
Abstract
A series of 4-arylamido 5-methylisoxazole derivatives with quinazoline core was designed and synthesised based on conformational rigidification of a previous type II FMS inhibitor. Most of quinazoline analogues displayed activity against FLT3 and FLT3-ITD. Compound 7d, 5-methyl-N-(2-(3-(4-methylpiperazin-1-yl)-5-(trifluoromethyl)phenyl)quinazolin-7-yl)isoxazole-4-carboxamide, exhibited the most potent inhibitory activity against FLT3 (IC50= 106 nM) with excellent selectivity profiles over 36 other protein kinases including cKit and FMS kinase. Compound 7d was also active in FLT-ITD, with an IC50 value of 301 nM, and other FLT3 mutants showing potential as an AML therapeutics.
1. Introduction
FLT3, a type of trans-membrane receptor tyrosine kinase expressed on lympho-hematopoietic cells, is an attractive target in acute myeloid leukaemia (AML)Citation1. When FLT3 ligand binds to FLT3 kinase, it is activated by auto-phosphorylation, which then activates its multiple downstream signalling pathways, including signal transducer and activator of transcription 5 (STAT5), ras/mitogen-activated protein kinase, and phosphatidylinositol 3-kinase/Akt pathways, playing a key role in cell proliferation, survival, and immune responseCitation2–5.
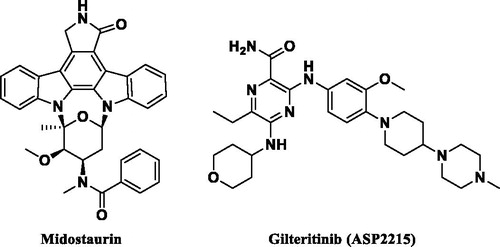
However, mutation of FLT3 causes its ligand-independent activation and can be classified by location and type, including internal tandem duplication (ITD) and point mutation in the tyrosine kinase domain (TKD). FLT3-ITD mutations are present in 20–30% of AML patients and are especially related to leucocytosis and poor prognosis and point mutation in TKD is found in 5% of AML patientsCitation6. Despite the significance of seeking AML therapeutics, only a few molecules have been approved, such as midostaurin and gilteritinib (ASP2215), which are depicted in Citation7,8.
Figure 1. FLT3 inhibitors approved by the FDA as AML treatment.
Protein kinase inhibitors can be classified as type I, II, III, IV and VCitation9 based on their binding mode. Among these inhibitors, type II kinase inhibitor can take advantage of selectivity acquisition and exhibits promising potency due to additional interaction with the DFG pocket adjacent to the ATP-binding pocket on top of the hinge hydrogen bond in the ATP siteCitation10. Previously, we reported very potent type II FMS inhibitorsCitation11 and selective benzimidazole-incorporated FLT3 inhibitorsCitation12. Here, we demonstrate that the amide or urea between the middle phenyl ring and the secondary hydrophobic aromatic ring can be rigidified further, introducing a quinazoline structure as a bioisostere (). The quinazoline structures are well-known privileged structures in medicinal chemistry, exhibiting diverse biological activitiesCitation13. However, this is the first attempt to incorporate one in type II PKI modification. Introducing quinazoline into type II FMS kinase inhibitor, we could discover a novel FLT3 inhibitor with excellent selective profile.
Figure 2. Design of quinazoline derivatives as bioisosteres of the middle phenyl ring-amide-bond.

2. Results and discussion
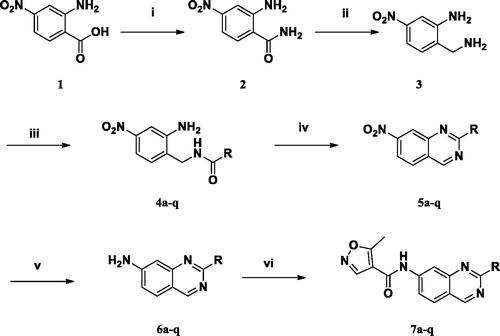
Scheme 1 shows the general synthetic route for quinazolinyl-isoxazole-4-carboxamide analogues (7a–q). First, 2-amino-4-nitro-benzoic acid (1) was converted to 2-amino-4-nitrobenzamide (2) and then reduced to 2-(aminomethyl)-5-nitroaniline (3) using borane-tetrahydrofuran. Then, the benzyl amine was coupled with various benzoyl chlorides to produce carboxamide derivatives (4a–q); subsequently, benzamide was treated with concentrated HCl in acetic and subjected to microwave irradiation at 150 °C for cyclisation to yield a dihydroquinazoline compound. Without further purification, they were treated with p-chloranil oxidising agent to obtain quinazoline derivatives (5a–q) as core intermediates. Next, the nitro group was reduced to amine (6a–q) using Fe catalyst and was then coupled with isoxazole chloride to produce the final quinazolinyl-isoxazole-4-carboxamides (7a–q).
Scheme 1. Syntheses of 1H-quinazolyl isoxazole-4-carboxamide derivatives. (i) EDC, HOBt, TEA, NH3 in MeOH, rt; (ii) BH3-THF, reflux; (iii) benzoyl chloride, CH2Cl2, 0 oC→ rt; (iv) (1) HCl/H2O/AcOH, μW, 150 oC, 10 min; (2) p-chloranil, toluene, reflux; (v) Fe, AcOH/H2O/EtOH, 60 oC; (vi) 5-methylisoxazole-4-carbonyl chloride, TEA, THF, rt.
All quinazoline compounds, 7a–q, were evaluated for their activity against FLT3 kinase and FLT3-ITD mutation and the results are shown in . Most of the synthesised compounds exhibited selective activity against FLT3, particularly those incorporating the piperazine moiety. Among the compounds evaluated, 7d showed the most potent activity against FLT3 with an IC50 value of 106 nM, and FLT3-ITD with an IC50 value of 301 nM. Structure activity relationships (SARs) were inferred from the data.
Table 1. Enzymatic activity of 5-methyl-N-(2-arylquinazolin-7-yl) isoxazole-4-carboxamide analogues.
In our previous work, benzimidazole compounds retained their activity against FLT3 regardless of presence of 1,3,5-substituted or 1,3,4-substituted benzoic acid, and the activity was determined as piperazine > imidazole > morpholine substituentsCitation12. We optimised quinazoline derivatives based on the observation of previous benzimidazole derivatives. Those with methyl piperazine or morpholine as the phenyl substitution group (7d and 7b) were more potent about 2- to 5-fold (IC50 values of 0.106 and 3.98 µM, respectively) compared to corresponding benzimidazole series (IC50 values of 0.495 and 7.94 µM), and 7c displayed better potency (IC50 value of 1.58 µM) than that of benzimidazole (IC50 value of 2.33 µM). Introduction of 3,5-disubstituted benzoic acid through quinazoline connection maintained the activity (7b, 7c, 7d, 7e, 7n), but quinazoline compound with 1,3,4-substituted benzoic acid (7a) and one with pyrazole (7h) caused loss of activity towards FLT3.
With the result of 7d, we synthesised compound 7e to optimise the linkage between the phenyl group and the piperazine moiety. Although inhibitory activity towards FLT3 was retained, 7e exhibited decreased activity, about 10-fold less than that of 7d. On the predicted binding mode of 7d, strong ionic interaction between the protonated nitrogen of the piperazine and Asp829 might enhance its binding affinity. Almost the same ionic interaction seems possible in case 7e, but the ionic interaction might push the whole compound slightly out of the active site due to its length, resulting in loss of multiple interactions such as hydrogen bonding with Asp829, π–π interaction with Phe691, and π–cation interaction with Lys644 (). We also replaced piperazine with a piperidine moiety (7n) to investigate the role of nitrogen in the piperazine structure. The IC50 value of 7n was 3.59 µM, similar but weaker than 7e despite their similar structures. Our docking study showed that one conformer of 7n with equatorial O linkage bound with FLT3 similarly to compound 7e, but the other conformer with axial O linkage was not suitable to bind tightly to the active site because of its non-linear piperidine moiety ().
Figure 3. (Left) Compound 7d (green) at the active site of FLT3 (PDB: 4RT7); (right) 7e (yellow) at the active site of FLT3 (PDB: 4RT7).
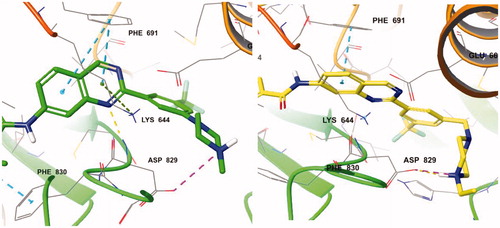
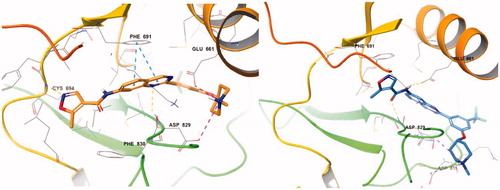
Figure 4. (Left) Compound 7n with equatorial O linkage (orange) at the active site of FLT3 (PDB: 4RT7); (right) compound 7n with axial O linkage (azure) at the active site of FLT3 (PDB: 4RT7).
We diversified our quinazoline compounds for further optimisation, introducing a halogen group (7f, 7g, and 7i), tert-butyl isoxazole (7l), and styrenyl group (7j and 7k). However, only 7j exhibited competitive activity against FLT3, with an IC50 value of 4.7 µM. In addition, we tried to introduce isoxazole, indazole, acetyl piperidine, and pyridine (7l–q). Compound 7m showed activity against FLT3, with an IC50 value of 0.79 µM. Compounds 7l, 7o, 7p, and 7q did not show inhibitory activity towards FLT3 or were very weak. The tert-butyl isoxazole, acetyl piperidine, and pyridine moieties were less basic than other moieties (piperazine, morpholine, imidazole, indazole, and piperidine). For inhibition against FLT3, the ionic interaction with Asp829 seems to play a crucial. Compounds incorporating basic moieties (7b, 7c, 7d, 7e, 7j, 7m, and 7n) maintained inhibitory activity towards FLT3, while compounds that were substituted with less basic moieties (7o, 7p, and 7q) lost the activity. The quinazoline derivatives were also tested for their activity against FLT3-ITD, and they displayed inhibitory activity against FLT3-ITD similar to level of activity on FLT3 (7b, 7c, 7d, 7j, 7m, and 7n).
Moreover, the most potent 7d was examined further for inhibitory activity against other FLT3 mutants and the result was impressive (). The 7d was a potent inhibitor towards FLT3 (F594_R_R595) (IC50 = 0.524 µM), FLT3 (F594_EY_R595) (IC50 = 0.495 µM), FLT3 (Y591_ VDFREYEYD_V592) (IC50 = 0.728 µM), and FLT3 (D835Y), with an IC50 value of 0.228 µM. Since FLT3-ITD mutation occur mainly in the juxtamembrane domain away from the ATP binding site, the FLT3 inhibitors binding in ATP site exhibit potency against FLT3-ITD mutantCitation14. On the other hand, a mutation in active site such as FLT3 (D835Y) induces active conformation of kinase and type II inhibitors generally could not bind well enough. This is also known to be a mechanism of secondary resistance of type II FLT3 inhibitor.Citation13 Nevertheless, our compound 7d, which is a type II FLT3 inhibitor, exhibits a potent inhibitory activity (IC50 = 228 nM) in FLT3-TKD (D835Y) similar to that of FLT3 wild type also. FLT3-TKD, especially D835Y, D835H mutant was identified as a main drug resistance mechanism of FLT3 inhibitor in the clinical trial.Citation15–18 Like quizartinib, 7d displays potent inhibitory activity against FLT3-TKD (D835Y) with value of IC50 228 nM, while decreased about 2-fold compared to that of FLT3 wild type.
Table 2. Enzymatic activities of compound 7d against FLT3 mutants.
Next, we investigated kinase panel screening of 7d and 7b over 36 different kinases at a single dose of 10 µM (). Both compounds 7d and 7b (data in Supplementary material) showed an excellent selectivity profile, having almost no activity on other kinases including FMS and cKit. High selectivity over cKit might be an opportunity to avoid myelosuppression toxicity, which was reported from a dual FLT3/cKit inhibitorCitation19–22. It is a valuable result to secure such a kinase selectivity profile considering the similarities of the same type III receptor tyrosine kinase (FMS, cKit and FLT3).
Figure 5. Kinase profiling according to chemical scaffold. The profiles of the FMS kinase inhibitor [13] and quinazoline derivative 7d (10 μM) are shown.
![Figure 5. Kinase profiling according to chemical scaffold. The profiles of the FMS kinase inhibitor [13] and quinazoline derivative 7d (10 μM) are shown.](/cms/asset/9b010760-9995-4cd8-97e3-0eec80929fa2/ienz_a_1758689_f0005_c.jpg)
For better understanding of the interactions between our compounds and FLT3, molecular docking of compound 7d into the ATP binding pocket of FLT3 (PDB: 4RT7) was performed using Glide software (Schrödinger, Version 14.2). The docking result is depicted in . In the binding model of 7d, 5-methyisoxazole forms a π-π interaction with Phe830 instead of hydrogen bonding with Cys694, contrast to the co-crystal structure of quirzatinibCitation23. Instead, the quinazoline ring of 7d showed possibilities of multiple interactions: one hydrogen bond with Asp829, π–π interaction with Phe691, and π-cation interaction with Lys644. As it was designed as a hybrid of the middle phenyl ring and urea (or amide) linker, the quinazoline seems tightly bound with ATP-binding site and adjacent region like other type II inhibitors. However, instead of fitting into a hydrophobic pocket surrounded by hydrophobic residue, the strong ionic interaction between the protonated nitrogen of the piperazine ring and the Asp796 might enhance the binding affinity, resulting in potent inhibitory activity and considerable selectivity about FLT3. Due to its strong ionic interaction, the fused ring system of quinazoline might make additional interactions in the active site of FLT3 unlike benzimidazole derivativeCitation12. The quinazoline derivative 7d interacts with not only Lys644 (π–cation interaction between guanidine group of Lys644 and quinazoline ring) also gatekeeper residue, Phe691, (π–π interaction between phenyl group of side chain and quinazoline structure) properly, which could contribute to enhanced activity of compound 7d presumably compared with benzimidazole compoundCitation12.
Figure 6. (left) Docking structures of 7d in FLT3 (PDB: 4RT7) and 7d in FMS (PDB:3LCO).
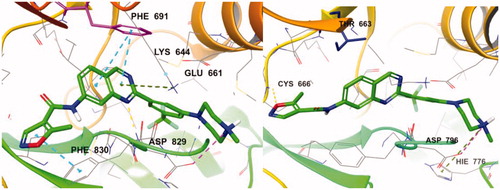
We also tried to explain the selectivity of quinazoline compound for FMS and cKit. As shown in , FMS kinase has Thr663 instead of Phe691 in the active site. 7d could not bind well in the FMS kinase without multiple interaction which quinazoline forms. Although 7d forms hydrogen bonding with Cys666 and strong ionic interaction with Asp796, the quinazoline moiety does not form any interaction with Thr663 or other residue. The difference of only one residue in the highly conserved active site seems to play crucial role in selectivity profile and the interaction between quinazoline structure and Phe691 also seems to be important to binding affinity. In the case of cKit kinase, threonine residue locates at the same position, and same explanation could be applied.
3. Conclusions
We designed and synthesised a series of 4-arylamido 5-methylisoxazole derivatives containing quinazoline using a conformational rigidification strategy. Various analogues were synthesised and tested for inhibitory activity against FLT3. Compound 7d displayed the most potent inhibitory activity against FLT3, with an IC50 of 106 nM, and was also active against FLT3-ITD and FLT3-TKD (D835Y), with an IC50 of 301 nM and 228 nM respectively. The novel type II FLT3 inhibitors showed an excellent selectivity profile having 20% or less activity towards other kinases including similar FMS, cKit. Specifically, the selectivity over FMS and cKit potentially relieve the myelosuppression toxicity observed in the dual FLT3/cKit kinase inhibitor. Molecular docking of 7d into the ATP binding site of FLT3 kinase was performed and the result disclosed that the quinazoline structure could perform an adequate or even better bioisostere of benzimidazole in previous report. Considering that FLT3 and FLT3 mutants have been significantly associated with FLT3 mutant-mediated AML, compound 7d could be a valuable molecule for FLT3-mutant positive physiology and pathology research and a potential therapeutic agent. Even if further optimisation would be necessary in terms of potency, our research towards a novel selective FLT3 inhibitor suggests the potential of quinazoline compounds as AML therapeutics.
Supplemental Material
Download PDF (1.3 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Swords R, Freeman C, Giles F. Targeting the FMS-like tyrosine kinase 3 in acute myeloid leukemia. Leukemia 2012;26:2176–85.
- Drexler HG. Expression of FLT3 receptor and response to FLT3 ligand by leukemic cells. Leukemia 1996;10:588–99.
- Zhang S, Fukuda S, Lee Y, et al. essential role of signal transducer and activator of transcription (Stat)5a but not Stat5b for Flt3-dependent signaling. J Exp Med 2000;192:719–28.
- Takahashi S. Downstream molecular pathways of FLT3 in the pathogenesis of acute myeloid leukemia: Biology and therapeutic implications. J Hematol Oncol 2011;1:1–10.
- Gary Gilliland D, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood 2002;100:1532–42.
- Larrosa-Garcia M, Baer MR. FLT3 inhibitors in acute myeloid leukemia: current status and future directions. Mol Cancer Ther 2017;16:991–1001.
- Mori M, Kaneko N, Ueno Y, et al. Gilteritinib, a FLT3/AXL inhibitor, shows antileukemic activity in mouse models of FLT3 mutated acute myeloid leukemia. Invest New Drugs 2017;35:556–65.
- Perl AE, Altman JK, Cortes J, et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: a multicentre, first-in-human, open-label, phase 1–2 study. Lancet Oncol 2017;18:1061–75.
- Wu P, Nielsen TE, Clausen MH. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci 2015;36:422–39.
- Versele M, Haefner B, Wroblowski B, et al. Covalent Flt3-Cys828 inhibition represents a novel therapeutic approach for the treatment of Flt3- ITD and Flt3-D835 mutant acute myeloid leukemia. Cancer Res 2016;76:4800.
- Im D, Jung K, Yang S, et al. Discovery of 4-arylamido 3-methyl isoxazole derivatives as novel FMS kinase inhibitors. Eur J Med Chem 2015;102:600–10.
- Im D, Moon H, Kim J, et al. Conformational restriction of a type II FMS inhibitor leading to discovery of 5-methyl- N-(2-aryl-1H-benzo[d]imidazo-5-yl)isoxazole-4-carboxamide analogues as selective FLT3 inhibitors. J Enzyme Inhib Med Chem 2019;34:1716–21.
- Kamal A, Reddy K, Devaiah V, et al. Recent advances in the solid-phase combinatorial synthetic strategies for the quinoxaline, quinazoline and benzimidazole based privileged structures. Mini-Rev Med Chem 2006;6:71–89.
- Chen Y, Pan Y, Guo Y, et al. Tyrosine kinase inhibitors targeting FLT3 in the treatment of acute myeloid leukemia. Stem Cell Investig 2017;4:48–13.
- Smith CC, Lin K, Stecula A, et al. FLT3 D835 mutations confer differential resistance to type II FLT3 inhibitors. Leukemia 2015;29:2390–2.
- Levis M. FLT3 mutations in acute myeloid leukemia?: What is the best approach in 2013? Hematology Am Soc Hematol Educ Program 2013;2013:220–6.
- Sun D, Yang Y, Lyu J, et al. Discovery and rational design of pteridin-7(8H)-one-based inhibitors targeting FMS-like tyrosine kinase 3 (FLT3) and its mutants. J Med Chem 2016;59:6187–200.
- Roskoski R. Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol Res 2019;144:19–50.
- Fathi AT, Chen YB. The role of FLT3 inhibitors in the treatment of FLT3-mutated acute myeloid leukemia. Eur J Haematol 2017;98:330–6.
- Chen Y, Pan Y, Guo Y, et al. Tyrosine kinase inhibitors targeting FLT3 in the treatment of acute myeloid leukemia. Stem Cell Invest 2017;4:48–52.
- Warkentin AA, Lopez MS, Lasater EA, et al. Overcoming myelosuppression due to synthetic lethal toxicity for FLT3-targeted acute myeloid leukemia therapy. Elife 2014;3:e03445.
- Liang X, Wang B, Chen C, et al. Discovery of N-(4-(6-acetamidopyrimidin-4-yloxy)phenyl)-2-(2-(trifluoromethyl)phenyl)acetamide (CHMFL-FLT3-335) as a potent Fms-like tyrosine kinase 3 internal tandem duplications (FLT3-ITD) mutant selective inhibitor for acute myeloid leukemia. J Med Chem 2019;62:875–892.
- Smith CC, Lin KC, Zhang Y, et al. Characterizing and overriding the structural mechanism of the quizartinib-resistant FLT3 “gatekeeper” F691L mutation with PLX3397. Cancer Discov 2015;5:668–79.