
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
The Carbonic Anhydrase (CA, EC 4.2.1.1) activating properties of histamine have been known for a long time. This compound has been extensively modified but only in few instances the imidazole ring has been replaced with other heterocycles. It was envisaged that the imidazoline ring could be a bioisoster of the imidazole moiety. Indeed, we report that clonidine, a 2-aminoimidazoline derivative, was found able to activate several human CA isoforms (hCA I, IV, VA, VII, IX, XII and XIII), with potency in the micromolar range, while it was inactive on hCA II. A series of 2-aminoimidazoline, structurally related to clonidine, was then synthesised and tested on selected hCA isoforms. The compounds were inactive on hCA II while displayed activating properties on hCA I, VA, VII and XIII, with KA values in the micromolar range. Two compounds (10 and 11) showed some preference for the hCA VA or VII isoforms.
1. Introduction
Carbonic anhydrases (CAs, EC 4.2.1.1) are metallo-enzymes widespread in all life kingdoms. These enzymes catalyse a plethora of reactions, among which the reversible hydration of CO2 is the most important oneCitation1. The active site contains the cofactor, a metal ion (usually Zn2+) which coordinates a water molecule responsible, once activated as hydroxide ion, of the nucleophilic attack onto carbon dioxide. Eight genetically different families have been found (α-ι); 15 isoforms belonging to the α class have been characterised in humansCitation1,Citation2.
CAs have been drug targets since more than 70 years; inhibitors of these enzyme are used for the treatment of oedema, glaucoma and epilepsy but several new therapeutic applications are under studyCitation3. In recent years, the attention has been focussed also on activators of these enzymes, despite the fact that CA are among the most efficient enzymes known. In fact, genetic deficiencies of several CA isoforms were reported in the last decades (reviewed in Refs. [Citation3,Citation4]), and in principle a loss of function of these enzymes could be treated with CA selective activators (CAAs). In addition, there is evidence that CA activation improves cognitive performanceCitation5–10. However, the influence of CA on these processes is complex since also inhibitors have been found to improve memory deficits in animal models (reviewed in Ref. [Citation11]); these findings point out the need for isoform selective inhibitors or activators to elucidate the role of CA isoforms in cognitive processes. Other possible applications of CAAs could be in the formation of artificial tissuesCitation12 and in CO2 capture and sequestration processesCitation13.
Histamine (HST, ) was among the first reported activators, whose interaction mode was elucidated by means of X-ray crystallographyCitation14. The adduct with hCA II revealed a complex network of H-bonds involving the Zn-bound water molecule, His64 and the imidazole ring of the activator, which is located far away from the metal ion, in a region approaching the edge of the active site cavity. X-ray crystallographic studies have later shown that also other activators bind in this areaCitation4.
Chart 1. Chemical structure of CA activators.
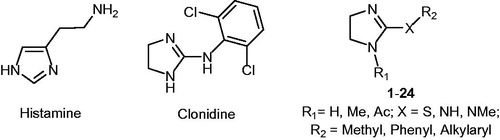
As common structural feature, CAAs possess flexible tails decorated with protonable moieties, with pKa values spanning between 6 and 8. The molecule of histamine has been extensively modified, placing substituents on the imidazole C atoms and on the NH2 group, showing that the latter is not essential, since it can be largely modified to keep or improve potency (reviewed in Ref. [Citation4]). Only in few instances the imidazole ring has been replaced by another heterocycle, such as a thiadiazole ringCitation15.
In search for bioisosters of the imidazole moiety, our attention was attracted by the imidazoline ring. This feature is present in a well-known drug, Clonidine (CLO, ), which is clinically used as an antihypertensive agent being an agonist at the central α2-adrenergic receptor, but it is able to interact with other targets, such as the imidazoline binding sites and the hyperpolarization-activated cyclic nucleotide gated channelsCitation16,Citation17. Therefore, we decided to measure the potential CA activating properties of this compound, finding that CLO behaves as CAA on several CA isoforms (). Encouraged by this positive outcome, we synthesised a series of 2-substituted imidazolines (compounds 1–24, ) and tested their activity on five different hCA isoforms. The ubiquitous cytosolic enzymes CA I and II, the mitochondrial CA VA, which is associated with the glucose homeostasis,Citation18 the cytosolic CA VII which is particularly abundant in the CNS and has been recently demonstrated to have a protective role against oxidative damage,Citation19 and the cytosolic CA XIII, which is particularly expressed in the reproductive organsCitation20,Citation21 were selected.
Table 1. Activation constants of Clonidine (CLO) and histamine (HST) on selected human CA isoforms, measured by means of a stopped-flow, CO2 hydrase assay.Table Footnotea
2. Materials and methods
2.1. Chemistry
All melting points were taken on a Büchi apparatus and are uncorrected. NMR spectra were recorded on a Brucker Avance 400 spectrometer (400 MHz for 1H NMR, 100 MHz for 13 C). Chromatographic separations were performed on a silica gel column by gravity chromatography (Kieselgel 40, 0.063–0.200 mm; Merck) or flash chromatography (Kieselgel 40, 0.040–0.063 mm; Merck). Yields are given after purification, unless differently stated. When reactions were performed under anhydrous conditions, the mixtures were maintained under nitrogen. High-resolution mass spectrometry (HR-MS) analyses were performed with a Thermo Finnigan LTQ Orbitrap mass spectrometer equipped with an electrospray ionisation source (ESI). Analyses were carried out in positive ion mode monitoring protonated molecules, [M + H]+ species, and a proper dwell time acquisition was used to achieve 60,000 units of resolution at Full Width at Half Maximum (FWHM). Elemental composition of compounds were calculated on the basis of their measured accurate masses, accepting only results with an attribution error less than 5 ppm and a not integer RDB (double bond/ring equivalents) value, in order to consider only the protonated speciesCitation22. Compounds were named following IUPAC rules by means of ChemDraw 14.0.
2.1.1. General procedure for the preparation of 2-amino-imidazoline 2–20
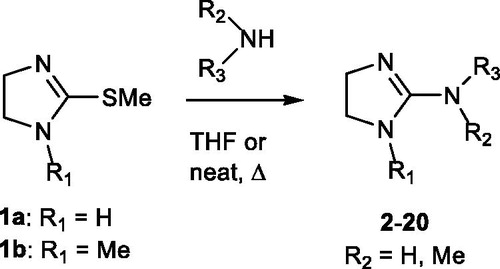
A mixture of the appropriate intermediate (1aCitation23 or 1bCitation24, 0.05 g) and the amine (1 eq) was suspended in THF (5 ml) and the mixture was heated at 70 °C until reaction completion (TLC monitoring, eluent CH2Cl2/CH3OH/NH4OH 90:10:1); alternatively, an excess of amine (5 eq) was used as solvent. Volatiles were evaporated under vacuum and the residue was triturated with Et2O to remove the unreacted amine, affording the desired imidazoline derivative. In some instances, additional purification by means of flash chromatography was necessary. The conditions for each compound are reported in . With this method, the following compounds were prepared.
Table 2. Synthetic details for the preparation of compounds 2–20 (see general formula in Scheme 1) starting from 1a (R1 = H) or 1 b (R1 = Me) and amines R2R3NH.
N-benzyl-4,5-dihydro-1H-imidazol-2-amine hydroiodide 2Citation23. White solid; m.p. 143–146 °C ESI-LC-MS (m/z) 176.0 [M + H]+ [1H]-NMR (D2O) δ: 3.56 (s, 4H, CH2CH2); 4.31 (s, 2H, CH2); 7.24–7.35 (m, 5H, Ar) ppm. [13C]-NMR (D2O) δ: 42.70 (CH2Ph); 45.65 (2CH2); 126.93 (CHAr); 128.00 (CHAr); 128.95 (CHAr); 136.25 (CAr); 159.91 (C=N) ppm.
N-Phenethyl-4,5-dihydro-1H-imidazol-2-amine hydroiodide 3Citation25. White solid; m.p 82–83 °C; ESI-LCMS (m/z) 189.7 [M + H]+. [1H]-NMR (D2O) δ: 2.75 (t, J = 6.6 Hz, 2H, CH2); 3.33 (t, J = 6.6 Hz, 2H, CH2); 3.43 (s, 4H, CH2CH2); 7.14–7.31 (m, 5H, Ar) ppm. [13C]-NMR (D2O) δ: 34.76 (CH2); 42.63 (2CH2); 43.78 (CH2); 126.94 (CHAr); 128.84 (CHAr); 129.13 (CHAr); 138.50 (CAr); 159.83 (C=N) ppm.
(S) N-(1-Phenylethyl)-4,5-dihydro-1H-imidazol-2-amine hydroiodide (S)-4Citation26. Purification by flash chromatography (CH2Cl2/CH3OH/NH3 87:13:1.3 as eluent); m.p. 103–107 °C; ESI-LCMS (m/z) 190.2 [M + H]+. [1H]-NMR (D2O) δ: 1.41 (d, J = 6.8 Hz, 3H, CH3); 3.49–3.53 (m, 4H, 2CH2); 4.55 (q, J = 6.8 Hz, 1H, CH); 7.23–7.35 (m, 5H, Ar) ppm. [13C]-NMR (D2O) δ: 22.52 (CH3); 42.62 (2CH2); 52.79 (CH); 125.59 (CHAr); 128.03 (CHAr); 129.08 (CHAr); 142.03 (CAr); 158.98 (C=N) ppm.
(R) N-(1-Phenylethyl)-4,5-dihydro-1H-imidazol-2-amine hydroiodide (R)-4Citation26. Purification by flash chromatography (CH2Cl2/CH3OH/NH3 87:13:1.3 as eluent); m.p. 105–109 °C; ESI-LCMS (m/z) 190.2 [M + H]+. [1H]-NMR (D2O) δ: 1.40 (d, J = 6.8 Hz, 3H, CH3); 3.47–3.52 (m, 4H, 2CH2); 4.54 (q, J = 6.8 Hz, 1H, CH); 7.21–7.40 (m, 5H, Ar) ppm. [13C]-NMR (D2O) δ: 22.54 (CH3); 42.67 (2CH2); 52.84 (CH); 125.63 (CHAr); 128.07 (CHAr); 129.13 (CHAr); 142.06 (CAr); 159.03 (C=N) ppm.
N-(3-Phenylpropyl)-4,5-dihydro-1H-imidazol-2-amine hydroiodide 5. White solid; m.p. 85–87 °C. [1H]-NMR (CDCl3) δ: 1.90 (p, J = 7.2 Hz, 2H, CH2); 2.70 (t, 2H, J = 7.9 Hz, PhCH2); 3.34 (apparent q, J = 6.4 Hz, 2H, NCH2); 3.60 (s, 4H, CH2CH2); 7.07 (bs, 1H, NH), 7.12–7.26 (m, 5H, Ar); 7.63 (s, 1H, NH) ppm. [13C]-NMR (CDCl3) δ: 30.68 (CH2); 32.63 (CH2); 42.57 (CH2); 43.39 (CH2); 126.22 (CHAr); 128.54 (CHAr); 140.62 (CAr); 159.67 (C=N) ppm. ESI-HRMS (m/z) [M + H]+: calculated for C12H18N3+ 204.1495; found 204.1498.
N-Benzyl-N-methyl-4,5-dihydro-1H-imidazol-2-amine hydroiodide 6. Purification by flash chromatography (CH2Cl2/CH3OH/NH3 80:20:1 as eluent). White solid; m.p. 150–153 °C. [1H]-NMR (D2O) δ: 2.89 (s, 3H, NCH3); 3.61 (s, 4H, CH2CH2); 4.42 (s, 2H, CH2Ph); 7.15–7.40 (m, 5H, Ar) ppm. [13C]-NMR (D2O) δ: 36.38 (CH3); 43.14 (2CH2); 54.29 (CH2Ph); 127.05 (CHAr); 128.25 (CHAr); 129.11 (CHAr); 134.71 (CAr); 160.548 (C=N) ppm. ESI-HRMS (m/z) [M + H]+: calculated for C11H16N3+ 190.1339; found 190.1342.
N-Methyl-N-phenethyl-4,5-dihydro-1H-imidazol-2-amine hydroiodide 7. White solid; m.p. 200–204 °C. [1H]-NMR (D2O) δ: 2.80 – 2.89 (m, 5H, NCH3 + PhCH2); 3.42 (s, 4H, 2CH2); 3.46 (t, J = 6.6 Hz, 2H, NCH2); 7.19–7.33 (m, 5H, Ar) ppm. [13C]-NMR (D2O) δ: 32.62 (CH2Ph); 36.19 (NCH3); 42.83 (N-CH2); 52.53 (CH2CH2); 126.98 (CHAr); 128.82 (CHAr); 129.05 (CHAr); 138.13 (CAr); 159.89 (C=N) ppm. ESI-HRMS (m/z) [M + H]+: calculated for C12H18N3+ 204.1495; found 204.1499.
1-Methyl-N-phenethyl-4,5-dihydro-1H-imidazol-2-amine hydroiodide 8. White solid; m.p. 118–220 °C. [1H]-NMR (CDCl3) δ: 3.00–3.08 (m, 5H, PhCH2 + NCH3); 3.48 (s, 4H, 2CH2); 3.65 (apparent q, J = 6.8 Hz, 2H, NCH2); 6.96 (s, 1H, NH), 7.12–7.35 (m, 5H, Ar), 7.49 (t, J = 5.6, 1H, NH) ppm. [13C]-NMR (CDCl3) δ: 33.78 (CH3); 35.49 (CH2); 41.04 (CH2); 45.22 (CH2); 50.11 (CH2); 126.79 (CHAr); 128.67 (CHAr); 129.32 (CHAr); 137.87 (CAr); 158.22 (C=N) ppm. ESI-HRMS (m/z) [M + H]+: calculated for C12H18N3+ 204.1495; found 204.1494.
N,1-Dimethyl-N-phenethyl-4,5-dihydro-1H-imidazol-2-amine hydroiodide 9. White solid; m.p. 109–110 °C. [1H]-NMR (CDCl3) δ: 2.82 (s, 3H, CH3); 2.97 (t, J = 6.8 Hz, 2H, PhCH2); 3.13 (s, 3H, CH3); 3.55–3.62 (m, 4H, CH2CH2); 3.77 (t, J = 6.9 Hz, 2H, CH2N); 7.32 (t, J = 7.5 Hz, 5H, Ar), 8.34 (bs, 1H, NH) ppm. [13C]-NMR (CDCl3) δ: 33.66 (CH2); 36.97 (CH3); 39.70 (CH3); 40.33 (CH2); 52.89 (CH2); 54.71 (CH2); 127.23 (CH-Ar); 128.92 (CH-Ar); 136.96 (C-Ar); 162.47 (C=N) ppm. ESI-HRMS (m/z) [M + H]+: calculated for C13H20N3+ 218.1652; found 218.1649.
N-Benzyl-N,1-dimethyl-4,5-dihydro-1H-imidazol-2-amine hydroiodide 10. Purification by flash chromatography (CH2Cl2/CH3OH/NH3 90:10:1 as eluent). Oil. [1H]-NMR (CDCl3) δ: 3.01 (s, 3H, CH3); 3.07 (s, 3H, CH3); 3.70–3.87 (m, 4H, CH2CH2); 4.63 (s, 2H, CH2Ph); 7.22–7.35 (m, 5H, Ar), 8.10 (s, 1H, NH) ppm. [13C]-NMR (CDCl3) δ: 37.03 (CH3); 39.72 (CH3); 40.53 (CH2); 53.14 (CH2); 56.60 (CH2); 127.20 (CHAr); 128.44 (CHAr); 129.25 (CHAr); 133.94 (CAr); 162.63 (C=N) ppm. ESI-HRMS (m/z) [M + H]+: calculated for C12H18N3+ 204.1495; found 204.1497.
N-Benzyl-1-methyl-4,5-dihydro-1H-imidazol-2-amine hydroiodide 11. White solid; m.p. 130–133 °C. [1H]-NMR (CDCl3) δ: 3.16 (s, 3H, CH3); 3.58–3.66 (m, 4H, CH2CH2); 4.67 (d, J = 5.6 Hz, 2H, CH2Ph); 6.87 (s, 1H, NH); 7.25–7.38 (m, 3H, Ar); 7.49 (d, J = 7.2 Hz, 2H, Ar); 8.21 (s, 1H, NH) ppm. [13C]-NMR (CDCl3) δ: 33.76 (CH3); 41.31 (CH2); 46.38 (CH2); 50.22 (CH2); 128.13 (CHAr); 128.34 (CHAr); 129.04 (CHAr); 135.63 (CAr); 158.35 (C=N) ppm. ESI-HRMS (m/z) [M + H]+: calculated for C11H16N3+ 190.1339; found 190.1338.
1-Methyl-N-(3-phenylpropyl)-4,5-dihydro-1H-imidazol-2-amine hydroiodide 12. Purification by flash chromatography (CH2Cl2/CH3OH/NH3 90:10:1 as eluent). Gum. [1H]-NMR (CDCl3) δ: 2.02 (p, J = 7.6 Hz, 2H); 2.69 (t, J = 8.0 Hz, 2H, CH2); 2.97 (s, 3H, CH3); 3.41–3.54 (m, 4H, CH2CH2); 3.60 (t, J = 8.0 Hz, 2H, CH2); 7.10–7.25 (m, 5H, Ar) ppm. [13C]-NMR (CDCl3) δ: 30.62 (CH2), 32.93 (CH2), 33.77 (NCH3), 40.97 (CH2), 43.83 (CH2), 50.11 (CH2), 126.01 (CHAr), 128.44 (CHAr), 128.58 (CHAr), 141.25 (CAr), 157.91 (C=N) ppm. ESI-HRMS (m/z) [M + H]+: calculated for C13H20N3+ 218.1652; found 218.1654.
N-(4-Chlorobenzyl)-1-methyl-4,5-dihydro-1H-imidazol-2-amine hydroiodide 13. White solid, m.p. 84 °C. [1H]-NMR (MeOD) δ: 2.96 (s, 3H, CH3), 3.61–3.76 (m, 4H, CH2CH2), 4.41 (s, 2H, CH2), 7.31 (d, J = 8.4 Hz, 2H, Ar), 7.35 (d, J = 8.4 Hz, 2H, Ar), 7.42 (s, 2H, NH) ppm. [13C]-NMR (MeOD) δ: 30.68 (CH3), 40.95 (CH2), 45.41 (CH2), 50.09 (CH2), 128.92 (CHAr), 130.37 (CHAr), 133.46 (CCl), 135.00 (CAr), 158.68 (C=N), ppm. ESI-HRMS (m/z) [M + H]+: calculated for C11H15ClN3+ 224.0949; found 224.0946.
N-(4-Methoxybenzyl)-1-methyl-4,5-dihydro-1H-imidazol-2-amine hydroiodide 14. White solid, m.p. 167 °C. [1H]-NMR (CDCl3) δ: 3.18 (s, 3H, CH3), 3.64 (s, 4H, CH2CH2), 3.76 (s, 3H, OCH3), 4.58 (d, J = 5.7 Hz, 2H, CH2), 6.38 (s, 1H, NH), 6.85 (d, J = 8.6 Hz, 2H, Ar), 7.39 (d, J = 8.6 Hz, 2H, Ar), 8.10 (s, 1H, NH), ppm. [13C]-NMR (CDCl3) δ: 33.77 (NCH3), 41.29 (CH2), 45.86 (CH2), 50.14 (CH2), 55.36 (OCH3), 114.38 (CHAr), 127.62 (CAr), 129.69 (CHAr), 158.19 (CAr), 159.59 (C=N) ppm. ESI-HRMS (m/z) [M + H]+: calculated for C12H18N3O+ 220.1444; found 220.1443.
N-(4-Fluorobenzyl)-1-methyl-4,5-dihydro-1H-imidazol-2-amine hydroiodide 15. White solid, m.p. 157 °C. [1H]-NMR (MeOD) δ: 2.97 (s, 3H, CH3), 3.61 – 3.75 (m, 4H, CH2CH2), 4.42 (s, 2H, CH2), 7.08 (t, J = 8.5 Hz, 2H, Ar), 7.42 (dd, J = 8.5, 5.4 Hz, 2H, Ar), ppm. [13C]-NMR (MeOD) δ: 30.81 (CH3), 40.98 (CH2), 45.44 (CH2), 50.11 (CH2), 115.26 (d, JC-F = 22 Hz, CHAr), 129.12 (d, JC-F = 8 Hz, CHAr), 132.25 (CAr), 158.62 (C=N), 162.48 (d, JC-F = 244 Hz, CF), ppm. ESI-HRMS (m/z) [M + H]+: calculated for C11H15FN3+ 208.1245; found 208.1245.
N-(3-Chlorobenzyl)-1-methyl-4,5-dihydro-1H-imidazol-2-amine hydroiodide 16. White solid, m.p. 190 °C. [1H]-NMR (MeOD) δ: 2.96 (s, 3H, CH3), 3.58–3.71 (m, 4H), 4.41 (s, 2H, CH2), 7.20–7.25 (m, 1H, Ar), 7.25–7.37 (m, 3H, Ar) ppm. [13C]-NMR (MeOD) δ: 30.56 (CH3), 40.93 (CH2), 45.45 (CH2), 50.06 (CH2), 125.25 (CHAr), 126.91 (CHAr), 127.74 (CHAr), 130.07 (CHAr), 134.35 (C-Cl), 138.59 (CAr), 154.10 (C=N) ppm. ESI-HRMS (m/z) [M + H]+: calculated for C11H15ClN3+ 224.0949; found 224.0949.
N-(3-Methoxybenzyl)-1-methyl-4,5-dihydro-1H-imidazol-2-amine hydroiodide 17. White solid, m.p. 147 °C. [1H]-NMR (CDCl3) δ: 2.95 (s, 3H, CH3), 3.61 (s, 4H, CH2CH2), 3.76 (s, 3H, OCH3), 4.59 (s, 2H, CH2), 6.77 (d, J = 6.4 Hz, 1H, NH), 7.06–7.66 (m, 4H, Ar), 8.31 (s, 1H, NH) ppm. [13C]-NMR (CDCl3) δ: 33.70 (CH3), 41.32 (CH2), 46.20 (CH2), 50.22 (CH2), 55.68 (OCH3), 113.67 (CHAr), 114.00 (CHAr), 120.27 (CHAr), 129.96 (CHAr), 137.43 (CAr), 158.22 (CAr), 159.94 (C=N), ppm. ESI-HRMS (m/z) [M + H]+: calculated for C12H18N3O+ 220.1444; found 220.1447.
N-(3-Fluorobenzyl)-1-methyl-4,5-dihydro-1H-imidazol-2-amine hydroiodide 18. White solid, m.p. 195 °C. [1H]-NMR (CDCl3) δ: 3.19 (s, 3H, CH3), 3.61–3.71 (m, 4H, CH2CH2), 4.68 (s, 2H, CH2), 6.95 (t, J = 7.7 Hz, 1H, NH), 7.09 – 7.41 (m, 4H, Ar), 8.27 (bs, 1H, NH) ppm. [13C]-NMR (CDCl3) δ: 33.86 (CH3), 41.19 (CH2), 45.42 (CH2), 50.20 (CH2), 115.05 (d, JCF= 25 Hz, CHAr), 123.94 (CHAr), 130.45 (d, J = 8 Hz, CHAr), 138.48 (CAr), 158.11 (C=N), 162.78 (d, J = 246 Hz, CF) ppm.
N1-Benzyl-N2-(4,5-dihydro-1H-imidazol-2-yl)ethane-1,2-diamine hydroiodide 19. White solid, m.p. 230 °C (dec). [1H]-NMR (MeOD) δ: 3.37 (t, J = 6.2 Hz, 2H, CH2), 3.64 (t, J = 6.2 Hz, 2H, CH2), 3.70 (s, 4H, CH2CH2), 4.34 (s, 2H, PhCH2), 7.40–7.47 (m, 3H, Ar), 7.59–7.64 (m, 2H, Ar) ppm. [13C]-NMR (MeOD) δ: 38.87 (CH3), 42.82 (CH2CH2), 45.73 (CH2), 51.20 (PhCH2), 128.92 (CHAr), 129.49 (CHAr), 129.99 (CHAr), 130.69 (CAr), 159.88 (C=N) ppm. ESI-HRMS (m/z) [M + H]+: calculated for C12H19N4+ 219.1604; found 219.1606.
N1-Benzyl-N2-(1-methyl-4,5-dihydro-1H-imidazol-2-yl)ethane-1,2-diamine hydroiodide 20. White solid, m.p. 235 °C (dec). [1H]-NMR (MeOD) δ: 2.93 (s, 3H, CH3), 3.33 (t, J = 6.0 Hz, 2H, CH2), 3.63–3.72 (m, 6H, 3CH2), 4.28 (s, 2H, PhCH2), 7.38–7.44 (m, 3H, Ar), 7.48 – 7.54 (m, 2H, Ar) ppm. [13C]-NMR (MeOD) δ: 30.59 (CH3), 39.07 (CH3), 40.94 (CH2), 45.61 (CH2), 50.04 (PhCH2), 51.17 (CH2), 128.56 (CHAr), 129.37 (CHAr), 129.80 (CHAr), 130.81 (CAr), 156.18 (C=N) ppm. ESI-HRMS (m/z) [M + H]+: calculated for C13H21N4+ 233.1761; found 233.1760.
2.1.2. N1-(4,5-Dihydro-1H-imidazol-2-yl)ethane-1,2-diamine 21Citation27
The compound was prepared following the procedure reported by A. Bucio-Cano et al.Citation27 Yellow oil, isolated then as oxalate salt. Yield: 61%; m.p. (oxalate salt): 153–154 °C. [1H] NMR (D2O) δ: 3.10 (t, J = 6 Hz, 2H, CH2); 3.44 (t, J = 6 Hz, 2H, CH2); 3.59 (s, 4H, NHCH2CH2NH2) ppm. [13C] NMR (D2O) δ: 38.28 (CH2); 39.70 (CH2); 42.82 (2CH2); 159.80 (C); 165.0 (CO Oxalate) ppm.
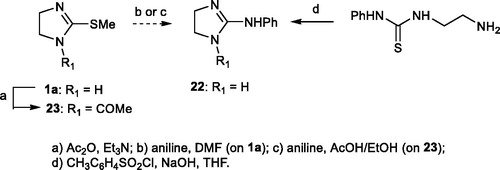
2.1.3. N-Phenyl-4,5-dihydro-1H-imidazol-2-amine 22Citation28
1-(2-Aminoethyl)-3-phenylthioureaCitation29 (110 mg, 0.56 mmol) was dissolved in THF (5 ml) and a 1 M sodium hydroxide/water solution (1 eq) was added while stirring. After 0.5 h, a solution of TsCl (1–1.1 equiv) in THF (5 ml) was slowly added and the mixture was stirred for 5 h at room T. The solvent was evaporated and diethyl ether and brine were added to the residue. The layers were separated and the aqueous layer was extracted three times with diethyl ether. The combined organic layers were dried (Na2SO4), filtered and evaporated, leaving a residue which was purified by flash chromatography (CH2Cl2: MeOH: NH3 80:20:1 as eluent). Compound 22 was obtained as a white solid, m.p. 137–140 °C, in 50% yields. [1H]-NMR (D2O) δ: 3.40 (s, 4H, CH2CH2), 6.94 (d, J = 7.2 Hz, 2H), 7.03 (t, J = 7.2 Hz, 1H), 7.27 (t, J = 8.0 Hz, 2H) ppm. [13C]-NMR (D2O) δ: 42.37 (CH2), 123.34 (CHAr), 123.40 (CHAr), 129.52 (CHAr), 146.90 (CAr), 161.03 (C=N) ppm.
2.1.4. 1–(2-(Methylthio)-4,5-dihydro-1H-imidazol-1-yl) ethanone 23Citation30
The imidazoline 1a was acylated on one imidazoline-nitrogen atom to obtain 23 as reported by Gomez-San Juan et al.Citation30 Yellow solid, yield: 100%; m.p.= 107–110 °C. [1H]-NMR (D2O) δ: 2.07 (s, 3H, SCH3); 2.23 (s, 3H, CH3); 3.76 (t, J = 8.8 Hz, 2H, CH2); 3.92 (t, J = 8.8 Hz, 2H, CH2) ppm. [13C]-NMR (D2O) δ: 14.5 (SCH3); 23.5 (CH3); 48.0 (2CH2); 161.5 (C=N); 171.9 (CO) ppm.
2.1.5. 2-(Benzylthio)-4,5-dihydro-1H-imidazole 24 hydrobromideCitation31
A mixture of commercially-available 2-imidazolidinethione (0.1 g, 0.98 mmol) and benzyl bromide (1.1 eq) in anhydrous methanol (5 ml) was heated at 65 °C for 2 h. After cooling, the solvent was removed under vacuum and the residue was treated with Et2O until it became solid; filtration and drying under vacuum gave the desired compound as a white solid, m.p. 181–183 °C. ESI-LC-MS (m/z) 193.1 [M + H]+ [1H]-NMR (D2O) δ: 3.81 (s, 4H, CH2CH2); 4.35 (s, 2H, CH2); 7.31– 7.42 (m, 5H, Ar) ppm. [13C]-NMR (D2O) δ: 35.15 (CH2Ph); 45.24 (2CH2); 128.31 (CHAr); 128.72 (CHAr); 129.10 (CHAr); 134.12 (CAr); 169.69 (C=N) ppm.
2.2. CA activation
An Sx.18Mv-R Applied Photophysics (Oxford, UK) stopped-flow instrument has been used to assay the catalytic activity of various CA isozymes for CO2 hydration reactionCitation32. Phenol red (at a concentration of 0.2 mM) was used as indicator, working at the absorbance maximum of 557 nm, with 10 mM Hepes (pH 7.5, for α-CAs)Citation33–37 as buffers, 0.1 M NaClO4 (for maintaining constant ionic strength), following the CA-catalysed CO2 hydration reaction for a period of 10 s at 25 °C. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each activator at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The uncatalysed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of activators (at 0.1 mM) were prepared in distilled-deionised water and dilutions up to 1 nM were made thereafter with the assay buffer. Enzyme and activator solutions were pre-incubated together for 15 min prior to assay, in order to allow for the formation of the enzyme–activator complexes. The activation constant (KA), defined similarly with the inhibition constant KI, can be obtained by considering the classical Michaelis–Menten equation (EquationEquation (1)(1)
(1) , which has been fitted by non-linear least squares by using PRISM 3:
(1)
(1)
where [A]f is the free concentration of activator.
Working at substrate concentrations considerably lower than KM ([S] ≪KM), and considering that [A]f can be represented in the form of the total concentration of the enzyme ([E]t) and activator ([A]t), the obtained competitive steady-state equation for determining the activation constant is given by EquationEquation (2)(2)
(2) :
(2)
(2)
where v0 represents the initial velocity of the enzyme-catalysed reaction in the absence of activatorCitation33–39. This type of approach to measure enzyme–ligand interactions is in excellent agreement with recent results from native mass spectrometry measurementsCitation40.
3. Results and discussion
3.1. Chemistry
2-Amino imidazolines 2–20 were prepared by reacting primary and secondary amines with 2-(methylthio)-4,5-dihydro-1H-imidazole hydroiodide 1aCitation23 or its N1-methyl derivative 1bCitation24 using tetrahydrofuran or an excess of amine as solvents (Scheme 1). The final compounds were obtained as hydroiodide salts in different yields. Synthetic details are reported in the experimental section. Reactions with 3- or 4-nitrobenzyl amine or with dibenzyl amine were unsuccessful.
Scheme 1. Synthesis of compounds 2-20.
This method failed also to give compound 22 (step b, Scheme 2), whose synthesis was therefore attempted according to the procedure of Gomez Saint-Juan (step c, Scheme 2);Citation30 however, also this method was abandoned since the reaction of 1-acetyl-2-(methylthio)-4,5-dihydro-1H-imidazole 23 with aniline was not successful. Finally, 22 was prepared from 1–(2-aminoethyl)-3-phenylthioureaCitation29 according to Heinelt et al.Citation28 Compound 21Citation27 was synthesised through condensation of guanidine and ethylenediamine while 24Citation31 was prepared by reaction of imidazoline-2-thione with benzyl bromide.
Scheme 2. Synthesis of compounds 22 and 23.
3.2. CA activating profile
The stopped-flow methodCitation32 has been used for assaying the CO2 hydration activity catalysed by different CA isoforms; the results are expressed as KA (activation constant, µM). The activating profile of CLO is reported in , in comparison with HST. CLO behaved as an activator on several CA isoforms (I, IV, VA, VII, IX, XII and XIII), with KA values in the range 7.8–136 µM, while on CA II it was inactive up to a dose of 200 µM. In particular, the isoforms most sensitive to CLO were CA VII and CA XIII.
The KA values of the synthesised compounds are reported in . All compounds have been tested as hydroiodide salts, with the exception of 21 (oxalate), 22 and 23 (free bases), and 24 (as HBr salt). As CLO, none of the compounds was active on hCA II at the highest tested concentration, while on the other isoforms the compounds showed KA values mainly in the low-medium range, allowing to derive the following structure–activity relationships.
Table 3. Activation constant (KA) of the synthesised compounds and Clonidine (CLO) for human I, II, VA, VII and XIII Carbonic Anhydrase isoforms. 
3.2.1. hCA I
The KA value of CLO on this isoform was 76.3 µM. Removal of both chlorine atoms abolished activity, since 22 (R2 = Ph) was inactive when tested up to a 150 µM concentration. On the contrary, inserting a CH2 unit between the exocyclic N atom and the Ph ring of 22 improved the activity: in fact, 2 (KA 4.18 µM), 18 times more potent than CLO, was one of the most potent compounds on this isoform among the newly synthesised analogues. The elongation of the methylene chain gave compounds less active than 2; interestingly, a chain formed by 3 CH2 units was tolerated (5, KA 36.7 µM) while a CH2CH2 chain was not (3, KA >150 µM). Side-chain branching abolished activity (S-4, R-4: KA >150 µM). Methylation of the exocyclic Nα atom was tolerated when R2 was a phenethyl group (7: KA 68.6 µM) but not when R2 was a benzyl moiety (6: KA >150 µM). Also the methylation of the endocyclic N1 atom gave contrasting results, since compounds 8 and 12 were more active than their non-methylated analogues 3 and 5, but 11 (R2 = benzyl) showed a fourfold decrease of activity with respect to 2 (11: KA 16.9; 2: 4.18 µM). Double methylation was productive for the phenethyl analogues (compare 9 with 3, 7 and 8); for the benzyl analogue 10 this modification improved the activity only with respect to 6. The aminoethyl derivative 21 (KA 3.87 µM) was the most potent compound on the hCAI isoform; the addition of a benzyl moiety on the primary amine group abolished activity (19, KA > 100 µM), and also the N1-methyl analogue 20 was inactive.
Aromatic substitution on the benzyl moiety did not improve the potency: in fact, while the 4-Cl derivative 13 was equiactive with 11, a 4-OMe (14) or 4-F substituent (15) increased from 2 to 3.5 times the KA values. The same substituents in the meta position reduced to a higher extent or abolished the activity. As far as the sulphur analogues 1a, 23 and 24 are concerned, the small methyl group seemed tolerated, not the bulkier benzyl moiety (24, KA >150 µM). The basicity of the amidine group appeared to be not crucial, since the NH and the N-acetyl derivatives (1a and 23, respectively) were equipotent.
3.2.2. hCA VA
The KA value of CLO on this isoform was 42.6 µM. The removal of both chlorine atoms did not affect activity, since 22 (R2 = Ph, KA 52.7 µM) was almost equipotent with CLO. Also the insertion of a CH2 unit between the exocyclic N atom and the Ph ring of 22 did not substantially modified potency (2, KA 45.7 µM). On this isoform, the majority of the compounds showed good activating properties, with potency higher than CLO: the KA values of 24, 23, 3–5, 7–9, 11 and 12 were in the range 3.7–17.2 µM.
The most potent compound was 10 (KA 0.9 µM), a benzyl derivative carrying a methyl group on both N1 and Nα atoms; this compound was 47 times more active than CLO. The removal of the exocyclic Nα-Me group decreased 4 times the activity (11, KA 3.7 µM), while the removal of the N1-methyl group was more detrimental: as a matter of fact, compounds 6 (KA 40.5 µM) and 2 (KA 45.5 µM) were about 40 times less potent than 10 (KA 0.9 µM). On the contrary, the degree of methylation did not substantially affect the potency of the phenylethyl and phenyl propyl derivatives, since compounds 3, 5, 7, 8 and 12 had KA values in the range 9.9–17.2 µM. Similarly, aromatic substitution on the benzyl moiety slightly decreased the potency without substantial modulation, the KA values of compounds 13–18 being 2–4 times higher than 11. Side-chain branching (compounds S-4 and R-4) improved the activity on this isoform, and a small enantioselectivity was observed: the R-enantiomer was twice more potent as the S-isomer. A benzyl moiety on the terminal amino group of 21 (KA 31.2 µM) increased the activity, as analogue 19 and its N1-methyl derivative 20 were about 3 times more potent than the parent compound.
As far as the sulphur derivatives are concerned, the replacement of the NαH moiety of 2 (KA 45.5 µM) with S (24, KA 11.1 µM) brought a fourfold improvement in activity. Acetylation of the N1 nitrogen was also favourable, as 23 was twice more potent than 1a.
3.2.3. hCA VII
The KA value of CLO on this isoform was 8.4 µM. All the tested compounds showed activation properties on this isoform, with KA values between 0.9 and 91.6 µM. The least potent compound was the primary amine 21 (KA 91.6 µM), whose activity was however improved by adding a benzyl group on the terminal NH2 moiety (19, KA 41.7 µM) and a methyl group on the endocyclic N atom (20, KA 29.0 µM). The removal of chlorine atoms of CLO reduced 4 times the activity (22, R2 = Ph, KA 32.6 µM) while the separation of the phenyl and NαH moieties by means of a CH2 unit did not substantially modified potency (3, KA 35.2 µM). On the contrary, the potency increased by elongating the chain from 1 to 3 CH2 units (3, KA 35.2 µM; 5, KA 11.4 µM) and by adding a methyl group on the NαH moiety: with the latter modification the potency of 3 (KA 18.9 µM) and of 2 (KA 35.2 µM) were increased 3 (6, KA 11.0 µM) and 8 times (7, KA 2.4 µM), respectively. Side-chain branching did not substantially affect activity, since S-4 and R-4 were equipotent with 2. Methylation on the endocyclic N atom was the most effective modification in this set of molecules: as a matter of fact, with this structural change the KA value of 2 (KA 35.2 µM) was reduced 39 times, and 11 (KA 0.9 µM) resulted in the most potent compound on this isoform. The same modification was also effective on the phenylpropyl derivative 5 (KA 11.4 µM), whose activity was increased 4 times (12, KA 3.1 µM). The double methylation on the N1 and Nα atoms gave potent compounds (10, KA 6.5 µM and 9, KA 2.6 µM) even if the KA values are, respectively, 7 and 3 times lower than that of 11. Aromatic substitution on the benzyl moiety of 11 was detrimental for activity, as compounds 13–18 were 19–33 times less potent than 11. As far as the sulphur analogues 1a, 23 and 24 are concerned, their KA values were in the range 30.9–46.7 µM, not better that the other tested 2-aminoimidazoline derivatives. Attempts to crystallise adducts of 7, 11 and 12 with hCA VII are ongoing.
3.2.4. hCA XIII
This is the isoform most sensitive to CLO among those studied (KA 7.8 µM). As it happened on the hCA I isoform, the removal of both chlorine atoms, to give 22, abolished activity. Several other compounds resulted inactive when tested at concentrations up to 100 µM, i.e. the sulphur derivatives, the polar aminoethyl derivative 21, and all the compounds having both the N1 and Nα atoms as secondary amines, with the exception of the lipophilic phenylpropyl derivative 5 (KA 24.3 µM). The activity of 21 could be restored by adding a benzyl moiety on the primary amino-group (19, KA 20.1 µM) and a methyl group on the endocyclic N atom (20, KA 16.3 µM). Also the methylation of the phenethyl analogue 3 on the Nα atom re-established activity, giving 7 (KA 6.5 µM) which resulted the most potent compound of the series on this isoform. Methylation on the endocyclic N1 atom gave compounds 8, 11 and 12 whose potency ranged from 10.9 to 31.0 µM, the most potent being the derivative carrying a phenylpropylamino side chain (12). Methylation on both N1 and Nα atoms or aromatic substitution on the benzyl moiety did not improve activity.
As far as selectivity is concerned, the two compounds showing submicromolar KA values displayed also interesting selectivity profiles: 10 was more active on hCA VA with respect to hCA I (33 times), II (>100 times), VII (7 times), and XIII (19 times), while 11 showed a preference for hCA VII over hCA I (19 times), II (>100 times), VA (4 times), and XII (21 times).
4. Conclusions
We have synthesised a series of 2-aminoimidazolines, structurally related to Clonidine, and tested them on five different hCA isoforms (I, II, VA, VII and XIII). As the lead compound, none of the newly synthesised molecules was active on the ubiquitously expressed CA II; on the contrary, the compounds showed activity in the micromolar range on the other tested CA isoforms. Structure–activity relationships were derived, which were different on the various isoforms, suggesting that it could be possible, in this class of compounds, to find molecules, selective for a particular CA isoform. Indeed, from these preliminary modifications it has been possible to find two compounds, 10 and 11, with a promising preference towards, respectively, CA VA and VII. Work is underway to improve both potency and selectivity, in order to find new pharmacological tool to activate specific CA isoforms in pathologies characterised by their loss of functionality.
Disclosure statement
The authors report no conflict of interest.
Additional information
Funding
References
- Supuran CT, De Simone G, Carbonic anhydrases: an overview. In: Claudiu T, Supuran GDS, eds. Carbonic anhydrases as biocatalysts – from theory to medical and industrial applications. Amsterdam: Elsevier; 2015.
- Jensen EL, Clement R, Kosta A, et al. A new widespread subclass of carbonic anhydrase in marine phytoplankton. ISME J 2019;13:2094–106.
- Bozdag M, Altamimi ASA, Vullo D, et al. State of the art on carbonic anhydrase modulators for biomedical purposes. Curr Med Chem 2019;26:2558–73.
- Supuran CT. Carbonic anhydrase activators. Future Med Chem 2018;10:561–73.
- Sun MK, Alkon DL. Carbonic anhydrase gating of attention: memory therapy and enhancement. Trends Pharmacol Sci 2002;23:83–9.
- Canto de Souza L, Provensi G, Vullo D, et al. Carbonic anhydrase activation enhances object recognition memory in mice through phosphorylation of the extracellular signal-regulated kinase in the cortex and the hippocampus. Neuropharmacology 2017;118:148–56.
- Temperini C, Scozzafava A, Supuran CT. Carbonic anhydrase activators and the drug design. Curr Pharm. Des 2008;14:708–15.
- Akocak S, Supuran CT. Activation of α-, β-, γ- δ-, ζ- and η- class of carbonic anhydrases with amines and amino acids: a review. J Enz Inhib Med Chem 2019;34:1652–9.
- Supuran CT, Vullo D, Manole G, et al. Designing of novel carbonic anhydrase inhibitors and activators. Curr Med Chem Cardiovasc Hematol Agents 2004;2:49–68.
- Akocak S, Lolak N, Bua S, et al. α-Carbonic anhydrases are strongly activated by spinaceamine derivatives. Bioorg Med Chem 2019;27:800–4.
- Provensi G, Carta F, Nocentini A, et al. A new kid on the block? Carbonic anhydrases as possible new targets in Alzheimer’s disease. Int J Mol Sci 2019;20:4724.
- Wang X, Schröder HC, Schlossmacher U, et al. Modulation of the initial mineralization process of SaOS-2 cells by carbonic anhydrase activators and polyphosphate. Calcified Tissue Int 2014;94:495–509.
- Capasso C, Supuran CT, Targeting carbonic anhydrases in biotechnology. In: Capasso C, Supuran CT eds. Targeting carbonic anhydrases. Future Medicine; 2014:158–69.
- Briganti F, Mangani S, Orioli P, et al. Carbonic anhydrase activators: X-ray crystallographic and spectroscopic investigations for the interaction of isozymes I and II with histamine. Biochemistry 1997;36:10384–92.
- Supuran CT, Barboiu M, Luca C, et al. Carbonic anhydrase activators. Part 14. Syntheses of mono and bis pyridinium salt derivatives of 2-amino-5-(2-aminoethyl)- and 2-amino-5-(3-aminopropyl)-1,3,4-thiadiazole and their interaction with isozyme II. Eur J Med Chem 1996;31:597–606.
- Bousquet P, Hudson A, García-Sevilla JA, Li J-X. Imidazoline receptor system: the past, the present, and the future. Pharmacol Rev 2020;72:50–79.
- Romanelli MN, Sartiani L, Masi A, et al. HCN channels modulators: the need for selectivity. Curr Top Med Chem 2016;16:1764–91.
- Price TO, Sheibani N, Shah GN. Regulation of high glucose-induced apoptosis of brain pericytes by mitochondrial CA VA: a specific target for prevention of diabetic cerebrovascular pathology. Biochim Biophys Acta 2017;1863:929–35.
- Di Fiore A, Monti DM, Scaloni A, et al. Protective role of carbonic anhydrases III and VII in cellular defense mechanisms upon redox unbalance. Oxid Med Cell Longev 2018;2018: 2018306.
- José O, Torres-Rodríguez P, Forero-Quintero LS, et al. Carbonic anhydrases and their functional differences in human and mouse sperm physiology. Biochem Biophys Res Commun 2015;468:713–8.
- Karim K, Giribabu N, Muniandy S, Salleh N. Estrogen and progesterone differentially regulate carbonic anhydrase II, III, IX, XII, and XIII in ovariectomized rat uteri. Syst Biol Reprod Med 2016;62:57–68.
- Marshall AG, Hendrickson CL. High-resolution mass spectrometers. Ann Rev Anal Chem 2008;1:579–99.
- Aoyagi N, Endo T. Synthesis of five- and six-membered cyclic guanidines by guanylation with isothiouronium iodides and amines under mild conditions. Synth Commun 2017;47: 442–8.
- McKay AF, Kreling ME. Nitration of 1-substituted-2-iminoimidazolidines. J Org Chem 1957;22:1581–3.
- Tronche P, Amelot A, Bayard J, Laroussinie C. Synthesis of some N-substituted 2-aminoimidazolines. Ann Pharm Fr 1960; 18:726–35.
- Genc M, Servi S. Microwave-induced synthesis of 2-aminoimidazolines under neat conditions. Synth Commun 2009;39:3263–77.
- Bucio-Cano A, Reyes-Arellano A, Correa-Basurto J, et al. Targeting quorum sensing by designing azoline derivatives to inhibit the N-hexanoyl homoserine lactone-receptor CviR: synthesis as well as biological and theoretical evaluations. Bioorg Med Chem 2015;23:7565–77.
- Heinelt U, Lang HJ. Process for synthesizing heterocyclic compounds, Patent US2004/0242560 A1.
- Tilley JW, Levitan P, Kierstead RW, Cohen M. Antihypertensive (2-aminoethyl)thiourea derivatives. 1. J Med Chem 1980;23:1387–92.
- Gómez-SanJuan A, Botija JM, Méndez A, et al. C-N bond forming reactions in the synthesis of substituted 2-aminoimidazole derivatives. Arkivoc 2014;44–56.
- Aspinall SR, Bianco EJ. A synthesis of 2-alkylamino-4,5-dihydroimidazoles. J Am Chem Soc 1951;73:602–3.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73.
- Temperini C, Scozzafava A, Vullo D, Supuran CT. Carbonic anhydrase activators. Activation of isozymes I, II, IV, VA, VII, and XIV with L- and D-histidine and crystallographic analysis of their adducts with isoform II: engineering proton-transfer processes within the active site of an enzyme. Chem Eur J 2006;12:7057–66.
- Akocak S, Lolak N, Vullo D, et al. Synthesis and biological evaluation of histamine Schiff bases as carbonic anhydrase I, II, IV, VII, and IX activators. J Enz Inhib Med Chem 2017;32:1305–12.
- Akocak S, Lolak N, Bua S, et al. Activation of human α-carbonic anhydrase isoforms I, II, IV and VII with bis-histamine schiff bases and bis-spinaceamine substituted derivatives. J Enz Inhib Med Chem 2019;34:1193–8.
- Angeli A, Del Prete S, Osman SM, et al. Activation studies with amines and amino acids of the β-carbonic anhydrase encoded by the Rv3273 gene from the pathogenic bacterium Mycobacterium tuberculosis. J Enz Inhib Med Chem 2018;33:364–9.
- Angeli A, Alasmary FAS, Del Prete S, et al. The first activation study of a δ-carbonic anhydrase: TweCAδ from the diatom Thalassiosira weissflogii is effectively activated by amines and amino acids. J Enz Inhib Med Chem 2018;33:680–5.
- Angeli A, Buonanno M, Donald WA, et al. The zinc – but not cadmium – containing ζ-carbonic from the diatom Thalassiosira weissflogii is potently activated by amines and amino acids. Bioorg Chem 2018;80:261–5.
- Vistoli G, Aldini G, Fumagalli L, et al. Activation effects of carnosine- and histidine-containing dipeptides on human carbonic anhydrases: a comprehensive study. Int J Mol Sci 2020;21:1761.
- Nguyen GTH, Tran TN, Podgorski MN, et al. Nanoscale ion emitters in native mass spectrometry for measuring ligand–protein binding affinities. ACS Central Sci 2019;5:308–18.