
Abstract
The purpose of this review article is to provide an overview of recent achievements in the synthesis of novel steroid sulphatase (STS) inhibitors. STS is a crucial enzyme in the biosynthesis of active hormones (including oestrogens and androgens) and, therefore, represents an extremely attractive molecular target for the development of hormone-dependent cancer therapies. The inhibition of STS may effectively reduce the availability of active hormones for cancer cells, causing a positive therapeutic effect. Herein, we report examples of novel STS inhibitors based on steroidal and nonsteroidal cores that contain various functional groups (e.g. sulphamate and phosphorus moieties) and halogen atoms, which may potentially be used in therapies for hormone-dependent cancers. The presented work also includes examples of multitargeting agents with STS inhibitory activities. Furthermore, the fundamental discoveries in the development of the most promising drug candidates exhibiting STS inhibitory activities are highlighted.
1. Introduction
Cancer is among the leading causes of death. According to the International Agency for Research on Cancer estimates in 2018, there were more than 18 million new cases and 9.5 million tumour-related deaths worldwideCitation1. Additionally, the National Cancer Institute (NCI) expects that the number of new cancer cases will have risen to approximately 23.6 million per year by 2030. The NCI warns that this disease will be diagnosed in approximately 38.4% of men and women during their lifetimes. The most common types are breast, lung, and bronchus, prostate and colorectal tumours, and they account for almost 50% of all new cancer cases. Moreover, lung and bronchus, colorectal, pancreatic, and breast cancers are responsible for nearly 50% of all deaths. The estimates for 2019 indicate that almost 270,000 and 175,000 patients will be diagnosed with breast and prostate tumours, respectively, and more than 41,000 (breast) and 31,000 (prostate) deaths will occur from these diseases in the United StatesCitation2. It is known that most cancers show a hormone-dependent nature in their early stages (e.g. more than 90% of breast cancer cases are initially hormone-dependent)Citation3. Therefore, the World Health Organisation (WHO) describes biologically active hormones (androgens and oestrogens) as the main cancer growth stimulants. Considering the aforementioned facts, the application of drugs that can effectively reduce concentrations of active hormones should be the basis of modern therapiesCitation4.
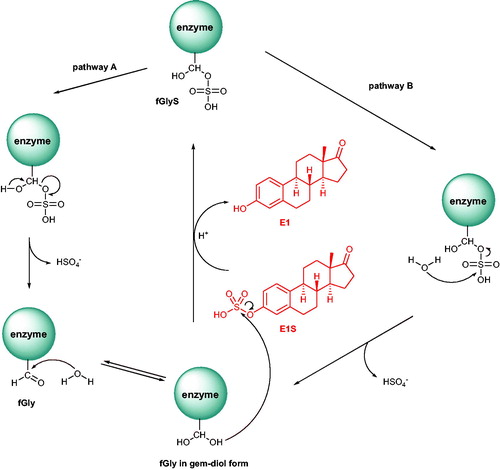
The hormone signalling pathway is a well-established target for the development of hormone-dependent cancer drugs (e.g. breast cancer)Citation5. For example, the clinically used drug Tamoxifen 1 () acts as a selective oestrogen receptor modulator (SERM). In contrast, chemotherapeutics, which may influence the hormone formation process, are also of high therapeutic importance. The biosynthesis of active steroids (e.g. oestradiol [E2] and androstenediol [Adiol]) in cancer tissues mainly depends on the following three enzymatic pathways: aromatase (AROM), 17β-hydroxysteroid dehydrogenase (17β-HSD), and steroid sulphatase (STS) (Scheme 1)Citation6. For example, currently used Letrozole 2 and Anastrozole 3 () block the conversion of androgens to oestrogens via inhibition of the AROM complex. However, therapies using the described above drugs often turn out to be unsatisfactory and result in the development of resistance, leading to relapses in tumour progressionCitation7–10. In light of recent research indicating that sulphation/desuphfation process disorders may be responsible for numerous pathologiesCitation11, another enzyme implicated in the steroidogenesis process, STS, is becoming a new interesting molecular target in the development of novel and effective hormone-dependent cancer treatment methods. In contrast to aromatase, STS activity is present in most cancer cases (e.g. STS expression is detected in 90% of breast tumours)Citation12. Furthermore, it has been noticed that STS mRNA levels in malignant tissues have been higher than in normal breast tissues in 87% of tested patientsCitation13.
Scheme 1. The biosynthesis pathway for steroids with oestrogenic properties.
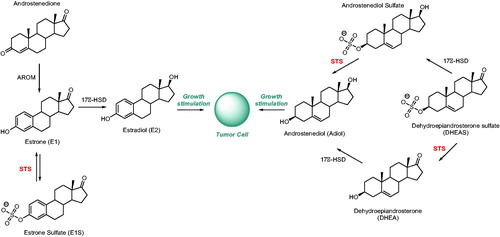
Figure 1. Chemical structures of Tamoxifen 1, Letrozole 2, and Anastrozole 3.
STS belongs to a group of 15 human sulphatasesCitation6. This protein consists of 587 amino acid residues and is encoded by the STS gene. STS is found ubiquitously throughout the body, what is strictly related to its involvement in numerous physiological and pathological processesCitation14. This enzyme is mainly localised in skin, fallopian tubes, testis, ovary, adrenal glands, brain, foetal lung, endometrium, aorta, kidneys, bones, placenta, and breastsCitation15. STS catalyses the hydrolysis of steroid sulphates (including oestrone sulphate [E1S] and dehydroepiandrosterone sulphate [DHEAS]) to their unsulphated derivatives (oestrone [E1] and dehydroepiandrosterone [DHEA], respectively) (Scheme 1)Citation16,Citation17. E1 and DHEA may be subsequently transformed into bioactive oestrogens and androgens (e.g. E2 and Adiol, respectively), which are responsible for the stimulation of hormone-dependent cancer cell proliferationCitation18. Considering the aforementioned facts, STS plays a pivotal role in breast cancer tumourigenesis and is, therefore, an extremely attractive molecular target for the development of hormone-dependent cancer therapies.
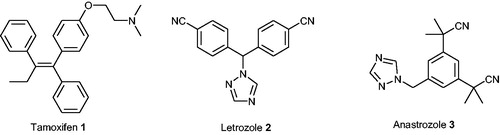
The crystallographic structure of STS is knownCitation19. It is composed of a globular domain with polar characteristics and a stem domain consisting of two antiparallel hydrophobic helices that resemble a mushroom structure. The active site is located in a cavity on the border of polar and hydrophobic domains of the enzymeCitation20. STS demonstrates a high similarity to arylsulphatase A (ARSA) and B (ARSB). The topology of active sites of all three enzymes is very similar. One of the characteristic features of all sulphatases is a posttranslational modification within the active site involving the conversion of cysteine to a formylglycine residue (fGly)Citation21. In the absence of substrate, the catalytic region of human STS consists of a sulphated fGly residue in its gem-diol form, which is coordinated to a calcium ion. Moreover, the enzyme’s active site is composed of nine other catalytically important amino acid residues, including Asp35, Asp36, Arg79, Lys134, His136, His290, Asp342, Gln343, and Lys368. However, when a natural substrate (e.g. E1S) associates with the STS active site (), its steroidal core becomes surrounded by other amino acid residues located in the binding region (Leu74, Arg98, Thr99, Val101, Leu103, Leu167, Val177, Phe178, Thr180, Gly181, Thr484, and Phe488) and interacts with them via hydrophobic interactionsCitation14.
Figure 2. The structure of STS with its natural substrate (E1S) bound to the active site.
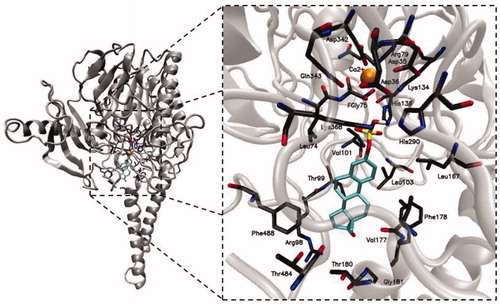
Because the fGly residue, in its gem-diol form, is involved in the hydrolysis of sulphate substrates in ARSA and ARSB, a mechanism of enzymatic reaction catalysed by STS may proceed according to two putative pathways (Scheme 2). Pathway A starts with the decomposition of formylglycine sulphate (fGlyS) into a sulphate anion and fGly, which then reacts with a water molecule furnishing the hydrated formylglycine form. In contrast, pathway B assumes the straightforward nucleophilic attack of a water molecule on the fGlyS sulphur atom, giving rise to the fGly gem-diol form, which subsequently reacts with E1S to release E1 via SN2 attack of one of the hydroxyl groupsCitation14.
Scheme 2. Two putative mechanisms of action for STS (pathways A and B).
Studies focussed on the development of effective STS inhibitors lacking adverse side effects as drug candidates have been carried out for over 30 years. The first fruitful 1990–1999 decade was successfully continued during the years of 2000–2010. In that time, many scientific papers devoted to the design, synthesis, and biological evaluation of compounds based on steroidal or nonsteroidal cores were published. Those years’ achievements have been well summarised in a few scientific reviewsCitation6,Citation12,Citation14,Citation18,Citation22–25. The unique significance of these two decades highlights the fact that almost thorough clinical progress is strictly associated with a few STS inhibitors discovered at that time (e.g. E2MATE, Irosustat [also known as 667-COUMATE, STX64 and BN-83495])Citation26–29.
The results of some clinical trials dedicated to known STS inhibitors seem to be promising and such compounds have currently a great chance to be used in the treatment of several hormone-dependent types of cancer (especially, hormone-dependent breast cancer [HDBC]). However, more clinical trials involving significantly greater number of patients are definitely required. Furthermore, it is worth noting that to date, none of the developed STS inhibitors has reached the pharmaceutical market and therefore, there is still place for the development of novel drug candidates (e.g. for compounds, which mechanism of action is based on some innovative multi-targeting approaches). The need for these studies results from the unique and extremely important role of STS in biosynthesis of active steroids mentioned above. Furthermore, according to the opinions presented in many scientific papers and evidences confirming the key role of STS enzyme in the process of growth and development of hormone-dependent cancers, the intensive search for new and effective STS inhibitors seems to be fully justified. Therefore, a summary of the last years’ achievements in the identification and synthesis of novel derivatives demonstrating STS inhibitory properties may be valuable for future researchers.
2. Steroidal STS inhibitors
Initially, the development of STS inhibitors was based on the synthesis of steroid analogues. For example, replacement of the sulphate group in E1S with phosphonate and thiophosphonate moieties led to obtain one of the first potent STS inhibitors, oestrone 3-O-methylthiophosphonate 4 – E1-3-MTP ()Citation30. In MCF-7 breast cancer cells, E1-3-MTP competitively inhibited STS activity by 52 and >98% at 100 and 10 µM concentrations, respectivelyCitation31. In the early 90 s, the research group of Prof. Michael Reed and Dr. Atul Purohit at Imperial College London, and Prof. Barry Potter at Bath University pioneered the STS inhibitors field including a new series of STS inhibitors based on an oestrone core containing a different pharmacophore – a sulphamate moietyCitation32. The discovery of these compounds based on aryl sulphamate esters became a milestone in the development of derivatives exhibiting extremely high inhibitory activities against STS. Some of them have entered clinical trials with very promising results, especially in the treatment of cancer. Despite the fact that the mechanism of inhibition is still not completely confirmed, it is currently accepted that the inactivation of STS by compounds containing the sulphamate-aryl system occurs via a sulphamoyl moiety transfer to the catalytic fGly residue. The current knowledge about a mechanism of STS inhibition by sulphamate-containing compounds has been recently well summarised in the review of Prof. Barry PotterCitation26.
Figure 3. Chemical structures of E1-3-MTP 4, EMATE 5, and E2MATE 6.
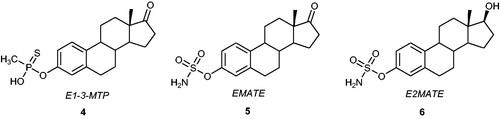
One of the most active sulphamoylated aryl derivatives, oestrone-3-O-sulphamate 5 (EMATE) (), inhibits the STS enzyme in MCF-7 cells with an IC50 value of 65 pM. EMATE is a time-, concentration-, and pH-dependent STS inhibitor that acts in an irreversible and active-site-directed mannerCitation33. Despite the promising STS inhibitory activities of steroid sulphamates (e.g. EMATE)Citation34,Citation35, their applications as therapeutic agents in the treatment of hormone-dependent cancers were unfortunately limited because of their very potent oestrogenic properties in ratsCitation36. Nevertheless, oestradiol 3-O-sulphamate 6 () (E2MATE), was clinically evaluated (phase II clinical trials) in hormone replacement therapy as an oestradiol prodrugCitation26,Citation27. Furthermore, E2MATE is still being considered as a drug candidate for hormone-dependent endometriosis therapyCitation37. It was noticed that the treatment of mice with E2MATE resulted in a reduction of the development of endometriosis. E2MATE inactivated STS in the murine uterus in dose-dependent manner with the lack of an effect on plasma oestradiol levelsCitation38 and demonstrated clinical advantages in randomised and proof-of-principle phase I clinical trialsCitation37. Administration of E2MATE in monotherapy and in combination with norethindrone acetate (NETA) to healthy women led to a reduction in STS activity in endometrium tissues. Further investigation on its efficacy, safety, pharmacokinetics, and pharmacodynamics are ongoing in phase II clinical trialsCitation26.
Considering its high STS inhibitory potency, EMATE became a lead compound for the development of numerous derivatives with improved biological properties. Woo et al.Citation39 synthesised a large library of EMATE analogues substituted at the 2- and/or 4-positions (e.g. with halogen atoms, nitro, propenyl, n-propyl, and cyano groups) and its derivative with the removed 17-carbonyl motif. An extensive structure–activity relationship (SAR) analysis has been performed – including the influence of electronic effects of substituents on the A-ring and modification of the sulphamate pharmacophore on STS inhibitory activity. It has been noticed, that the presence of electron-withdrawing groups (EWG) in the A-ring affects the potency in a positive way. However, the STS inhibitory effect depended on the type and location of the substituents. For example, higher inhibition was observed for derivatives containing halogen atom at the 2-position. On the other hand, the location of nitro groups at the 4-position was much more favourable. 4-Nitro-EMATE 7 () was found to be the most potent derivative with IC50 values of 0.8 and 0.01 nM in assays with placental microsomes and MCF-7 cells, respectively. Due to a very high STS inhibitory activity, this compound seems to be very promising in the context of further research. Subsequent appropriate in vivo investigations should verify the existing potential of 4-nitro-EMATE 7 and its usefulness in further clinical trials.
Table 1. Examples of steroidal STS inhibitors 7–12.
Other STS inhibitors based on a steroidal core have been developed recently. A modification of oestrone’s structure by the replacement of a 17-carbonyl group with N-17β-arylsulphonamide and N-17β-alkylbenzenesulphonamide moieties led to the series of 17β-arylsulphonamides and 17β-alkylbenzenesulphonamides of 17β-aminoestra-1,3,5(10)-trien-3-ol as reversible STS inhibitorsCitation40. Interestingly, despite the absence of the sulphamate functional group connected to the A-ring in a steroidal core, the obtained compounds exhibited extremely high STS inhibitory properties. The most potent analogue 8 () inhibited STS with an IC50 value of 9 nM (when evaluated in an assay with purified enzyme). According to the presented SAR analysis, the introduction of alkyl substituents into the arylsulphonamide group resulted in the improvement of STS inhibitory potency – the presence of an n-butyl chain was the most favourable (the introduction of an n-pentyl substituent resulted in a decrease in the potency). Furthermore, the alteration of an n-butyl chain with a tert-butyl group occurred to be beneficial for STS inhibitory effect. On the other hand, studies with 3′- and 4′-substituted benzenesulphonamides bearing EWG and electron-donating groups (EDG) showed that 3′-bromo and 3′-trifluoromethyl derivatives were the most potent.
In 201541, the same research group prepared and examined a series of A-ring-substituted 17β-arylsulphonamides of 17β-aminoestra-1,3,5(10)-trien-3-ol as STS inhibitors. The authors noticed that the presence of a nitro group or a fluorine atom at the 4-position of the A-ring resulted in an increase in the potency. In contrast, the derivatives containing these substituents at the 2-position occurred to be less active than A-ring-unsubstituted analogue. The most active derivative 9 () demonstrated STS inhibition with Ki of 1 nM. Furthermore, the antiproliferative activities (examined using the NCI 60 cell-line panel) of obtained derivatives indicated, that these compounds are promising drug candidates in the treatment of breast and ovarian cancers. Further research dedicated to studies of oestrogenic properties may be useful in the evaluation of their clinical potential.
Recently, Grienke et al.Citation42 applied interesting in silico method to identify STS inhibitors from natural sources. According to the pharmacophore-based virtual screening of the Traditional Chinese Medicine (TCM) database, triterpenes based on the lanostane structure were taken into account as potential STS ligands. Evaluated compounds were isolated from traditionally used polypores, which are rich in lanostane derivatives. (i.e. Ganoderma lucidum Karst., Gleophyllum odoratum Imazeki, and Fomitopsis pinicola Karst). After sophisticated purification protocols, 3 out of 18 isolated lanostane analogues exhibited STS inhibitory activities (when evaluated in an assay with JEG-3 cells). Piptolinic acid D 10, Pinicolic acid B 11, and Ganadrol A 12 () inhibited STS with IC50 values of 10.5, 12.4, and 15.7 µM, respectively. Despite the moderate STS inhibitory potency, authors indicated that reported compounds are the first STS inhibitors isolated from natural sources. Further application of in silico methods might be useful in the identification of more potent natural compounds demonstrating STS inhibitory properties.
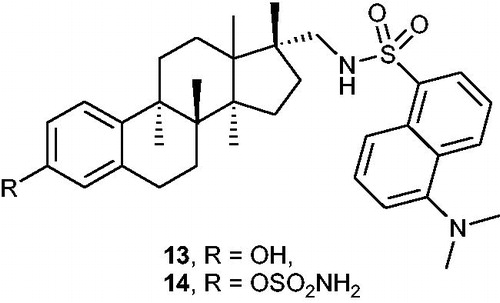
Maltais et al.Citation43 designed fluorescent STS inhibitors based on the steroidal scaffold using quantitative SAR (QSAR) and molecular modelling studies. Based on the previously undertaken researchCitation44,Citation45, two newly compounds: 17α-dansylaminomethyl-oestradiol 13 and its sulphamoylated derivative 14 () were designed and synthesised. Both compounds occurred to be effective STS inhibitors, demonstrating IC50 values of 69 and 2.1 nM (for compounds 13 and 14, respectively) in an assay with HEK-293 cells. Interestingly, it was shown that the sulphamoylated derivative 14 may be applicable as an optical imaging tool to investigate intracellular sub-localisation of STS enzyme and inhibitory mechanisms. According to the confocal microscopy analysis, good penetration of the fluorescent inhibitor 14 in cells and its localisation in the endoplasmic reticulum (where STS is localised) have been confirmed.
Figure 4. Chemical structures of 17α-dansylaminomethyl-oestradiol 13 and its sulphamoylated derivative 14.
3. Nonsteroidal STS inhibitors
Although the application of steroid derivatives in the development of potential STS inhibitors seemed to be reasonable, the clinical application may be limited in some cases due to their other undesired properties (e.g. the estrogenicity of EMATE occurred to be problematicCitation36). However, there is insufficient evidence to disparage the use of steroid derivatives in clinical practice. Fortunately, the potential limitations of STS inhibitors based on the steroidal scaffolds can be overcome by the development of nonsteroidal analoguesCitation14. Theoretically, the change of the steroid core (present in the chemical structure of STS natural substrates) might affect the reduction of the binding abilities of nonsteroidal analogues to the enzyme’s active site. However, a large number of steroid system mimics with promising biological activities has been reported, and until now, the synthesis of nonsteroidal STS inhibitors based on different cores has become a priority in the development of drug candidates with greater clinical applications. To date, derivatives of tyramine, triazole, piperazinyl-ureides, biphenyl, flavones, carboranes, and many others have been reported as potent STS inhibitors (see sections below). Among them, the most promising results have been achieved with the application of coumarin-based analoguesCitation26. The tricyclic coumarin derivatives effectively mimicked the steroid core of natural STS substrates, and one of them – Irosustat – proved to be extremely promising STS inhibitor without having in vitro and in vivo oestrogenic propertiesCitation27. The discovery of Irosustat occurred to be a milestone in the development of potential anticancer therapies based on STS inhibitorsCitation26.
3.1. Nonsteroidal STS inhibitors containing a sulphamate moiety
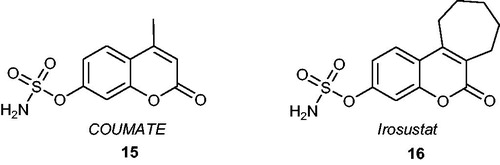
An important class of compounds that proved to be promising mimics of known steroidal STS inhibitors are sulphamoylated coumarin derivatives. It has been noticed that these analogues exhibited high inhibitory potency against STS and fewer adverse effects, such as much weaker oestrogenic properties than EMATECitation46. Similar to EMATE, the sulphamoylated coumarin analogues, e.g. 4-methylcoumarin-7-O-sulphamate (also known as COUMATE) 15 (), are classified as irreversible inhibitors that act in a time- and concentration-dependent manner. COUMATE exhibited good activity with an IC50 value of 380 nM (when evaluated against placental microsomes)Citation47. Its modification through the introduction of an additional aliphatic ring gave rise to a wide range of tricyclic coumarin derivatives that mimicked the ABC rings of the natural substrate and demonstrated significantly higher inhibitory effects than COUMATE. For example, one of the most promising drug candidates based on a sulphamoylated coumarin core, Irosustat 16 (), exhibited very potent activity towards STS (IC50 value of 8 nM) without having in vitro and in vivo oestrogenic propertiesCitation48,Citation49. Woo et al.Citation50 published a key paper dedicated to the SAR studies for this first-in-class clinical STS inhibitor. The compound was characterised by favourable bioavailability (however, the bioavailability decreases with increasing doses)Citation51, and was orally active and well-tolerated in patientsCitation52. It has reached clinical trials, and its high therapeutic potential has been proven in several clinical studiesCitation53,Citation54. A combination therapy of Irosustat with a first-line aromatase inhibitor (phase II clinical studies) in patients with advanced ER-positive breast cancer resulted in clinical benefits with an acceptable safety profileCitation55,Citation56. Furthermore, Irosustat has recently shown clinical advantages in early breast cancerCitation57 and ER-positive advanced endometrial cancer treatmentsCitation58.
Figure 5. Chemical structures of STS inhibitors based on sulphamoylated coumarin derivatives –COUMATE 15 and Irosustat 16.
The synthesis of sulphamoylated coumarin derivatives is still one of the main directions in the development of compounds with STS inhibitory activities. Ganeshapillai et al.Citation59 synthesised a series of novel STS inhibitors based on bicyclic coumarin sulphamates. Based on the chemical structure of COUMATE, a large library of C-3- and C-4-substituted derivatives containing a sulphamate moiety was obtained and a wide SAR analysis was performed. In the case of many synthesised compounds, high STS inhibitory potency was achieved (with the measured IC50 values within the nanomolar range). Some of the reported analogues were found to inhibit the STS activity approximately 100–500-fold more effectively than the parent COUMATE, with the best examples close in the potency to the clinically evaluated Irosustat. Among the synthesised analogues, compounds 17 (a COUMATE derivative with an n-hexyl chain at C-3 position) and 18 (a COUMATE derivative with a benzyl motif at C-3 position) () were particularly effective with IC50 values of 0.68 and 1 nM in intact MCF-7 cells and 8 and 32 nM in an assay with placental microsomal STS, respectively. Presented results showed that these compounds could represent new leads for potential development after further preclinical studies.
Table 2. Examples of nonsteroidal STS inhibitors with sulphamate moiety 17–31.
A wide range of novel sulphamoylated derivatives of coumarin has been also synthesised by Hng et al. recentlyCitation60. In the course of undertaken research, the modifications at the 3-position of a coumarin core have been evaluated. SAR analysis indicated, that the attachment of a benzylamino group at the 3-position of sulphamoylated coumarin improved the STS inhibitory activity. Furthermore, the presence of methoxy substituents in the structure of a terminal phenyl ring was significant for STS inhibition. It has been noticed that methoxy para-substitution is not associated with the improvement in STS inhibitory potency. On the other hand, the presence of the methoxy substituents at the ortho- and meta-positions is much more favourable. Among the obtained set of compounds, analogue 19 () is found to exhibit the highest STS inhibitory activity against enzyme isolated from human placenta (IC50 value of 0.13 µM) and MCF-7 cell lines (IC50 value of 1.35 µM). However, this derivative occurs to be still 13 times less potent than Irosustat (used as a reference). Despite the moderate STS inhibitory potency of reported compounds, authors indicated that the further modifications of the benzylamino group of compound 19 might be worth exploring.
A strategy involving the introduction of fluorine atoms into the structure of biologically active compounds has been utilised to design new STS inhibitors based on the coumarin core. It is generally known that the introduction of a fluorine atom may affect nearly all physical properties of the drug, as well as its absorption, distribution, metabolism, and excretion. Since 1955, more than 150 fluorinated molecules have succeeded in reaching the market and now are approximately 20–30% of all pharmaceuticalsCitation68. In 2016, fluorinated 3-phenylcoumarin sulphamates were obtained as STS inhibitorsCitation61. It was noticed that the presence of a fluorinated phenyl ring at the 3-position of a coumarin core is beneficial for STS inhibitory activity. The most favourable effects were observed for derivatives containing fluorine atoms at the meta position in a phenyl substituent. For example, two compounds, 20 and 21 (), demonstrated the highest activity in an STS enzyme assay with IC50 values of 270 nM in both cases. Further investigation on this type of STS inhibitors, involving the synthesis of analogues with an additional aromatic ring attached via the amide moiety, led to the development of their more potent N-benzoyl and N-phenylacetoyl derivativesCitation62. The most active compounds, 22 and 23 (), inhibited a purified STS enzyme with IC50 values of 180 nM in both cases. Furthermore, compounds 20–23 also demonstrated cytotoxic properties. For example, analogue 23 demonstrated the most potent antiproliferative activity with GI50 values of 15.9 and 8.7 µM for MCF-7 and T47D cell lines, respectively. However, 23 was not selective towards oestrogen-dependent cells and effectively inhibited the growth of ER- and PR-negative cell lines (the GI50 values for the SkBr3 and MDA-MB-231 cell lines were 18.8 and 8.1 µM, respectively). In contrast, despite the slightly lower activities of compounds 20 and 21 compared with 23, analogues 20 and 21 demonstrated selective inhibition of ER- and PR-positive cell line growth. Despite the promising in vitro activities of these compounds, their clinical potential should be confirmed in in vivo models.
Interestingly, the design of detailed structures of STS inhibitors based on fluorinated 3-phenylcoumarin sulphamates was supported by molecular modelling techniques. Currently, computational approaches have become a key step in the rational development of new potential therapeutics. They are effective in the hit identification and lead optimisation processes as well as structure- or ligand-based virtual screening studies. The molecular docking of small ligands to enzyme/receptor binding sites has been intensively developed since the 1980s (and still remains widely utilised by scientists). Docking calculations may be used as a hit-identification tool, when the structure of a molecular target (or its active/binding site alone) is known as well as during the computational optimisation of the chemical structure of lead compounds before the synthesis of their derivativesCitation69. In case of STS inhibitors based on fluorinated 3-phenylcoumarin sulphamates, molecular docking techniques allowed the determination of binding modes of synthesised compounds to the STS structure and the identification of potential interactions between inhibitors and amino acid residues located in the STS active siteCitation61,Citation62. For example, the fluorine atoms from some docked derivatives are within short distances to the Arg98 or Thr484 residues, suggesting the presence of additional stabilising interactions that may influence the binding of a potential drug molecule to the enzyme’s active site. Additionally, computational research on geometric optimisation methods for STS inhibitors based on fluorinated 3-phenylcoumarin sulphamates and their influence on the free binding energy with STS has been performedCitation70. The obtained results indicated that the MM + and PM7 geometry optimisation methods could be successfully employed for the geometric optimisation of STS inhibitors before their docking procedure and for molecular descriptor calculations.
A fluorination strategy has been recently utilised in the development of novel STS inhibitors based on a tyramine coreCitation63. The series of obtained N-acylated tyramine sulphamates inhibited the STS enzyme at micromolar levels. In the course of undertaken research, the introduction of fluorinated N-benzoyl and N-phenylacetoyl groups as well as N-perfluoroalkanoyl chains into a sulphamoylated tyramine core has been evaluated. Generally, it has been noticed that most of the fluorinated derivatives demonstrated higher STS inhibitory activity than their nonfluorinated counterparts. The IC50 value of the most active inhibitor 24 () was determined to be 2.18 µM in an enzymatic assay.
Recently, Daśko et al.Citation64 developed a new series of potent STS inhibitors based on the fluorinated 4-(1-phenyl-1H-1,2,3-triazol-4-yl)phenyl sulphamate core. Almost all of the synthesised compounds inhibited STS at the nanomolar level. During the course of the investigation, the highest inhibitory activities were exhibited by derivatives containing a fluorine atom at the meta position of a terminal aromatic ring. The most active analogues, 4-[1-(3,5-difluorophenyl)-1H-1,2,3-triazol-4-yl]phenyl sulphamate 25 and 4-[1-(2,3,4-trifluorophenyl)-1H-1,2,3-triazol-4-yl]phenyl sulphamate 26 (), inhibited STS enzyme with IC50 values of 36 and 58 nM, respectively, when evaluated in an enzymatic assay (in comparison, the IC50 value measured for Irosustat in the same assay was 25 nM). According to the molecular docking calculations, the fluorine atoms of compounds 25 and 26 may interact with the Arg98 residue located in the STS active site. This additional interaction may stabilise the inhibitor-enzyme complex resulting in improved inhibitory activity. The presented results indicated that fluorinated 4-(1-phenyl-1H-1,2,3-triazol-4-yl)phenyl sulphamates are promising STS inhibitors, however, their clinical potential should be proved in further in vivo evaluation.
Kajita et al.Citation65 obtained a series of silicon derivatives of diphenylmethane as potent STS inhibitors, which also served as pro-oestrogen antagonists. In the course of the undertaken research, the derivatives with a modified Si-alkyl motif and an aromatic ring system were synthesised. It was noticed that the presence of ethyl groups on a silicon atom was the most favourable (the derivatives containing methyl, n-propyl and n-butyl were much less active). Among the obtained derivatives, silicon-containing compound 27 () showed high STS inhibitory effect (IC50 of 0.17 µM when evaluated in an assay with MCF-7 cells). Interestingly, the STS inhibitory activity of compound 27 was significantly higher than the corresponding carbon analogues. Furthermore, the putative metabolite of 27, compound 28 (), showed potent ERα-antagonistic activity (IC50 value of 29.7 nM) and lacked ERα-agonistic activity.
El-Gamal et al.Citation66 reported a new series of arylamides containing sulphonate and sulphamate moieties as novel STS inhibitors. The structures of obtained analogues differed in a pharmacophore region (sulphonate, sulphamate, and N-substituted sulphamate groups were evaluated) and in the size of an aliphatic ring (five and six-membered rings were introduced). As it had been predicted, the most potent derivative 29 () containing free sulphamate moiety inhibited STS activity by 72.0% and 55.7% at 20 and 10 µM, respectively, in a cell-free assay. Further investigation showed that 29 inhibited (in a dose-dependent manner) 93.9% and 86.1% of STS activity in JEG-3 placental carcinoma cells at 20 and 10 µM, respectively (IC50 value of 0.421 µM). Among the derivatives containing aliphatic sulphonate groups, the presence of the ethanesulphonate moiety occurred to be the most favourable. On the other hand, the para-tosylate derivative was more active than the other aromatic sulphonates. It was also noticed that the derivatives containing a cyclohexyl ring were significantly more active than the corresponding analogues bearing a cyclopentyl ring in general. The authors indicated that the newly obtained compounds based on a relatively simple structure were promising candidates for future lead optimisation, which could be carried out by an appropriate substitution of the two ring systems, although no article have been published up to date.
Recently, Moi et al.Citation67 reported two new groups of piperazinyl-ureido sulphamates as potent STS inhibitors. Initially, the undertaken studies led to the development of a series of 4-(piperazinocarbonyl)-aminosulphamates that demonstrated promising STS inhibitory potency. The development of a few lead compounds resulted in the synthesis of derivatives containing halogen atoms (fluorine or chlorine) within the aryl-sulphamate pharmacophore. The most active derivatives 30 and 31 () demonstrated high STS inhibitory activities in an assay with JEG-3 cell lysate (IC50 values of 5.1 and 8.8 nM, respectively). In spite of the fact that further inhibitor structure optimisation efforts have been made (such as the introduction of an additional pyrimidinyl ring), enhancement of the inhibitory potency has not been achieved.
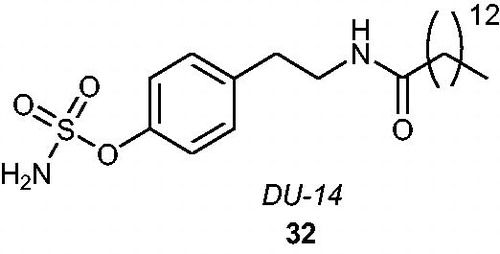
Recently, an interesting in vivo study focussed on a derivative of p-O-sulphamoyl-N-alkanoyl tyramine, namely, DU-14 32 (containing an N-tetradecanoyl chain) (), has been described as a potential treatment method of neurodegenerative disorders (e.g. Alzheimer’s disease) in ratsCitation71. Historically, a series of p-O-sulphamoyl-N-alkanoyl tyramines was synthesised as STS inhibitors by Li et al.Citation72. The most active analogue, DU-14, promoted an STS inhibitory effect with an IC50 value of 55.8 nM when evaluated in an assay with placental microsomes as the enzyme sourceCitation72 and an IC50 value of 350 nM when evaluated in an assay with MDA-MB-231 cellsCitation73. It has been shownCitation74 that DU-14 can enhance the reversal of amnesia by DHEAS, suggesting that STS inhibition could promote the memory-enhancing properties of DHEAS. Yue et al.Citation71 reported neuroprotective properties of DU-14 against neurotoxic amyloid β protein (Aβ), suggesting that upregulation of endogenous DHEAS by DU-14 may be responsible for a beneficial alleviation of impairments in spatial memory and synaptic plasticity induced by Aβ in rats.
Figure 6. The chemical structure of DU-14 32.
3.2. Nonsteroidal STS inhibitors containing phosphorus moieties
The initial strategy applied in the designing of STS inhibitors involving the replacement of the sulphate group in natural STS substrates has been recently extended to numerous phosphorus moietiesCitation75. Because methods for the chemical introduction of phosphorus moieties are widely utilisedCitation76, medical applications of organophosphorus compounds as drugs or drug candidates in the treatment of various diseases have expanded greatly in recent years. Compounds containing phosphorus groups are used in clinical practice as agents with anticancer and antiviral properties as well as in the treatment of metabolic bone disorders such as osteoporosisCitation75,Citation77. It is known that various phosphorus moieties may react with amino acid functional groups and create electrostatic interactions inside the enzyme active site that may influence the binding of potential drugs to the enzyme’s active site.
Recently, Kozak et al. have synthesised new tricyclic coumarin derivatives containing phosphoric acid residues, dimethyl- and diethylphosphate groups as well as phosphoroamidates and phosphorodiamidates that were able to inhibit STS activity in the micromolar rangeCitation78. The most active derivative 33 () inhibited STS with the IC50 value of 21.5 µM in an enzymatic assay. Although the mechanism of inhibition is unknown, docking studies suggest the possibility of a phosphate group transfer to fGly75 during the inactivation process. Further development led to the synthesis of thiophosphate analogues with slightly improved inhibitory potenciesCitation79. The highest activity (in an assay with an isolated enzyme) was exhibited by compound 34 (), which contained a chlorothiophosphate group (IC50 value of 13.3 µM).
Table 3. Examples of nonsteroidal STS inhibitors containing phosphorus moieties 33–46.
In 2015, the same research group synthesised a series of bicoumarin thiophosphate derivatives as STS inhibitorsCitation80. Although the mechanism of inhibition remains unclear, molecular modelling studies suggest a completely different manner of binding to the STS active site compared to previously evaluated phosphorus STS inhibitors. The docking experiments indicated that these compounds were able to bind to the STS active site and to fill the whole cavity, preventing the substrate’s access to the catalytic amino acid residues. Both reversible and irreversible STS inhibitors were found within this class of compounds. For example, the hydrogenthiophosphate derivative 35 () inhibited STS reversibly, whereas chlorothiophosphate 36 () and methylthiophosphate 37 () were irreversible inhibitors (measured IC50 values of 860 nM, 7.76 µM, and 6.77 µM, respectively, when evaluated in an enzymatic assay).
The same research approach for designing phosphorus STS inhibitors has been utilised by Demkowicz et al. for biphenyl and flavone derivatives. Initially, a series of biphenyl phosphates and thiophosphates was synthesisedCitation81. Among them, compound 38 () inhibited purified STS enzyme with an IC50 value of 46.8 µM. When the biphenyl core was replaced by the flavone framework, the inhibitory activities of the obtained compounds were enhanced. For example, compound 39 () inhibited STS activity with an IC50 value of 29.0 µM in an enzymatic assayCitation82. However, when the thiophosphate group was connected to two biphenyl or flavone residues, the inhibitory activities of the obtained compounds were significantly greater; e.g. IC50 values of 22.1 and 3.2 µM were measured for compounds 40 and 41 (), respectively (measured in an assay with STS).
In 2015, a series of N-alkanoyl tyramine phosphates and thiophosphates were obtained as potent STS inhibitorsCitation83. In the course of undertaken research, it was noticed that the highest STS inhibitory effects were exhibited by compounds containing a hydrophobic dodecanoyl carbon chain in the structure of an N-alkanoyl tyramine core. In general, the highest STS inhibitory effects were exhibited by analogues containing phosphate moieties. For example, compounds 42 and 43 () containing dimethylphosphate or methylphosphoroamidate groups demonstrated the highest inhibitory activities in an enzymatic assay (IC50 values of 0.39 and 1.31 µM, respectively). Their mechanism of action is unconfirmed; however, the alkylating properties of methylphosphates suggest the possible methylation of the catalytic fGly75 residue. Interestingly, compound 42 also demonstrated high cytotoxic activity and effectively inhibited the proliferation of MCF-7, MDA-MB-231 and SkBr3 cancer cells (GI50 values of 8.80, 6.48 and 5.76 µM, respectively).
Recently, the synthesis of compounds with both sulphamate and phosphorus groups has been proceded by Daśko et al.Citation84. A 3-phenyl-coumarin derivative was used as the structural core. Among the obtained series, compounds 44 and 45 () exhibited the highest inhibitory activities against purified STS enzyme (IC50 values of 190 and 240 nM, respectively). The molecular docking studies indicated that sulphamate moieties in compounds 44 and 45 were located within the STS catalytic region, while phosphoroamidate groups were within a short distance of the Arg98 residue, suggesting the presence of additional stabilising interactions. When the phosphoroamidates were replaced with thiophosphoroamidates, the next group of STS inhibitors was obtainedCitation85. In this case, the activity of the most active compound 46 () (IC50 value of 200 nM) was comparable with derivatives 44 and 45 (when evaluated in an enzymatic assay). A more detailed biological evaluation indicated that derivative 44 inhibited STS enzyme by 99.9% in an assay with MCF-7 cells (at 100 nM). Furthermore, toxicology analysis revealed that compounds 44–46 were safe for zebrafish embryos at concentrations exceeding IC50 values by up to 12-fold. Further in vivo studies might prove their development potential.
4. Multitargeting agents with STS inhibitory activities
The development of inhibitors demonstrating multitargeting properties is an alternative approach to STS inhibition. To date, this strategy has allowed scientists to develop novel STS inhibitors able to inhibit other enzymes implicated in the hormone biosynthesis process (e.g. AROM and 17β-HSD). For example, multitargeting agents demonstrating both STS and AROM inhibitory activities are known as dual aromatase-sulphatase inhibitors (DASIs). Initially, the synthesis of the most promising DASIs was based on the already known procedures for AROM inhibitors substituted with a sulphamate pharmacophore. The presence of the sulphamate-aryl system (responsible for STS inhibition) and the 1,2,4-triazole ring in the structure of a single molecule led to compounds exhibiting high STS and AROM inhibitory properties simultaneously. One of the first DASIs, STX681 47 (), a sulphamoylated derivative of the AROM inhibitor YM511, was synthesised by Woo et al.Citation86,Citation94 STX681 inhibited STS and AROM activities in JEG-3 cells with IC50 values of 590 and 0.77 nM, respectively. Furthermore, its high STS and AROM inhibitory activities and promising therapeutic advantages have been also proven in in vivo studiesCitation7,Citation95.
Table 4. Examples of multitargeting agents with STS and/or AROM and/or 17β-HSD1 inhibitory properties 47–60.
In 201089, the same research group reported the first highly potent examples of DASIs based on a biphenyl core. The most active derivative, STX1983 48 (), effectively inhibited STS and AROM activities in a cellular assay with JEG-3 cells (IC50(STS) value of 5.5 nM and an IC50(AROM) value of 0.5 nM) and was nonestrogenic. Furthermore, STX1983 strongly reduced plasma oestradiol levels and potently inhibited liver STS in vivo. Further development of DASIs based on the STX681 and STX1983 structures has led to a series of hybrid structures with improved dual STS and AROM inhibitory activities (in the picomolar range) in an assay using JEG-3 cellsCitation88. For example, the extremely active derivative 49 () (IC50(STS) value of 830 pM and an IC50(AROM) value of 15 pM) appears to be a very promising candidate for in vivo evaluation in hormone-dependent cancer treatments.
Woo et al.Citation89 reported extensive research on the synthesis of novel DASIs based on the STX681 template. The range of chemical modifications in the STX681 structure was far-reaching and included the relocation and replacement of a halogen atom, introduction of more halogens and replacement of a methylene linker with a difluoromethylene motif. Replacements of a p-cyano-phenyl ring with other ring structures and the replacement of the triazolyl group with an imidazolyl group were performed too. Eventually, a series of seventeen new DASIs was obtained. Among them, the fluorinated compound 50 () containing an imidazole ring exhibited the highest STS and AROM inhibitory activities (IC50(STS) value of 2.5 nM and an IC50(AROM) value of 0.2 nM when evaluated in an assay with JEG-3 cells). Interestingly, the parent phenol 51 () demonstrated an even higher AROM inhibitory activity (IC50(AROM) value of 0.028 nM).
There has recently been extensive progress in the development of novel DASIs, and several other series of compounds exhibiting dual aromatase-sulphatase inhibitory activities have been obtained – specifically sulphamoylated Letrozole (e.g. compound 52, [])Citation90,Citation96–98 and Anastrozole derivatives (e.g. compound 53, [])Citation91,Citation99.
Recently, the other enzyme implicated in the hormone biosynthesis process, 17β-HSD, has reached potential therapeutic importance in the treatment of hormone-dependent diseases. Salah et al.Citation92 reported the first dual inhibitors of STS and 17β-hydroxysteroid dehydrogenase type 1 (17β-HSD1) as promising therapeutics for oestrogen-dependent diseases. The design of dual STS/17β-HSD1 inhibitors was based on combining structural elements, essential for 17β-HSD1 inhibition, with the sulphamate-aryl pharmacophore (crucial in STS inactivation). Among 12 synthesised compounds, derivative 54 () demonstrated the most promising biological properties with well-balanced activities against both the STS and 17β-HSD1 enzymes (IC50(STS) value of 15.6 nM and an IC50(17β-HSD1) value of 22.2 nM when evaluated in an assay with T47D cells). Compound 54 did not exhibit cytotoxic properties or oestrogen receptor interference. Moreover, 54 was also able to effectively reverse E1S/E1-stimulated proliferation of T47D cells (at 400 nM). Authors noticed, that the activity towards STS required an additional substituent within the sulphamate-aryl system and EWG substituents turned out to be the most favourable. On the other hand, strong EWGs impaired STS inhibition by reducing the chemical stability of the sulphamate pharmacophore.
Bacsa et al.Citation93 synthesised a series of 2- and/or 4-halogenated (chlorinated, brominated, or iodinated) 13β- and 13α-oestrone derivatives as interesting multitargeting agents. Among the obtained analogues, a few derivatives demonstrated dual STS/17β-HSD1 inhibitory properties. For example, two 4-halo-17-oxo-13β compounds 55 and 56 () inhibited STS with IC50(STS) values of 0.23 and 0.89 µM, respectively, and 17β-HSD1 with IC50(17β-HSD1) values of 0.36 and 0.30 µM, respectively. Interestingly, two other compounds, namely, 2-bromo- and 2-chloro-13β-estrones 57 and 58 (), caused inhibitory effects towards three investigated enzymes, becoming triple STS/17β-HSD1/AROM inhibitors (IC50(STS) values of 2.0 and 2.4 µM; IC50(17β-HSD1) values of 0.095 and 0.18 µM; and IC50(AROM) values of 8.7 and 6.0 µM for compounds 57 and 58, respectively). The authors conducted extensive SAR analysis regarding the influence of type and position of halogen substitution on the inhibitory properties. For example, it was noticed that the inhibitory potential of iodine-containing derivatives of 13β-oestrone depended on the position of an iodine atom. The 2-iodo derivative 59 () was a highly specific 17β-HSD1 inhibitor, whereas its 4-iodo analogue 55 demonstrated dual STS/17β-HSD1 inhibitory properties. In contrast, the 2,4-diiodo derivative exhibited weak inhibitory effect against all of the investigated enzymes.
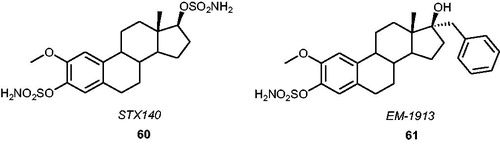
An extremely interesting drug candidate based on a 2-methoxyestradiol (2ME) structure, STX140 60 (), has become a very promising multitargeted antitumour agent with a wide range of potential medicinal applicationsCitation26,Citation100,Citation101. In addition to its high STS inhibitory potency in vitro and in vivo (IC50 value of 39 nM when evaluated in an assay with placental microsomes)Citation102, STX140 demonstrated numerous interesting properties (e.g. antiproliferative and antiangiogenic activities as well as the ability to induce cell cycle arrest and apoptosis in human tumour xenografts)Citation103. It has been observed that STX140 can inhibit the proliferation of oestrogen receptor-positive (MCF-7) and -negative (MDA-MB-231) cells (with the GI50 values of 0.25 and 0.29 µM, respectively)Citation104. Therefore, it may be effectively utilised as an STS inhibitor in the treatment of hormone-dependent cancers, however, due to its multitargeting activities, it may be considered in the treatment of tumours with hormone-independent nature as well. Furthermore, favourable pharmacokinetic properties of STX140, including good bioavailability and low metabolism in rodentsCitation105, have proved its major clinical advantagesCitation106 also for chemotherapy-resistant tumoursCitation107. In 2013, in vivo studies with three breast cancer modelsCitation108 demonstrated that STX140 had greater anticancer efficacy and therapeutic index as well as reduced neurotoxicity compared to paclitaxel (clinically used for hormone-refractory breast cancer), which may bring significant benefits to patients with breast cancer. Furthermore, a 2-[Citation11C]methoxy derivative of STX140 has been reported as a new potential imaging agent of STS in tumours in the positron emission tomography techniqueCitation109. To date, more sulphamoylated derivatives of 2ME have been developedCitation110,Citation111, including EM-1913 61 ()Citation112, which shows to be a potent STS inhibitor able to block tumour growth (MCF-7 xenograft) in nude miceCitation113.
Figure 7. Chemical structures of STX140 60 and EM-1913 61.
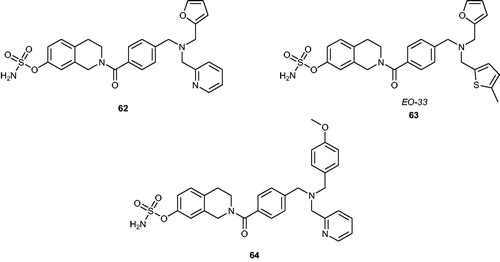
Recently, Ouellet et al.Citation114,Citation115 have reported a new group of STS inhibitors based on N-substituted tetrahydroisoquinoline derivatives containing the sulphamate pharmacophore. The most recent researchCitation116 has demonstrated that such derivatives exhibit both STS inhibitory and selective oestrogen receptor modulating properties. Initially, three parent phenolic compounds, devoid of undesirable oestrogenic activity and toxicity, have been chosen and modified with a sulphamate moiety. These sulphamoylated analogues 62–64 () were potent STS inhibitors (IC50 values of 3.9, 8.9 and 16.6 nM, when evaluated in an assay with STS-transfected HEK-293 cells, respectively) without undesirable oestrogenic properties. However, it was noticed that they exhibited moderate antiestrogenic activity. Recent in vivo mouse model studiesCitation117 of one of them (63, also known as EO-33) have indicated that EO-33 exhibits a SERM effect and blocks the changes in uterine weight, stimulated by E1S in ovariectomised mice. It has been determined that EO-33 effectively inhibits STS activity by 81% in the liver and blocks 90% of tumour growth induced by oestradiol sulphate (E2S). Furthermore, no toxic effects (by assessing body weight, liver weight, and liver appearance) have been reported, proving that EO-33 is a promising nonsteroidal STS inhibitor with SERM activity.
Figure 8. Chemical structures of STS inhibitors with SERM activity 62–64.
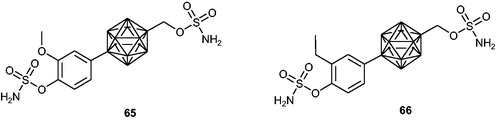
Kaise et al.Citation118,Citation119 designed and synthesised two series of novel p-carborane-containing sulphamates as multitargeting anticancer agents. Obtained compounds possessed both sulphamoylated phenol and alcohol systems at the same time. In the course of undertaken research, it was noticed that the former are much more effective in STS inhibition than the latter. Among the obtained compounds, a few analogues demonstrated high STS inhibitory properties with IC50 values in the nanomolar range (when evaluated in an assay with MCF-7 cells). Two of the most promising ones, 65 and 66 (), displayed high STS inhibitory effects with IC50 values of 0.3 and 1.8 nM, respectively. Interestingly, these compounds demonstrated high antiproliferative activities against significant number of human cancer cell lines, making them promising therapeutic agents for anticancer treatment (a screening panel of 39 human cancer cell lines [e.g. breast, central nervous system, colon, lung, melanoma, ovary, kidney, and stomach] was utilised). Furthermore, both compounds 65 and 66 exhibited tubulin-polymerisation-inhibitory (TPI) activities.
Figure 9. Chemical structures of STS inhibitors based on p-carborane-containing sulphamates 65 and 66.
5. Conclusion
Despite the development of many treatment strategies, cancer remains one of the most important medical problems that modern medicine has to face. Considering that numerous carcinomas exhibit a hormone-dependent nature, it is rational to design agents that can block hormone biosynthesis processes. The administration of small molecules, exhibiting inhibitory activities against enzymes implicated in hormone biosynthesis may reduce the concentrations of these in tumour tissues and consequently may limit the oestrogenic stimulation of cancer cell growth. Currently, STS is recognised as an extremely promising molecular target in the development of effective agents with high therapeutic potential in the treatment of hormone-dependent cancers. Scientists announce discoveries of new molecules demonstrating STS inhibitory properties every year. Most of them are based on nonsteroidal cores and contain a sulphamate moiety. However, current studies have demonstrated that derivatives with other functional groups (e.g. phosphates and thiophosphates) also exhibit high STS inhibitory activities. Despite the fact that organic synthesis is the most important source of novel STS inhibitors, it is worth noting that a few of recently reported compounds are of natural origin. The research on the natural-based compounds and their modified synthetic analogues may lead to identification of new ones with interesting physicochemical and biological properties. Furthermore, the design process as well as the identification of the potential STS inhibitors are currently more often supported by the computational approaches, e.g. virtual screening, QSAR analysis, and molecular docking studies.
To date, some of the reported compounds have been evaluated in clinical trials (e.g. Irosustat and E2MATE) and they seem to be very promising as drug candidates. However, none of them has reached pharmacies. Therefore, the identification and synthesis of novel compounds demonstrating STS inhibitory activity as well as the evaluation of the biological influence and safety of known STS inhibitors are crucial for the development of effective treatment methods of hormone-dependent cancers. The discoveries in the synthesis of compounds, which mechanism of action is based on some innovative multi-targeting approaches, have reached the key importance. The recent research has indicated that it is possible to design multitargeting agents that can effectively inhibit STS and other enzymes involved in the hormone biosynthesis pathway (e.g. AROM and 17β-HSD1) or SERM activity. This direction in the development of novel drug candidates with multitargeted biological properties may enhance the efficacy of novel treatment methods. On the other hand, a combination therapy of STS inhibitors with agents targeting other enzymes involved in the hormone biosynthesis process may affect potential clinic advantages (as it has been noticed in the combination therapy of Irosustat and AROM inhibitor). Potentially, further clinical benefits may be received in the administration of the STS inhibitors with other anticancer agents, which mechanism of action is not associated with the steroidogenesis process. In this matter, the wide possibilities for improving the efficacy of potential therapies seem to be still achievable. For example, novel compounds chemically constructed of STS inhibitors and other anticancer agents (additionally linked to natural or synthetic carriers providing the selective uptake by the cancer cells) may increase efficacy, selectivity of the therapies and may reduce potential side effects connected with the administration of some agents. On the other hand, the most recent studies have indicated that newly developed compounds (e.g. derivatives exhibiting fluorescent properties) may be applied not only as STS inhibitors but also as optical imaging tools to investigate intracellular sub-localisation of STS enzyme and inhibitory mechanisms.
Acknowledgements
The authors gratefully acknowledge the National Science Centre (Poland) for their financial support.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- IARC: CANCER FACT SHEETS [Internet]. Lyon (France): International Agency for Research on Cancer [cited 2019 July 16]. Available from: https://gco.iarc.fr/today/fact-sheets-cancers.
- NCI: Cancer Statistics [Internet]. Bethesda (MD): National Cancer Institute [cited 2019 July 16]. Available from: https://www.cancer.gov/about-cancer/understanding/statistics.
- Pasqualini JR. The selective estrogen enzyme modulators in breast cancer: a review. Biochim Biophys Acta 2004; 1654:123–43.
- Poirier D. Recent patents on new steroid agents targeting the steroidogenesis for endocrine cancer treatments. Recent Pat Endocr Metab Immune Drug Discov 2015;9:15–23.
- Saha T, Makar S, Swetha R, et al. Estrogen signaling: an emanating therapeutic target for breast cancer treatment. Eur J Med Chem 2019;177:116–43.
- Shah R, Singh J, Singh D, et al. Sulfatase inhibitors for recidivist breast cancer treatment: a chemical review. Eur J Med Chem 2016;114:170–90.
- Foster PA, Chander SK, Newman SP, et al. A new therapeutic strategy against hormone-dependent breast cancer: the preclinical development of a dual aromatase and sulfatase inhibitor. Clin Cancer Res 2008;14:6469–77.
- Dixon JM. Endocrine resistance in breast cancer. New J Sci 2014;2014:1–27.
- Reinert T, Saad ED, Barrios CH, et al. Clinical implications of ESR1 mutations in hormone receptor-positive advanced breast cancer. Front Oncol 2017;7:26.
- Augusto TV, Correia-da-Silva G, Rodrigues CMP, et al. Acquired resistance to aromatase inhibitors: where we stand. ! Endocr Relat Cancer 2018;25:R283–301.
- Mueller JW, Gilligan LC, Idkowiak J, et al. The regulation of steroid action by sulfation and desulfation. Endocr Rev 2015;36:526–63.
- Foster PA, Reed MJ, Purohit A. Recent developments of steroid sulfatase inhibitors as anti-cancer agents. Anti-Cancer Agents Med Chem 2008;8:732–8.
- Smith HJ, Simons C, Development of enzyme inhibitors as drugs. In: Smith HJ, Simons C, eds. Enzymes and their inhibition: drug development. London: CRC Press; 2005;171–250.
- Reed MJ, Purohit A, Woo LWL, et al. Steroid sulfatase: molecular biology, regulation and inhibition. Endocr Rev 2005;26:171–202.
- Miki Y, Nakata T, Suzuki T, et al. Systemic distribution of steroid sulfatase and estrogen sulfotransferaze in human adult and fetal tissues. J Clin Endocrinol Metabol 2002;87:5760–8.
- Hanson SR, Best MD, Wong CH. Sulfatases: structure, mechanism, biological activity, inhibition, and synthetic utility. Angew Chem Ind Ed 2004;43:5736–63.
- Thomas MP, Potter B. The structure biology of estrogen metabolites. J Steroid Biochem Mol Biol 2013;137:27–49.
- Purohit A, Foster PA. Steroid sulfatase inhibitors for estrogen- and androgen-dependent cancers. J Endocrinol 2012;212:99–110.
- Hernandez-Guzman FG, Higashiyama T, Pangborn W, et al. Structure of human estrone sulfatase suggests functional roles of membrane association. J Biol Chem 2003;278:22989–97.
- Ghosh D. Human sulfatases: a structural perspective to catalysis. Cell Mol Life Sci 2007;64:2013–22.
- Selmer T, Hallmann A, Schmidt B, et al. The evolutionary conservation of a novel protein modification, the conversion of cysteine o serine-semialdehyde in arylsulfatase from Volvox carteri. Eur J Biochem 1996;238:341–5.
- Maltais R, Poirier D. Steroid sulfatase inhibitors: a review covering the promising 2000–2010 decade. Steroids 2011;76:929–48.
- Mostafa YA, Taylor SD. Steroid derivatives as inhibitors of steroid sulfatase. J Steroid Biochem Mol Biol 2013;137:183–98.
- Woo LWL, Purohit A, Potter B. Development of steroid sulfatase inhibitors. Mol Cell Endocrinol 2011;340:176–85.
- Geisler J, Sasano H, Chen S, et al. Steroid sulfatase inhibitors: promising new tools for breast cancer therapy? J Steroid Biochem Mol Biol 2011;125:39–45.
- Potter B, SULFATION P. Steroid sulphatase inhibition via aryl sulphamates: clinical progress, mechanism and future prospects. J Mol Endocrinol 2018;61:T233–52.
- Thomas MP, Potter B. Discovery and development of the aryl O-sulfamate pharmacophore for oncology and women’s health. J Med Chem 2015;58:7634–58.
- Rizner TL. The important roles of steroid sulfatase and sulfotransferases in gynecological diseases. Front Pharmacol 2016;7:1–16.
- Sadozai H. Steroid sulfatase inhibitors: promising new therapy for breast cancer. J Pak Med Assoc 2013;63:509–15.
- Howarth NM, Cooper G, Purohit A, et al. Phosphonates and thiophosphonates as sulfate surrogates: synthesis of estrone-3-methylthiophosphonates, a potent inhibitor of estrone sulfatase. Bioorg Med Chem Lett 1993;3:313–8.
- Duncan L, Purohit A, Howarth NM, et al. Inhibition of estrone sulfatase activity by estrone-3-methylthiophosphonate: a potential therapeutic agent in breast cancer. Cancer Res 1993;53:298–303.
- Howarth NM, Purohit A, Reed MJ, et al. Estrone sulfamates: potent inhibitors of estrone sulfatase with therapeutic potential. J Med Chem 1994;37:219–21.
- Purohit A, Williams GJ, Howarth NM, et al. Inactivation of sulfatase by an active site-directed inhibitor, estrone-3-O-sulfamate. Biochemistry 1995;34:11508–14.
- Elger W, Barth A, Hedden A, et al. Estrogen sulfamates: a new approach to oral estrogen therapy. Reprod Fertil Dev 2001;13:297–305.
- Hidalgo Aragones MI, Purohit A, Parish D, et al. Pharmacokinetics of oestrone-3-O-sulphamate. J Steroid Biochem Mol Biol 1996;58:611–7.
- Elger W, Schwarz S, Hedden A, et al. Sulfamates of various estrogens are prodrugs with increased systemic and reduced hepatic estrogenicity at oral application. J Steroid Biochem Mol Biol 1995;55:395–403.
- Pohl O, Bestel E, Gotteland JP. Synergistic effects of E2MATE and norethindrone acetate on steroid sulfatase inhibition: a randomized phase I proof-of-principle clinical study in women of reproductive age. Reprod Sci 2014;21:1256–65.
- Colette S, Defrère S, Lousse JC, et al. Inhibition of steroid sulfatase decreases endometriosis in an in vivo murine model. Hum Reprod 2011;26:1362–70.
- Woo LWL, Leblond B, Purohit A, et al. Synthesis and evaluation of analogues of estrone-3-O-sulfamate as potent steroid sulfatase inhibitors. Bioorg Med Chem 2012;20:2506–19.
- Mostafa YA, Taylor SD. 17β-arylsulfonamides of 17β-aminoestra-1,3,5(10)-trien-3-ol as highly potent inhibitors of steroid sulfatase. Bioorg Med Chem 2012;20:1535–44.
- Mostafa YA, Kralt B, Rao PPN, et al. A-ring substituted 17β-arylsulfonamides of 17β-aminoestra-1,3,4,(10)-trien-3-ol as highly potent reversible inhibitors of steroid sulfatase. Bioorg Med Chem 2015;23:5681–92.
- Grienke U, Kaserer T, Kirchweger B, et al. Steroid sulfatase inhibiting lanostane triterpenes – Structure activity relationship and in silico insights. Bioorg Chem 2020;95:103495.
- Maltais R, Djiemeny AN, Roy J, et al. Design and synthesis of dansyl-labeled inhibitors of steroid sulfatase for optical imaging. Bioorg Med Chem 2020;28:115368.
- Fournier D, Poirier D. Chemical synthesis and evaluation of 17alpha-alkylated derivatives of estradiol as inhibitors of steroid sulfatase. Eur J Med Chem 2011;46:4227–37.
- Boivin RP, Luu-The V, Lachance R, et al. Structure-activity relationships of 17alpha-derivatives of estradiol as inhibitors of steroid sulfatase. J Med Chem 2000;43:4465–78.
- Purohit A, Woo LWL, Singh A, et al. In vivo activity of 4-methylcoumarin-7-O-sulfamate, a nonsteroidal, nonestrogenic steroid sulfatase inhibitor. Canc Res 1996;56:4950–5.
- Woo LWL, Howarth NM, Purohit A, et al. Steroidal and nonsteroidal sulfamates as potent inhibitors of steroid sulfatase. J Med Chem 1998;41:1068–83.
- Malini B, Purohit A, Ganeshapillai D, et al. Inhibition of steroid sulphatase activity by tricyclic coumarin sulphamates. J Steroid Biochem Mol Biol 2000;75:253–8.
- Woo LWL, Purohit A, Malini B, et al. Potent active site-directed inhibition of steroid sulphatase by tricyclic coumarin-based sulphamates. Chem Biol 2000;7:773–91.
- Woo LWL, Ganeshapillai D, Thomas MP, et al. Structure-activity relationship for the first-in-class clinical steroid sulfatase inhibitor Irosustat (STX64, BN83495). Chem Med Chem 2011;6:2019–34.
- Parra-Guillen ZP, Cendrós Carreras JM, Peraire C, et al. Population pharmacokinetic modelling of Irosustat in postmenopausal women with Oestrogen-receptor positive breast cancer incorporating non-linear red blood cell uptake. Pharm Res 2015;32:1493–504.
- Coombes RC, Cardoso F, Nicolas I, et al. A phase I dose escalation study to determine the optimal biological dose of irosustat, an oral steroid sulfatase inhibitor, in postmenopausal women with estrogen receptor-positive breast cancer. Breast Cancer Res Treat 2013;140:73–82.
- Stanway SJ, Purohit A, Woo LWL, et al. Phase I study of STX64 (667 Coumate) in breast cancer patients: the first study of a steroid sulfatase inhibitor. Clin Cancer Res 2006;12:1585–92.
- Palmieri C, Januszewski A, Stanway S, et al. Irosustat: a first-generation steroid sulfatase inhibitor in breast cancer. Expert Rev Anticancer Ther 2011;11:179–83.
- Palmieri C, Stein RC, Liu X, et al. IRIS study: a phase II study of the steroid sulfatase inhibitor Irosustat when added to an aromatase inhibitor in ER-positive breast cancer patients. Breast Cancer Res Treat 2017;165:343–53.
- Palmieri C, Stein RC, Liu X, et al. A phase II study to assess the safety and efficacy of the steroid sulfatase inhibitor Irosustat when added to an aromatase inhibitor in ER positive locally advanced or metastatic breast cancer patient (IRIS) – trial results. J Clin Oncol 2016;34:549.
- Palmieri C, Szydlo R, Miller M, et al. IPET study: an FLT-PET window study to assess the activity of the steroid sulfatase inhibitor irosustat in early breast cancer. Breast Cancer Res Treat 2017;166:527–39.
- Pautier P, Vergote I, Joly F, et al. A phase 2, randomized, open-label study of Irosustat versus megestrol acetate in advanced endometrial cancer. Int J Gynecol Cancer 2017;27:258–66.
- Ganeshapillai D, Woo LWL, Thomas MP, et al. C-3- and C-4-substituted bicyclic coumarin sulfamates as potent steroid sulfatase inhibitors. ACS Omega 2018;3:10748–72.
- Hng Y, Lin MH, Lin TS, et al. Design and synthesis of 3-benzylaminocoumarin-7-O-sulfamate derivatives as steroid sulfatase inhibitors. Bioorg Chem 2020;96:103618.
- Demkowicz S, Daśko M, Kozak W, et al. Synthesis and biological evaluation of fluorinated 3-phenylcoumarin-7-O-sulfamate derivative as steroid sulfatase inhibitors. Chem Biol Drug Des 2016;87:233–8.
- Daśko M, Przybyłowska M, Rachon J, et al. Synthesis and biological evaluation of fluorinated N-benzoyl and N-phenylacetoyl derivatives of 3-(4-aminophenyl)-coumarin-7-O-sulfamate as steroid sulfatase inhibitors. Eur J Med Chem 2017;128:79–87.
- Daśko M, Rachon J, Masłyk M, et al. Synthesis and biological evaluation of N-acylated tyramine sulfamates containing C-F bonds as steroid sulfatase inhibitors. Chem Biol Drug Des 2017; 90:156–61.
- Daśko M, Demkowicz S, Rachon J, et al. New potent STS inhibitors based on fluorinated 4-(1-phenyl-1H-[1,2,3]triazol-4-yl)-phenyl sulfamates. J Asian Nat Prod Res 2019. doi: 10.1080/10286020.2019.1680642.
- Kajita D, Nakamura M, Matsumoto Y, et al. Design and synthesis of silicon-containing steroid sulfatase inhibitors possessing pro-estrogen antagonistic character. Bioorg Med Chem 2014;22:2244–52.
- El-Gamal MI, Semreen MH, Foster PA, et al. Design, synthesis, and biological evaluation of new arylamide derivatives possessing sulfonate or sulfamate moieties as steroid sulfatase enzyme inhibitors. Bioorg Med Chem 2016;24:2762–7.
- Moi D, Foster PA, Rimmer LG, et al. Synthesis and in vitro evaluation of piperazinyl-ureido sulfamates as steroid sulfatase inhibitors. Eur J Med Chem 2019;182:111614.
- Zhou Y, Wang J, Gu Z, et al. Next generation of fluorine-containing pharmaceuticals, compounds currently in phase II-III clinical trials of major pharmaceutical companies: new structural trends and therapeutic areas. Chem Rev 2016;116:422–518.
- Kitchen DB, Decornez H, Furr JR, et al. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov 2004;3:935–49.
- Jagiello K, Sosnowska A, Kar S, et al. Geometry optimization of steroid sulfatase inhibitors – the influence on the free binding energy with STS. Struct Chem 2017;28:1017–32.
- Yue XH, Tong JQ, Wang ZJ, et al. Steroid sulfatase inhibitor DU-14 protects spatial memory and synaptic plasticity from disruption by amyloid β protein in male rats. Horm Behav 2016;83:83–92.
- Li PK, Milano S, Kluth L, et al. Synthesis and sulfatase inhibitory activities of non-steroidal estrone sulfatase inhibitors. J Steroid Biochem Mol Biol 1996;59:41–48.
- Selcer KW, Hegde PV, Li PK. Inhibition of estrone sulfatase and proliferation of human breast cancer cells by nonsteroidal (p-O-sulfamoyl)-N-alkanoyl tyramines. Cancer Res 1997;57:702–7.
- Li PK, Rhodes ME, Burke AM, et al. Memory enhancement mediated by the steroid sulfatase inhibitor (p-O-sulfamoyl)-N-tetradecanoyl tyramine. Life Sci 1997;60:45–51.
- Demkowicz S, Rachon J, Daśko M, et al. Selected organophosphorus compounds with biological activity. Applications in medicine. RSC Adv 2016;6:7101–7112.
- Kozak W, Rachon J, Daśko M, et al. Selected methods for the chemical phosphorylation and thiophosphorylation of phenols. Asian J Org Chem 2018;7:314–23.
- Demkowicz S, Kozak W, Daśko M, et al. Phosphoroorganic metal complexes in therapeutics. Mini Rev Med Chem 2016;16:1359–73.
- Kozak W, Daśko M, Masłyk M, et al. Phosphate tricyclic coumarin analogs as steroid sulfatase inhibitors: synthesis and biological activity. RSC Adv 2014;4:44350–8.
- Kozak W, Daśko M, Masłyk M, et al. Synthesis and biological evaluation of thiophosphate tricyclic coumarin derivatives as steroid sulfatase inhibitors. J Asian Nat Prod Res 2015;17:1091–6.
- Demkowicz S, Kozak W, Daśko M, et al. Synthesis of bicoumarin thiopshosphate derivatives as steroid sulfatase inhibitors. Eur J Med Chem 2015;101:358–66.
- Demkowicz S, Kozak W, Daśko M, et al. Phosphate and thiophosphate biphenyl analogs as steroid sulfatase inhibitors. Drug Dev Res 2015;76:94–104.
- Kozak W, Daśko M, Masłyk M, et al. Steroid sulfatase inhibitors based on phosphate and thiophosphate flavone analogs. Drug Dev Res 2015;76:450–62.
- Kozak W, Daśko M, Wołos A, et al. Synthesis and steroid sulfatase inhibitory activities of N-alkanoyl tyramine phosphates and thiophosphates. RSC Adv 2015;5:32594–603.
- Daśko M, Masłyk M, Kubiński K, et al. Synthesis and steroid sulfatase inhibitory activities of N-phosphorylated 3-(4-aminophenyl)-coumarin-7-O-sulfamates. Med Chem Commun 2016;7:1146–50.
- Daśko M, Demkowicz S, Biernacki K, et al. Novel steroid sulfatase inhibitors based on N-thiophosphorylated 3-(4-aminophenyl)-coumarin-7-O-sulfamates. Drug Dev Res 2019;80:857–866.
- Woo LWL, Sutcliffe OB, Bubert C, et al. First dual aromatase-steroid sulfatase inhibitors. J Med Chem 2003;46:3193–6.
- Woo LWL, Jackson T, Putey A, et al. Highly potent first examples of dual aromatase-steroid sulfatase inhibitors based on biphenyl template. J Med Chem 2010;53:2155–70.
- Woo LWL, Bubert C, Purohit A, et al. Hybrid dual aromatase-steroid sulfatase inhibitors with exquisite picomolar inhibitory activity. ACS Med Chem Lett 2011;2:243–7.
- Woo L, Wood PM, Bubert C, et al. Synthesis and structure-activity relationship studies of derivatives of the dual aromatase-sulfatase inhibitor 4-{[(4-cyanophenyl)(4H-1,2,4-triazol-4-yl)amino]methyl}phenyl sulfamate. ChemMedChem 2013;8:779–99.
- Wood PM, Woo LWL, Labrosse JR, et al. Chiral aromatase and dual aromatase-steroid sulfatase inhibitors from the letrozole template: synthesis, absolute configuration, and in vitro activity. J Med Chem 2008;51:4226–38.
- Jackson T, Woo LWL, Trusselle MN, et al. Dual aromatase-sulfatase inhibitors based on the anastrozole template: synthesis, in vitro SAR, molecular modeling and in vivo activity. Org Biomol Chem 2007;5:2940–52.
- Salah M, Abdelsamie AS, Frotscher M. First dual inhibitors of steroid sulfatase (STS) and 17β-hydroxysteroid dehydrogenase type 1 (17β-HSD1): designed multiple ligands as novel potential therapeutics for estrogen-dependent diseases. J Med Chem 2017;60:4086–92.
- Bacsa I, Herman BE, Jójárt R, et al. Synthesis and structure-activity relationships of 2- and/or 4-genated 13β- and 13α-estrone derivatives as enzyme inhibitors of estrogen biosynthesis. J Enzyme Inhib Med Chem 2018;33:1271–82.
- Potter BVL, Reed LWL, Woo LWL, et al. Sulfamic acid ester compounds useful in the inhibition of steroid sulphatase activity and aromatase activity. US patent 20070213383; 2007.
- Woo LWL, Bubert C, Sutcliffe OB, et al. Dual aromatase-steroid sulfatase inhibitors. J Med Chem 2007;50:3540–60.
- Wood PM, Woo LWL, Labrosse JR, et al. Bicyclic derivatives of the potent dual aromatase- steroid sulfatase inhibitor 2-bromo-4-{[(4-cyanophenyl)(4H-1,2,4-triazol-4-yl)amino]methyl}phenylsulfamate: synthesis, SAR, crystal structure, and in vitro and in vivo activities. ChemMedChem 2010;5:1577–93.
- Wood PM, Woo LWL, Thomas MP, et al. Aromatase and dual aromatase-steroid sulfatase inhibitors from the Letrozole and Vorozole templates. ChemMedChem 2011;6:1423–38.
- Lafay J, Rondot B, Bonnet P, et al. 1-N-phenyl-amino-1h-imidazole derivatives and pharmaceutical compositions containing them. US patent 20070112009; 2007.
- Woo LWL, Jackson T, Bubert C, et al. Phenyl-sulfamates as aromatase inhibitors. US patent 7763642; 2010.
- Potter BVL, Reed MJ, Woo LWL, et al. Oestrogen-17-suphamates as inhibitors of steroid sulphatase. US Patent 8030296; 1996.
- Tagg SLC, Foster PA, Leese MP, et al. 2-Methoxyestradiol-3,17-O,O-bis-sulphamate and 2-deoxy-D-glucose in combination: a potential treatment for breast and prostate cancer. Br J Cancer 2008;99:1842–48.
- Raobaikady B, Purohit A, Chander SK, et al. Inhibition of MCF-7 breast cancer cell proliferation and in vivo steroid sulphatase activity by 2-methoxyoestradiol-bis-sulphamate. J Steroid Biochem Mol Biol 2003;84:351–8.
- Leese MP, Leblond B, Smith A, et al. 2-substituted estradiol bis-sulfamates, multitargeted antitumor agents: synthesis in vitro SAR, protein crystallography, and in vivo activity. J Med Chem 2006;49:7683–96.
- Utsumi T, Leese MP, Chander SK, et al. The effects of 2-methoxyestrogen sulphamates on the in vitro and in vivo proliferation of breast cancer cells. J Steroid Biochem Mol Biol 2005;94:219–27.
- Ireson CR, Chander SK, Purohit A, et al. Pharmacokinetics and efficacy of 2-methoxyoestradiol and 2-methoxyoestradiol-bis-sulphamate in vivo in rodents. Br J Cancer 2004;90:932–7.
- Thomas MP, Potter B. Estrogen O-sulfamates and their analogues: clinical steroid sulfatase inhibitors with broad potential. J Steroid Biochem Mol Biol 2015;153:160–9.
- Newman SP, Foster PA, Stengel C, et al. STX140 is efficacious in vitro and in vivo in taxane-resistant breast carcinoma cells. Clin Cancer Res 2008;14:597–606.
- Meyer-Losic F, Newman SP, Day JM, et al. STX140, but not Paclitaxel, inhibits mammary tumor initiation and progression in C3(1)/SV40T/t-antigen transgenic mice. PLoS One 2013;8:e80305.
- Wang M, Xu L, Mingzhang G, et al. Synthesis of 2-[11C]methoxy-3,17β-O,O-bis(sulfamoyl)estradiol as a new potential PET agent for imaging of steroid sulfatase (STS) in cancers. Steroids 2012;77:864–70.
- Jourdan F, Leese MP, Dohle W, et al. Synthesis, antitubulin and antiproliferative SAR of analogues of 2-methoxyestradiol-3,17-O,O-bis sulfamate. J Med Chem 2010;53:2942–51.
- Jourdan F, Leese MP, Dohle W, et al. Structure-activity relationships of C-17-substituted estratriene-3-O-sulfamates as anticancer agents. J Med Chem 2011;54:4863–79.
- Potter BVL, Reed MJ, Lebrond B, et al. Aryl linker derivatised estrogen 3-sulfamates as inhibitors of steroid sulfatase. US patent 7067503; 2006.
- Poirier D, Roy J, Maltais R, et al. A potent inhibitor of steroid sulfatase (EM-1913) blocks tumor growth in nude mice (MCF-7 xenograft). Curr Enzyme Inhib 2015;11:65–73.
- Ouellet É, Maltais R, Ouellet C, et al. Investigation of a tetrahydroisoquinoline scaffold as dual-action steroid sulfatase inhibitors generated by parallel solid phase synthesis. Med Chem Commun 2013;4:681–92.
- Ouellet C, Ouellet É, Poirier D. In vitro evaluation of a tetrahydroisoquinoline derivative as a steroid sulfatase inhibitor and a selective estrogen receptor modulator. Invest New Drugs 2015;33:95–103.
- Ouellet C, Maltais R, Ouellet É, et al. Discovery of a sulfamate-based steroid sulfatase inhibitor with intrinsic selective estrogen receptor modulator properties. Eur J Med Chem 2016;119:169–82.
- Poirier D, Roy J, Maltais R, et al. Antisulfatase, osteogenic, and anticancer activities of steroid sulfatase inhibitor EO-33 in mice. J Med Chem 2019;62:5512–21.
- Kaise A, Ohta K, Endo Y. Novel p-carborane-containing multitarget anticancer agents inspired by the metabolism of 17β-estradiol. Bioorg Med Chem 2017;25:6371–78.
- Kaise A, Ohta K, Shirata C, et al. Design and synthesis of p-carborane-containing sulfamates as multitarget anti-breast cancer agents. Bioorg Med Chem 2017;25:6417–26.