
Abstract
The human sirtuin silent information regulator 1 (SIRT1) is a NAD+-dependent deacetylase enzyme. It deacetylates many protein substrates, including histones and transcription factors, thereby controlling many physiological and pathological processes. Several synthetic inhibitors and activators of SIRT1 have been developed, and some therapeutic applications have been explored. The indole EX-527 and its derivatives are among the most potent and selective SIRT1 inhibitors. EX-527 has been often used as a pharmacological tool to explore the effect of SIRT1 inhibition in various cell types. Its therapeutic potential has, therefore, been evaluated in animal models for several pathologies, including cancer. It has also been tested in phase II clinical trial for the treatment of Huntington’s disease (HD). In this review, we will provide an overview of the literature on EX-527, including its mechanism of inhibition and biological studies.
1. Introduction
Human silent information regulator 1 (SIRT1) belongs to the sirtuin family of enzymes, which constitute class III of the histone deacetylase family (HDAC). It is the most studied of the seven human sirtuins known to date. It is a NAD+-dependent deacetylase, which deacetylates many protein substrates, including histones and transcription factorsCitation1. SIRT1 has been linked to type 2 diabetesCitation2, cancerCitation3, Alzheimer diseaseCitation4, and more generally diseases of ageingCitation5,Citation6. In particular, the contradictory roles of human SIRT1 in cancer have been reviewed and are still a subject of debateCitation7,Citation8. To study these biological activities, the modulation of SIRT1 expression and activity by bioengineering (mutations, overexpression, siRNA, or knockout for example) has been largely employedCitation7,Citation9,Citation10.
In addition to these genetic manipulation studies, pharmacological modulation of SIRT1 has been the subject of intense research. SIRT1 modulators in general and their roles in cancer in particular have been often reviewed, usually giving an overview of several inhibitors and activators, but limited information on each oneCitation11–14. We present here an overview of the literature data on the SIRT1 selective and potent inhibitor EX-527 (SEN0014196 or selisistat) since its first disclosure in 2005Citation15. Key data are reported, regarding its mechanism of inhibition and inhibitory potency in vitro, its effect on various cell types (used alone or in combination with other molecules), biological studies in animal models, and results of a clinical trial. This review primarily describes studies in which EX-527 is the main compound of interest, but we also included selected studies using EX-527 as a control and/or pharmacological tool to explore SIRT1 related pathways. To complete this overview, we also included some examples in which the inhibitor EX-527 was used to counteract the effects of other molecules, such as SIRT1 activators.
2. In vitro assays of EX-527 on isolated enzymes and mechanism of inhibition
2.1. Discovery, properties, IC50 values, and structure/activity relationship studies
EX-527 was identified in 2005 by high throughput screening of libraries of compounds on the enzyme SIRT1 ()Citation15. It has now been the subject of more than 200 articles.
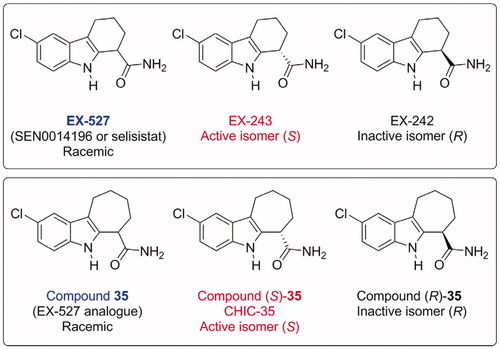
Figure 1. Structures of SIRT1 inhibitors EX-527 and its analogue Compound 35, indicating their absolute stereochemistry and the corresponding names used in the literatureCitation15. EX-527 and CHIC-35 are now commercially available from suppliers.
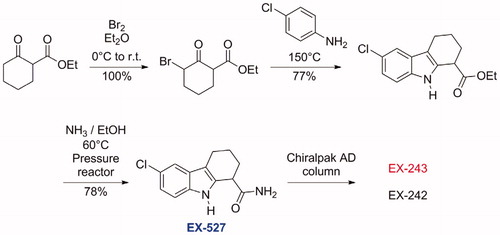
A typical synthesis of this family of compounds is depicted in Scheme 1. These compounds were obtained by a Bischler indole synthesis. In the first step, a β-keto ester was brominated on α to the ketone, affording a bromo keto ester, which was heated in the second step with an aniline, affording the tetrahydrocarbazole ester. The ester was then converted to the primary amide under pressure. In case enantiomerically pure material was needed, separation by chiral column chromatography was achievedCitation15.
Scheme 1. Chemical synthesis of EX-527Citation15.
EX-527 is a potent and selective SIRT1 inhibitor, with IC50 values as low as 38 nM, depending on assay conditionsCitation16. In the first report, it was shown to be more selective for SIRT1 than for SIRT2 or SIRT3 (200–500-fold)Citation15. EX-527 does not inhibit class I/II HDAC activity at concentrations up to 100 µM. EX-527 is racemic, the active isomer (designated EX-243) being (S), whereas the other (R) isomer (designated EX-242) is inactive. IC50 values for sirtuin inhibition by EX-527 have been measured in several studies, using a variety of assay methods and peptide substrates (). They range from 0.038 to 3 µM, usually between 0.1 and 1 µM. They depend mostly on the nature and concentration of the peptide substrates and on NAD+ concentration, because of the uncompetitive inhibition mechanism of EX-527 (see below). Very stringent structure/activity relationships were described in the original articleCitation15 and were later explained in light of the crystal structure published in 2013 (see below)Citation31. Compound 35 () is an analogue of EX-527, very potent inhibitor of SIRT1: the IC50 of the (S) isomer is 60 nM, and the IC50 of the racemic mixture is 124 nM. It is selective for SIRT1, with an IC50 for SIRT2 of 2.77 µMCitation15.
Table 1. In vitro assays of EX-527 and its analogue 35 on isolated recombinant sirtuins expressed in bacteria.
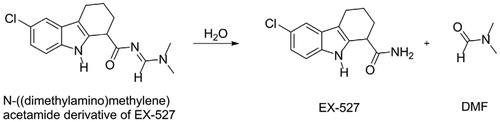
EX-527 was also identified independently in 2006 from another high throughput screening. The screened compound was in fact the N-((dimethylamino)methylene)acetamide derivative (a dimethylformamide adduct), which was rapidly hydrolysed in aqueous solution to form EX-527 and dimethylformamide (Scheme 2)Citation32.
Scheme 2. Spontaneous hydrolysis of the DMF adduct of EX-527.
EX-527 is also able to block the protein-protein interaction taking place between deleted in breast cancer 1 (DBC1) and SIRT1Citation33. DBC1 is an endogenous protein shown to interact with SIRT1 and to inhibit its catalytic activityCitation34,Citation35. The regulation of this interaction is complex. For example, DBC1 itself is a substrate of SIRT1, and deacetylated DBC1 does not bind to SIRT1Citation33. However, the team of Sinclair showed that EX-527 blocks the interaction via an acetylation-independent mechanism in vitro. They also demonstrated, using a luciferase complementation assay, that the inhibitor is able to block the SIRT1-DBC1 interaction in cells with an IC50 of approximately 1 µMCitation33.
In addition to sirtuins, EX-527 and racemic 35 (rac-35) have been tested in vitro on other isolated enzyme and receptor targets. Overall, they displayed very little to no activity. They did not inhibit class I and II HDACs and NAD+ glycohydrolase at 100 µMCitation15. PARP are enzymes using the NAD+ as cosubstrate for ADP-ribosyl transfer, producing nicotinamide, like sirtuins. Therefore, inhibitors targeting the nicotinamide binding pocket like EX-527 could have an inhibitory effect on PARP enzymes. No inhibition was observed on PARP1 and PARP10Citation29,Citation36. On cardiac potassium channels (hERG/IKr), EX-527 had an IC50 of 43 µM, with 0% inhibition at 10 µMCitation37, and rac-35 displayed only 10% inhibition at 10 µMCitation15. Cytochrome P450 are key enzymes involved in metabolism of drugs. They are largely evaluated in screening panels of new biologically active molecules, to identify P450 substrates or inhibitors. On cytochromes P450 (3A4, 2D6, 2C9, 2C19, 1A2, 2C8, and 2E1), both molecules had weak or no inhibitory potency at 1 µM, the highest values being 23% inhibition for 2C19 and 1A2 with rac-35. IC50 values determined for EX-527 were higher than 100 µM for all cytochromes P450 except 2C9 (62.4 µM), 2C19 (72.2 µM), and A2 (8.7 µM)Citation15,Citation37.
2.2. Mechanism of inhibition and crystal structures
A simplified mechanism of deacetylation of a substrate catalysed by sirtuins is represented in Citation38. The acetylated substrate makes a nucleophilic substitution on the C1′ of the NAD+ cofactor, releasing nicotinamide. The 1′-O-alkylimidate intermediate formed reacts intramolecularly to generate a bicyclic intermediate. This intermediate is subsequently hydrolysed to form the deacetylated product and the 2′-O-AcADPr coproduct.
Figure 2. (A) Mechanism of sirtuin-catalysed deacetylation of a peptide (or protein) substrate Ac-Pep (acetylated peptide). For simplicity, acidic and basic general catalysis is not represented in this mechanism. (B) Proposed simplified mechanism of sirtuin inhibition by EX-243, adapted from Gertz et al.Citation25. E: enzyme. Note that former studies of SIRT1 inhibition by substrate analogues suggested (i) a random addition of substrates (therefore, Ac-Pep could be added first to the enzyme, not represented here for simplification) and (ii) a departure of the peptide product from the enzyme in the last step (which would disagree here with the existence of the crystallised complex E/2′-O-AcADPr/EX-243)Citation39.
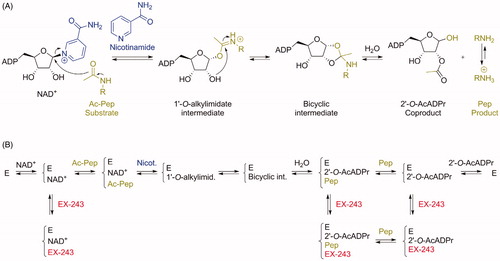
The mechanism of SIRT1 inhibition by EX-527 is represented in , adapted from Gertz et al.Citation25. Mechanistic studies on SIRT1, SIRT3, and Sir2Tm (sirtuin from Thermotoga maritima) demonstrated in all three cases that the inhibition by EX-527 was non-competitive with substrate and uncompetitive with NAD+. Therefore, the inhibition potency depends on the NAD+ concentration. Binding parameters are summarised in . Kd values for EX-527 measured for the apo enzymes and in the presence of NAD+ confirmed the uncompetitive nature of the inhibition. Indeed, EX-527 does not bind to the apo enzyme, but binds with low micromolar affinity in the presence of NAD+.
Table 2. Binding parameters of EX-527 with sirtuins.
Another interesting aspect of these mechanistic studies concerns the specificity of EX-527 for sirtuin isoforms. The authors propose that the difference between EX-527-sensitive enzymes (like SIRT1 and Sir2Tm) and less sensitive ones (like SIRT2 and SIRT3) comes from differences in their kinetics of catalysis, and not from differences in the binding pockets, which are very similarCitation25. Indeed, they suggest that binding of EX-527 either after or before the rate-limiting step leads to differences in inhibition potency.
Six crystal structures of sirtuins in complex with the active enantiomers of the inhibitors, compound (S)-35 and EX-243, have been described. The first one was between SIRT1, NAD+, and compound (S)-35 (PDB 4I5I)Citation31. The others were part of a series of structures solved to study the mechanism of sirtuin inhibition by EX-243 (the active enantiomer of EX-527), with human SIRT3 and Sir2Tm: SIRT3/NAD+/EX-243 (4BV3), SIRT3/ADPr/EX-243 (4BVB), SIRT3/2′-O-AcADPr/EX-243 (4BVH), Sir2Tm soaking (4BUZ, partially with substrates Ac-p53 peptide and NAD+, and partially with product 2′-O-AcADPr and EX-243), and Sir2Tm/2′-O-AcADPr/deacetyl p53 peptide/EX-243 (4BV2)Citation25.
In all these structures, the inhibitors occupy the nicotinamide binding pocket (the so-called C-pocket) of the sirtuin, and one of the following molecules is also co-crystallised, forming a ternary complex: NAD+, the coproduct 2′-O-AcADPr, or ADPr (). This observation is in agreement with the uncompetitive nature of the inhibition with the cofactor NAD+, which is required for efficient inhibition, as mentioned above. The inhibitors are deeply buried in the C-pocket and make hydrogen bonds contacts and hydrophobic interactions with the enzyme, which explain the stringent structure/activity relationships observedCitation15.
Figure 3. Crystal structures of sirtuins in complex with indole inhibitors EX-243 and its analogue (S)-35. Left: SIRT1/NAD+/(S)-35 (4I5I)Citation31; middle: SIRT3/ADPr/EX-243 (4BVB); right: Sir2Tm/2′-O-AcADPr/deacetyl p53 peptide/EX-243 (4BV2)Citation25. Active site close-up representations are displayed below the full structures. Pep: deacetyl p53 peptide.
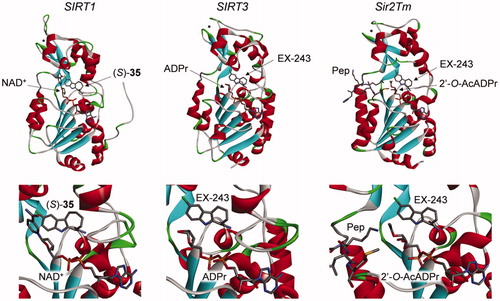
Moreover, the mechanistic studies showed that sirtuin inhibition with EX-527 allows the formation of one molecule of product per molecule of enzyme, indicating that the inhibitor binds most efficiently after bicyclic intermediate formation and allows coproduct formationCitation25. The authors proposed that EX-243 inhibits sirtuins mostly by binding in the presence of the coproduct 2′-O-AcADPr. Finally, from the comparison of crystals structures with and without the inhibitor, it appears that a flexible cofactor-binding loop moves towards the inhibitor and the coproduct during inhibition, resulting in a “closed” conformation preventing product releaseCitation25.
3. Cellular assays of EX-527
EX-527 has been tested on several cell lines, either as the main molecule of interest for potential therapeutic applications, or as a control experiment for comparison with other sirtuin modulators (inhibitors or activators). Often, it has been used as a pharmacological tool to demonstrate the involvement of SIRT1 in a biological response. An overview of literature data is summarised in .
Table 3. Representative examples of cellular effects of EX-527.
On tumour cell lines, several reports demonstrated the ability of EX-527 to increase p53 acetylation from 1 to 25 µM concentrations, when used either alone or in combination with cytotoxic moleculesCitation16,Citation23,Citation44,Citation46,Citation51,Citation56,Citation63. EX-527 was shown to improve the efficiency of cytotoxic agents on cancer cells, with several chemotherapeutic and genotoxic agentsCitation40,Citation42,Citation60. However, in few cases, EX-527 administered alone increased cell proliferation of cancer cell linesCitation49,Citation71. The conclusion of one of these studies on the role of SIRT1 in cancer cells is a simple summary of these apparently contradictory results:
In summary, our results suggest that both activators and inhibitors of SirT1 have therapeutic potential as anti-tumor agents. A simple scenario is that SirT1 activators may impart cancer prevention effects by enhancing the growth-inhibitory effect of SirT1 in benign tumors. Its effect on advanced stage tumors may be heterogeneous, depending on whether a tumor has evolved to rely on SirT1 for survival. However, when tumors are being treated with chemotherapy, SirT1 inhibitors may be useful for enhancing apoptotic responseCitation40.
Ten years after this report, the list of EX-527 studies has grown to reinforce this view (). For example, a decrease in cell survival and migration and an increase in apoptosis was recently observed on hepatocellular carcinoma (HCC: HepG2 and Huh7) cell lines with EX-527 aloneCitation63. Moreover, the same study demonstrated that EX-527 induced the downregulation of ABC transporters P-gp and MRP3 in HepG2 cells, suggesting an additional potential application of this SIRT1 inhibitor in combination with conventional therapeutic drugs to overcome multi-drug resistance (MDR) during HCC therapyCitation63. Indeed, one of the most potent effect was obtained when EX-527 was used in combination with Hsp-90 inhibitors on CSCs (cancer stem-like cells) or MDR variants, with a potent increase in cytotoxicity of the Hsp-90 inhibitor with only 10 nM EX-527Citation52,Citation53. Moreover, EX-527 at 1 µM decreased colony formation of ovarian carcinoma cells, with or without overexpression of SIRT1Citation72. At 600 nM, it suppressed cell migration and inhibited the occurrence of epithelial–mesenchymal transition (EMT) in chemotherapy resistant oesophageal cancer cellsCitation71. Overall, several factors are important to consider to understand the effect of EX-527 on cancer cells: (i) the type of cell line and the cancer stage, from benign to advanced, (ii) the presence of other agents, conventional chemotherapy, or additional HDAC inhibitors for example, and (iii) the dose, because at higher doses (ex. 40 µM or above), EX-527 may significantly inhibit SIRT2 and may have other targets. For potential anti-cancer therapeutic applications, aiming for a specific SIRT1 inhibition at low concentrations of EX-527 (ex. 1 µM or below) in combination with cytotoxic agents may be the most promising strategy.
On non-cancer cell lines, fewer studies were published than on cancer-cell lines. For example on HUVEC, EX-527 was shown to protect from H2O2 damageCitation49, but to abolish the protective effect of resveratrol under high-glucose conditionsCitation67. Several articles described effects on cells involved in the immune system, macrophages, and T cells. Beneficial effects on autoimmune diseases and graft rejection problems can be envisioned from these cell assays, for example through reduction of effector T cell proliferation and differentiationCitation57,Citation69, and increase in the number and suppressive function of T regulatory cells Tregs (see Chapter undefined for in vivo results)Citation64.
Many of the studies evaluating the role of EX-527 in cells summarised in this review incorporated control experiments with SIRT1 knockdown, mostly with anti-SIRT1 siRNA. These studies, in which the same effects were obtained with anti-SIRT1 siRNA or with its pharmacological inhibition with EX-527, make a strong case for the use of EX-527 as a pharmacological tool to study SIRT1 activity. However, the fact that EX-527 only targets SIRT1 must be tempered. Indeed, in vitro studies show that the extent of its specificity, in particular towards SIRT2, depends on the assay types (nature of the substrate and concentration of NAD+ for example) and may not be so high under certain conditions (). Consequently, its specificity inside cells or in vivo is even less predictable and quantifiable. Therefore, the results of studies concluding that SIRT1 is involved in the observed effect must be taken with caution, if they are solely based on the effect of EX-527 as a pharmacological control. SIRT2 and other unknown potential protein targets may be involved.
4. In vivo assays of EX-527
EX-527 has been tested in several organisms, mostly mice and rats, but also in the nematode C. elegans, in Drosophila melanogaster (D. melanogaster) and in humans in exploratory clinical trials ( and ).
Table 4. Selected pharmacokinetics parameters of EX-527 (in plasma).
Table 5. Representative examples of in vivo assays of EX-527.
Pharmacokinetic data were obtained in mice and human, both in female and male. Selected parameters are given in . In R6/2 mice model of Huntington’s disease (HD) with 10–20 mg/kg dosing, average plasma concentrations over 24 h were in the low micromolar range (1.5–3.2 µM)Citation47. In healthy male human volunteers with 150–300 mg doses, average plasma concentrations over 24 h were also in the low micromolar range (1.6–3.9 µM)Citation37. However, a higher than proportional concentration (11.8 µM) was observed with 600 mg dosing, suggesting that one or more clearance mechanisms are approaching saturation at this dose. For multiple oral doses (for ex. 300 mg daily for 7 d for male), the data suggested that the pharmacokinetic steady-state was reached within 4 d, with an exposure higher than predicted from single-dose data.
The fraction of unchanged EX-527 excreted in the urine was very low for all doses in male subjects (<0.02% up to 24 h post-dose). The compound was transformed in vivo by hydroxylation and oxidative deamination followed by glucuronic acid conjugation, across all species studied (mouse, rat, dog, and human)Citation37.
Pharmacogenomics studies suggested that EX-527 treatment in human was associated with a specific transcriptional signature in blood cells, with genes involved in mechanisms of signal transduction and transmembrane transport, as well as metabolic and redox processesCitation37.
The conclusion of the safety study in healthy volunteers indicated that EX-527 was safe and well tolerated by female and male subjects after single doses up to 600 mg and multiple doses up to 300 00/d for 7 d. Moreover, no meaningful cardiovascular effects were observed in beagle dogs up to 100 mg/kgCitation37.
In vivo, numerous studies have been carried out to explore the effect of EX-527 under physiological or pathological conditions (see for representative examples). Although most cell-based assays used cancer cells, in vivo, EX-527 was assayed in a more diverse set of pathologies, and only in a small number of cancer models on mice xenograft. Overall, it appeared very well tolerated when administered alone, in agreement with the phase I clinical trial described aboveCitation37.
Apparent detrimental effects of EX-527 often consisted in inhibition of beneficial effects induced by additional compounds. For example, mice and rats suffering from ischaemia, sepsis, or chronic obstructive pulmonary disease were treated with several natural products including melatoninCitation76,Citation87–89, diallyl trisulphideCitation90, and punicalaginCitation86. Other examples include the effects of ghrelinCitation82,Citation84, hydrogen-rich salineCitation83, carbon monoxideCitation62, the SIRT1 activators resveratrolCitation67,Citation79,Citation91 and scopolinCitation92, and the PARP inhibitor 3-aminobenzamideCitation36. In all these cases, EX-527 was used as a pharmacological tool to demonstrate that SIRT1 activation was involved in the beneficial effects of the compounds under study. When used alone, a detrimental effect of EX-527 on pancreatic tumour xenograft was observed in one study, which gave surprising resultsCitation77. Indeed, EX-527 increased the cytotoxic effect of gemcitabine in vitro in PANC-1 cells, in agreement with another studyCitation50, but it activated the tumour xenograft of the same cells in vivoCitation77. The activity of EX-527 on other cell types in the tumour microenvironment is a possible explanation for this discrepancy. We note that in this xenograft study, the addition of EX-527 at 10 mg/kg with gemcitabine apparently did not have any effect, but the tumour growth in the control experiments with gemcitabine alone was already very limited.
Beneficial effects were observed in several pathologies. In cancer, EX-527 decreased the tumour growth of xenografted mice with endometrial and lung cancer cellsCitation60,Citation55. In immunity-related diseases, a first report in 2011 indicated that, when used in combination with rapamycin, it prolonged heart allograft survival in miceCitation74. The involvement of Tregs through increased expression of Foxp3 was proposed. Other studies confirmed these beneficial effects of EX-527 on Tregs through increased Foxp3 expression and acetylation, and the possible involvement of another SIRT1 substrate, NF-κBCitation69,Citation75,Citation93. In a mouse model of multiple sclerosis, an immune disorder, it strongly suppressed the number of paralysed mice, through an effect of Th17 effector cellsCitation57.
In a phase II clinical trial involving HD patients, EX-527 was found to be safe and well-toleratedCitation73. However, no clinical benefit was observed after the two weeks treatment. For this slowly progressive neurodegenerative disease, longer treatment durations of 2 years may be required to observe clinical benefits. In addition, and maybe for the same reason, no effects on the levels of soluble mutated huntingtin (mHtt) in healthy peripheral blood mononuclear cells (PBMCs) were observed.
5. Conclusion
EX-527 has been tested on many cell lines, alone or in combination with other molecules, resulting in a variety of cellular effects. Moreover, it displayed several biological effects in vivo in various pathological conditions. These results are in agreement with the fact that its specific target SIRT1 is a key regulator of cell fate, through its deacetylation action on a large number of protein substrates. The expression and the activity of SIRT1 can be either up- or down-regulated, depending on the cellular state in the physiological or pathological conditions under study. The administration of EX-527 appears to be beneficial in cases where the activity of SIRT1 is upregulated. Perhaps the most promising in vivo results have been obtained on mice and rats in autoimmune diseases and allograft tolerance, with a significant increase in survival.
Although the results of a phase II clinical trial in HD did not provide the expected beneficial effects, the safety of EX-527 was demonstrated with patients in phase I clinical trials. Therefore, further preclinical and clinical studies in other pathologies appear attractive. In this way, the SIRT1 Antagonism For Endometrial Receptivity (SAFER) clinical trial with EX-527 (Selisistat) will enrol around 30 women with unexplained failure after embryo transfer with euploid embryos. This phase II trial will start on 1 January 2021, and finish on 31 December 2022. The drug will be administered daily for 5 d, beginning with the start of progesterone therapy, and ending 24 h before embryo transfer. Pregnancy rates and pregnancy outcome will be monitored (trial number NCT04184323).
New derivatives of EX-527 with greater activity and selectivity for SIRT1, as well as improved pharmacokinetic and pharmacodynamic properties, may lead to results that are even more promising, and reach further advanced clinical trials.
Disclosure statement
The authors report no conflict of interest.
Additional information
Funding
References
- Jing H, Lin H. Sirtuins in epigenetic regulation. Chem Rev 2015;115:2350–75.
- Milne JC, Lambert PD, Schenk S, et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 2007;450:712–6.
- Han J, Hubbard BP, Lee J, et al. Analysis of 41 cancer cell lines reveals excessive allelic loss and novel mutations in the SIRT1 gene. Cell Cycle 2013;12:263–70.
- Kim D, Nguyen DM, Dobbin MM, et al. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J 2007;26:3169–79.
- Westphal CH, Dipp MA, Guarente L. A therapeutic role for sirtuins in diseases of aging? Trends Biochem Sci 2007;32:555–60.
- Hubbard BP, Gomes AP, Dai H, et al. Evidence for a common mechanism of SIRT1 regulation by allosteric activators. Science 2013;339:1216–9.
- Lin Z, Fang D. The roles of SIRT1 in cancer. Genes Cancer 2013;4:97–104.
- Rifaï K, Idrissou M, Penault-Llorca F, et al. Breaking down the contradictory roles of histone deacetylase SIRT1 in human breast cancer. Cancers 2018;10:409.
- Herranz D, Serrano M. Sirt1: recent lessons from mouse models. Nat Rev Cancer 2010;10:819–23.
- Fujita Y, Yamashita T. Sirtuins in neuroendocrine regulation and neurological diseases. Front Neurosci 2018;12:778.
- Blum CA, Ellis JL, Loh C, et al. SIRT1 modulation as a novel approach to the treatment of diseases of aging. J Med Chem 2011;54:417–32.
- Hu J, Jing H, Lin H. Sirtuin inhibitors as anticancer agents. Future Med Chem 2014;6:945–66.
- Kozako T, Suzuki T, Yoshimitsu M, et al. Anticancer agents targeted to sirtuins. Molecules 2014;19:20295–313.
- Kumar A, Chauhan S. How much successful are the medicinal chemists in modulation of SIRT1: a critical review. Eur J Med Chem 2016;119:45–69.
- Napper AD, Hixon J, McDonagh T, et al. Discovery of indoles as potent and selective inhibitors of the deacetylase SIRT1. J Med Chem 2005;48:8045–54.
- Solomon JM, Pasupuleti R, Xu L, et al. Inhibition of SIRT1 catalytic activity increases p53 acetylation but does not alter cell survival following DNA damage. Mol Cell Biol 2006;26:28–38.
- Huhtiniemi T, Suuronen T, Rinne VM, et al. Oxadiazole-carbonylaminothioureas as SIRT1 and SIRT2 Inhibitors. J Med Chem 2008;51:4377–80.
- Liu Y, Gerber R, Wu J, et al. High-throughput assays for sirtuin enzymes: a microfluidic mobility shift assay and a bioluminescence assay. Anal Biochem 2008;378:53–9.
- Smith BC, Hallows WC, Denu JM. A continuous microplate assay for sirtuins and nicotinamide-producing enzymes. Anal Biochem 2009;394:101–9.
- Peck B, Chen CY, Ho KK, et al. SIRT inhibitors induce cell death and p53 acetylation through targeting both SIRT1 and SIRT2. Mol Cancer Ther 2010;9:844–55.
- Pasco MY, Rotili D, Altucci L, et al. Characterization of sirtuin inhibitors in nematodes expressing a muscular dystrophy protein reveals muscle cell and behavioral protection by specific sirtinol analogues. J Med Chem 2010;53:1407–11.
- Rotili D, Tarantino D, Carafa V, et al. Identification of tri- and tetracyclic pyrimidinediones as sirtuin inhibitors. ChemMedChem 2010;5:674–7.
- Mellini P, Carafa V, Di Rienzo B, et al. Carprofen analogues as sirtuin inhibitors: enzyme and cellular studies. ChemMedChem 2012;7:1905–8.
- Disch JS, Evindar G, Chiu CH, et al. Discovery of thieno[3,2-d]pyrimidine-6-carboxamides as potent inhibitors of SIRT1, SIRT2, and SIRT3. J Med Chem 2013;56:3666–79.
- Gertz M, Fischer F, Nguyen GTT, et al. Ex-527 inhibits Sirtuins by exploiting their unique NAD+-dependent deacetylation mechanism. Proc Natl Acad Sci USA 2013;110:E2772–81.
- Kokkonen P, Rahnasto-Rilla M, Mellini P, et al. Studying SIRT6 regulation using H3K56 based substrate and small molecules. Eur J Pharm Sci 2014;63:71–6.
- Yang T, Chen X, Jin H-X, et al. Functionalized tetrahydro-1H-pyrido[4,3-b]indoles: a novel chemotype with Sirtuin 2 inhibitory activity. Eur J Med Chem 2015;92:145–55.
- Therrien E, Larouche G, Nguyen N, et al. Discovery of bicyclic pyrazoles as class III histone deacetylase SIRT1 and SIRT2 inhibitors. Bioorg Med Chem Lett 2015;25:2514–8.
- Ekblad T, Schüler H. Sirtuins are unaffected by PARP inhibitors containing planar nicotinamide bioisosteres. Chem Biol Drug Des 2016;87:478–82.
- Schnekenburger M, Goffin E, Lee J-Y, et al. Discovery and characterization of R/S-N-3-cyanophenyl-N′-(6-tert-butoxycarbonylamino-3,4-dihydro-2,2-dimethyl-2H-1-benzopyran-4-yl)urea, a new histone deacetylase class III inhibitor exerting antiproliferative activity against cancer cell lines. J Med Chem 2017;60:4714–33.
- Zhao X, Allison D, Condon B, et al. The 2.5 Å crystal structure of the SIRT1 catalytic domain bound to nicotinamide adenine dinucleotide (NAD+) and an indole (EX527 analogue) reveals a novel mechanism of histone deacetylase inhibition. J Med Chem 2013;56:963–9.
- Nayagam VM, Wang X, Cheng Tan Y, et al. SIRT1 modulating compounds from high-throughput screening as anti-inflammatory and insulin-sensitizing agents. J Biomol Screen 2006;11:959–67.
- Hubbard BP, Loh C, Gomes AP, et al. Carboxamide SIRT1 inhibitors block DBC1 binding via an acetylation-independent mechanism. Cell Cycle 2013;12:2233–40.
- Kim JE, Chen J, Lou Z. DBC1 is a negative regulator of SIRT1. Nature 2008;451:583–6.
- Zhao W, Kruse J-P, Tang Y, et al. Negative regulation of the deacetylase SIRT1 by DBC1. Nature 2008;451:587–90.
- Gueguen C, Palmier B, Plotkine M, et al. Neurological and histological consequences induced by in vivo cerebral oxidative stress: evidence for beneficial effects of SRT1720, a sirtuin 1 activator, and sirtuin 1-mediated neuroprotective effects of poly(ADP-ribose) polymerase inhibition. PLoS One 2014;9:e87367.
- Westerberg G, Chiesa JA, Andersen CA, et al. Safety, pharmacokinetics, pharmacogenomics and QT concentration − effect modelling of the SirT1 inhibitor selisistat in healthy volunteers. Br J Clin Pharmacol 2015;79:477–91.
- Sauve AA, Wolberger C, Schramm VL, Boeke JB. The biochemistry of sirtuins. Annu Rev Biochem 2006;75:435–65.
- Stein RL, Kinetics of enzyme action. New York, NY: Wiley; 2011.
- Kabra N, Li Z, Chen L, et al. SirT1 is an inhibitor of proliferation and tumor formation in colon cancer. J Biol Chem 2009;284:18210–7.
- Cea M, Soncini D, Fruscione F, et al. Synergistic interactions between HDAC and sirtuin inhibitors in human leukemia cells. PLoS One 2011;6:e22739.
- Zhu H, Xia L, Zhang Y, et al. Activating transcription factor 4 confers a multidrug resistance phenotype to gastric cancer cells through transactivation of SIRT1 expression. PLoS One 2012;7:e31431.
- Rotili D, Tarantino D, Nebbioso A, et al. Discovery of salermide-related sirtuin inhibitors: binding mode studies and antiproliferative effects in cancer cells including cancer stem cells. J Med Chem 2012;55:10937–47.
- Suzuki T, Khan MNA, Sawada H, et al. Design, synthesis, and biological activity of a novel series of human sirtuin-2-selective inhibitors. J Med Chem 2012;55:5760–73.
- Lugrin J, Ciarlo E, Santos A, et al. The sirtuin inhibitor cambinol impairs MAPK signaling, inhibits inflammatory and innate immune responses and protects from septic shock. Biochim Biophys Acta 2013;1833:1498–510.
- Schuster S, Penke M, Gorski T, et al. Resveratrol differentially regulates NAMPT and SIRT1 in hepatocarcinoma cells and primary human hepatocytes. PLoS One 2014;9:e91045.
- Smith MR, Syed A, Lukacsovich T, et al. A potent and selective Sirtuin 1 inhibitor alleviates pathology in multiple animal and cell models of Huntington’s disease. Hum Mol Genet 2014;23:2995–3007.
- Valle C, Salvatori I, Gerbino V, et al. Tissue-specific deregulation of selected HDACs characterizes ALS progression in mouse models: pharmacological characterization of SIRT1 and SIRT2 pathways. Cell Death Dis 2014;5:e1296–e1296.
- Li Y, Wang K, Feng Y, et al. Novel role of silent information regulator 1 in acute endothelial cell oxidative stress injury. Biochim Biophys Acta 2014;1842:2246–56.
- Zhang JG, Hong DF, Zhang CW, et al. Sirtuin 1 facilitates chemoresistance of pancreatic cancer cells by regulating adaptive response to chemotherapy-induced stress. Cancer Sci 2014;105:445–54.
- Gollavilli PN, Kanugula AK, Koyyada R, et al. AMPK inhibits MTDH expression via GSK3b and SIRT1 activation: potential role in triple negative breast cancer cell proliferation. FEBS Journal 2015; 282:3971–85.
- Kim HB, Lee SH, Um JH, et al. Sensitization of chemo-resistant human chronic myeloid leukemia stem-like cells to Hsp90 inhibitor by SIRT1 inhibition. Int J Biol Sci 2015;11:923–34.
- Kim HB, Lee SH, Um JH, et al. Sensitization of multidrug-resistant human cancer cells to Hsp90 inhibitors by down-regulation of SIRT1. Oncotarget 2015;6:36202–18.
- Hu Y, Zhang N, Fan Q, et al. Protective efficacy of carnosic acid against hydrogen peroxide induced oxidative injury in HepG2 cells through the SIRT1 pathway. Can J Physiol Pharmacol 2015;93:1–7.
- Asaka R, Miyamoto T, Yamada Y, et al. Sirtuin 1 promotes the growth and cisplatin resistance of endometrial carcinoma cells: a novel therapeutic target. Lab Investig 2015;95:1363–73.
- Kumari S, Chaurasia SN, Nayak MK, et al. Sirtuin inhibition induces apoptosis-like changes in platelets and thrombocytopenia. J Biol Chem 2015;290:12290–9.
- Lim HW, Kang SG, Ryu JK, et al. SIRT1 deacetylates RORgt and enhances Th17 cell generation. J Exp Med 2015;212:607–17.
- Kim HW, Kim SA, Ahn SG. Sirtuin inhibitors, EX527 and AGK2, suppress cell migration by inhibiting HSF1 protein stability. Oncol Rep 2016;35:235–42.
- Kim BS, Lee CH, Chang GE, et al. A potent and selective small molecule inhibitor of sirtuin 1 promotes differentiation of pluripotent P19 cells into functional neurons. Sci Rep 2016;6:34324.
- Chen G, Zhang B, Xu H, et al. Suppression of Sirt1 sensitizes lung cancer cells to WEE1 inhibitor MK-1775-induced DNA damage and apoptosis. Oncogene 2017;36:6863–10.
- Yang X, Wei J, He Y, et al. SIRT1 inhibition promotes atherosclerosis through impaired autophagy. Oncotarget 2017;8:51447–61.
- Sun J, Guo E, Yang J, et al. Carbon monoxide ameliorates hepatic ischemia/reperfusion injury via sirtuin 1-mediated deacetylation of high-mobility group box 1 in rats. Liver Transplant 2017;23:510–26.
- Ceballos MP, Decándido G, Quiroga AD, et al. Inhibition of sirtuins 1 and 2 impairs cell survival and migration and modulates the expression of P-glycoprotein and MRP3 in hepatocellular carcinoma cell lines. Toxicol Lett 2018;289:63–74.
- Jiang H, Xin S, Yan Y, et al. Abnormal acetylation of FOXP3 regulated by SIRT-1 induces Treg functional deficiency in patients with abdominal aortic aneurysms. Atherosclerosis 2018;271:182–92.
- Shokri Afra H, Zangooei M, Meshkani R, et al. Is a potent bioactivator that activates SIRT1-AMPK signaling pathway in HepG2 cells. J Physiol Biochem 2019;75:125–33.
- Yan S, Miao L, Lu Y, Wang L. Sirtuin 1 inhibits TNF-α-mediated osteoclastogenesis of bone marrow-derived macrophages through both ROS generation and TRPV1 activation. Mol Cell Biochem 2019;455:135–45.
- Huang X, Sun J, Chen G, et al. Z. Resveratrol promotes diabetic wound healing via SIRT1-FOXO1-c-Myc signaling pathway-mediated angiogenesis. Front Pharmacol 2019;10:1–10.
- Zhang Y, Li Y, Li J, et al. SIRT1 alleviates isoniazid-induced hepatocyte injury by reducing histone acetylation in the IL-6 promoter region. Int Immunopharmacol 2019;67:348–55.
- Daenthanasanmak A, Iamsawat S, Chakraborty P, et al. Targeting Sirt-1 controls GVHD by inhibiting T-cell allo-response and promoting Treg stability in mice. Blood 2019;133:266–79.
- Wang Y, Zeng J, Wu W, et al. Nicotinamide N-methyltransferase enhances chemoresistance in breast cancer through SIRT1 protein stabilization. Breast Cancer Res 2019;21:17.
- Qin T, Liu W, Huo J, et al. SIRT1 expression regulates the transformation of resistant esophageal cancer cells via the epithelial-mesenchymal transition. Biomed Pharmacother 2018;103:308–16.
- Mvunta DH, Miyamoto T, Asaka R, et al. SIRT1 regulates the chemoresistance and invasiveness of ovarian carcinoma cells. Transl Oncol 2017;10:621–31.
- Süssmuth SD, Darpo B, Westerberg G. et al. An exploratory double-blind, randomized clinical trial with selisistat, a SirT1 inhibitor, in patients with Huntington’s disease. Br J Clin Pharmacol 2015;79:465–76.
- Beier UH, Wang L, Bhatti TR, et al. Sirtuin-1 targeting promotes Foxp3+ T-regulatory cell function and prolongs allograft survival. Mol Cell Biol 2011;31:1022–9.
- Kwon HS, Lim HW, Wu J, et al. Three novel acetylation sites in the Foxp3 transcription factor regulate the suppressive activity of regulatory T cells. J Immunol 2012;188:2712–21.
- Zhao L, An R, Yang Y, et al. Melatonin alleviates brain injury in mice subjected to cecal ligation and puncture via attenuating inflammation, apoptosis, and oxidative stress: the role of SIRT1 signaling. J Pineal Res 2015;59:230–9.
- Oon CE, Strell C, Yeong KY, et al. SIRT1 inhibition in pancreatic cancer models: contrasting effects in vitro and in vivo. Eur J Pharmacol 2015;757:59–67.
- Kim HD, Hesterman J, Call T, et al. SIRT1 mediates depression-like behaviors in the nucleus accumbens. J Neurosci 2016;36:8441–52.
- Guo YJ, Dong SY, Cui XX, et al. Resveratrol alleviates MPTP-induced motor impairments and pathological changes by autophagic degradation of a-synuclein via SIRT1-deacetylated LC3. Mol Nutr Food Res 2016;60:2161–75.
- Huang J, Tian R, Yang Y, et al. The SIRT1 inhibitor EX-527 suppresses mTOR activation and alleviates acute lung injury in mice with endotoxiemia. Innate Immun 2017;23:678–86.
- Çakir I, Perello M, Lansari O, et al. Hypothalamic Sirt1 regulates food intake in a rodent model system. PLoS One 2009;4:e8322.
- Velásquez DA, Martínez G, Romero A, et al. The central sirtuin 1/p53 pathway is essential for the orexigenic action of ghrelin. Diabetes 2011;60:1177–85.
- Qi LS, Yao L, Liu W, et al. Sirtuin type 1 mediates the retinal protective effect of hydrogen-rich saline against light-induced damage in rats. Invest Ophthalmol Vis Sci 2015;56:8268–79.
- Ugwu FN, Yu AP, Sin TK, et al. Protective effect of unacylated ghrelin on compression-induced skeletal muscle injury mediated by SIRT1-signaling. Front Physiol 2017;8:962.
- Nikseresht S, Khodagholi F, Ahmadiani A. Protective effects of ex‐527 on cerebral ischemia–reperfusion injury through necroptosis signaling pathway attenuation. J Cell Physiol 2019;234:1816–26.
- Yu LM, Dong X, Xue XD, et al. Protection of the myocardium against ischemia/reperfusion injury by punicalagin through an SIRT1-NRF-2-HO-1-dependent mechanism. Chem Biol Interact 2019;306:152–62.
- Yu L, Sun Y, Cheng L, et al. Melatonin receptor-mediated protection against myocardial ischemia/reperfusion injury: role of SIRT1. J Pineal Res 2014;57:228–38.
- Yang Y, Jiang S, Dong Y, et al. Melatonin prevents cell death and mitochondrial dysfunction via a SIRT1-dependent mechanism during ischemic-stroke in mice. J Pineal Res 2015;58:61–70.
- Peng Z, Zhang W, Qiao J, He B. Melatonin attenuates airway inflammation via SIRT1 dependent inhibition of NLRP3 inflammasome and IL-1β in rats with COPD. Int Immunopharmacol 2018;62:23–8.
- Yu L, Li S, Tang X, et al. Diallyl trisulfide ameliorates myocardial ischemia-reperfusion injury by reducing oxidative stress and endoplasmic reticulum stress-mediated apoptosis in type 1 diabetic rats: role of SIRT1 activation. Apoptosis 2017;22:942–54.
- Yao X, Chen W, Liu J, et al. Deep vein thrombosis is modulated by inflammation regulated via sirtuin 1/NF-κB signalling pathway in a rat model. Thromb Haemost 2019;119:421–30.
- Yoo A, Narayan VP, Hong EY, et al. Scopolin ameliorates high-fat diet induced hepatic steatosis in mice: potential involvement of SIRT1-mediated signaling cascades in the liver. Sci Rep 2017;7:2251.
- Zhang D, Qiu X, Li J, et al. MiR-23a-3p-regulated abnormal acetylation of FOXP3 induces regulatory T cell function defect in Graves disease. Biol Chem 2019;400:639–50.