
Abstract
The clinically used antibiotic Furagin and its derivatives possess inhibitory activity on human (h) carbonic anhydrases (CA, EC 4.2.1.1), some of which are highly expressed in various tissues and malignancies (hCA IX/XII). Furagin exhibited good hCA IX and XII inhibition with KIs of 260 and 57 nM, respectively. It does not inhibit off-target CA I and poorly inhibited CA II (KI = 9.6 μM). Some synthesised Furagin derivatives with aminohydantoin moieties as zinc binding group exhibited weak inhibition of CA I/II, and good inhibition of CA IX/XII with KIs ranging from 350 to 7400 and 150 to 5600 nM, respectively. Docking and molecular dynamics simulations suggest that selectivity for the cancer-associated CA IX/XII over CA II is due to strong H-bond interactions in CA IX/XII, involving the tail orientated towards hydrophobic area of the active site. These results suggest a possible drug repurposing of Furagin as anti-cancer agent.
1. Introduction
Carbonic anhydrases (CAs, EC 4.2.1.1) are ubiquitous metalloenzymes, being encoded by at least eight different genetic families, which have been found in organisms all over the phylogenetic treeCitation1–10. CAs catalyse a crucial physiologic reaction, where by hydratation of CO2 is formed a weak base (bicarbonate) and a strong acid (hydronium ions). These enzymes are involved in a multitude of physiologic processes, starting with pH regulation and ending with metabolismCitation1–3,Citation7–10,Citation11–22.
CAs are also involved in various pathological processes and therefore are drug targets for decades, with their inhibitors having pharmacological applications in many fieldsCitation1–3,Citation7–19. The primary sulphonamides were discovered as CA inhibitors (CAIs) already in the 40 s, and most of the drugs that were launched in the next decades as diuretics, antiepileptics, or antiglaucoma agents targeting CAs belonged to this class of compoundsCitation1–3,Citation7–19. Although highly potent as CAIsCitation1–3, the sulphonamides generally non-selectively inhibit most α-CA isoforms present in humans and mammals in generalCitation1–3 as well as CAs from the other genetic families (β-, γ-, δ-, ζ-, η-, θ- and ι-CAs)Citation4–19, therefore alternative, isoform selective CAI classes were searched. A multitude of new chemotypes as well as novel CA inhibition mechanisms were reported in the last decadeCitation1–3,Citation11–14,Citation23–30.
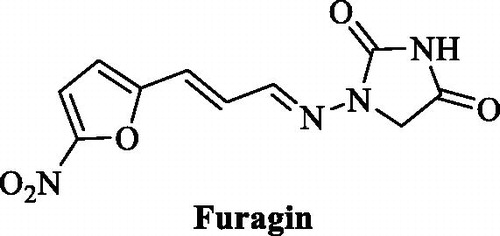
That has highly enriched our understanding of these enzymes and also allowed obtaining of isoform-selective CAIs targeting physiologically relevant isoformsCitation11–14,Citation23–27. Among the new chemotypes, which also exhibited the highest levels of isoform selectivity, were the coumarinsCitation27, the sulfocoumarinsCitation23–26 and their congeners, homosulfocoumarins (3H-1,2-benzoxathiepine 2,2-dioxides)Citation31, and saccharin derivativesCitation32–34. Considering the fact that this last chemotype was somewhat chemically similar to hydantoin (imidazolidine-2,4-dione) that may serve as zinc binding group (ZBG) we investigated clinically used antibiotic Furagin (), also known under names Furazidine, Furamags or FurazidinCitation35, that contains hydantoin moiety, as well as newly prepared its derivatives.
Figure 1. Structure of Furagin.
2. Materials and methods
2.1. Chemical syntheses – general
Reagents, starting materials and solvents were obtained from commercial sources and used as received. Thin-layer chromatography was performed on silica gel, spots were visualised with UV light (254 and 365 nm). Melting points were determined on an OptiMelt automated melting point system. IR spectra were recorded on Shimadzu FTIR IR Prestige-21 spectrometer. NMR spectra were recorded on Bruker Avance Neo (400 MHz) spectrometer with chemical shifts values (δ) in ppm relative to TMS using the residual DMSO-d6 signal (1H 2.50; 13C 39.52) or CDCl3 signal (1H 7.26; 13C 77.16) as an internal standard, or D2O signal and dioxane (1H 4.79; 13C 67.19). High-resolution mass spectra (HRMS) were recorded on a mass spectrometer with a Q-TOF micro mass analyser using the ESI technique. Examples of spectral data are furnished in the Supporting Information to the aricle.
2.2. General procedure for compound 2–17 synthesis
To a solution of 1-aminoimidazolidine-2,4-dione hydrochloride (1) (1.0 eq.) in EtOH (15 ml per 1 mmol of compound 1) appropriate aldehyde (1.05 eq.) was added. The resulting mixture was stirred at room temperature overnight.
The solvent was removed under vacuum and the crude product was re-crystallized form EtOH to afford product.
2.2.1. 1-(Benzylideneamino)-imidazolidine-2,4-dione (2)Citation36
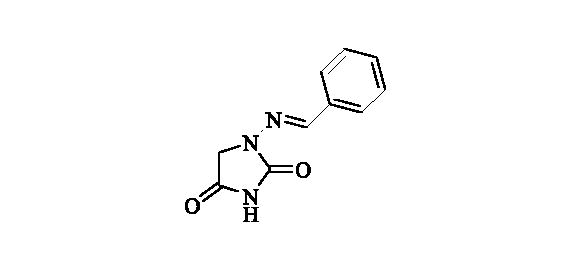 Compound 2 was prepared according to the general procedure from compound 1 (0.5 g; 3.30 mmol) and benzaldeyde (0.35 ml; 3.46 mmol) as white solid (0.60 g; 90%). Mp 252 – 253 °C. IR (film, cm−1) νmax= 1778 (C=O), 1717 (C=O); 1H NMR (400 MHz, DMSO-d6) δ = 4.36 (s, 2H), 7.38–7.48 (m, 3H), 7.68–7.72 (m, 2H), 7.80 (s, 1H), 11.25 (s, 1H) ppm 13C NMR (100 MHz, DMSO-d6) δ = 48.9, 126.8, 128.8, 129.8, 134.3, 143.0, 153.4, 169.0 ppm HRMS (ESI) [M + H]+: m/z calcd for (C10H10N3O2) 204.0773. Found 204.0783.
Compound 2 was prepared according to the general procedure from compound 1 (0.5 g; 3.30 mmol) and benzaldeyde (0.35 ml; 3.46 mmol) as white solid (0.60 g; 90%). Mp 252 – 253 °C. IR (film, cm−1) νmax= 1778 (C=O), 1717 (C=O); 1H NMR (400 MHz, DMSO-d6) δ = 4.36 (s, 2H), 7.38–7.48 (m, 3H), 7.68–7.72 (m, 2H), 7.80 (s, 1H), 11.25 (s, 1H) ppm 13C NMR (100 MHz, DMSO-d6) δ = 48.9, 126.8, 128.8, 129.8, 134.3, 143.0, 153.4, 169.0 ppm HRMS (ESI) [M + H]+: m/z calcd for (C10H10N3O2) 204.0773. Found 204.0783.
2.2.2. 1-((4-Methoxybenzylidene)amino)imidazolidine-2,4-dione (3)
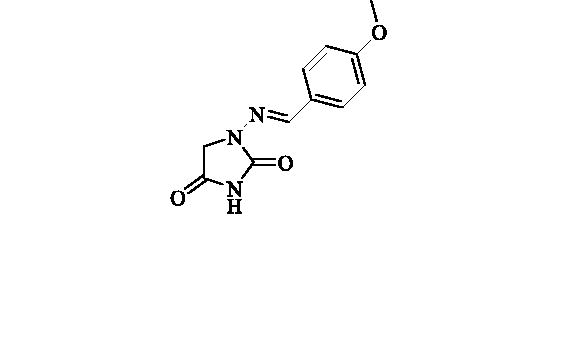 Compound 3 was prepared according to the general procedure from compound 1 (0.5 g; 3.30 mmol) and 4-methoxybenzaldehyde (0.42 ml; 3.46 mmol) as white solid (0.62 g; 80%). Mp 242 – 244 °C. IR (film, cm−1) νmax= 1768 (C=O), 1718 (C=O); 1H NMR (400 MHz, DMSO-d6) δ = 3.80 (s, 3H), 4.33 (s, 2H), 6.99–7.04(m, 2H), 7.62–7.66 (m, 2H), 7.75 (s, 1H), 11.18 (s, 1H) ppm 13C NMR (100 MHz, DMSO-d6) δ = 48.9, 55.3, 114.3, 126.9, 128.4, 142.9, 153.4, 160.6, 169.1 ppm HRMS (ESI) [M + H]+: m/z calcd for (C11H12N3O3) 234.0879. Found 234.0885.
Compound 3 was prepared according to the general procedure from compound 1 (0.5 g; 3.30 mmol) and 4-methoxybenzaldehyde (0.42 ml; 3.46 mmol) as white solid (0.62 g; 80%). Mp 242 – 244 °C. IR (film, cm−1) νmax= 1768 (C=O), 1718 (C=O); 1H NMR (400 MHz, DMSO-d6) δ = 3.80 (s, 3H), 4.33 (s, 2H), 6.99–7.04(m, 2H), 7.62–7.66 (m, 2H), 7.75 (s, 1H), 11.18 (s, 1H) ppm 13C NMR (100 MHz, DMSO-d6) δ = 48.9, 55.3, 114.3, 126.9, 128.4, 142.9, 153.4, 160.6, 169.1 ppm HRMS (ESI) [M + H]+: m/z calcd for (C11H12N3O3) 234.0879. Found 234.0885.
2.2.3. 1-((4-Nitrobenzylidene)amino)imidazolidine-2,4-dione (4)Citation37
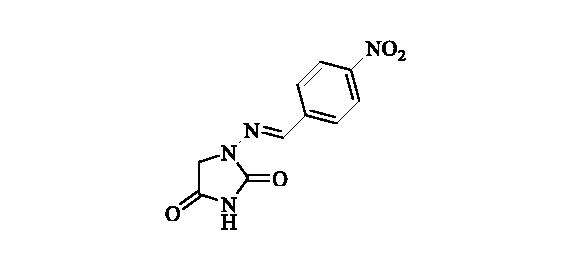 Compound 4 was prepared according to the general procedure from compound 1 (0.5 g; 3.30 mmol) and 4-nitrobenzaldehyde (0.52 g; 3.46 mmol) as yellowish solid (0.68 g; 82%). Mp 280 °C dec. IR (film, cm−1) νmax= 1780 (C=O), 1714 (C=O); 1H NMR (400 MHz, DMSO-d6) δ = 4.38 (s, 2H), 7.90–7.96 (m, 3H), 8.28–8.33 (m, 2H), 11.39 (s, 1H) ppm 13C NMR (100 MHz, DMSO-d6) δ = 49.1, 124.2, 127.7, 140.6, 140.7, 147.6, 153.4, 168.9 ppm HRMS (ESI) [M + H]+: m/z calcd for (C10H9N4O4) 249.0624. Found 249.0616.
Compound 4 was prepared according to the general procedure from compound 1 (0.5 g; 3.30 mmol) and 4-nitrobenzaldehyde (0.52 g; 3.46 mmol) as yellowish solid (0.68 g; 82%). Mp 280 °C dec. IR (film, cm−1) νmax= 1780 (C=O), 1714 (C=O); 1H NMR (400 MHz, DMSO-d6) δ = 4.38 (s, 2H), 7.90–7.96 (m, 3H), 8.28–8.33 (m, 2H), 11.39 (s, 1H) ppm 13C NMR (100 MHz, DMSO-d6) δ = 49.1, 124.2, 127.7, 140.6, 140.7, 147.6, 153.4, 168.9 ppm HRMS (ESI) [M + H]+: m/z calcd for (C10H9N4O4) 249.0624. Found 249.0616.
2.2.4. Methyl 4-(((2,4-dioxoimidazolidin-1-yl)imino)methyl)benzoate (5)
 Compound 5 was prepared according to the general procedure from compound 1 (0.5 g; 3.30 mmol) and methyl 4-formylbenzoate (0.57 g; 3.46 mmol) as white solid (0.82 g; 95%). Mp 280 °C dec. IR (film, cm−1) νmax= 1763 (C=O), 1717 (C=O); 1H NMR (400 MHz, DMSO-d6) δ = 3.86 (s, 3H), 4.37 (s, 2H), 7.80–7.87 (m, 3H), 8.00–8.05 (m, 2H), 11.33 (s, 1H) ppm 13C NMR (100 MHz, DMSO-d6) δ = 49.0, 52.2, 127.0, 129.7, 130.2, 138.8, 141.6, 153.4, 165.9, 168.9 ppm HRMS (ESI) [M + H]+: m/z calcd for (C12H12N3O4) 262.0828. Found 262.0834.
Compound 5 was prepared according to the general procedure from compound 1 (0.5 g; 3.30 mmol) and methyl 4-formylbenzoate (0.57 g; 3.46 mmol) as white solid (0.82 g; 95%). Mp 280 °C dec. IR (film, cm−1) νmax= 1763 (C=O), 1717 (C=O); 1H NMR (400 MHz, DMSO-d6) δ = 3.86 (s, 3H), 4.37 (s, 2H), 7.80–7.87 (m, 3H), 8.00–8.05 (m, 2H), 11.33 (s, 1H) ppm 13C NMR (100 MHz, DMSO-d6) δ = 49.0, 52.2, 127.0, 129.7, 130.2, 138.8, 141.6, 153.4, 165.9, 168.9 ppm HRMS (ESI) [M + H]+: m/z calcd for (C12H12N3O4) 262.0828. Found 262.0834.
2.2.5. 1,1’-((Pentane-1,5-diylidene)bis(azaneylylidene))bis(imidazolidine-2,4-dione) (6)
 Compound 6 was prepared according to the general procedure from compound 1 (0.5 g; 3.30 mmol) and glutaraldehyde 50 wt % solution in H2O (0.31 ml; 3.46 mmol) as white solid (0.49 g; 50%). Mp 237 °C dec. IR (film, cm−1) νmax= 1768 (C=O), 1734 (C=O); 1H NMR (400 MHz, DMSO-d6) δ = 1.72 (p, 2H, J = 7.4 Hz), 2.28–2.36 (m, 4H), 4.17 (s, 4H), 7.06 (t, 2H, J = 5.2 Hz), 11.07 (s, 2H) ppm 13C NMR (100 MHz, DMSO-d6) δ = 23.1, 31.3, 48.5, 146.7, 153.4, 169.1 ppm HRMS (ESI) [M + Na]+: m/z calcd for (C11H14N6O4Na) 317.0974. Found 317.0978.
Compound 6 was prepared according to the general procedure from compound 1 (0.5 g; 3.30 mmol) and glutaraldehyde 50 wt % solution in H2O (0.31 ml; 3.46 mmol) as white solid (0.49 g; 50%). Mp 237 °C dec. IR (film, cm−1) νmax= 1768 (C=O), 1734 (C=O); 1H NMR (400 MHz, DMSO-d6) δ = 1.72 (p, 2H, J = 7.4 Hz), 2.28–2.36 (m, 4H), 4.17 (s, 4H), 7.06 (t, 2H, J = 5.2 Hz), 11.07 (s, 2H) ppm 13C NMR (100 MHz, DMSO-d6) δ = 23.1, 31.3, 48.5, 146.7, 153.4, 169.1 ppm HRMS (ESI) [M + Na]+: m/z calcd for (C11H14N6O4Na) 317.0974. Found 317.0978.
2.2.6. 1-((Furan-3-ylmethylene)amino)imidazolidine-2,4-dione (7)
 Compound 7 was prepared according to the general procedure from compound 1 (0.5 g; 3.30 mmol) and 3-furaldehyde (0.33 g; 3.46 mmol) as yellowish solid (0.57 g; 89%). Mp 235 °C dec. IR (film, cm−1) νmax= 1780 (C=O), 1714 (C=O);
Compound 7 was prepared according to the general procedure from compound 1 (0.5 g; 3.30 mmol) and 3-furaldehyde (0.33 g; 3.46 mmol) as yellowish solid (0.57 g; 89%). Mp 235 °C dec. IR (film, cm−1) νmax= 1780 (C=O), 1714 (C=O);
1H NMR (400 MHz, DMSO-d6) δ = 4.30 (s, 2H), 6.74–6.76 (m, 1H), 7.73–7.77 (m, 2H), 8.05–8.07 (m, 1H), 11.18 (s, 1H) ppm 13C NMR (100 MHz, DMSO-d6) δ = 48.8, 107.0, 122.5, 136.1, 144.8, 144.9, 153.3, 169.1 ppm HRMS (ESI) [M + H]+: m/z calcd for (C8H8N3O3) 194.0566. Found 194.0570.
2.2.7. 1-((4-(Benzyloxy)benzylidene)amino)imidazolidine-2,4-dione (8)
 Compound 8 was prepared according to the general procedure from compound 1 (0.5 g; 3.30 mmol) and 4-benzyloxybenzaldehyde (0.73 g; 3.46 mmol) as white solid (0.92 g; 90%). Mp 258–260 °C. IR (film, cm−1) νmax= 1790 (C=O), 1730 (C=O); 1H NMR (400 MHz, DMSO-d6) δ = 4.33 (s, 2H), 5.15 (s, 2H), 7.07–7.12 (m, 2H), 7.31–7.36 (m, 1H), 7.37–7.43 (m, 2H), 7.44–7.49 (m, 2H), 7.62–7.67 (m, 2H), 7.75 (s, 1H), 11.19 (s, 1H) ppm 13C NMR (100 MHz, DMSO-d6) δ = 48.9, 69.4, 115.1, 127.1, 127.8, 127.9, 128.4, 128.5, 136.8, 142.8, 153.4, 159.7, 169.1 ppm HRMS (ESI) [M + H]+: m/z calcd for (C17H16N3O3) 310.1192. Found 310.1194.
Compound 8 was prepared according to the general procedure from compound 1 (0.5 g; 3.30 mmol) and 4-benzyloxybenzaldehyde (0.73 g; 3.46 mmol) as white solid (0.92 g; 90%). Mp 258–260 °C. IR (film, cm−1) νmax= 1790 (C=O), 1730 (C=O); 1H NMR (400 MHz, DMSO-d6) δ = 4.33 (s, 2H), 5.15 (s, 2H), 7.07–7.12 (m, 2H), 7.31–7.36 (m, 1H), 7.37–7.43 (m, 2H), 7.44–7.49 (m, 2H), 7.62–7.67 (m, 2H), 7.75 (s, 1H), 11.19 (s, 1H) ppm 13C NMR (100 MHz, DMSO-d6) δ = 48.9, 69.4, 115.1, 127.1, 127.8, 127.9, 128.4, 128.5, 136.8, 142.8, 153.4, 159.7, 169.1 ppm HRMS (ESI) [M + H]+: m/z calcd for (C17H16N3O3) 310.1192. Found 310.1194.
2.2.8. Ethyl (2E)-4-((2,4-dioxoimidazolidin-1-yl)imino)but-2-enoate (9)
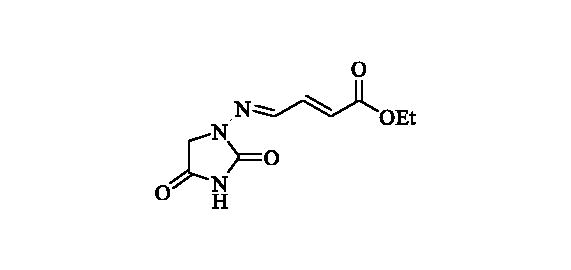 Compound 9 was prepared according to the general procedure from compound 1 (0.5 g; 3.30 mmol) and ethyl trans-4-oxo-2-butenoate (0.42 ml; 3.46 mmol) as white solid (0.60 g; 81%). Mp 210–211 °C. IR (film, cm−1) νmax= 1772 (C=O), 1721 (C=O); 1H NMR (400 MHz, DMSO-d6) δ = 1.24 (t, 3H, J = 7.1 Hz), 4.17 (q, 2H, J = 7.1 Hz), 4.27 (s, 2H), 7.37 (d, 1H, J = 15.6 Hz), 7.16–7.24 (m, 1H), 7.60 (d, 1H, J = 9.3 Hz), 11.39 (s, 1H) ppm 13C NMR (100 MHz, DMSO-d6) δ = 14.1, 49.0, 60.4, 126.5, 140.3, 141.7, 153.2, 165.4, 168.7 ppm HRMS (ESI) [M + H]+: m/z calcd for (C9H12N3O4) 226.0828. Found 226.0834.
Compound 9 was prepared according to the general procedure from compound 1 (0.5 g; 3.30 mmol) and ethyl trans-4-oxo-2-butenoate (0.42 ml; 3.46 mmol) as white solid (0.60 g; 81%). Mp 210–211 °C. IR (film, cm−1) νmax= 1772 (C=O), 1721 (C=O); 1H NMR (400 MHz, DMSO-d6) δ = 1.24 (t, 3H, J = 7.1 Hz), 4.17 (q, 2H, J = 7.1 Hz), 4.27 (s, 2H), 7.37 (d, 1H, J = 15.6 Hz), 7.16–7.24 (m, 1H), 7.60 (d, 1H, J = 9.3 Hz), 11.39 (s, 1H) ppm 13C NMR (100 MHz, DMSO-d6) δ = 14.1, 49.0, 60.4, 126.5, 140.3, 141.7, 153.2, 165.4, 168.7 ppm HRMS (ESI) [M + H]+: m/z calcd for (C9H12N3O4) 226.0828. Found 226.0834.
2.2.9. 1-((3-Methylbut-2-en-1-ylidene)amino)imidazolidine-2,4-dione (10)
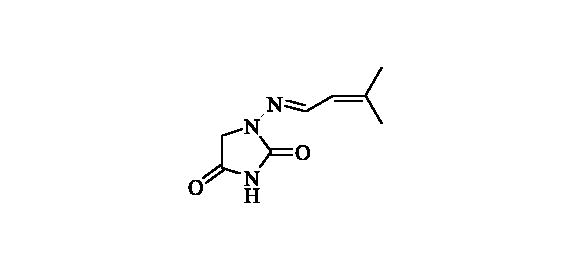 Compound 10 was prepared according to the general procedure from compound 1 (0.5 g; 3.30 mmol) and 3-methyl-2-butenal (0.33 ml; 3.46 mmol) as white solid (0.43 g; 72%). Mp 186–187 °C. IR (film, cm−1) νmax= 1768 (C=O), 1717 (C=O); 1H NMR (400 MHz, DMSO-d6) δ = 1.84–1.89 (m, 6H), 4.28 (s, 2H), 5.93–5.99 (m, 1H), 7.57 (d, 1H, J = 9.5 Hz), 11.11 (s, 1H) ppm 13C NMR (100 MHz, DMSO-d6) δ = 18.7, 26.2, 48.9, 121.9, 142.4, 144.3, 153.3, 169.2 ppm HRMS (ESI) [M + H]+: m/z calcd for (C8H12N3O2) 182.0930. Found 182.0938.
Compound 10 was prepared according to the general procedure from compound 1 (0.5 g; 3.30 mmol) and 3-methyl-2-butenal (0.33 ml; 3.46 mmol) as white solid (0.43 g; 72%). Mp 186–187 °C. IR (film, cm−1) νmax= 1768 (C=O), 1717 (C=O); 1H NMR (400 MHz, DMSO-d6) δ = 1.84–1.89 (m, 6H), 4.28 (s, 2H), 5.93–5.99 (m, 1H), 7.57 (d, 1H, J = 9.5 Hz), 11.11 (s, 1H) ppm 13C NMR (100 MHz, DMSO-d6) δ = 18.7, 26.2, 48.9, 121.9, 142.4, 144.3, 153.3, 169.2 ppm HRMS (ESI) [M + H]+: m/z calcd for (C8H12N3O2) 182.0930. Found 182.0938.
2.2.10 1-(((2e)-3–(4-methoxyphenyl)allylidene)amino)imidazolidine-2,4-dione (11)
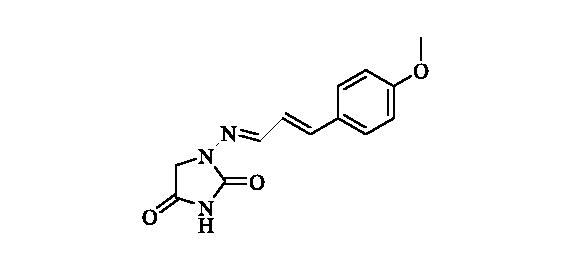 Compound 11 was prepared according to the general procedure from compound 1 (0.5 g; 3.30 mmol) and trans-4-methoxycinnamaldehyde (0.56 g; 3.46 mmol) as white solid (0.61 g; 71%). Mp 250 °C dec. IR (film, cm−1) νmax= 1770 (C=O), 1731 (C=O); 1H NMR (400 MHz, DMSO-d6) δ = 3.78 (s, 3H), 4.29 (s, 2H), 6.85–7.00 (m, 4H), 7.51–7.59 (m, 3H), 11.18 (s, 1H) ppm 13C NMR (100 MHz, DMSO-d6) δ = 48.8, 55.2, 114.3, 123.1, 128.5, 128.6, 138.5, 145.5, 153.3, 159.9, 169.1 ppm HRMS (ESI) [M + H]+: m/z calcd for (C13H14N3O3) 260.1035. Found 260.1047.
Compound 11 was prepared according to the general procedure from compound 1 (0.5 g; 3.30 mmol) and trans-4-methoxycinnamaldehyde (0.56 g; 3.46 mmol) as white solid (0.61 g; 71%). Mp 250 °C dec. IR (film, cm−1) νmax= 1770 (C=O), 1731 (C=O); 1H NMR (400 MHz, DMSO-d6) δ = 3.78 (s, 3H), 4.29 (s, 2H), 6.85–7.00 (m, 4H), 7.51–7.59 (m, 3H), 11.18 (s, 1H) ppm 13C NMR (100 MHz, DMSO-d6) δ = 48.8, 55.2, 114.3, 123.1, 128.5, 128.6, 138.5, 145.5, 153.3, 159.9, 169.1 ppm HRMS (ESI) [M + H]+: m/z calcd for (C13H14N3O3) 260.1035. Found 260.1047.
2.2.11. 1-((2,4-Dihydroxybenzylidene)amino)imidazolidine-2,4-dione (12)
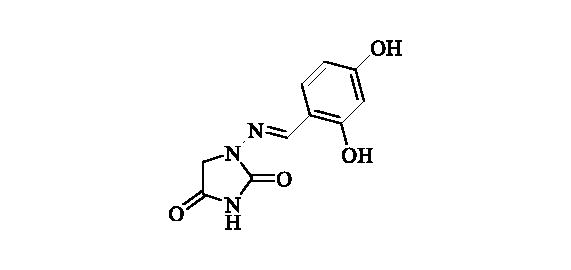 Compound 12 was prepared according to the general procedure from compound 1 (0.5 g; 3.30 mmol) and 2,4-dihydroxybenzaldehyde (0.48 g; 3.46 mmol) as white solid (0.72 g; 93%). Mp >300 °C. IR (film, cm−1) νmax= 3260 (OH), 3188 (OH), 1780 (C=O), 1717 (C=O); 1H NMR (400 MHz, DMSO-d6) δ = 4.34 (s, 2H), 6.31 (d, 1H, J = 2.3 Hz), 6.35 (dd, 1H, J = 8.5, 2.3 Hz), 7.33 (d, 1H, J = 8.5 Hz), 7.90 (s, 1H), 9.90 (br s, 1H), 10.73 (s, 1H), 11.23 (br s, 1H) ppm 13C NMR (100 MHz, DMSO-d6) δ = 48.5, 102.6, 107.8, 110.7, 130.5, 144.0, 153.3, 158.6, 160.5, 169.1 ppm HRMS (ESI) [M + H]+: m/z calcd for (C10H10N3O4) 236.0671. Found 236.0677.
Compound 12 was prepared according to the general procedure from compound 1 (0.5 g; 3.30 mmol) and 2,4-dihydroxybenzaldehyde (0.48 g; 3.46 mmol) as white solid (0.72 g; 93%). Mp >300 °C. IR (film, cm−1) νmax= 3260 (OH), 3188 (OH), 1780 (C=O), 1717 (C=O); 1H NMR (400 MHz, DMSO-d6) δ = 4.34 (s, 2H), 6.31 (d, 1H, J = 2.3 Hz), 6.35 (dd, 1H, J = 8.5, 2.3 Hz), 7.33 (d, 1H, J = 8.5 Hz), 7.90 (s, 1H), 9.90 (br s, 1H), 10.73 (s, 1H), 11.23 (br s, 1H) ppm 13C NMR (100 MHz, DMSO-d6) δ = 48.5, 102.6, 107.8, 110.7, 130.5, 144.0, 153.3, 158.6, 160.5, 169.1 ppm HRMS (ESI) [M + H]+: m/z calcd for (C10H10N3O4) 236.0671. Found 236.0677.
2.2.12. 4-(((2,4-Dioxoimidazolidin-1-yl)imino)methyl)phenyl)boronic acid (13)
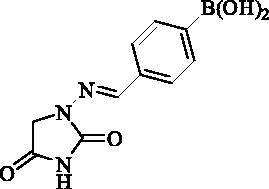 Compound 13 was prepared according to the general procedure from compound 1 (0.5 g; 3.30 mmol) and 4-formylphenylboronic acid (0.52 g; 3.46 mmol) as white solid (0.72 g; 88%). Mp >300 °C. IR (film, cm−1) νmax= 3349 (OH), 3173 (OH), 1780 (C=O), 1716 (C=O); 1H NMR (400 MHz, DMSO-d6) δ = 4.37 (s, 2H), 7.64–7.68 (m, 2H), 7.79 (s, 1H), 7.83–7.87 (m, 2H), 8.12 (s, 2H), 11.26 (s, 1H) ppm 13C NMR (100 MHz, DMSO-d6) δ = 48.9, 125.8, 134.5, 135.7, 136.0 (br) 143.0, 153.4, 169.1 ppm HRMS (ESI) [M + H]+: m/z calcd for (C10H11BN3O4) 248.0843. Found 248.0847.
Compound 13 was prepared according to the general procedure from compound 1 (0.5 g; 3.30 mmol) and 4-formylphenylboronic acid (0.52 g; 3.46 mmol) as white solid (0.72 g; 88%). Mp >300 °C. IR (film, cm−1) νmax= 3349 (OH), 3173 (OH), 1780 (C=O), 1716 (C=O); 1H NMR (400 MHz, DMSO-d6) δ = 4.37 (s, 2H), 7.64–7.68 (m, 2H), 7.79 (s, 1H), 7.83–7.87 (m, 2H), 8.12 (s, 2H), 11.26 (s, 1H) ppm 13C NMR (100 MHz, DMSO-d6) δ = 48.9, 125.8, 134.5, 135.7, 136.0 (br) 143.0, 153.4, 169.1 ppm HRMS (ESI) [M + H]+: m/z calcd for (C10H11BN3O4) 248.0843. Found 248.0847.
2.2.13. 1-((Pyridin-2-ylmethylene)amino)imidazolidine-2,4-dione (14)
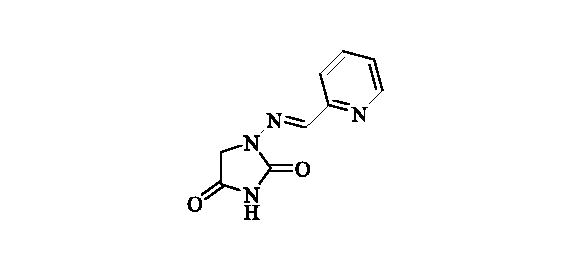 Compound 14 was prepared according to the general procedure from compound 1 (0.5 g; 3.30 mmol) and pyridine-2-carbaldehyde (0.33 ml; 3.46 mmol) as white solid (0.64 g; 95%). Mp 280 °C dec. IR (film, cm−1) νmax= 1770 (C=O), 1730 (C=O); 1H NMR (400 MHz, DMSO-d6) δ = 4.43 (s, 2H), 7.61–7.66 (m, 1H), 7.89 (s, 1H), 8.02–8.06 (m, 1H), 8.16 (dt, 1H, J = 7.7, 1.4 Hz), 8.68–8.72 (m, 1H), 11.50 (s, 1H) ppm 13C NMR (100 MHz, DMSO-d6) δ = 49.0, 121.6, 125.2, 139.3, 140.6, 146.8, 150.5, 153.3, 168.7 ppm HRMS (ESI) [M + H]+: m/z calcd for (C9H9N4O2) 205.0726. Found 205.0732.
Compound 14 was prepared according to the general procedure from compound 1 (0.5 g; 3.30 mmol) and pyridine-2-carbaldehyde (0.33 ml; 3.46 mmol) as white solid (0.64 g; 95%). Mp 280 °C dec. IR (film, cm−1) νmax= 1770 (C=O), 1730 (C=O); 1H NMR (400 MHz, DMSO-d6) δ = 4.43 (s, 2H), 7.61–7.66 (m, 1H), 7.89 (s, 1H), 8.02–8.06 (m, 1H), 8.16 (dt, 1H, J = 7.7, 1.4 Hz), 8.68–8.72 (m, 1H), 11.50 (s, 1H) ppm 13C NMR (100 MHz, DMSO-d6) δ = 49.0, 121.6, 125.2, 139.3, 140.6, 146.8, 150.5, 153.3, 168.7 ppm HRMS (ESI) [M + H]+: m/z calcd for (C9H9N4O2) 205.0726. Found 205.0732.
2.2.14. 1-((Pyridin-3-ylmethylene)amino)imidazolidine-2,4-dione (15)
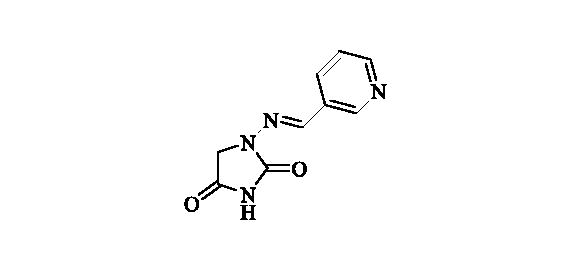 Compound 15 was prepared according to the general procedure from compound 1 (0.5 g; 3.30 mmol) and pyridine-3-carbaldehyde (0.33 ml; 3.46 mmol) as white solid (0.60 g; 90%). Mp 280 °C dec. IR (film, cm−1) νmax= 1764 (C=O), 1722 (C=O); 1H NMR (400 MHz, D2O + NaOH + dioxane) δ = 7.46–7.51 (m, 1H), 7.56 (s, 1H), 8.18 (td, 1H, J = 8.0, 1.8 Hz), 8.48 (dd, 1H, J = 4.9, 1.6 Hz), 8.74 (d, 1H, J = 1.8 Hz) ppm 13C NMR (100 MHz, D2O + NaOH + dioxane) δ = 49.3 (br), 125.1, 131.6, 135.1, 138.9, 148.1, 149.7, 170.0, 186.3 ppm HRMS (ESI) [M + H]+: m/z calcd for (C9H9N4O2) 205.0726. Found 205.0731.
Compound 15 was prepared according to the general procedure from compound 1 (0.5 g; 3.30 mmol) and pyridine-3-carbaldehyde (0.33 ml; 3.46 mmol) as white solid (0.60 g; 90%). Mp 280 °C dec. IR (film, cm−1) νmax= 1764 (C=O), 1722 (C=O); 1H NMR (400 MHz, D2O + NaOH + dioxane) δ = 7.46–7.51 (m, 1H), 7.56 (s, 1H), 8.18 (td, 1H, J = 8.0, 1.8 Hz), 8.48 (dd, 1H, J = 4.9, 1.6 Hz), 8.74 (d, 1H, J = 1.8 Hz) ppm 13C NMR (100 MHz, D2O + NaOH + dioxane) δ = 49.3 (br), 125.1, 131.6, 135.1, 138.9, 148.1, 149.7, 170.0, 186.3 ppm HRMS (ESI) [M + H]+: m/z calcd for (C9H9N4O2) 205.0726. Found 205.0731.
2.2.15. 1-((Pyridin-4-ylmethylene)amino)imidazolidine-2,4-dione (16)
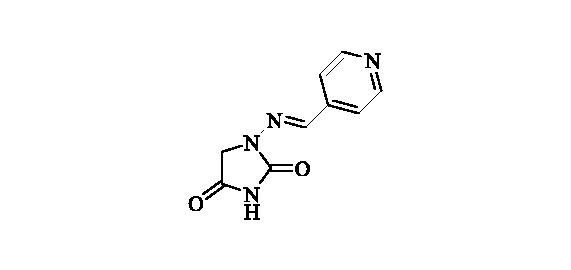 Compound 16 was prepared according to the general procedure from compound 1 (0.5 g; 3.30 mmol) and pyridine-4-carbaldehyde (0.33 ml; 3.46 mmol) as white solid (0.61 g; 91%). Mp 280 °C dec. IR (film, cm−1) νmax= 1750 (C=O), 1723 (C=O); 1H NMR (400 MHz, D2O + NaOH + dioxane) δ = 7.46 (s, 1H), 7.62–7.66 (m, 2H), 8.47–8.51 (m, 2H) ppm 13C NMR (100 MHz, D2O + NaOH + dioxane) δ = 49.3 (br), 121.9, 139.2, 143.5, 149.7, 170.0, 186.4 ppm HRMS (ESI) [M + H]+: m/z calcd for (C9H9N4O2) 205.0726. Found 205.0730.
Compound 16 was prepared according to the general procedure from compound 1 (0.5 g; 3.30 mmol) and pyridine-4-carbaldehyde (0.33 ml; 3.46 mmol) as white solid (0.61 g; 91%). Mp 280 °C dec. IR (film, cm−1) νmax= 1750 (C=O), 1723 (C=O); 1H NMR (400 MHz, D2O + NaOH + dioxane) δ = 7.46 (s, 1H), 7.62–7.66 (m, 2H), 8.47–8.51 (m, 2H) ppm 13C NMR (100 MHz, D2O + NaOH + dioxane) δ = 49.3 (br), 121.9, 139.2, 143.5, 149.7, 170.0, 186.4 ppm HRMS (ESI) [M + H]+: m/z calcd for (C9H9N4O2) 205.0726. Found 205.0730.
2.2.16. 1-(((1 h-Imidazol-5-yl)methylene)amino)imidazolidine-2,4-dione (17)
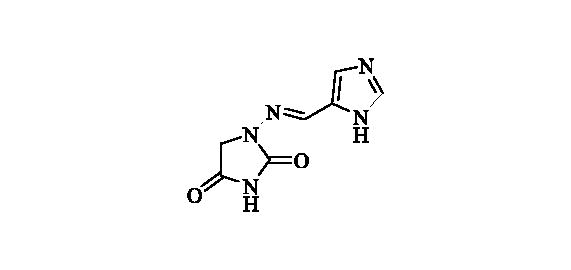 Compound 17 was prepared according to the general procedure from compound 1 (0.5 g; 3.30 mmol) and 1H-imidazole-5-carbaldehyde (0.33 g; 3.46 mmol) as white solid (0.62 g; 97%). Mp 270 °C dec. IR (film, cm−1) νmax= 1764 (C=O), 1715 (C=O); 1H NMR (400 MHz, D2O + NaOH + dioxane) δ = 7.45–7.48 (m, 1H), 7.67 (s, 1H), 7.72–7.76 (m, 1H) ppm 13C NMR (100 MHz, D2O + NaOH + dioxane) δ = 49.5 (br), 125.1, 134.2, 136.5, 140.8, 170.3, 186.7 ppm HRMS (ESI) [M + H]+: m/z calcd for (C7H8N5O2) 194.0678. Found 194.0687
Compound 17 was prepared according to the general procedure from compound 1 (0.5 g; 3.30 mmol) and 1H-imidazole-5-carbaldehyde (0.33 g; 3.46 mmol) as white solid (0.62 g; 97%). Mp 270 °C dec. IR (film, cm−1) νmax= 1764 (C=O), 1715 (C=O); 1H NMR (400 MHz, D2O + NaOH + dioxane) δ = 7.45–7.48 (m, 1H), 7.67 (s, 1H), 7.72–7.76 (m, 1H) ppm 13C NMR (100 MHz, D2O + NaOH + dioxane) δ = 49.5 (br), 125.1, 134.2, 136.5, 140.8, 170.3, 186.7 ppm HRMS (ESI) [M + H]+: m/z calcd for (C7H8N5O2) 194.0678. Found 194.0687
2.3. Ca inhibitory assay
An Applied Photophysics stopped-flow instrument has been used for assaying the CA catalysed CO2 hydration activity, as reported earlierCitation38,Citation39. The inhibition constants were obtained by non-linear least-squares methods using PRISM 3 and the Cheng-Prusoff equation as reported earlierCitation40 and represent the mean from at least three different determinations. The four tested CA isoforms were recombinant ones obtained in-house as reported earlierCitation41–43.
2.4. Computational studies
The crystal structure of CA II (pdb 5LJT)Citation43, CA IX (pdb 5FL4)Citation44 and CA XII (pdb JLD0)Citation45 were prepared using the Protein Preparation Wizard tool implemented in Maestro - Schrödinger suite, assigning bond orders, adding hydrogens, deleting water molecules, and optimising H-bonding networksCitation46. Energy minimisation protocol with a root mean square deviation (RMSD) value of 0.30 was applied using an Optimised Potentials for Liquid Simulation (OPLS3e) force field. 3D ligand structures were prepared by MaestroCitation46a and evaluated for their ionisation states at pH 7.4 ± 0.5 with EpikCitation46b. Additionally, the imidic nitrogen of the hydantoin nucleus was negatively charged in simulations. OPLS3e force field in MacromodelCitation46e was used for energy minimisation for a maximum number of 2500 conjugate gradient iteration and setting a convergence criterion of 0.05 kcal mol−1 Å−1. The docking grid was centred on the centre of mass of the co-crystallized ligands and Glide used with default settings. Ligands were docked with the standard precision mode (SP) of GlideCitation46e and the best 5 poses of each molecule retained as output. The best pose for each compound, evaluated in terms of coordination, hydrogen bond interactions and hydrophobic contacts, was refined with PrimeCitation46d with a VSGB solvation model considering the target flexible within 3 Å around the ligandCitation47–49.
The best poses of Furagin and 12 to CA II, CA IX and CA XII were submitted to a MD simulation using DesmondCitation50 and the OPL3e force field. Specifically, the system was solvated in an orthorhombic box using TIP4PEW water molecules, extended 15 Å away from any protein atom. It was neutralised adding chlorine and sodium ions. The simulation protocol included a starting relaxation step followed by a final production phase of 100 ns. In particular, the relaxation step comprised the following: (a) a stage of 100 ps at 10 K retaining the harmonic restraints on the solute heavy atoms (force constant of 50.0 kcal mol−1 Å−2) using the NPT ensemble with Brownian dynamics; (b) a stage of 12 ps at 10 K with harmonic restraints on the solute heavy atoms (force constant of 50.0 kcal mol−1 Å−2), using the NVT ensemble and Berendsen thermostat; (c) a stage of 12 ps at 10 K and 1 atm, retaining the harmonic restraints and using the NPT ensemble and Berendsen thermostat and barostat; (f) a stage of 12 ps at 300 K and 1 atm, retaining the harmonic restraints and using the NPT ensemble and Berendsen thermostat and barostat; (g) a final 24 ps stage at 300 K and 1 atm without harmonic restraints, using the NPT Berendsen thermostat and barostat. The final production phase of MD was run using a canonical NPT Berendsen ensemble at temperature 300 K. During the MD simulation, a time step of 2 fs was used while constraining the bond lengths of hydrogen atoms with the M-SHAKE algorithm. The atomic coordinates of the system were saved every 100 ps along the MD trajectory. Protein and ligand RMSD values, ligand torsions evolution and occupancy of intermolecular hydrogen bonds and hydrophobic contacts were computed along the production phase of the MD simulation with the Simulation Interaction Diagram tools implemented in Maestro.
3. Results and discussion
3.1. Chemistry
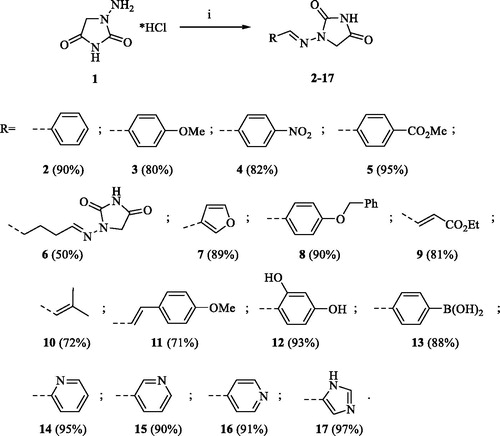
A series of Furagin derivatives 2–17 were prepared in reaction between 1-aminohydantoin hydrochloride (1) and various aldehydes (Scheme 1). Compounds 2–17 were isolated in good to excellent yields, all new structure were proven by 1H and 13C NMR and IS spectroscopy as well as high-resolution mass spectra. The purity of all compounds was greater than 95% according UPLC analysis.
Scheme 1. Reagents and conditions: i. RCHO, EtOH, RT, 16 h
3.2. Biological evaluation
The CA inhibitory profiles of Furagin and synthesised aminohydantoin derivatives were evaluated by applying a stopped flow carbon dioxide hydrase assayCitation51, in comparison to acetazolamide (AAZ) as a standard CAI against four physiologically significant isoforms CA I, II, IX, and XII. The following structure–activity relationship (SAR) can be concluded from the inhibition data presented in .
Table 1. Inhibition data of human CA isoforms CA I, II, IX, and XII with aminohydantoines (2–17, Furagin) using AAZ as a standard inhibitor.
All the tested aminohydantoins exhibited weak inhibitory effect on the slow cytosolic isoform, hCA I, where the binding affinity constant (KI) values fluctuating in the thousands nM range (KI 16 800->100 000 nM).
The physiologically relevant isoform, hCA II, was better inhibited by most of the tested compounds (KIs: 620-59 000 nM). It is observed that, the aminohydantoin compounds (2, 7, 8 and 15) were more potent hCA II inhibitors with KIs in range from 540-900 nM. These compounds have unsubstituted Ph or hetaryl moieties. Rest of the compounds showed weaker inhibitory effect of CA II with KIs in range from 3100–59 900 nM. It is interesting to note, that compound 12 having dihydroxyphenyl substituent stood out by nearly three times weaker inhibition compare to the second weakest inhibitor 9.
The tumour associated isoform hCA IX was inhibited in nano-molar range by compounds 7-9, 11, 17 and Furagin (KIs: 260–850 nM), where the strongest inhibition was observed for Furagin. Rest of the aminohydantoin derivatives showed one order weaker inhibition with KIs in range from 1100-7 300 nM. Certain pattern can be observed, where better CA IX inhibition can be observed for compounds with vinyl substituents (9, 11, 17 and Furagin) or small hetaryl substituents (7 and 17), with exception in case of compound 8, containing ester moiety on phenyl ring.
The other tumour associated isoform hCA XII was best inhibited among all isoforms studied. The best compound of this series was Furagin with KI = 57 nM. It was followed by vinyl substituted aminohydantoin derivatives 6, 9 and 10 with KIs 160, 360 and 880 nM, respectively. One order weaker CA XII inhibition compare to Furagin was also observed for aryl (5, 8 and 12) and hetaryl (16 and 17) derivatives ranging KIs from 150 to 930 nM.
In general good selectivity against cancer associated CA isoforms (CA IX and CA XII) compare to off-target ones (CA I and CA II) was observed for three compounds Furagin, 9 and 12.
3.3. Computational studies
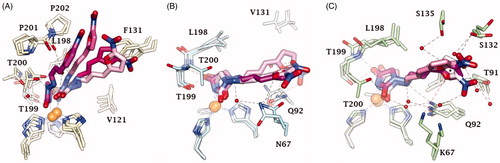
Docking studies were used to investigate the binding mode of Furagin and aminohydantoines 2-17 within the active site of CA II (pdb 5LJT)Citation44, IX (pdb 5FL4)Citation43 and XII (pdb JLD0)Citation45. Similarly to benzenesulfonamides (pKa 10.1) which binds to the CA Zn ion in the deprotonated form, the imidic nitrogen of the hydantoin nucleus as well was considered negatively charged (pKa 9.16)Citation52 in the docking experiments and resulted to coordinate the zinc ion in all the obtained poses with CAs II, IX and XII. Furthermore, the oxygen atom of the CO in position 4 of the hydantoin core acts as a bifurcated acceptor establishing two H-bonds with T199, that is, O…(H-N, HG1-O), whereas overall the heterocycle forms VdW contacts with residues H94, H96, H119, L198, T200 and W209 (.
Figure 2. Predicted docking orientations of 7 (green) and Furagin (pink) to (A) CA II, (B) CA IX and (C) CA XII.
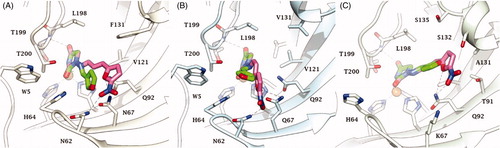
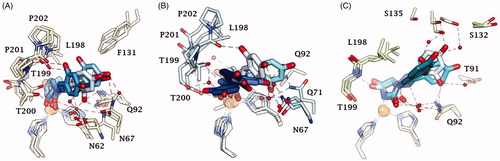
In CA II and CA IX, the N1 pendants of all ligands are oriented towards a hydrophilic cleft defined by H4, W5, N62, N67 and H64, except 8 and 9, whose N1 tails are housed, in CA II, into a hydrophobic pocket formed by I91, V121 and F131 (). Amino acids T91, Q92, A131, S132 and S135 are instead targeted by the pendants on the aminohydantoin of the ligands in all docking solutions with CA XII (). The docking procedure was complemented with 100 ns long molecular dynamic (MD) simulations on the predicted binding conformations of Furagin and 12, the most potent CA XII inhibitors also showing significant CA XII over CA II selectivity. The structure of the three investigated CA isoforms was stable during the computation with the backbone atom RMSDs exhibiting small fluctuations over the 100 ns ( and ). Additionally, the ZBG of the ligands remains stably anchored to the metal ion all over the MD, with the hydantoin core receiving H-bonds by the amidic NH and side chain OH of Thr199 ( and ).
Figure 3. RMSD analysis of Furagin heavy atoms and (A) CA II, (B) CAIX and (C) CA XII backbone over the 100 ns MD simulation. The ligand colour darkens over the dynamic simulation.
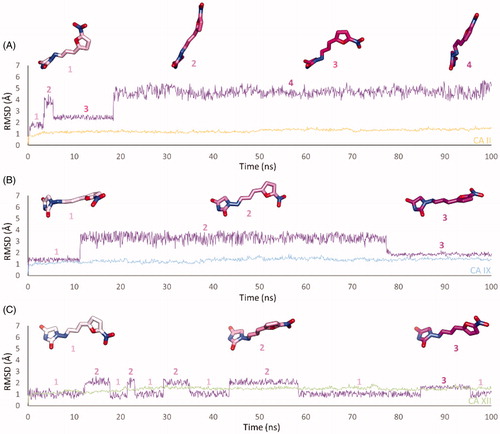
Figure 4. Dynamics evolution of the binding mode of Furagin to (A) CA II, (B) CA IX and (C) CA XII over the course of 100 ns. Water molecules are represented as red spheres. The ligand colour darkens over the dynamic simulation.
After an initial equilibration, mainly occurring in CA II and IX, the molecular tail of Furagin undergoes minor conformational fluctuations during simulation approaching to stable binding conformations within the three CA isoforms ( and ). In CA IX and XII, the ligand accommodates the N1-pendant in the hydrophilic half of the active sites where it makes VdW contacts and both direct and water mediated H-bond interactions with the enzymes (). In the CA II, the ligand-bound conformation of Furagin orients the tail towards the hydrophobic area of the target and does not form persistent H-bond interactions over the 100 ns (). The hydrogen bond persistence within the three CA isoforms is in good agreement with the inhibitory profile of the ligand (CAXII > CA IX > CA II).
An ensemble of few conformations is representative of the binding of 12 within CA II and IX ( and ). Here, the ligand approaches the hydrophobic regions of the enzymes and, coming next to the end of the simulation, the N1 tails lose direct or water-bridged H-bonds with glutamine and asparagine residues, progressively moving towards T199 or T200, that is, the area of the enzyme that undergoes to the greatest residue displacement. In CA XII, the docked pose of 12 remains firmly anchored to the residues of the hydrophilic portion of the enzyme throughout the dynamic. A wide network of direct and water mediated H-bonds stabilise the binding of the ligand. This is consistent with the inhibition profile exhibited by 12 in CA XII as compared with the other two CA isoforms.
Figure 5. RMSD analysis of 12 heavy atoms and (A) CA II, (B) CAIX and (C) CA XII backbone over the 100 ns MD simulation. The ligand colour darkens over the dynamic simulation.
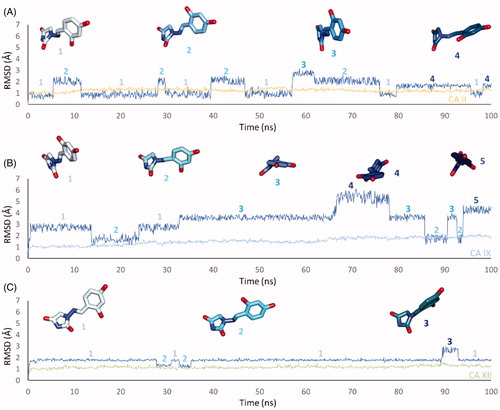
Figure 6. Dynamics evolution of the binding mode of 12 to (A) CA II, (B) CA IX and (C) CA XII over the course of 100 ns. Water molecules are represented as red spheres. The ligand colour darkens over the dynamic simulation.
4. Conclusions
In summary, we have demonstrated that clinically used antibiotic – Furagin and its derivatives 2-17 are potential CA inhibitors. Furagin and all newly synthesised hydantoin derivative were examined for their inhibitory activities towards hCA I, II, IX and XII. The four studied hCA isoforms were inhibited by Furagin and its derivatives at various degrees. In particular, Furagin and prepared compounds 2-17 did not inhibit or poorly inhibited off-target hCA I with KIs ranging from >100 μM (compounds 4, 5, 8, 11, 12 and Furagin) to 9.6 μM. Ubiquitous hCA II was poorly inhibited by compounds 3-6, 9-14, 16, 17 and Furagin (KIs from 59.9 to 3.1 μM). Rest of the compounds significantly inhibited hCA II (KIs from 900 nM to 540 nM). Remarkable inhibition of cancer associated hCA IX was observed for Furagin (KI=260 nM) and compounds 7–9, 11 and 17 with KIs ranging from 350 to 850 nM. The rest of compounds exhibited slightly weaker inhibition of hCA IX with KIs ranging from 1100 to 7400 nM. Other cancer associated isoform – hCA XII also was significantly inhibited by Furagin (KI=57 nM) and compounds 5, 6, 8-13, 16 and 17 (KIs from 160 to 910 nM). The rest of the compounds exhibited slightly weaker inhibition with KIs ranging from 1300 to 5600 nM. Docking and molecular dynamics simulations shed light on the ligands selectivity for the cancer-associated CAs over ubiquitous CA II. The significant inhibition activity and especially selectivity of Furagin against hCA IX and XII was attributed due to the strong H-bond interactions, whereas in case of hCA II no persistent H-bond interactions are formed due to Furagin’s tails orientation towards hydrophobic area of the enzyme.
The knowledge obtained gives the solid base for both – investigation of drug repurposing of clinically used antibiotic Furagin for anti-cancer therapy and further studies of new chemotype of inhibitors of CAs.
Supplemental Material
Download PDF (509.8 KB)Disclosure statement
The authors have no relevant affiliations of financial involvement with any organisation or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript.
Related Research Data
References
- Supuran CT. Structure and function of carbonic anhydrases. Biochem J 2016;473:2023–32.
- Alterio V, Di Fiore A, D’Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms. ? Chem Rev 2012;112:4421–68.
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nature Rev Drug Discov 2008;7:168–81.
- Jensen EL, Clement R, Kosta A, et al. A new widespread subclass of carbonic anhydrase in marine phytoplankton. Isme J 2019;13:2094–106.
- Del Prete S, Vullo D, Fisher GM, et al. Discovery of a new family of carbonic anhydrases in the malaria pathogen Plasmodium falciparum – the η-carbonic anhydrases. Bioorg Med Chem Lett 2014;24:4389–96.
- Xu Y, Feng L, Jeffrey PD, et al. Structure and metal exchange in the cadmium carbonic anhydrase of marine diatoms. Nature 2008;452:56–61.
- Supuran CT, Capasso C. Biomedical applications of prokaryotic carbonic anhydrases. Expert Opin Ther Pat 2018;28:745–54.
- Capasso C, Supuran CT. An overview of the alpha-, beta- and gamma-carbonic anhydrases from bacteria: can bacterial carbonic anhydrases shed new light on evolution of bacteria?. J Enzyme Inhib Med Chem 2015;30:325–32.
- Capasso C, Supuran CT. Bacterial, fungal and protozoan carbonic anhydrases as drug targets. Expert Opin Ther Targets 2015;19:1689–704.
- Capasso C, Supuran CT. Anti-infective carbonic anhydrase inhibitors: a patent and literature review. Expert Opin Ther Pat 2013;23:693–704.
- Nocentini A, Supuran CT. Advances in the structural annotation of human carbonic anhydrases and impact on future drug discovery. Expert Opin Drug Discov 2019;14:1175–97.
- Supuran CT. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin Drug Discov 2017;12:61–88.
- Supuran CT. How many carbonic anhydrase inhibition mechanisms exist?. J Enzyme Inhib Med Chem 2016;31:345–60.
- De Simone G, Supuran CT. (In)organic anions as carbonic anhydrase inhibitors. J Inorg Biochem 2012;111:117–29.
- Supuran CT. Carbonic anhydrase inhibitors as emerging agents for the treatment and imaging of hypoxic tumors. Expert Opin Investig Drugs 2018;27:963–70.
- Supuran CT. Carbonic anhydrase inhibitors and their potential in a range of therapeutic areas. Expert Opin Ther Pat 2018;28:709–12.
- Supuran CT. Applications of carbonic anhydrases inhibitors in renal and central nervous system diseases. Expert Opin Ther Pat 2018;28:713–21.
- Supuran CT, Alterio V, Di Fiore A, et al. Inhibition of carbonic anhydrase IX targets primary tumors, metastases, and cancer stem cells: three for the price of one. Med Res Rev 2018;38:1799–836.
- Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77.
- Da’dara AA, Angeli A, Ferraroni M, et al. Crystal structure and chemical inhibition of essential schistosome host-interactive virulence factor carbonic anhydrase SmCA. Commun Biol 2019;2:333.
- Supuran CT. Carbonic anhydrases and metabolism. Metabolites 2018;8:25.
- Supuran CT. Carbonic anhydrase inhibition and the management of hypoxic tumors. Metabolites 2017;7:E48.
- Tars K, Vullo D, Kazaks A, et al. Sulfocoumarins (1,2-benzoxathiine 2,2-dioxides): a class of potent and isoform-selective inhibitors of tumor-associated carbonic anhydrases. J Med Chem 2013;56:293–300.
- Nocentini A, Ceruso M, Carta F, et al. 7-Aryl-triazolyl-substituted sulfocoumarins are potent, selective inhibitors of the tumorassociated carbonic anhydrase IX and XII. J Enzyme Inhib Med Chem 2016;31:1226–33.
- Grandane A, Tanc M, Di Cesare Mannelli L, et al. Substituted sulfocoumarins are selective carbonic anhdydrase IX and XII inhibitors with significant cytotoxicity against colorectal cancer cells. J Med Chem 2015;58:3975–83.
- Tanc M, Carta F, Bozdag M, et al. 7-Substituted-sulfocoumarins are isoform-selective, potent carbonic anhydrase II inhibitors. Bioorg Med Chem 2013;21:4502–10.
- Touisni N, Maresca A, McDonald PC, et al. Glycosylcoumarin carbonic anhydrase IX and XII inhibitors strongly attenuate the growth of primary breast tumors. J Med Chem 2011;54:8271–7.
- Maresca A, Temperini C, Pochet L, et al. Deciphering the mechanism of carbonic anhydrase inhibition with coumarins and thiocoumarins. J Med Chem 2010;53:335–44.
- Temperini C, Innocenti A, Scozzafava A, et al. The coumarin binding site in carbonic anhydrase accommodates structurally diverse inhibitors: the antiepileptic lacosamide as an example. J Med Chem 2010;53:850–4.
- Maresca A, Temperini C, Vu H, et al. Non-zinc mediated inhibition of carbonic anhydrases: coumarins are a new class of suicide inhibitors. J Am Chem Soc 2009;131:3057–62.
- Pustenko A, Stepanovs D, Žalubovskis R, et al. 3H-1,2-benzoxathiepine 2,2-dioxides: a new class of isoform-selective carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2017;32:767–75.
- Ivanova J, Carta F, Vullo D, et al. N-Substituted and ring opened saccharin derivatives selectively inhibit transmembrane, tumor-associated carbonic anhydrases IX and XII. Bioorg Med Chem 2017;25:3583–9.
- Alterio V, Tanc M, Ivanova J, et al. X-ray crystallographic and kinetic investigations of 6-sulfamoyl-saccharin as a carbonic anhydrase inhibitor. Org Biomol Chem 2015;13:4064–9.
- Ivanova J, Leitans J, Tanc M, et al. X-ray crystallography-promoted drug design of carbonic anhydrase inhibitors. Chem Commun (Camb) 2015;51:7108–11.
- Chernov NM, Koshevenko AS, Yakovlev IP, et al. Synthesis and antimicrobial activity of 4-hydroxy-2-[5-nitrofuran(thien)-2-yl]-6h-1,3-oxazin-6-ones. Pharm Chem J 2017;51:644–7.
- Gut J, Novacek A, Fiedler P. Reaction of six-membered cyclic hydrazides with aromatic aldehydes. Collect Czechoslovak Chem Comm 1968;33:2087–96.
- Wang Q, Liu Y-C, Chen Y-J, et al. Development of a direct competitive chemiluminescent ELISA for the detection of nitrofurantoin metabolite 1-amino-hydantoin in fish and honey. Anal Methods 2014;6:4414–20.
- Nocentini A, Ceruso M, Bua S, et al. Discovery of β-adrenergic receptors blocker–carbonic anhydrase inhibitor hybrids for multitargeted antiglaucoma therapy. J Med Chem 2018;61:5380–94.
- Köhler K, Hillebrecht A, Schulze Wischeler J, et al. Saccharin inhibits carbonic anhydrases: possible explanation for its unpleasant metallic aftertaste. Angew Chem Int Ed Engl 2007;46:7697–9.
- Eldehna WM, Nocentini A, Bonardi A, et al. 3-Hydrazinoisatin-based benzenesulfonamides as novel carbonic anhydrase inhibitors endowed with anticancer activity: Synthesis, in vitro biological evaluation and in silico insights. Eur J Med Chem 2019;184:111768.
- Eldehna WM, Nocentini A, Al-Rashood ST, et al. Tumor-associated carbonic anhydrase isoform IX and XII inhibitory properties of certain isatin-bearing sulfonamides endowed with in vitro antitumor activity towards colon cancer. Bioorg Chem 2018;81:425–32.
- Al-Sanea MM, Elkamhawy A, Paik S, et al. Synthesis and biological evaluation of novel 3-(quinolin-4- ylamino)benzenesulfonamides as carbonic anhydrase isoforms I and II inhibitors. J Enzyme Inhib Med Chem 2019;34:1457–64.
- Leitans J, Kazaks A, Balode A, et al. Efficient expression and crystallization system of cancer-associated carbonic anhydrase isoform IX. J Med Chem 2015;58:9004–9.
- Nocentini A, Ferraroni M, Carta F, et al. Benzenesulfonamides incorporating flexible Triazole moieties are highly effective carbonic anhydrase inhibitors: synthesis and kinetic, crystallographic, computational, and intraocular pressure lowering investigations. J Med Chem 2016;59:10692–704.
- Whittington DA, Waheed A, Ulmasov B, et al. Crystal structure of the dimeric extracellular domain of human carbonic anhydrase XII, a bitopic membrane protein overexpressed in certain cancer tumor cells. Proc Natl Acad Sci USA 2001;98:9545–50.
- Schrödinger Suite Release 2019-1, Schrödinger, LLC, New York, NY, 2019: (a) Prime, v.5.5; Maestro v.11.9; (b) Epik, v.4.7; (c) Impact, v.8.2; (d) Macromodel v.12.3. (e) Glide, v.8.2.
- (a) Nocentini A, Gratteri P, Supuran CT. Phosphorus versus Sulfur: Discovery of Benzenephosphonamidates as Versatile Sulfonamide-Mimic Chemotypes Acting as Carbonic Anhydrase Inhibitors. Chemistry 2019;25:1188–92. (b) Supuran CT. Carbon- versus sulphur-based zinc binding groups for carbonic anhydrase inhibitors? J Enzyme Inhib Med Chem 2018;33:485–95. 33:
- Nocentini A, Bonardi A, Gratteri P, et al. Steroids interfere with human carbonic anhydrase activity by using alternative binding mechanisms. J Enzyme Inhib Med Chem 2018;33:1453–9.
- Nocentini A, Carta F, Tanc M, et al. Deciphering the mechanism of human carbonic anhydrases inhibition with sulfocoumarins: computational and experimental studies. Chemistry 2018;24:7840–4.
- Schrödinger Release 2019-2: Desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY, 2019, v.5.7.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73.
- Verdolino V, Cammi R, Munk BH, et al. Calculation of pKa values of nucleobases and the guanine oxidation products guanidinohydantoin and spiroiminodihydantoin using density functional theory and a polarizable continuum model. J Phys Chem B 2008;112:16860–73.