134.16 (CH arom.); 129.18 (CH arom.); 125.71 (C); 120.57 (CH arom.); 114.67 (C); 111.27 (CH arom.); 109.38 (CH arom.); 105.65 (CH arom.); 98.74 (CH arom.); 59.80 (CH2); 56.20 (OCH3); 55.93 (OCH3); 55.89 (OCH3); 55.59 (OCH3); 55.30 (CH2); 50.85 (CH2); 33.11 (CH2); 28.16 (CH2) ppm. ESI-HRMS (m/z) calculated for [M + H]+ ion species C28H33N2O5= 477.2384, found 477.2379.
Hydrochloride: mp 233–235 °C.
N-(4–(2-(6,7-dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl)-2,6-dimethoxybenzamide (4)
Procedure B in CHCl3 (free of ethanol), starting from aniline 27 and 2,6-dimethoxybenzoic acid.
Free base: chromatographic eluent: CH2Cl2/MeOH 95:5. Yield: 75.0%.
1H NMR (400 MHz, CDCl3) δ: 7.53 (d, J = 8.4 Hz, 2H, CH arom.); 7.50 (s, 1H, NH); 7.24 (t, J = 8.4 Hz, 1H, CH arom.); 7.15 (d, J = 8.4 Hz, 2H, CH arom.); 6.55 (s, 1H, CH arom.); 6.53 (d, J = 8.4 Hz, 2H, CH arom.); 6.48 (s, 1H, CH arom.); 3.79 (s, 3H, OCH3); 3.78 (s, 3H, OCH3); 3.76 (s, 6H, OCH3); 3.63 (s, 2H, NCH2Ar); 2.90–2.70 (m, 8H, CH2) ppm. 13C NMR (100 MHz, CDCl3) δ: 163.65 (C=O); 157.55 (C); 147.64 (C); 147.31 (C); 136.58 (C); 135.85 (C); 131.03 (CH arom.); 129.13 (CH arom.); 125.91 (C); 119.82 (CH arom.); 116.00 (C); 111.39 (CH arom.); 109.52 (CH arom.); 104.11 (CH arom.); 59.92 (CH2); 56.02 (OCH3); 55.94 (OCH3); 55.90 (OCH3); 55.42 (CH2); 50.90 (CH2); 33.17 (CH2); 28.34 (CH2) ppm. ESI-HRMS (m/z) calculated for [M + H]+ ion species C28H33N2O5= 477.2384, found 477.2384.
Hydrochloride: mp 212–214 °C.
N-(4–(2-(6,7-dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl)-2,3-dimethoxybenzamide (5)
Procedure B in CHCl3 (free of ethanol), starting from aniline 27 and 2,3-dimethoxybenzoic acid.
Free base: chromatographic eluent: CH2Cl2/MeOH/NH4OH 97:3:0.3. Yield: 65.6%.
1H NMR (400 MHz, CDCl3) δ: 9.93 (s, 1H, NH); 7.73 (dd, J = 8.0, 1.6 Hz, 1H, CH arom.); 7.58 (d, J = 8.4 Hz, 2H, CH arom.); 7.20 (d, J = 8.4 Hz, 2H, CH arom.); 7.15 (t, J = 8.0 Hz, 1H, CH arom.); 7.03 (dd, J = 8.0, 1.6 Hz, 1H, CH arom.); 6.56 (s, 1H, CH arom.); 6.50 (s, 1H, CH arom.); 3.93 (s, 3H, OCH3); 3.87 (s, 3H, OCH3); 3.80 (s, 3H, OCH3); 3.79 (s, 3H, OCH3); 3.62 (s, 2H, NCH2Ar); 2.90–2.84 (m, 2H, CH2); 2.83–2.69 (m, 6H, CH2) ppm. 13C NMR (100 MHz, CDCl3) δ: 162.90 (C=O); 152.61 (C); 147.57 (C); 147.25 (C); 147.20 (C); 136.51 (C); 136.28 (C); 129.29 (CH arom.); 126.93 (C); 126.40 (C); 126.10 (C); 124.74 (CH arom.); 122.93 (CH arom.); 120.28 (CH arom.); 115.70 (CH arom.); 111.40 (CH arom.); 109.52 (CH arom.); 61.65 (OCH3); 60.13 (CH2); 56.14 (OCH3); 55.94 (OCH3); 55.91 (OCH3); 55.64 (CH2); 51.00 (CH2); 33.40 (CH2); 28.62 (CH2) ppm. ESI-HRMS (m/z) calculated for [M + H]+ ion species C28H33N2O5= 477.2384, found 477.2375.
Hydrochloride: mp 230–233 °C.
N-(4–(2-(6,7-dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl)-1-naphthamide (6)
Procedure B in CHCl3 (free of ethanol), starting from aniline 27 and 1-naphthoic acid.
Free base: chromatographic eluent: CH2Cl2/MeOH/NH4OH 97:3:0.3. Yield: 53.6%.
1H NMR (400 MHz, CDCl3) δ: 8.29–8.27 (m, 1H, CH arom.); 7.95 (s, 1H, NH); 7.87 (d, J = 7.6 Hz, 1H, CH arom.); 7.84–7.81 (m, 1H, CH arom.); 7.62 (d, J = 7.6 Hz, 1H, CH arom.); 7.56 (d, J = 8.4 Hz, 2H, CH arom.); 7.51–7.46 (m, 2H, CH arom.); 7.38 (t, J = 7.6 Hz, 1H, CH arom.); 7.19 (d, J = 8.4 Hz, 2H, CH arom.); 6.56 (s, 1H, CH arom.); 6.49 (s, 1H, CH arom.); 3.79 (s, 3H, OCH3); 3.78 (s, 3H, OCH3); 3.63 (s, 2H, NCH2Ar); 2.92–2.85 (m, 2H, CH2); 2.84–2.68 (m, 6H, CH2) ppm. 13C NMR (100 MHz, CDCl3) δ: 167.58 (C=O); 147.60 (C); 147.28 (C); 136.67 (C); 136.24 (C); 134.49 (C); 133.72 (C); 130.91 (CH arom.); 130.10 (C); 129.31 (CH arom.); 128.40 (CH arom.); 127.27 (CH arom.); 126.53 (CH arom.); 126.28 (C); 126.07 (C); 125.30 (CH arom.); 125.10 (CH arom.); 124.71 (CH arom.); 120.29 (CH arom.); 111.42 (CH arom.); 109.54 (CH arom.); 60.05 (CH2); 55.94 (OCH3); 55.90 (OCH3); 55.61 (CH2); 51.00 (CH2); 33.36 (CH2); 28.56 (CH2) ppm. ESI-HRMS (m/z) calculated for [M + H]+ ion species C30H31N2O3= 467.2329, found 467.2320.
Hydrochloride: mp 218–220 °C.
N-(4–(2-(6,7-dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl)-2-methoxy-1-naphthamide (7)
Procedure A in an. CH2Cl2, starting from aniline 27 and 2-methoxy-1-naphthoic acid.
Free base: chromatographic eluent: CH2Cl2/MeOH 95:5. Yield: 30.5%.
1H NMR (400 MHz, CDCl3) δ: 7.99 (d, J = 8.0 Hz, 1H, CH arom); 7.84 (d, J = 8.8 Hz, 1H, CH arom.); 7.81 (s, 1H, NH); 7.75 (d, J = 8.0 Hz, 1H, CH arom.); 7.60 (d, J = 8.4 Hz, 2H, CH arom.); 7.44 (t, J = 8.0 Hz, 1H, CH arom.); 7.33 (t, J = 8.0 Hz, 1H, CH arom.); 7.22 (d, J = 8.8 Hz, 1H, CH arom.); 7.21 (d, J = 8.4 Hz, 2H, CH arom.); 6.56 (s, 1H, CH arom.); 6.50 (s, 1H, CH arom.); 3.91 (s, 3H, OCH3); 3.80 (s, 3H, OCH3); 3.79 (s, 3H, OCH3); 3.63 (s, 2H, NCH2Ar); 2.95–2.86 (m, 2H, CH2); 2.85–2.69 (m, 6H, CH2) ppm. 13C NMR (100 MHz, CDCl3) δ: 165.45 (C=O); 153.71 (C); 147.60 (C); 147.28 (C); 136.45 (C); 131.57 (CH arom.); 129.28 (CH arom.); 128.85 (C); 128.03 (CH arom.); 127.66 (CH arom.); 126.38 (C); 126.11 (C); 124.34 (CH arom.); 124.25 (CH arom.); 120.51 (C); 120.07 (CH arom.); 113.08 (CH arom.); 111.44 (CH arom.); 109.57 (CH arom.); 60.12 (CH2); 56.77 (OCH3); 55.95 (OCH3); 55.91 (OCH3); 55.63 (CH2); 51.01 (CH2); 33.37 (CH2); 28.60 (CH2) ppm. ESI-HRMS (m/z) calculated for [M + H]+ ion species C31H33N2O4= 497.2435, found 497.2425.
Hydrochloride: mp 239–241 °C.
N-(4–(2-(6,7-dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl)-2,3-dimethoxy-1-naphthamide (8)
Procedure A in an. CH2Cl2, starting from aniline 27 and 2,3-dimethoxy-1-naphthoic acid.
Free base: chromatographic eluent: CH2Cl2/MeOH 95:5. Yield: 24.1%.
1H NMR (400 MHz, CDCl3) δ: 7.98 (d, J = 8.0 Hz, 1H, CH arom.); 7.90 (s, 1H, NH), 7.67 (d, J = 8.0 Hz, 1H, CH arom.); 7.61 (d, J = 8.4 Hz, 2H, CH arom.); 7.42–7.33 (m, 2H, CH arom.); 7.24 (d, J = 8.4 Hz, 2H, CH arom.); 7.10 (s, 1H, CH arom.); 6.57 (s, 1H, CH arom.); 6.51 (s, 1H, CH arom.); 3.93 (s, 3H, OCH3); 3.88 (s, 3H, OCH3); 3.82 (s, 3H, OCH3); 3.81 (s, 3H, OCH3); 3.66 (s, 2H, NCH2Ar); 2.95–2.88 (m, 2H, CH2); 2.87–2.74 (m, 6H, CH2) ppm. 13C NMR (100 MHz, CDCl3) δ: 164.75 (C=O); 151.32 (C); 147.63 (C); 147.30 (C); 146.22 (C); 136.57 (C); 136.27 (C); 131.39 (C); 129.38 (CH arom.); 127.22 (C); 126.73 (CH arom.); 126.03 (C); 125.96 (CH arom.); 125.12 (CH arom.); 124.71 (CH arom.); 120.17 (CH arom.); 111.37 (CH arom.); 109.49 (CH arom.); 108.85 (CH arom.); 62.40 (OCH3); 60.01 (CH2); 55.95 (OCH3); 55.91 (OCH3); 55.76 (OCH3); 55.55 (CH2); 50.97 (CH2); 33.32 (CH2); 28.47 (CH2) ppm. ESI-HRMS (m/z) calculated for [M + H]+ ion species C32H35N2O5= 527.2541, found 527.2534.
Hydrochloride: mp 245–248 °C.
N-(4–(2-(6,7-dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl)nicotinamide (9)
Procedure B in an. CH3CN, starting from aniline 27 and nicotinic acid.
Free base: chromatographic eluent: CH2Cl2/MeOH/NH4OH 93:7:0.3. Yield: 40.5%.
1H NMR (400 MHz, CDCl3) δ: 9.02 (s, 1H, NH); 8.70 (s, 1H, CH arom.); 8.62 (d, J = 4.4 Hz, 1H, CH arom.); 8.13 (d, J = 8.0 Hz, 1H, CH arom.); 7.52 (d, J = 8.4 Hz, 2H, CH arom.); 7.30 (dd, J = 8.0, 4.4 Hz, 1H, CH arom.); 7.16 (d, J = 8.4 Hz, 2H, CH arom.); 6.54 (s, 1H, CH arom.); 6.48 (s, 1H, CH arom.); 3.77 (s, 3H, OCH3); 3.76 (s, 3H, OCH3); 3.60 (s, 2H, NCH2Ar); 2.87–2.67 (m, 8H, CH2) ppm. 13C NMR (100 MHz, CDCl3) δ: 164.18 (C=O); 152.07 (CH arom.); 148.30 (CH arom.); 147.52 (C); 147.19 (C); 136.87 (C); 135.95 (C); 135.48 (CH arom.); 130.80 (C); 129.21 (CH arom.); 126.26 (C); 126.04 (C); 123.49 (CH arom.); 120.97 (CH arom.); 111.36 (CH arom.); 109.48 (CH arom.); 59.96 (CH2); 55.88 (OCH3); 55.84 (OCH3); 55.57 (CH2); 50.96 (CH2); 33.30 (CH2); 28.53 (CH2) ppm. ESI-HRMS (m/z) calculated for [M + H]+ ion species C25H28N3O3= 418.2125, found 418.2128.
Hydrochloride: mp 151–154 °C.
N-(4–(2-(6,7-dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl)-6-methoxynicotinamide (10)
Procedure B in an. CH3CN, starting from aniline 27 and 6-methoxynicotinic acid.
Free base: chromatographic eluent: CH2Cl2/MeOH/NH4OH 97:3:0.3. Yield: 29.3%.
1H NMR (400 MHz, CDCl3) δ: 8.65 (s, 1H, CH arom.); 8.03 (d, J = 8.4 Hz, 1H, CH arom.); 7.89 (s, 1H, NH); 7.50 (d, J = 6.8 Hz, 2H, CH arom.); 7.18 (d, J = 6.8 Hz, 2H, CH arom.); 6.75 (d, J = 8.4 Hz, 1H, CH arom.); 6.56 (s, 1H, CH arom.); 6.50 (s, 1H, CH arom.); 3.95 (s, 3H, OCH3); 3.80 (s, 3H, OCH3); 3.79 (s, 3H, OCH3); 3.63 (s, 2H, NCH2Ar); 2.91–2.70 (m, 8H, CH2) ppm. 13C NMR (100 MHz, CDCl3) δ: 166.07 (C=O); 164.08 (C); 147.67 (C); 147.33 (C); 146.84 (CH arom.); 137.93 (CH arom.); 136.31 (C); 136.15 (C); 129.20 (CH arom.); 125.83 (C); 124.04 (C); 120.79 (CH arom.); 111.38 (CH arom.); 110.78 (CH arom.); 109.50 (CH arom.); 59.69 (CH2); 55.92 (OCH3); 55.89 (OCH3); 55.34 (CH2); 53.94 (OCH3); 50.82 (CH2); 33.08 (CH2); 28.25 (CH2) ppm. ESI-HRMS (m/z) calculated for [M + H]+ ion species C26H30N3O4= 448.2231, found 448.2236.
Hydrochloride: mp 123–126 °C.
N-(4–(2-(6,7-dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl)pyrazine-2-carboxamide (11)
Procedure A in an. CH2Cl2, starting from aniline 27 and pyrazine-2-carboxylic acid.
Free base: chromatographic eluent: CH2Cl2/MeOH/NH4OH 97:3:0.3. Yield: 63.8%.
1H NMR (400 MHz, CDCl3) δ: 9.59 (s, 1H, NH); 9.45 (d, J = 1.2 Hz, 1H, CH arom.); 8.73 (d, J = 2.4 Hz, 1H, CH arom.); 8.51 (dd, J = 2.4, 1.2 Hz, 1H, CH arom.); 7.64 (d, J = 8.4 Hz, 2H, CH arom.); 7.22 (d, J = 8.4 Hz, 2H, CH arom.); 6.55 (s, 1H, CH arom.); 6.48 (s, 1H, CH arom.); 3.79 (s, 3H, OCH3); 3.78 (s, 3H, OCH3); 3.61 (s, 2H, NCH2Ar); 2.90–2.83 (m, 2H, CH2); 2.82–2.69 (m, 6H, CH2) ppm. 13C NMR (100 MHz, CDCl3) δ: 160.55 (C=O); 147.60 (C); 147.47 (CH arom.); 147.28 (C); 144.62 (CH arom.); 144.44 (C); 142.35 (CH arom.); 136.95 (C); 135.35 (C); 129.40 (CH arom.); 126.32 (C); 126.07 (C); 119.96 (CH arom.); 111.42 (CH arom.); 109.53 (CH arom.); 59.98 (CH2); 55.94 (OCH3); 55.90 (OCH3); 55.62 (CH2); 50.99 (CH2); 33.41 (CH2); 28.57 (CH2) ppm. ESI-HRMS (m/z) calculated for [M + H]+ ion species C24H27N4O3= 419.2078, found 419.2079.
Hydrochloride: mp 246–249 °C.
N-(4–(2-(6,7-dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl)-6-methoxypyrazine-2-carboxamide (12)
Procedure B in an. CH3CN, starting from aniline 27 and 6-methoxypyrazine-2-carboxylic acid.
Free base: chromatographic eluent: CH2Cl2/MeOH/NH4OH 93:7:0.3. Yield: 89.5%.
1H NMR (400 MHz, CDCl3) δ: 9.32 (s, 1H, NH); 9.02 (s, 1H, CH arom.); 8.41 (s, 1H, CH arom.); 7.62 (d, J = 8.4 Hz, 2H, CH arom.); 7.24 (d, J = 8.4 Hz, 2H, CH arom.); 6.57 (s, 1H, CH arom.); 6.50 (s, 1H, CH arom.); 4.06 (s, 3H, OCH3); 3.81 (s, 3H, OCH3); 3.80 (s, 3H, OCH3); 3.65 (s, 2H, NCH2Ar); 2.95–2.87 (m, 2H, CH2); 2.86–2.73 (m, 6H, CH2) ppm. 13C NMR (100 MHz, CDCl3) δ: 160.76 (C=O); 140.43 (C); 139.35 (CH arom.); 138.65 (C); 136.53 (C); 136.14 (CH arom.); 135.37 (C); 132.67 (C); 132.13 (C); 129.45 (CH arom.); 125.67 (C); 125.45 (C); 120.13 (CH arom.); 111.31 (CH arom.); 109.42 (CH arom.); 59.58 (CH2); 55.95 (OCH3); 55.91 (OCH3); 55.29 (CH2); 53.89 (OCH3); 50.82 (CH2); 33.10 (CH2); 28.04 (CH2) ppm. ESI-HRMS (m/z) calculated for [M + H]+ ion species C25H29N4O4= 449.2183, found 449.2187.
Hydrochloride: mp 224–226 °C.
N-(4–(2-(6,7-dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl)-6-methoxyquinoline-4-carboxamide (13)
Procedure B in CHCl3 (free of ethanol), starting from aniline 27 and 6-methoxyquinoline-4-carboxylic acid.
Free base: yield: 100.0%.
1H NMR (400 MHz, CDCl3) δ: 9.24 (s, 1H, NH); 8.35 (d, J = 4.0 Hz, 1H, CH arom.); 7.78 (d, J = 9.2 Hz, 1H, CH arom.); 7.68 (d, J = 8.0 Hz, 2H, CH arom.); 7.30 (d, J = 2.0 Hz, 1H, CH arom.); 7.24–7.20 (m, 3H, CH arom.); 7.13 (d, J = 4.0 Hz, 1H, CH arom.); 6.55 (s, 1H, CH arom.); 6.49 (s, 1H, CH arom.); 3.78 (s, 6H, OCH3); 3.76 (s, 3H, OCH3); 3.63 (s, 2H, NCH2Ar); 2.93–2.87 (m, 2H, CH2); 2.85–2.73 (m, 6H, CH2) ppm. 13C NMR (100 MHz, CDCl3) δ: 165.86 (C=O); 158.41 (C); 147.50 (C); 147.16 (C); 146.62 (CH arom.); 144.34 (C); 140.14 (C); 136.87 (C); 136.27 (C); 130.51 (CH arom.); 129.32 (CH arom.); 126.09 (C); 125.95 (C); 125.45 (C); 122.94 (CH arom.); 120.57 (CH arom.); 118.92 (CH arom.); 111.31 (CH arom.); 109.43 (CH arom.); 102.57 (CH arom.); 59.92 (CH2); 55.87 (OCH3); 55.83 (OCH3); 55.52 (OCH3); 55.48 (CH2); 50.88 (CH2); 33.21 (CH2); 28.42 (CH2) ppm. ESI-HRMS (m/z) calculated for [M + H]+ ion species C30H32N3O4= 498.2387, found 498.2395.
Hydrochloride: mp 228–230 °C.
General procedures for the synthesis of the ester compounds 14–26
Compounds were synthesised using two different general procedures.
General procedure A. To a solution of the phenol 28Citation34 (0.25 mmol) in 5 ml of an. CH2Cl2 or an. CH3CN at 0 °C, the proper carboxylic acid (0.30 mmol), DMAP (0.20 mmol) and EDC hydrochloride (0.45 mmol) were added. The reaction mixture was stirred at 0 °C for 1 h then at room temperature for 48 h. The mixture was treated with CH2Cl2 and the organic layer was washed with water and with a saturated solution of NaHCO3. After drying with Na2SO4, the solvent was removed under reduced pressure. The residue was then purified by flash chromatography using the appropriate eluting system, obtaining the desired compound as an oil.
General procedure B. The proper carboxylic acid (0.30 mmol) was transformed into the acyl chloride by reaction with SOCl2 (4 eq) in 5 ml of CHCl3 (free of ethanol) or an. CH3CN at 60 °C for 6–8 h. The reaction mixture was cooled to rt, and the solvent was removed under reduced pressure. The mixture was then treated twice with cyclohexane and the solvent removed under reduce pressure. The acyl chloride obtained was dissolved in CHCl3 (free of ethanol), and the phenol 28Citation34 (0.25 mmol) was added. The mixture was kept at rt for 24 h, then the organic layer was washed twice with a saturated solution of NaHCO3. After drying with Na2SO4, the solvent was removed under reduced pressure. The residue obtained was purified by flash chromatography using the appropriate eluting system, yielding the desired compound as an oil.
In both procedures A and B, all the compounds were transformed into the corresponding hydrochloride as solid. The salts were crystallised from abs. ethanol/petroleum ether.
4–(2-(6,7-Dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl 2,3,4-trimethoxybenzoate (14)
Procedure B in CHCl3 (free of ethanol), starting from phenol 28 and 2,3,4-trimethoxybenzoic acid.
Free base: chromatographic eluent: CH2Cl2/MeOH/NH4OH 97:3:0.3. Yield: 17.2%.
1H NMR (400 MHz, CDCl3) δ: 7.79 (d, J = 8.8 Hz, 1H, CH arom.); 7.28 (d, J = 8.4 Hz, 2H, CH arom.); 7.13 (d, J = 8.4 Hz, 2H, CH arom.); 6.75 (d, J = 8.8 Hz, 1H, CH arom.); 6.60 (s, 1H, CH arom.); 6.53 (s, 1H, CH arom.); 3.97 (s, 3H, OCH3); 3.93 (s, 3H, OCH3); 3.89 (s, 3H, OCH3); 3.84 (s, 3H, OCH3); 3.83 (s, 3H, OCH3); 3.68 (s, 2H, NCH2Ar); 2.97–2.91 (m, 2H, CH2); 2.90–2.76 (m, 6H, CH2) ppm. 13C NMR (100 MHz, CDCl3) δ: 163.97 (C=O); 157.79 (C); 155.34 (C); 149.32 (C); 147.66 (C); 147.33 (C); 143.19 (C); 137.61 (C); 129.67 (CH arom.); 127.50 (CH arom.); 126.03 (C); 125.94 (C); 121.76 (CH arom.); 117.10 (C); 111.39 (CH arom.); 109.51 (CH arom.); 107.03 (CH arom.); 61.90 (OCH3); 61.06 (OCH3); 59.88 (CH2); 56.15 (OCH3); 55.95 (OCH3); 55.91 (OCH3); 55.50 (CH2); 50.96 (CH2); 33.25 (CH2); 28.40 (CH2) ppm. ESI-HRMS (m/z) calculated for [M + H]+ ion species C29H34NO7= 508.2330, found 508.2326.
Hydrochloride: mp 207–210 °C.
4–(2-(6,7-Dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl 2-methoxybenzoate (15)
Procedure B in CHCl3 (free of ethanol), starting from phenol 28 and 2-methoxybenzoic acid.
Free base: chromatographic eluent: CH2Cl2/MeOH/NH4OH 97:3:0.3. Yield: 44.6%.
1H NMR (400 MHz, CDCl3) δ: 7.95 (dd, J = 8.0, 1.8 Hz, 1H, CH arom.); 7.48 (td, J = 8.0, 1.8 Hz, 1H, CH arom.); 7.23 (d, J = 8.4 Hz, 2H, CH arom.); 7.11 (d, J = 8.4 Hz, 2H, CH arom.); 7.02–6.96 (m, 2H, CH arom.); 6.56 (s, 1H, CH arom.); 6.50 (s, 1H, CH arom.); 3.87 (s, 3H, OCH3); 3.79 (s, 3H, OCH3); 3.78 (s, 3H, OCH3); 3.61 (s, 2H, NCH2Ar); 2.93–2.85 (m, 2H, CH2); 2.84–2.70 (m, 6H, CH2) ppm. 13C NMR (100 MHz, CDCl3) δ: 164.53 (C=O); 159.79 (C); 149.31 (C); 147.62 (C); 147.30 (C); 137.70 (C); 134.22 (CH arom.); 132.09 (CH arom.); 129.59 (CH arom.); 126.31 (C); 126.07 (C); 121.72 (CH arom.); 120.21 (CH arom.); 119.28 (C); 112.27 (CH arom.); 111.46 (CH arom.); 109.58 (CH arom.); 59.97 (CH2); 56.05 (OCH3); 55.95 (OCH3); 55.92 (OCH3); 55.59 (CH2); 51.00 (CH2); 33.34 (CH2); 28.55 (CH2) ppm. ESI-HRMS (m/z) calculated for [M + H]+ ion species C27H30NO5= 448.2119, found 448.2123.
Hydrochloride: mp 220–222 °C.
4–(2-(6,7-Dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl 2,4-dimethoxybenzoate (16)
Procedure B in CHCl3 (free of ethanol), starting from phenol 28 and 2,4-dimethoxybenzoic acid.
Free base: chromatographic eluent: CH2Cl2/MeOH/NH4OH 97:3:0.3. Yield: 18.7%.
1H NMR (400 MHz, CDCl3) δ: 8.02 (d, J = 8.8 Hz, 1H, CH arom.); 7.23 (d, J = 8.4 Hz, 2H, CH arom.); 7.09 (d, J = 8.4 Hz, 2H, CH arom.); 6.57 (s, 1H, CH arom.); 6.52 (dd, J = 8.8, 2.2 Hz, 1H, CH arom.); 6.51 (s, 1H, CH arom.); 6.49 (d, J = 2.2 Hz, 1H, CH arom.); 3.87 (s, 3H, OCH3); 3.85 (s, 3H, OCH3); 3.81 (s, 3H, OCH3); 3.80 (s, 3H, OCH3); 3.66 (s, 2H, NCH2Ar); 2.96–2.89 (m, 2H, CH2); 2.88–2.74 (m, 6H, CH2) ppm. 13C NMR (100 MHz, CDCl3) δ: 164.90 (C=O); 163.83 (C); 162.15 (C); 149.52 (C); 147.80 (C); 147.44 (C); 136.84 (C); 134.43 (CH arom.); 129.55 (CH arom.); 125.51 (C); 121.97 (CH arom.); 111.33 (CH arom.); 109.44 (CH arom.); 104.79 (CH arom.); 99.04 (CH arom.); 59.45 (CH2); 56.03 (OCH3); 55.96 (OCH3); 55.92 (OCH3); 55.57 (OCH3); 55.11 (CH2); 50.77 (CH2); 32.91 (CH2); 27.87 (CH2) ppm. ESI-HRMS (m/z) calculated for [M + H]+ ion species C28H32NO6= 478.2224, found 478.2230.
Hydrochloride: mp 221–223 °C.
4–(2-(6,7-Dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl 2,6-dimethoxybenzoate (17)
Procedure B in CHCl3 (free of ethanol), starting from phenol 28 and 2,6-dimethoxybenzoic acid.
Free base: chromatographic eluent: CH2Cl2/MeOH/NH4OH 97:3:0.3. Yield: 37.4%.
1H NMR (400 MHz, CDCl3) δ: 7.29 (t, J = 8.4 Hz, 1H, CH arom.); 7.24 (d, J = 8.4 Hz, 2H, CH arom.); 7.14 (d, J = 8.4 Hz, 2H, CH arom.); 6.56 (d, J = 8.4 Hz, 2H, CH arom.); 6.56 (s, 1H, CH arom.); 6.50 (s, 1H, CH arom.); 3.83 (s, 6H, OCH3); 3.80 (s, 3H, OCH3); 3.79 (s, 3H, OCH3); 3.65 (s, 2H, NCH2Ar); 2.94–2.87 (m, 2H, CH2); 2.86–2.70 (m, 6H, CH2) ppm. 13C NMR (100 MHz, CDCl3) δ: 165.12 (C=O); 157.63 (C); 149.47 (C); 147.71 (C); 147.37 (C); 137.59 (C); 131.58 (CH arom.); 129.62 (CH arom.); 125.87 (C); 121.75 (CH arom.); 112.52 (C); 111.42 (CH arom.); 109.55 (CH arom.); 104.04 (CH arom.); 59.73 (CH2); 56.13 (OCH3); 55.96 (OCH3); 55.92 (OCH3); 55.39 (CH2); 50.90 (CH2); 33.19 (CH2); 28.28 (CH2) ppm. ESI-HRMS (m/z) calculated for [M + H]+ ion species C28H32NO6= 478.2224, found 478.2224.
Hydrochloride: mp 213–215 °C.
4–(2-(6,7-Dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl 2,3-dimethoxybenzoate (18)
Procedure A in an. CH2Cl2, starting from phenol 28 and 2,3-dimethoxybenzoic acid.
Free base: chromatographic eluent: CH2Cl2/MeOH/NH4OH 97:3:0.3. Yield: 65.5%.
1H NMR (400 MHz, CDCl3) δ: 7.47 (dd, J = 7.2, 2.2 Hz, 1H, CH arom.); 7.26 (d, J = 8.4 Hz, 2H, CH arom.); 7.15–7.07 (m, 4H, CH arom.); 6.57 (s, 1H, CH arom.); 6.51 (s, 1H, CH arom.); 3.92 (s, 3H, OCH3); 3.87 (s, 3H, OCH3); 3.81 (s, 3H, OCH3); 3.80 (s, 3H, OCH3); 3.66 (s, 2H, NCH2Ar); 2.96–2.89 (m, 2H, CH2); 2.88–2.68 (m, 6H, CH2) ppm. 13C NMR (100 MHz, CDCl3) δ: 164.74 (C=O); 153.76 (C); 149.71 (C); 149.27 (C); 147.66 (C); 147.33 (C); 137.78 (C); 129.72 (CH arom.); 125.93 (C); 125.43 (C); 123.95 (CH arom.); 122.54 (CH arom.); 121.68 (CH arom.); 116.44 (CH arom.); 111.38 (CH arom.); 109.50 (CH arom.); 61.60 (OCH3); 59.88 (CH2); 56.14 (OCH3); 55.95 (OCH3); 55.92 (OCH3); 55.51 (CH2); 50.97 (CH2); 33.27 (CH2); 28.42 (CH2) ppm. ESI-HRMS (m/z) calculated for [M + H]+ ion species C28H32NO6= 478.2224, found 478.2221.
Hydrochloride: mp 237–239 °C.
4–(2-(6,7-Dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl 1-naphthoate (19)
Procedure A in an. CH2Cl2, starting from phenol 28 and 1-naphthoic acid.
Free base: chromatographic eluent: CH2Cl2/MeOH/NH4OH 97:3:0.3. Yield: 77.5%.
1H NMR (400 MHz, CDCl3) δ: 9.00 (d, J = 8.0 Hz, 1H, CH arom.); 8.44 (d, J = 7.6 Hz, 1H, CH arom.); 8.07 (d, J = 8.0 Hz, 1H, CH arom.); 7.89 (d, J = 7.6 Hz, 1H, CH arom.); 7.61 (t, J = 7.6 Hz, 1H, CH arom.); 7.54 (t, J = 8.0 Hz, 2H, CH arom.); 7.31 (d, J = 8.4 Hz, 2H, CH arom.); 7.19 (d, J = 8.4 Hz, 2H, CH arom.); 6.59 (s, 1H, CH arom.); 6.53 (s, 1H, CH arom.); 3.82 (s, 6H, OCH3); 3.67 (s, 2H, NCH2Ar); 2.99–2.91 (m, 2H, CH2); 2.90–2.76 (m, 6H, CH2) ppm. 13C NMR (100 MHz, CDCl3) δ: 165.97 (C=O); 149.32 (C); 147.67 (C); 147.35 (C); 137.93 (C); 134.30 (CH arom.); 133.95 (C); 131.70 (C); 131.19 (CH arom.); 129.82 (CH arom.); 128.71 (CH arom.); 128.17 (CH arom.); 126.42 (CH arom.); 126.14 (C); 126.00 (C); 125.76 (CH arom.); 124.54 (CH arom.); 121.81 (CH arom.); 111.42 (CH arom.); 109.54 (CH arom.); 59.94 (CH2); 55.97 (OCH3); 55.94 (OCH3); 55.59 (CH2); 51.01 (CH2); 33.35 (CH2); 28.50 (CH2) ppm. ESI-HRMS (m/z) calculated for [M + H]+ ion species C30H30NO4= 468.2169, found 468.2174.
Hydrochloride: mp 227–230 °C.
4–(2-(6,7-Dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl 2-methoxy-1-naphthoate (20)
Procedure A in an CH2Cl2, starting from phenol 28 and 2-methoxy-1-naphthoic acid.
Free base: chromatographic eluent: CH2Cl2/MeOH/NH4OH 95:5:0.5. Yield: 73.3%.
1H NMR (400 MHz, CDCl3) δ: 7.92 (d, J = 8.8 Hz, 2H, CH arom); 7.80 (d, J = 7.6 Hz, 1H, CH arom.); 7.52 (t, J = 7.6 Hz, 1H, CH arom.); 7.38 (t, J = 7.6 Hz, 1H, CH arom.); 7.34–7.27 (m, 3H, CH arom.); 7.25 (d, J = 8.4 Hz, 2H, CH arom.); 6.58 (s, 1H, CH arom.); 6.52 (s, 1H, CH arom.); 4.00 (s, 3H, OCH3); 3.83 (s, 3H, OCH3); 3.82 (s, 3H, OCH3); 3.65 (s, 2H, NCH2Ar); 3.00–2.90 (m, 2H, CH2); 2.89–2.75 (m, 6H, CH2) ppm. 13C NMR (100 MHz, CDCl3) δ: 166.67 (C=O); 155.02 (C); 149.38 (C); 147.67 (C); 147.34 (C); 137.85 (C); 132.26 (CH arom.); 131.02 (C); 129.79 (CH arom.); 128.55 (C); 128.24 (CH arom.); 127.92 (CH arom.); 125.86 (C); 124.29 (CH arom.); 123.58 (CH arom.); 121.71 (CH arom.); 116.70 (C); 113.17 (CH arom.); 111.36 (CH arom.); 109.49 (CH arom.); 59.78 (CH2); 56.92 (OCH3); 55.95 (OCH3); 55.92 (OCH3); 55.43 (CH2); 50.92 (CH2); 33.24 (CH2); 28.32 (CH2) ppm. ESI-HRMS (m/z) calculated for [M + H]+ ion species C31H32NO5= 498.2275, found 498.2272.
Hydrochloride: mp 208–210 °C.
4–(2-(6,7-Dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl 2,3-dimethoxy-1-naphthoate (21)
Procedure A in an. CH2Cl2, starting from phenol 28 and 2,3-dimethoxy-1-naphthoic acid.
Free base: chromatographic eluent: CH2Cl2/MeOH/NH4OH 95:5:0.5. Yield: 26.5%.
1H NMR (400 MHz, CDCl3) δ: 7.89–7.83 (m 1H, CH arom.); 7.75–7.69 (m, 1H, CH arom.); 7.44–7.38 (m, 2H, CH arom.); 7.32 (d, J = 8.0 Hz, 2H, CH arom.); 7.26 (s, 1H, CH arom.); 7.25 (d, J = 8.0 Hz, 2H, CH arom.); 6.58 (s, 1H, CH arom.); 6.52 (s, 1H, CH arom.); 4.02 (s, 3H, OCH3); 3.98 (s, 3H, OCH3); 3.82 (s, 3H, OCH3); 3.81 (s, 3H, OCH3); 3.65 (s, 2H, NCH2Ar); 2.98–2.90 (m, 2H, CH2); 2.89–2.74 (m, 6H, CH2) ppm. 13C NMR (100 MHz, CDCl3) δ: 166.08 (C=O); 151.66 (C); 149.22 (C); 147.66 (C); 147.48 (C); 147.33 (C); 138.08 (C); 131.14 (C); 129.90 (CH arom.); 126.90 (CH arom.); 125.90 (CH arom.); 125.47 (C); 125.31 (CH arom.); 123.95 (CH arom.); 123.59 (C); 121.66 (CH arom.); 111.36 (CH arom.); 109.61 (CH arom.); 109.48 (CH arom.); 61.94 (OCH3); 59.85 (CH2); 55.95 (OCH3); 55.91 (OCH3); 55.87 (OCH3); 55.49 (CH2); 50.95 (CH2); 33.27 (CH2); 28.39 (CH2) ppm. ESI-HRMS (m/z) calculated for [M + H]+ ion species C32H34NO6= 528.2381, found 528.2384.
Hydrochloride: mp 183–185 °C.
4–(2-(6,7-Dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl nicotinate (22)
Procedure A in an. CH3CN, starting from phenol 28 and nicotinic acid.
Free base: chromatographic eluent: CH2Cl2/MeOH/NH4OH 93:7:0.3. Yield: 16.7%.
1H NMR (400 MHz, CDCl3) δ: 9.36 (s, 1H, CH arom.); 8.82 (d, J = 4.8, 1H, CH arom.); 8.41 (d, J = 8.0, 1H, CH arom.); 7.43 (dd, J = 8.0, 4.8 Hz, 1H, CH arom.); 7.28 (d, J = 8.4 Hz, 2H, CH arom.); 7.12 (d, J = 8.4 Hz, 2H, CH arom.); 6.57 (s, 1H, CH arom.); 6.51 (s, 1H, CH arom.); 3.81 (s, 3H, OCH3); 3.80 (s, 3H, OCH3); 3.65 (s, 2H, NCH2Ar); 2.97–2.90 (m, 2H, CH2); 2.88–2.74 (m, 6H, CH2) ppm. 13C NMR (100 MHz, CDCl3) δ: 164.00 (C=O); 153.97 (CH arom.); 151.37 (CH arom.); 148.87 (C); 147.73 (C); 147.39 (C); 138.14 (C); 137.57 (CH arom.); 129.85 (CH arom.); 125.84 (C); 125.65 (C); 123.45 (CH arom.); 121.46 (CH arom.); 111.40 (CH arom.); 109.52 (CH arom.); 59.64 (CH2); 55.96 (OCH3); 55.92 (OCH3); 55.40 (CH2); 50.88 (CH2); 33.16 (CH2); 28.26 (CH2) ppm. ESI-HRMS (m/z) calculated for [M + H]+ ion species C25H27N2O4= 419.1965, found 419.1958.
Hydrochloride: mp 98–100 °C.
4–(2-(6,7-Dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl 6-methoxynicotinate (23)
Procedure B in an. CH3CN, starting from phenol 28 and 6-methoxynicotinic acid.
Free base: chromatographic eluent: CH2Cl2/MeOH/NH4OH 95:5:0.5. Yield: 21.0%.
1H NMR (400 MHz, CDCl3) δ: 8.97 (d, J = 2.0 Hz, 1H, CH arom.); 8.25 (dd, J = 8.8, 2.0 Hz, 1H, CH arom.); 7.27 (d, J = 8.4 Hz, 2H, CH arom.); 7.10 (d, J = 8.4 Hz, 2H, CH arom.); 6.80 (d, J = 8.8 Hz, 1H, CH arom.); 6.58 (s, 1H, CH arom.); 6.51 (s, 1H, CH arom.); 4.00 (s, 3H, OCH3); 3.82 (s, 3H, OCH3); 3.81 (s, 3H, OCH3); 3.69 (s, 2H, NCH2Ar); 3.00–2.92 (m, 2H, CH2); 2.91–2.77 (m, 6H, CH2) ppm. ESI-HRMS (m/z) calculated for [M + H]+ ion species C26H29N2O5= 449.2071, found 449.2063.
Hydrochloride: mp 254–256 °C.
4–(2-(6,7-Dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl pyrazine-2-carboxylate (24)
Procedure B in CHCl3 (free of ethanol), starting from phenol 28 and pyrazine-2-carboxylic acid.
Free base: chromatographic eluent: CH2Cl2/MeOH/NH4OH 97:3:0.3. Yield: 21.8%.
1H NMR (400 MHz, CDCl3) δ: 9.40 (d, J = 1.2 Hz, 1H, CH arom.); 8.78 (d, J = 2.4 Hz, 1H, CH arom.); 8.75 (dd, J = 2.4, 1.2 Hz, 1H, CH arom.); 7.26 (d, J = 8.8 Hz, 2H, CH arom.); 7.14 (d, J = 8.8 Hz, 2H, CH arom.); 6.55 (s, 1H, CH arom.); 6.49 (s, 1H, CH arom.); 3.79 (s, 3H, OCH3); 3.78 (s, 3H, OCH3); 3.61 (s, 2H, NCH2Ar); 2.92–2.85 (m, 2H, CH2); 2.83–2.72 (m, 6H, CH2) ppm. 13C NMR (100 MHz, CDCl3) δ: 162.66 (C=O); 148.80 (C); 148.16 (CH arom.); 147.71 (C); 147.36 (C); 146.77 (CH arom.); 144.67 (CH arom.); 142.96 (C); 138.18 (C); 129.93 (CH arom.); 125.62 (C); 125.36 (C); 121.40 (CH arom.); 111.29 (CH arom.); 109.40 (CH arom.); 59.38 (CH2); 55.93 (OCH3); 55.89 (OCH3); 55.17 (CH2); 50.73 (CH2); 32.97 (CH2); 27.97 (CH2) ppm. ESI-HRMS (m/z) calculated for [M + H]+ ion species C24H26N3O4= 420.1918, found 420.1911.
Hydrochloride: mp 96–99 °C.
4–(2-(6,7-Dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl 6-methoxypyrazine-2-carboxylate (25)
Procedure B in an. CH3CN, starting from phenol 28 and 6-methoxypyrazine-2-carboxylic acid.
Free base: chromatographic eluent: CH2Cl2/MeOH/NH4OH 93:7:0.3. Yield: 75.6%.
1H NMR (400 MHz, CDCl3) δ: 8.95 (s, 1H, CH arom.); 8.41 (s, 1H, CH arom.); 7.28 (d, J = 8.4 Hz, 2H, CH arom.); 7.14 (d, J = 8.4 Hz, 2H, CH arom.); 6.57 (s, 1H, CH arom.); 6.50 (s, 1H, CH arom.); 4.05 (s, 3H, OCH3); 3.81 (s, 3H, OCH3); 3.80 (s, 3H, OCH3); 3.63 (s, 2H, NCH2Ar); 2.95–2.90 (m, 2H, CH2); 2.89–2.73 (m, 6H, CH2) ppm. 13C NMR (100 MHz, CDCl3) δ: 162.79 (C=O); 159.99 (C); 148.90 (C); 147.62 (C); 147.29 (C); 140.08 (CH arom.); 139.08 (C); 138.38 (CH arom.); 138.33 (C); 129.82 (CH arom.); 126.09 (C); 125.96 (C); 121.34 (CH arom.); 111.37 (CH arom.); 109.49 (CH arom.); 59.80 (CH2); 55.93 (OCH3); 55.90 (OCH3); 55.52 (CH2); 54.13 (OCH3); 50.94 (CH2); 33.27 (CH2); 28.44 (CH2) ppm. ESI-HRMS (m/z) calculated for [M + H]+ ion species C25H28N3O5= 450.2024, found 450.2029.
Hydrochloride: mp 214–216 °C.
4–(2-(6,7-Dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl 6-methoxyquinoline-4-carboxylate (26)
Procedure B in CHCl3 (free of ethanol), starting from phenol 28 and 6-methoxyquinoline-4-carboxylic acid.
Free base: chromatographic eluent: CH2Cl2/MeOH/NH4OH 97:3:0.3. Yield: 32.5%.
1H NMR (400 MHz, CDCl3) δ: 8.93 (d, J = 4.8 Hz, 1H, CH arom.); 8.33 (d, J = 2.4 Hz, 1H, CH arom.); 8.19 (d, J = 4.8 Hz, 1H, CH arom.); 8.09 (d, J = 9.2 Hz, 1H, CH arom.); 7.44 (dd, J = 9.2, 2.4 Hz, 1H, CH arom.); 7.36 (d, J = 8.4 Hz, 2H, CH arom.); 7.20 (d, J = 8.4 Hz, 2H, CH arom.); 6.61 (s, 1H, CH arom.); 6.55 (s, 1H, CH arom.); 3.93 (s, 3H, OCH3); 3.85 (s, 3H, OCH3); 3.84 (s, 3H, OCH3); 3.71 (s, 2H, NCH2Ar); 3.02–2.97 (m, 2H, CH2); 2.92–2.80 (m, 6H, CH2) ppm. 13C NMR (100 MHz, CDCl3) δ: 165.03 (C=O); 159.57 (C); 148.91 (C); 147.79 (C); 147.42 (C); 146.93 (CH arom.); 145.80 (C); 137.98 (C); 131.57 (CH arom.); 131.20 (C); 130.04 (CH arom.); 127.04 (C); 125.50 (C); 123.37 (CH arom.); 123.15 (CH arom.); 121.68 (CH arom.); 111.26 (CH arom.); 109.37 (CH arom.); 102.97 (CH arom.); 59.36 (CH2); 55.95 (OCH3); 55.91 (OCH3); 55.65 (OCH3); 55.12 (CH2); 50.74 (CH2); 32.91 (CH2); 27.90 (CH2) ppm. ESI-HRMS (m/z) calculated for [M + H]+ ion species C30H31N2O5= 499.2228, found 499.2226.
Hydrochloride: mp 203–206 °C.
2-Methoxy-1-naphthoic acid (29)Citation42
To a stirred solution of CuBr2 (0.04 mmol) and 2-methoxy-1-naphthaldehyde (0.80 mmol) in 3.0 ml of an CH3CN was added 70% t-BuOOH in water (1.60 mmol). When all the aldehyde had been consumed, the solvent was removed under reduced pressure. The reaction mixture was treated with a saturated solution of NaHCO3 and extracted with ethyl acetate. The aqueous layer was acidified using 2 M HCl and then extracted with ethyl acetate. The organic phase was dried with Na2SO4, then the solvent was removed under reduced pressure. 2-methoxy-1-naphthoic acid was obtained pure as a pale yellow solid.
Yield: 67.6%. 1H NMR (400 MHz, CDCl3) δ: 8.38 (d, J = 8.4 Hz, 1H, CH arom.); 7.95 (d, J = 9.2 Hz, 1H, CH arom.); 7.78 (d, J = 8.4 Hz, 1H, CH arom.); 7.55 (t, J = 8.4 Hz, 1H, CH arom.); 7.39 (t, J = 8.4 Hz, 1H, CH arom.); 7.30 (d, J = 9.2 Hz, 1H, CH arom.); 4.05 (s, 3H, OCH3).
2,3-Dimethoxy-1-naphthoic acid (30)Citation43
To a solution of 2,3-dimethoxy-1-naphthaldehyde (0.23 mmol) in 3.0 ml of acetone was added Na2CO3 (0.23 mmol) in 1.0 ml of water. Then KMnO4 (0.23 mmol) was added portion wise. The reaction mixture was stirred at rt for 18 h then filtered. The filtrate was concentrated and extracted with ethyl acetate. The aqueous layer was acidified to pH 1 with 1 M HCl then extracted with ethyl acetate. After drying with Na2SO4, the solvent was removed under reduced pressure and 2,3-dimethoxy-1-naphthoic acid was obtained as a pale yellow solid.
Yield: 93.3%. 1H NMR (400 MHz, CDCl3) δ: 10.40 (s, 1H, OH); 7.89 (d, J = 7.6 Hz, 1H, CH arom.); 7.63 (d, J = 7.6 Hz, 1H, CH arom.); 7.35 (t, J = 7.6 Hz, 1H, CH arom.); 7.30 (t, J = 7.6 Hz, 1H, CH arom.); 7.14 (s, 1H, CH arom.); 3.92 (s, 3H, OCH3); 3.89 (s, 3H, OCH3).
6-Methoxyquinoline-4-carboxylic acid (31)Citation44
To a solution of quinine sulphate (0.90 mmol) in 12 ml of 10% sulphuric acid, MnO2 (1.70 mmol) was added. The mixture was raised to the boiling point, then CrO3 (14.30 mmol) dissolved in 3 ml of water was added dropwise during an hour. The refluxing was continued for 3 h, then 126 ml of water and 28 ml of 15 N ammonia were added. The reaction mixture was stirred at 100 °C for 18 h, then filtered with Celite. The residue was washed several times with hot 15 N ammonia solution. The combined filtrates were concentrated under reduced pressure, then acidified with acetic acid and filtered, to obtain 6-methoxyquinoline-4-carboxylic acid as pure yellow solid.
Yield: 89.4%. 1H NMR (400 MHz, DMSO-d6) δ: 8.83 (d, J = 2.0 Hz, 1H, CH arom.); 8.16 (s, 1H, CH arom.); 7.99 (d, J = 9.2 Hz, 1H, CH arom.); 7.88 (d, J = 2.0 Hz, 1H, CH arom.); 7.46 (d, J = 9.2 Hz, 1H, CH arom.); 3.88 (s, 3H, OCH3).
Biological assays
Cell lines and cultures
MDCK-MDR1, MDCK-MRP1 and MDCK-BCRP cells are a gift of Prof. P. Borst, NKI-AVL Institute, Amsterdam, The Netherlands. Caco-2 cells were a gift of Dr. Aldo Cavallini and Dr. Caterina Messa from the Laboratory of Biochemistry, National Institute for Digestive Diseases, “S. de Bellis”, Bari (Italy). HT29 and A549 were purchased from ATCC (Manassas, VA). The doxorubicin-resistant sublines, HT29/DX and A549/DX cells were generated by stepwise selection in medium with increasing concentrations of doxorubicinCitation45 and maintained in culture medium with 100 and 50 nM doxorubicin, respectively. MDCK and Caco-2 cells were grown in DMEM high glucose, HT29 and HT29/DX in RPMI-1640, A549 and A549/DX in HAM-F12 medium, all supplemented with 10% foetal bovine serum, 2 mM glutamine, 100 U/mL penicillin, 100 µg/mL streptomycin, in a humidified incubator at 37 °C with a 5% CO2 atmosphere.
Drugs and chemicals
Cell culture reagents were purchased from Celbio s.r.l. (Milano, Italy). CulturePlate 96/wells plates were purchased from PerkinElmer Life Science; Calcein-AM, bisBenzimide H 33342 trihydrochloride were obtained from Sigma-Aldrich (Milan, Italy).
Immunoblotting analysis of P-gp, MRP1 and BCRP expression in MDCK, MDCK-MDR1, MDCK-MRP1 and MDCK-BCRP cells
All MDCK cells were rinsed with lysis buffer (50 mM Tris-HCl, 1 mM EDTA, 1 mM EGTA, 150 mM NaCl, 1% v/v Triton-X100; pH 7.4), supplemented with the protease inhibitor cocktail III (Cabiochem, La Jolla, CA), sonicated and clarified at 13000 × g, for 10 min at 4 °C. Protein extracts (20 µg) were subjected to SDS-PAGE and probed with the following antibodies: anti-P-glycoprotein (P-gp; 1:250, rabbit polyclonal, #sc-8313, Santa Cruz Biotechnology Inc., Santa Cruz, CA), anti-multidrug resistant protein 1 (MRP1; 1:500, mouse clone MRPm5, Abcam, Cambridge, UK), anti-breast cancer resistance protein (BCRP; 1:500, mouse clone BXP-21, Santa Cruz Biotechnology Inc.), anti-β-tubulin (1:1000, mouse clone D10, Santa Cruz Biotechnology Inc.), followed by the horseradish peroxidase-conjugated secondary antibodies (Bio-Rad). The membranes were washed with Tris-buffered saline (TBS)/Tween 0.01% v/v. Proteins were detected by enhanced chemiluminescence (Bio-Rad Laboratories).
Calcein-AM experiments
These experiments were carried out as previously described by Teodori et al. with minor modificationsCitation34. Each cell line (30 000 cells per well) was seeded into black CulturePlate 96/wells plate with 100 µL medium and allowed to become confluent overnight. 100 µL of test compounds were solubilised in culture medium and added to monolayers, with final concentrations ranging from 1 nM to 100 µM. 96/Wells plate was incubated for 30 min in a humidified atmosphere 5% CO2 at 37 °C. Calcein-AM was added in 100 µL of Phosphate Buffered Saline (PBS) to yield a final concentration of 2.5 µM and plate was incubated for 30 min in a humidified atmosphere 5% CO2 at 37 °C. Each well was washed 3 times with ice cold PBS. Saline buffer was added to each well and the plate was read with Victor3 (PerkinElmer) at excitation and emission wavelengths of 485 and 535 nm, respectively. In these experimental conditions, Calcein cell accumulation in the absence and in the presence of tested compounds was evaluated and fluorescence basal level was estimated with untreated cells. In treated wells the increase of fluorescence with respect to basal level was measured. EC50 values were determined by fitting the fluorescence increase percentage versus log[dose].
Hoechst 33342 experiment
These experiments were carried out as described by Teodori et al.Citation34. Each cell line (30 000 cells per well) was seeded into black CulturePlate 96/wells plate with 100 µL medium and allowed to become confluent overnight. 100 µL of test compounds were solubilised in culture medium and added to monolayers, with final concentrations ranging from 1 to 100 µM. 96/Wells plate was incubated for 30 min in a humidified atmosphere 5% CO2 at 37 °C. Hoechst 33342 was added in 100 µl of phosphate-buffered saline (PBS) to yield a final concentration of 8 µM and plate was incubated for 30 min in a humidified atmosphere 5% CO2 at 37 °C. The supernatants were drained, and the cells were fixed for 20 min under light protection using 100 µL per well of a 4% PFA solution. Each well was washed three times with ice cold PBS. Saline buffer was added to each well and the plate was read with Victor3 (PerkinElmer) at excitation and emission wavelengths of 340/35 nm and 485/20 nm, respectively. In these experimental conditions, Hoechst 33342 accumulation in the absence and in the presence of tested compounds was evaluated and fluorescence basal level was estimated with untreated cells. In treated wells, the increase of fluorescence with respect to basal level was measured. EC50 values were determined by fitting the fluorescence increase percentage versus log[dose].
ATPlite assay
The MDCK-MDR1 cells were seeded into 96-well microplate in 100 µL of complete medium at a density 20,000 cells per well. The plate was incubated overnight (O/N) in a humidified atmosphere 5% CO2 at 37 °C. The medium was removed and 100 µL of complete medium either alone or containing different concentrations (ranging from 1 to 100 µM) of test compounds was added. The plate was incubated for 2 h in a humidified 5% CO2 atmosphere at 37 °C. 50 µL of mammalian cell lysis solution was added to all wells and the plate shacked for five minutes in an orbital shaker. 50 µL of substrate solution was added to all wells and the plate shacked for five minutes in an orbital shaker. The plate was dark adapted for ten minutes and the luminescence was measured.
Permeability experiments
Preparation of caco-2 monolayer
Caco-2 cells were seeded onto a Millicell® assay system (Millipore), where a cell monolayer is set in between a filter cell and a receiver plate, at a density of 10 000 cells/wellCitation46. The culture medium was replaced every 48 h and the cells kept for 21 days in culture. The Trans Epithelial Electrical Resistance (TEER) of the monolayers was measured daily, before and after the experiment, using an epithelial volt-ohmmeter (Millicell®-ERS). Generally, TEER values greater than 1000 Ω for a 21 days culture, are considered optimal.
Drug transport experiment
After 21 days of Caco-2 cell growth, the medium was removed from filter wells and from the receiver plate, which were filled with fresh HBSS buffer (Invitrogen). This procedure was repeated twice, and the plates were incubated at 37 °C for 30 min. After incubation time, the HBSS buffer was removed and drug solutions and reference compounds were added to the filter well at the concentration of 100 µM, while fresh HBSS was added to the receiver plate. The plates were incubated at 37 °C for 120 min. Afterwards, samples were removed from the apical (filter well) and basolateral (receiver plate) side of the monolayer to measure the permeability.
The apparent permeability (Papp), in units of nm/second, was calculated using the following equation:
VA = the volume (in mL) in the acceptor well;
Area = the surface area of the membrane (0.11 cm2 of the well);
time = the total transport time in seconds (7200s);
[drug]acceptor = the concentration of the drug measured by U.V. spectroscopy;
[drug]initial = the initial drug concentration (1 × 10−4 M) in the apical or basolateral wells.
Co-administration assay
The co-administration assay with doxorubicin was performed in MDCK-MDR1, HT29, HT29/DX, A549 and A549/DX cells at 48 h. On day 1, 10,000 cells/well were seeded into 96-well plates in a volume of 100 µL of fresh medium. On day 2, the tested drug was added alone to the cells at different concentrations (ranging from 40 nM to 10 µM). On day 3, the medium was removed and the drug at the same concentrations was added alone and in co-administration with 10 µM doxorubicin to the cells. After the established incubation time with the tested drug, MTT (0.5 mg/mL) was added to each well, and after 3 − 4 h incubation at 37 °C, the supernatant was removed. The formazan crystals were solubilised using 100 µL of DMSO/EtOH (1:1), and the absorbance values at 570 and 630 nm were determined on the microplate reader HTX Synergy (Bio-Tek Instruments).
Statistical analysis
All data in the text and figures are provided as means ± SEM. The results were analysed by a Student’s t-test and one-way ANOVA test, using Graph-Pad Prism (Graph-Pad software, San Diego, CA). p < 0.05 was considered significant.
Stability tests
Chemicals
Acetonitrile (Chromasolv), formic acid and ammonium formate (MS grade), NaCl, KCl, Na2HPO4 2H2O, KH2PO4 (Reagent grade), verapamil hydrochloride (analytical standard, used as internal standard) and ketoprofen (analytical standard) were purchased by Sigma-Aldrich (Milan, Italy). Ketoprofen ethyl ester (KEE) were obtained by Fisher’s reaction from ketoprofen and ethanol.
MilliQ water 18 MΩ cm−1 was obtained from Millipore’s Simplicity system (Milan, Italy).
Phosphate-buffered solution was prepared by adding 8.01 g L−1 of NaCl, 0.2 g L−1 of KCl, 1.78 g L−1 of Na2HPO4 2H2O and 0.27 g L−1 of KH2PO4. Human plasma was collected from healthy male volunteer and kept at −80 °C until use.
Preparation of samples
Each sample was prepared adding 10 µL of working solution 1 to 100 µL of tested matrix (PBS or human plasma) in micro centrifuge tubes. The obtained solutions correspond to 1 µM of analyte.
Each set of samples was incubated in triplicate at four different times, 0, 30, 60 and 120 min at 37 °C. Therefore, the degradation profile of each analyte was represented by a batch of 12 samples (4 incubation times × 3 replicates). After the incubation, the samples were added with 300 µL of ISTD solution and centrifuged (room temperature for 5 min at 10,000 rpm). The supernatants were transferred in auto sampler vials and dried under a gentle stream of nitrogen.
The dried samples were dissolved in 1.0 ml of 10 mM of formic acid in mQ water: acetonitrile 80:20 solution. The obtained sample solutions were analysed by LC-MS/MS methods described in Supplementary material.
Results and discussion
Chemistry
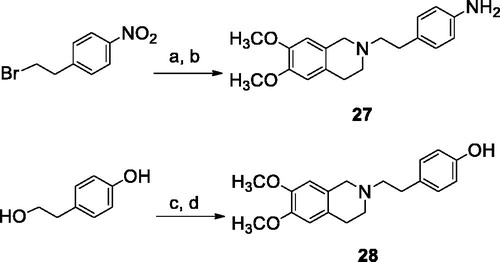
The reaction pathways used to synthesise derivatives 1–26 are reported in Schemes 1–4. The key intermediates needed to achieve amides and esters were the aniline 27 and the phenol 28, respectively, which were prepared (Scheme 1) as reported previouslyCitation34: aniline 27 was obtained by alkylation of 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline hydrochloride with 1–(2-bromoethyl)−4-nitrobenzene followed by catalytic reduction of the nitro group; phenol 28 was synthesised starting from the commercially available 4-(hydroxyethyl)phenol, transformed in the corresponding chloroethyl derivative by treatment with SOCl2 in ethanol-free CHCl3 and anhydrous triethylamine and then reacted with 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline hydrochloride in anhydrous CH3CN in the presence of K2CO3. Amides 1–13 and esters 14–26 were obtained by reaction of 27 and 28, respectively, with the proper carboxylic acid using EDC hydrochloride and DMAP in anhydrous CH2Cl2 or CH3CN, or with the acyl chloride obtained by treatment of the suitable acid with SOCl2 in ethanol-free CHCl3 or anhydrous CH3CN (Scheme 2, for details see Experimental Section). Most of the carboxylic acids were commercially available. 2-Methoxy-1-naphthoic acid 29 was synthetised starting from the corresponding aldehyde, following the general procedure described by DasCitation42 (Scheme 3), while 2,3-dimethoxy-1-naphthoic acid 30 was obtained from the corresponding aldehyde as described by OhnmachtCitation43 (Scheme 3). 6-Methoxyquinoline-4-carboxylic acid 31 was synthesised following the procedure described by KowankoCitation44 (Scheme 4).
Scheme 1. Reagents and conditions: (a) 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline HCl, K2CO3, an. CH3CN; (b) H2, Pd/C, EtOH; (c) SOCl2, ethanol-free CHCl3, an. Et3N; (d) 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline HCl, K2CO3, an. CH3CN.
Scheme 2. Reagents and conditions: (a) ArCOCl, ethanol-free CHCl3 or an. CH3CN; (b) ArCOOH, EDCI, DMAP, an. CH2Cl2 or CH3CN.
Scheme 3. Reagents and conditions: (a) R = H, CuBr2, t-BuOOH, an. CH3CN; R = OCH3, Na2CO3, KMnO4, acetone.
Scheme 4. Reagents and conditions: (a) H2SO4 (water sol. 10%), MnO2, CrO3, NH3 (15 N).
Biological activity: characterisation of P-gp interacting profile and ABC transporters selectivity
The interaction potency between the designed compounds and P-gp was evaluated by measuring the transport inhibition of the pro-fluorescent probe Calcein-AM, that is a P-gp substrate, in a cell line overexpressing P-gp (MDCK-MDR1)Citation47. The P-gp interacting profile was further investigated with two other assays: the Apparent Permeability (Papp) determination (BA/AB) in Caco-2 cell monolayerCitation48,Citation49, and the ATP cell depletion in the MDCK-MDR1 cell lineCitation50. Taking into account the results of the combined tests, an interacting compound can be classified as substrate or inhibitor. Papp determination measures the ratio between two fluxes: from the basolateral to apical compartments (BA, representative of passive diffusion) and from the apical to basolateral compartments (AB, representative of active transport). A (BA/AB) value <2 suggests that the compound can be considered an inhibitor. In the same manner, if (BA/AB) >2, the compound should probably be classified as a substrateCitation51. The other assay detects the consumption of ATP elicited by the transport mediated by the pump; in general, a substrate induces ATP cell depletion being transported by the pump (unambiguous substrate, category I), while a P-gp inhibitor does not induce ATP consumption. There is also a third substrate category (known as category IIB3) displaying a Papp value >2 but not inducing an ATP cell depletionCitation47. The selectivity of ligands 1–26 was evaluated by detecting their activity towards the other ABC transporters MRP1 and BCRP, by measuring the inhibition of the efflux of Calcein-AM (i.e., also a MRP1 substrate) in cells overexpressing MRP1 (MDCK-MRP1 cells), and the inhibition of the efflux of the fluorescent probe Hoechst 33342 (i.e. a BCRP substrate) in cells overexpressing BCRP (MDCK-BCRP cells), respectively.
Expression levels of P-gp, MRP1 and BCRP were periodically analysed by immunoblotting analysis in MDCK-MDR1, MDCK-MRP1 and MDCK-BCRP cells respectively, as described in the Experimental Section. Tubulin was used as control of equal protein loading. A representative western blot analysis is reported in the Supplemental Material.
The results of the three inhibition assays on compounds 1–26 are reported in together with those on tariquidar and elacridar used as reference compounds. The already described 3,4,5-trimethoxyphenyl derivatives I (amide) and II (ester)Citation34 have been added for comparison.
Table 1. MDR-reversing activity, P-gp interaction profile and calculated human plasma half-life of compounds 1–26.
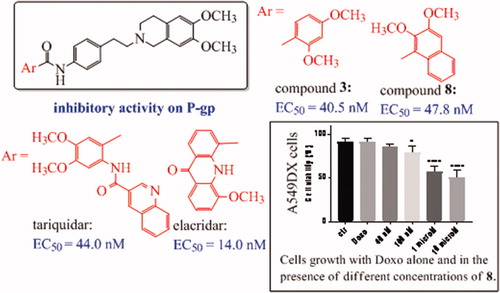
As shown in , all the compounds were active on P-gp. Indeed, most of them showed a high P-gp potency, with EC50 values below 1 µM, reaching also the nanomolar range, as in the case of compounds 3 and 8 (EC50= 40.5 nM and 47.8 nM, respectively). Only eight among the tested derivatives exhibited a lower P-gp activity, with EC50 values higher than 1 µM.
Our research started with the synthesis of 2,3,4-trimethoxyphenyl derivatives 1 (amide) and 14 (ester), which are isomers of the reference compounds I and IICitation34, reported in ; remarkably, both the 2,3,4-functionalized isosteres, bearing a substituent in ortho position, were more potent than the reference compounds. This result prompted us to synthesise a series of phenyl analogues carrying an ortho methoxy substituent, also combined with a second methoxy residue in different positions (amides 2–5 and esters 15–18). Moreover, since in a preceding work we had verified the positive effect of inserting a naphthalene ringCitation52, we also obtained compounds 7, 8, 20 and 21 and the non-substituted analogues 6 and 19 for comparison purposes.
As general trend, substituted benzene and naphthalene derivatives displayed activities in the submicromolar or nanomolar range (EC50 ranging from 40.5 to 0.67 µM for amides and from 0.10 to 0.84 µM for esters), with the exception of amide 4, carrying the 2,6-dimethoxy phenyl moiety, which showed an EC50 value of 7.20 µM. In this set of derivatives, it was not easy to define a relationship between potency values and structural characteristics: some compounds with interesting EC50 values could be found both among the amides and the esters, but the nature of the aryl moieties differently influenced the two groups of isosteres, without a definite trend. However, biological data confirmed that the presence of an ortho methoxy substituent is favourable for the activity. The most interesting result was the discovery of two amides endowed with an outstanding potency in the nanomolar range: the 2,4-dimethoxy phenyl derivative 3 (EC50 = 0.0405 µM) and the 2,3-dimethoxy naphthalene derivative 8 (EC50 = 0.0478 µM).
Compounds 9–13 and 22–26 instead carry a nitrogen-containing heterocycle: amides 9 and 11, and esters 22 and 24 are non-substituted pyridine or pyrazine derivatives, while 10, 12, 23 and 25 are the corresponding methoxylated analogues. The quinoline isosteres 13 and 26 were obtained only as methoxy-substituted molecules. These hetero aromatic derivatives displayed lower potency: only amides 12 and 13, and ester 25 showed a moderate/good activity (EC50 = 0.87, 0.66 and 0.22 µM, respectively). In the case of heterocycles, it was possible to verify that the presence of the OCH3 substituent improved the inhibiting activity on P-gp (compare amides 9 with 10, or 11 with 12, and the corresponding esters 22 with 23, or 24 with 25), and that the presence of an unsubstituted pyridine ring was detrimental for the activity. As regards the methoxy-quinoline derivatives, amide 13 was more potent than the corresponding ester 26.
With respect to the reference compounds tariquidar (EC50 = 0.044 µM) and elacridar (EC50 = 0.014 µM), compounds 3 and 8 showed a comparable activity on P-gp, endowed with an EC50 value in the nanomolar range (as above reported) and a P-gp maximum inhibition (90–95%) at 10 µM; the other compounds were less potent at a different extent, but more selective since almost all of them were inactive towards both MRP1 and BCRP. As regard BCRP, although previous SAR studies suggested that the 6,7-dimethoxy-2-phenethyl-1,2,3,4-tetrahydroisoquinoline nucleus is a requirement for inhibiting both P-gp and BCRP, only six of these derivatives showed a moderate activity versus BCRP (EC50 ranging from 1.0 to 21.2 µM), belonging to the amide and the ester series. Interestingly, the potent P-gp inhibitor 3 (2,4-dimethoxyphenyl amide) was also the most active in BCRP inhibition (EC50 = 1.0 µM). Regarding MRP1 inhibition, all the compounds could be considered inactive: only in the case of two ester derivatives, 14 and 16, an EC50 (69.7 and 87.0 µM) could be defined. It is noteworthy that these two compounds, the 2,3,4-trimethoxyphenyl ester 14 and the 2,4-dimethoxyphenyl ester 16, were active also on BCRP and highly potent vs P-gp, showing a profile endowed with a low selectivity.
Finally, as regard the P-gp interacting mechanism, all the compounds behaved as not transported substrates (category IIB3), since they had a BA/AB ratio >2 and were not able to induce ATP cell depletionCitation47.
On the bases of the obtained results, some very potent compounds were present both among the amide and the ester derivatives; however, the most interesting derivatives (3 and 8) belonged to amide series. These two compounds showed different selectivity profiles, since compound 3 was active also on BCRP, while compound 8 was not.
Co-administration assay
Compounds 3 and 8, endowing with the best P-gp activity profile in MDCK-MDR1, were tested alone and in co-administration with the antineoplastic drug, doxorubicin, that, as P-gp substrate, is usually inactive in tumours overexpressing the pump. A preliminary study was performed testing the two ligands 3 and 8 alone and in co-administration with 100 nM and 1 µM doxorubicin, without observing a significant reverting effect (data not shown). Therefore, we performed the same experiment using doxorubicin concentration of 10 µM.
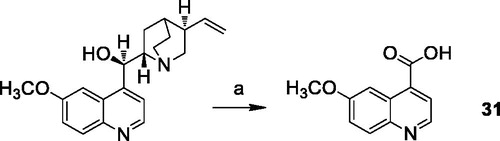
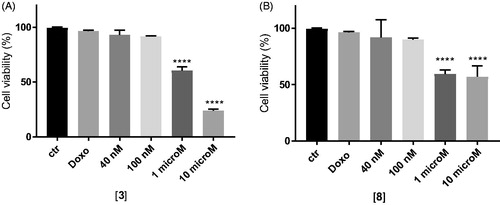
As depicted in , at their EC50 values (around 40 nM), the two compounds showed a very negligible cytotoxic effect at 48 h of incubation (around 20%), while at higher dose (10 µM) they reach 30% of cytotoxic effect, compared to untreated cells.
Figure 2. In vitro cell growth experiments performed on MDCK-MDR1 cells in the presence of different concentrations of the two compounds 3 (A) and 8 (B). Each bar represents the mean ± SEM of two experiments performed in triplicate. One-way ANOVA analysis: ****p < 0.0001.
In MDCK cells overexpressing P-gp, doxorubicin alone at 10 µM did not show cytotoxicity as expected, while the co-administration with the two compounds at 1 and 10 µM was able to rehabilitate the effect of the antineoplastic agent leading to high cytotoxicity. In particular, the co-administration of doxorubicin with compounds 3 and 8 at their higher dose (10 µM) showed cytotoxicity values of 77% for 3 and around 50% for 8 ().
Figure 3. In vitro cell growth experiments performed on MDCK-MDR1 cells in the presence of 10 μM doxorubicin (Doxo) alone and in the presence of different concentrations of the tested compounds 3 (A) and 8 (B). Each bar represents the mean ± SEM of two experiments performed in triplicate. One-way ANOVA analysis: ****p < 0.0001.
The efficacy of the compounds was next tested in cancer cell lines with various levels of P-gp, as the human adenocarcinoma colon cells (HT29) and non-small cell lung cancer cells (A549) displaying low amount of P-gp, and their resistant counterparts HT29/DX and A549/DX, with a high amount of P-gpCitation53. In preliminary assays, we verified that 3 and 8 were not cytotoxic at all the tested concentration (data not shown).
In HT29 cells, which express low levels of P-gpCitation53, neither 3 () nor 8 () increases the cytotoxicity, measured as reduced viability, of doxorubicin. By contrast, in the resistant counterpart HT29/DX, which have higher levels of the transporter, 3 significantly increased the reduction of cell viability induced by doxorubicin when used at 1 or 10 µM (). The same results were obtained in A549 and in A549/DX cells, characterised by high levels of P-gpCitation53. A similar trend was observed also for 8 (). More in details, at 10 µM, 3 reduced the cell viability of 5% and of 35% in doxorubicin-treated HT29 cells () and HT29/DX cells (), respectively. In A549 and A549/DX cells it decreased the viability of 25% () and 40% (), respectively. The co-administration with doxorubicin of compound 8, at 10 µM, reduced cell viability of 10% in HT29 cells (), 29% in HT29/DX cells (), 25% in A549 cells () and 49% in A549/DX cells ().
Figure 4. In vitro cell growth experiments performed on HT29 (A), HT29/DX cells (B), A549 (C) and A549/DX cells (D) in the presence of compound 3 alone at different concentrations and in co-administration of 10 μM doxorubicin (Doxo) (after the dotted line). Each bar represents the mean ± SEM of two experiments performed in triplicate. CTR represents the untreated cells, while ctr is the same cell lines treated with 10 μM Doxo alone. One-way ANOVA analysis: *p < 0.05; **p ≤ 0.005; ****p ≤ 0.0001.
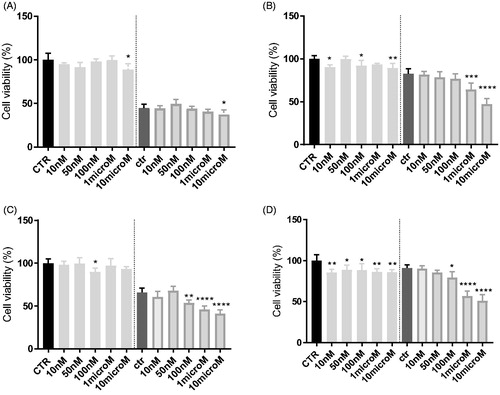
Figure 5. In vitro cell growth experiments performed on HT29 (A), HT29/DX cells (B), A549 (C) and A549/DX cells (D) in the presence of compound 8 alone at different concentrations and in co-administration of 10 μM doxorubicin (Doxo) (after the dotted line). Each bar represents the mean ± SEM of two experiments performed in triplicate. CTR represents the untreated cells, while ctr is the same cell lines treated with 10 μM Doxo alone. One-way ANOVA analysis: *p < 0.05; **p ≤ 0.005; ***p ≤ 0.001; ****p < 0.0001.
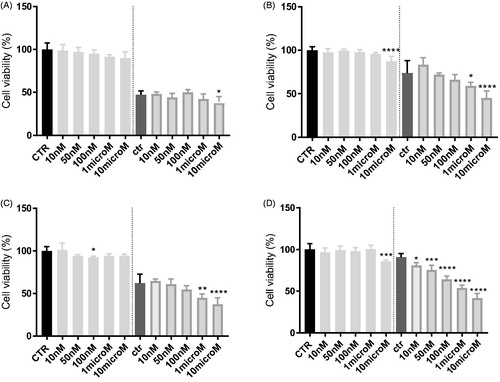
Notably, HT29 cells had very low/undetectable levels of P-gpCitation54, while A549 cells had slightly higher levels of P-gp; by contrast, the corresponding resistant cells display high (HT29/DX) and huge (A549/DX) amount of P-gpCitation54. The higher toxicity of the combination of compounds 3 or 8 plus doxorubicin in resistant cells, in particular in A549/DX subline, confirms that they potentiate cytotoxicity of doxorubicin by inhibiting P-gp activity and arousing a higher intracellular accumulation of the antineoplastic agent in P-gp expressing cells.
Chemical stability test
Due to the presence of ester or amide groups in the structure of compounds 1–26, a study aimed to evaluate their susceptibility to spontaneous or enzymatic hydrolysis, was planned. For this purpose, a series of experiments were carried out, adding a known amount of analyte in both phosphate buffer solution (PBS) and in human plasma. The samples were analysed by LC-MS/MS method operating in Multiple Reaction Monitoring (MRM) modeCitation55.
The solution stability of each studied compound was verified by monitoring the variation of the analyte concentration at different incubation times in PBS and human plasma samples. The processed raw data and the used LC-MS/MS methods are reported in Supplemental material.
The analyte concentration (1 µM) used during the stability tests is generally smaller than the corresponding Michaelis–Menten constant (KM); therefore, the enzymatic degradation rate is described by a first-order kinetic. Then, by plotting the natural logarithm of the quantitative data versus the incubation time, a linear function can be used and its slope represents the degradation rate (k). Accordingly with the linear function, the half-life (t1/2) of each tested compound can be calculated as follows:
The plots of the natural logarithm of the quantitative data versus the incubation time of all the studied compounds were analysed. The obtained results demonstrated that all the compounds were stable in PBS and most of them also in human plasma. In particular, all the amide derivatives were not susceptible to the enzymatic hydrolysis, while the degradation plots of the ester compounds 15, 16, 18, 24, 25, and 26 in human plasma showed a significant decay rate (t1/2 values between 6 and 124 min). The calculated half-life values of all tested compounds were reported in . The stable compounds showed the k values close to 0; consequently, for these derivatives, extremely high half-life can be calculated. Since under the proposed experimental conditions, a half-life over 240 min is not correctly evaluated, it is reasonable to consider that their half-life values could be equal or greater than 240 min. Furthermore, the half-life value of ketoprofene ethylester (KEE), used as reference compound, demonstrated that the employed human batch was enzymatically active (t1/2 < 2 h)Citation55.
The human plasma degradation profiles of the ester compound 25 and its amide homologous 12 are reported as an example in . All degradation plots of the studied compounds are reported in Supplementary material.
Figure 6. Degradation plots of the ester compound 25 (top) and the corresponding amide derivative 12 (bottom) in PBS (square blue) and human plasma (red triangle) samples. The ester compound 25 suffered a quickly enzymatic hydrolysis (t1/2 about 6 min), while the amide derivative 12 was stable over 120 min in human plasma samples. Both the concentration profiles resulted unmodified in PBS solutions.
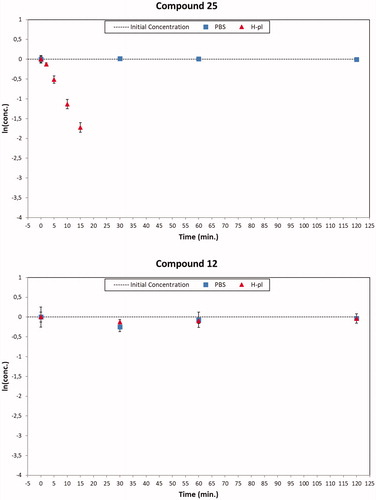
The obtained results indicate that the ester group is more susceptible to the enzymatic hydrolysis than the corresponding amide one, since some of the ester derivatives were not stable in human plasma samples.
Conclusions
In this work, a new series of amide and ester derivatives carrying a 6,7-dimethoxy-2-phenethyl-1,2,3,4-tetrahydroisoquinoline scaffold linked to different methoxy-substituted aryl moieties was synthesised. The obtained compounds were evaluated for their P-gp interaction profile and selectivity towards the two other ABC transporters, multidrug-resistance-associated protein-1 (MRP-1) and breast cancer resistance protein (BCRP). Most of the compounds displayed high activity versus P-gp: the presence of an amide or an ester function did not influence P-gp selectivity and interacting mechanism. In fact, among both amide and ester derivatives some very potent compounds could be found, which resulted in general to be more selective than the reference compounds tariquidar and elacridar. Moreover, in the current case the derivatives of both series appeared to be substrates belonging to the category IIB3, showing to interact in the same way with P-gp, differently from previous findingsCitation34.
Noteworthy, the design of the reported series allowed the identification of two potent P-gp substrates, amides 3 and 8, that are characterised by different selectivity profiles, since compound 3 was active also on BCRP, while compound 8 was not. These two aromatic derivatives carry an ortho methoxy substituent, combined with a second methoxy residue in different positions.
The presence of amide or ester groups in the compounds described in previous workCitation34 did not compromise their chemical stability to spontaneous or enzymatic hydrolysis, since they resulted stable in PBS and also in human plasma; interestingly, in the compounds described in the present work only the amides resulted stable both in PBS and in human plasma, while the degradation plots of some ester compounds in human plasma showed a significant decay rate.
Since amide derivatives 3 and 8 showed to be stable and endowed with the best P-gp activity profile, they were further tested alone and in co-administration with the antineoplastic drug doxorubicin in different cancer cell lines with various levels of P-gp. The compounds showed a significant sensitising activity towards doxorubicin in cell models that are similar to clinically observed doxorubicin-resistant cancers, indicating that the chemosensitizing effects were due to the inhibition of P-gp.
In summary, the design of the reported series allowed the identification of two potent P-gp substrates, stable both in PBS and in human plasma and able to rehabilitate the effect of the antineoplastic agent doxorubicin in different cancer cell lines.
Author contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.
Supplemental Material
Download PDF (3.1 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Related Research Data
References
- Mitscher LA, Pillai SP, Gentry EJ, et al. Multiple drug resistance. Med Res Rev 1999;19:477–96.
- Kaye SB. The multidrug resistance phenotype. Br J Cancer 1988;58:691–4.
- Chen Z, Shi T, Zhang L, et al. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family in multidrug resistance: a review of the past decade. Cancer Lett 2016;370:153–64.
- El-Awady R, Saleh E, Hashim A, et al. The role of eukaryotic and prokaryotic ABC transporter family in failure of chemotherapy. Front Pharmacol 2017;7:535.
- Altenberg GA. Structure of multidrug-resistance proteins of the ATP-binding cassette (ABC) superfamily. Curr Med Chem Anticancer Agents 2004;4:53–62.
- Loo TW, Clarke DM. Mutational analysis of ABC proteins. Arch Biochem Biophys 2008;476:51–64.
- Juliano RL, Ling V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta 1976;455:152–62.
- Sharom FJ. ABC multidrug transporters: structure, function and role in chemoresistance. Pharmacogenomics 2008;9:105–27.
- McGrath T, Center MS. Mechanisms of multidrug resistance in HL60 cells: evidence that a surface membrane protein distinct from P-glycoprotein contributes to reduced cellular accumulation of drug. Cancer Res 1988;48:3959–63.
- (a) Cole SP, Sparks KE, Fraser K, et al. Pharmacological characterization of multidrug resistant MRP-transfected human tumor cells. Cancer Res. 1994;54:5902–10. (b) Grant CE, Valdimarsson G, Hipfner DR, et al. Overexpression of multidrug resistance-associated protein (MRP) increases resistance to natural product drugs. Cancer Res. 1994;54:357–61.
- Doyle LA, Yang W, Abruzzo LV, et al. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc Natl Acad Sci USA 1998;95:15665–70.
- Nakanishi T, Ross DD. Breast cancer resistance protein (BCRP/ABCG2): its role in multidrug resistance and regulation of its gene expression. Chin J Cancer 2012;31:73–99.
- Li W, Zhang H, Assaraf YG, et al. Overcoming ABC transporter-mediated multidrug resistance: molecular mechanisms and novel therapeutic drug strategies. Drug Resist Updat 2016;27:14–29.
- Videira M, Reis RL, Brito MA. Deconstructing breast cancer cell biology and the mechanisms of multidrug resistance. Biochim Biophys Acta 2014;1846:312–25.
- Leslie EM, Deeley RG, Cole SP. Multidrug resistance proteins: role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol Appl Pharmacol 2005;204:216–37.
- Haber M, Smith J, Bordow SB, et al. Association of high-level MRP1 expression with poor clinical outcome in a large prospective study of primary neuroblastoma. J Clin Oncol 2006;24:1546–53.
- Yoh K, Ishii G, Yokose T, et al. Breast cancer resistance protein impacts clinical outcome in platinum-based chemotherapy for advanced non-small cell lung cancer. Clin Cancer Res 2004;10:1691–7.
- Choi YH, Yu AM. ABC transporters in multidrug resistance and pharmacokinetics, and strategies for drug development. Curr Pharm Des 2014;20:793–807.
- Kathawala RJ, Gupta P, Ashby CR Jr, et al. The modulation of ABC transporter-mediated multidrug resistance in cancer: a review of the past decade. Drug Resist Updat 2015;18:1–17.
- Teodori E, Dei S, Martelli C, et al. The functions and structure of ABC transporters: implications for the design of new inhibitors of Pgp and MRP1 to control multidrug resistance (MDR). Curr Drug Targets 2006;7:893–909.
- Yang K, Wu J, Li X. Recent advances in the research of P-glycoprotein inhibitors. Biosci Trends 2008;2:137–46.
- Lhommé C, Joly F, Walker JL, et al. Phase III study of valspodar (PSC 833) combined with paclitaxel and carboplatin compared with paclitaxel and carboplatin alone in patients with stage IV or suboptimally debulked stage III epithelial ovarian cancer or primary peritoneal cancer. J Clin Oncol 2008;26:2674–82.
- Kolitz JE, George SL, Marcucci G, et al.; for the Cancer and Leukemia Group B. P-glycoprotein inhibition using valspodar (PSC-833) does not improve outcomes for patients younger than age 60 years with newly diagnosed acute myeloid leukemia: Cancer and Leukemia Group B study 19808. Blood 2010;116:1413–21.
- Myer MS, Joone G, Chasen MR, et al. The chemosensitizing potential of GF120918 is independent of the magnitude of P-glycoprotein-mediated resistance to conventional chemotherapeutic agents in a small cell lung cancer line. Oncol Rep 1999;6:217–8.
- Fox E, Bates SE. Tariquidar (XR9576): a P-glycoprotein drug efflux pump inhibitor. Expert Rev Anticancer Ther 2007;7:447–59.
- Thomas H, Coley HM. Overcoming multidrug resistance in cancer: an update on the clinical strategy of inhibiting p-glycoprotein. Cancer Control 2003;10:159–65.
- Coley HM. Overcoming multidrug resistance in cancer: clinical studies of P-glycoprotein inhibitors. Methods Mol Biol 2010;596:341–58.
- Cripe LD, Uno H, Paietta EM, et al. Zosuquidar, a novel modulator of P-glycoprotein, does not improve the outcome of older patients with newly diagnosed acute myeloid leukemia: a randomized, placebo-controlled trial of the Eastern Cooperative Oncology Group 3999. Blood 2010;116:4077–85.
- Kelly RJ, Draper D, Chen CC, et al. A pharmacodynamic study of docetaxel in combination with the P-glycoprotein antagonist tariquidar (XR9576) in patients with lung, ovarian, and cervical cancer. Clin Cancer Res 2011;17:569–80.
- Palmeira A, Sousa E, Vasconcelos MH, et al. Three decades of P-gp inhibitors: skimming through several generations and scaffolds. Curr Med Chem 2012;19:1946–2025.
- Kühnle M, Egger M, Müller C, et al. Potent and selective inhibitors of breast cancer resistance protein (ABCG2) derived from the p-glycoprotein (ABCB1) modulator tariquidar. J Med Chem 2009;52:1190–7.
- Kakarla P, Inupakutika M, Devireddy AR, et al. 3D-QSAR and contour map analysis of tariquidar analogues as multidrug resistance protein-1 (MRP1) inhibitors. Int J Pharm Sci Res 2016;7:554–72.
- Sun YL, Chen JJ, Kumar P, et al. Reversal of MRP7 (ABCC10)-mediated multidrug resistance by tariquidar. PLoS One 2013;8:e55576.
- Teodori E, Dei S, Bartolucci G, et al. Structure-activity relationship studies on 6,7-dimethoxy-2-phenethyl-1,2,3,4-tetrahydroisoquinoline derivatives as multidrug resistance reversers. ChemMedChem 2017;12:1369–79.
- Orlandi F, Coronnello M, Bellucci C, et al. New structure-activity relationship studies in a series of N,N-bis(cyclohexanol)amine aryl esters as potent reversers of P-glycoprotein-mediated multidrug resistance (MDR). Bioorg Med Chem 2013;21:456–65.
- Teodori E, Dei S, Coronnello M, et al. N-alkanol-N-cyclohexanol amine aryl esters: multidrug resistance (MDR) reversing agents with high potency and efficacy. Eur J Med Chem 2017;127:586–98.
- Teodori E, Dei S, Floriddia E, et al. Arylamino esters As P-glycoprotein modulators: SAR studies to establish requirements for potency and selectivity. ChemMedChem 2015;10:1339–43.
- Dei S, Coronnello M, Floriddia E, et al. Multidrug resistance (MDR) reversers: High activity and efficacy in a series of asymmetrical N,N-bis(alkanol)amine aryl esters. Eur J Med Chem 2014;87:398–412.
- Martelli C, Dei S, Lambert C, et al. Inhibition of P-glycoprotein-mediated Multidrug Resistance (MDR) by N,N-bis(cyclohexanol)amine aryl esters: further restriction of molecular flexibility maintains high potency and efficacy. Bioorg Med Chem Lett 2011;21:106–9.
- Dei S, Budriesi R, Sudwan P, et al. Diphenylcyclohexylamine derivatives as new potent multidrug resistance (MDR) modulators. Bioorg Med Chem 2005;13:985–98.
- Marshall AG, Hendrickson CL. High-resolution mass spectrometers. Annu Rev Anal Chem (Palo Alto Calif) 2008;1:579–99.
- Das R, Chakraborty D. Cu(II) bromide catalyzed oxidation of aldehydes and alcohols. Appl Organometal Chem 2011;25:437–42.
- Ohnmacht CJ. N-(2-phenyl-4-piperidinybutyl)-5,6,7,8-tetrahydro-1-naphthalenecarboxamides and their use as neurokinin 1 (NK1) and/or neurokinin 2 (NK2) receptor antagonists. US 6403601B1. 2002.
- Kowanko N, Leete E. Biosynthesis of the cinchona alkaloids. I. The incorporation of tryptophan into quinine. J Am Chem Soc 1962;84:4919–21.
- Riganti C, Miraglia E, Viarisio D, et al. Nitric oxide reverts the resistance to doxorubicin in human colon cancer cells by inhibiting the drug efflux. Cancer Res 2005;65:516–25.
- Capparelli E, Zinzi L, Cantore M, et al. SAR Studies on tetrahydroisoquinoline derivatives: The role of flexibility and bioisosterism to raise potency and selectivity toward P-glycoprotein. J Med Chem 2014;57:9983–94.
- Polli JW, Wring SA, Humphreys JE, et al. Rational use of in vitro P-glycoprotein assays in drug discovery. J Pharmacol Exp Ther 2002;303:1029–8.
- Inglese C, Perrone MG, Berardi F, et al. Modulation and absorption of xenobiotics: the synergistic role of CYP450 and P-gp activities in cancer and neurodegenerative disorders. Curr Drug Metab 2011;12:702–12.
- Colabufo NA, Contino M, Cantore M, et al. Naphthalenyl derivatives for hitting P-gp/MRP1/BCRP transporters. Bioorg Med Chem 2013;21:1324–32.
- Kangas L, Grönroos M, Nieminen AL. Bioluminescence of cellular ATP: a new method for evaluating cytotoxic agents in vitro. Med Biol 1984;62:338–43.
- Feng B, Mills JB, Davidson RE, et al. In vitro P-glycoprotein assays to predict the in vivo interactions of P-glycoprotein with drugs in the central nervous system. Drug Metab Dispos 2008;36:268–75.
- Teodori E, Dei S, Garnier-Suillerot A, et al. Structure-activity relationship studies on the potent multidrug resistance (MDR) modulator 2-(3,4-dimethoxyphenyl)-2-(methylethyl)-5-[anthr-9-yl)methylamino]pentanenitrile (MM36). Med Chem Res 2001;10:563–76.
- Riganti C, Kopecka J, Panada E, et al. The role of C/EBP-β LIP in multidrug resistance. J Natl Cancer Inst 2015;107:pii: djv046.
- Kopecka J, Campia I, Jacobs A, et al. Carbonic anhydrase XII is a new therapeutic target to overcome chemoresistance in cancer cells. Oncotarget 2015;6:6776–93.
- Dei S, Braconi L, Trezza A, et al. Modulation of the spacer in N,N-bis(alkanol)amine aryl ester heterodimers led to the discovery of a series of highly potent P-glycoprotein-based multidrug resistance (MDR) modulators. Eur J Med Chem 2019;172:71–94.