
Abstract
Indoleamine 2,3-dioxygenase 1 (IDO1) as a key rate-limiting enzyme in the kynurenine pathway of tryptophan metabolism plays an important role in tumour immune escape. Herein, a variety of secondary sulphonamides were synthesised and evaluated in the HeLa cell-based IDO1/kynurenine assay, leading to the identification of new IDO1 inhibitors. Among them, compounds 5d, 5l and 8g exhibited the strongest inhibitory effect with significantly improved activity over the hit compound BS-1. The in vitro results showed that these compounds could restore the T cell proliferation and inhibit the differentiation of naïve CD4+ T cell into highly immunosuppressive FoxP3+ regulatory T (Treg) cell without affecting the viability of HeLa cells and the expression of IDO1 protein. Importantly, the pharmacodynamic assay showed that compound 5d possessed potent antitumour effect in both CT26 and B16F1 tumours bearing immunocompetent mice but not in immunodeficient mice. Functionally, subsequent experiments demonstrated that compound 5d could effectively inhibit tumour cell proliferation, induce apoptosis, up-regulate the expression of IFN-γ and granzyme B, and suppress FoxP3+ Treg cell differentiation, thereby activate the immune system. Thus, compound 5d could be a potential and efficacious agent for further evaluation.
1. Introduction
With the increasing recognition of the interaction between the immune system and tumour cells, immunotherapy has gained tremendous attention in the field of cancer researchCitation1,Citation2. The recent clinical successes of immune checkpoint inhibitors and chimeric antigen receptor T cell (CAR-T) therapy have dramatically changed the landscape of the cancer treatmentCitation3–6. To date, the US Food and Drug Administration (FDA) has approved six immune checkpoint inhibitors blocking cytotoxic T lymphocyte-associated antigen 4 (CTLA-4), programmed cell death protein 1 (PD-1) or programmed cell death ligand 1 (PD-L1), and the second CAR-T cell therapy for the treatment of solid and hematological malignancies. However, their limited response rate and immune-related adverse effects call for a need to develop other immunotherapy options to solve those problemsCitation5–7.
Indoleamine 2,3-dioxygenase 1 (IDO1) is a haem-containing enzyme that catalyses the initial and rate-limiting step in the catabolism of the essential amino acid tryptophan along the kynurenine pathwayCitation8. IDO1 plays an important role in the regulation of local inflammation and immune toleranceCitation9. In cancer, aberrant activation of IDO1 results in suppression of antitumour immunity. The depletion of tryptophan and the accumulation of kynurenine and its downstream metabolites in the tumour microenvironment have been demonstrated to induce effector T cell anergy and enhance regulatory T (Treg) cell function, which eventually lead to an immunosuppressive milieu that helps tumour cells evade detection and killing by the immune systemCitation10. Tryptophan depletion has been implicated in the activation of general control non-derepressible 2 (GCN2) and suppression of the mechanistic target of rapamycin kinase (mTOR) pathwaysCitation11,Citation12. Furthermore, kynurenine and other tryptophan metabolites can activate the aryl hydrocarbon receptor (AHR)Citation13. Indeed, high expression of IDO1 in tumour cells has been shown to correlate with worse clinical prognosis in patients with a variety of cancers, including colorectal, ovarian, endometrial, and hepatocellular carcinomasCitation14–18. Moreover, preclinical studies have demonstrated that IDO inhibitors can effectively restore antitumour immunity and have the potential to synergize with chemotherapy, radiotherapy, and other immunotherapies, including anti-CTLA-4, anti-PD-1 and anti-PD-L1 antibodiesCitation19–22. Hence, these findings have motivated interest in developing IDO1 inhibitors for cancer immunotherapy.
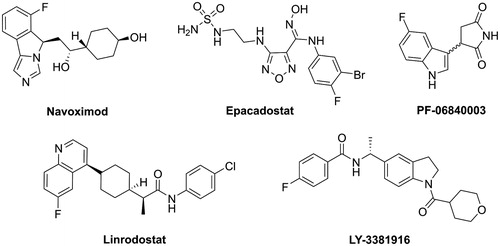
So far, a number of IDO1 inhibitors have been identified from high-throughput screeningCitation23,Citation24, structure-based designCitation25–27, and natural product screeningCitation28,Citation29. Several of them, including, navoximod, epacadostat, PF-06840003, linrodostat, LY-3381916 and KHK2455 (structure not disclosed), have entered clinical development as monotherapy and in combination with other immune therapies or chemotherapy ()Citation30. However, the FDA has not yet approved IDO inhibitors for the treatment of malignancies to date. We therefore need to continue to identify novel IDO1 inhibitors.
Figure 1. Structure of representative IDO1 clinical candidates.
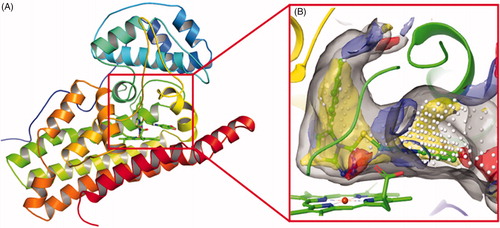
In recent years, we have been working on the development of novel IDO1 inhibitorsCitation31–34. From our previous study on the interactions between IDO1 and imidazole derivatives, it has been found that metal chelation, hydrogen bonding, and hydrophobic interactions between haem and the ligand contribute a large proportion to the binding affinity of the inhibitorCitation31. Furthermore, the human IDO1 crystal structures in complex with the ligand Amg-1 (PDB ID: 4PK5) revealed that IDO1 is characterised by a small and highly lipophilic active site including two pockets (A and B), implying that the hydrophobic interaction may account for a large part of protein-ligand nonbonding interactions (). In addition, Muller’s team demonstrated that as the charge on the coordinating atom decreased, the binding affinity to the haem iron increased, which would be possible to improve IDO1 inhibitory activityCitation35. These findings provide directions for improving the binding affinity of novel IDO1 inhibitors.
Figure 2. (A) The co-crystal of IDO1 (PDB ID: 4PK5). (B) Site maps generated using Sitemap: the ligand was Amg-1, white points represented the new identified active region, as well as yellow, blue and red solid maps represented hydrophobic regions, hydrogen bond donors and acceptors, respectively.
In this study, we screened our in-house compound library using the HeLa cell-based IDO1/kynurenine assay, and found that the secondary sulphonamide compound BS-1 showed inhibitory activity against IDO1 with an IC50 value of 48.42 µM. As well known, bezenesulfonamide is a common pharmacophore that is the basis of several groups of drugs, such as sulpha antibiotics, thiazide diuretics and nonsteroidal antiinflammatory drug (NSAID) celecoxibCitation36,Citation37. Moreover, the sulphonyl is a heme-binding function that coordinates to the haem iron of IDO1Citation38. Therefore, the hit compound BS-1 was further modified, as well as a variety of secondary sulphonamides were synthesised and evaluated in the HeLa cell-based IDO1/kynurenine assay. The resulting compounds 5d, 5l and 8g showed high inhibitory activity without affecting the cell viability and the IDO1 protein expression. Subsequent in vitro and in vivo experiments demonstrated that compound 5d could exert potent antitumour effects by activating the mouse immune system.
2. Material and methods
2.1. Chemistry
Melting points were determined on a RDCSY-I capillary apparatus and were uncorrected. Allmaterials used were commercially available and used as supplied. HG/T2354-92 silica gel 60 F254 sheets were used for analytical thin-layer chromatography (TLC). Column chromatography was performed on silica gel (300–400 mesh). 1H NMR spectra were recorded on a Bruker AV-300 spectrometer. Chemical shifts (δ) were given in parts per million (ppm) relative to the solvent peak. J values are in Hz. Chemical shifts are expressed in ppm downfield from internal standard TMS. Mass spectra (MS) were measured using a Thermo Scientific iCAP RQ ICP-MS. All the reagents and solvents were reagent grade and were used without further purification unless otherwise specified.
2.1.1. General preparation of compounds 3a-i
To a solution of substituted aniline (0.97 mmol) in DCM (15 ml) was added triethylamine (1.22 mmol)Citation39. A solution of 4-acrylamidobenzenesulfonyl chloride (0.81 mmol) in DCM (10 ml) was added dropwise to the mixture at 0 °C. The reaction was stirred at room temperature for 4 h. The solvent was evaporated under reduced pressure and the crude product was recrystallization to afford target compounds 3a-i.
2.1.1.1. N-(4-(N-Phenylsulfamoyl)phenyl)acetamide (3a)
White solid. Yield 90%. Mp 204–206 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.17 (s, 1H), 10.04 (s, 1H), 7.54 (s, 4H), 7.08 (t, J = 7.8 Hz, 2H), 6.94–6.84 (m, 3H), 1.91 (s, 3H). MS (EI) m/z 289.1 [M-H]−.
2.1.1.2. N-(4-(N-(p-Tolyl)sulfamoyl)phenyl)acetamide (3b)
White solid. Yield 87%. Mp > 250 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.45 (s, 1H), 9.89 (s, 1H), 7.69 (d, J = 8.1 Hz, 2H), 7.57 (d, J = 8.4 Hz, 2H), 7.08 (d, J = 7.8 Hz, 2H), 6.73 (d, J = 7.8 Hz, 2H), 2.19 (s, 3H), 1.92 (s, 3H). MS (EI) m/z 303.1 [M-H]−.
2.1.1.3. N-(4-(N-(4-Isopropylphenyl)sulfamoyl)phenyl)acetamide (3c)
Light yellow solid, Yield 90%, Mp 186–188 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.16 (s, 1H), 9.91 (s, 1H), 7.54 (s, 4H), 6.95 (d, J = 8.1 Hz, 2H), 6.94 (d, J = 8.1 Hz, 2H), 2.65–2.58 (m, 1H),1.92 (d, J = 2.4 Hz, 3H), 0.97 (dd, J = 2.1, 2.7 Hz, 6H). MS (EI) m/z 331.1 [M-H]−.
2.1.1.4. N-(4-(N-(3-Isopropylphenyl)sulfamoyl)phenyl)acetamide (3d)
White solid. Yield 90%. Mp 158–160 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.17 (s, 1H), 9.95 (s, 1H), 7.55 (s, 4H), 6.98 (t, J = 7.7 Hz, 1H), 6.79–6.72 (m, 3H), 2.63–2.58 (m, 1H), 1.92 (s, 3H), 0.95 (d, J = 6.9 Hz, 6H). MS (EI) m/z 331.1 [M-H]−.
2.1.1.5. N-(4-(N-Benzylsulfamoyl)phenyl)acetamide (3e)
White solid. Yield 90%. Mp 140–142 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.20 (s, 1H), 7.89 (t, J = 6.3 Hz, 1H), 7.64–7.57 (m, 4H), 7.18–7.07 (m, 5H), 3.81 (d, J = 6.3 Hz, 2H), 1.96 (s, 3H). MS (EI) m/z 303.1 [M-H]−.
2.1.1.6. N-(4-(N-(4-Chlorobenzyl)sulfamoyl)phenyl)acetamide (3f)
White solid. Yield 90%. Mp 172–174 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.20 (s, 1H), 7.95 (t, J = 6.5 Hz, 1H), 7.63–7.56 (m, 4H), 7.17 (dd, J = 8.1, 8.4 Hz, 4H), 3.81 (d, J = 6.3 Hz, 2H), 1.95 (s, 3H). MS (EI) m/z 337.1 [M-H]−.
2.1.1.7. N-(4-(N-(4-(Trifluoromethyl)benzyl)sulfamoyl)phenyl)acetamide (3g)
White solid. Yield 89%. Mp 186–188 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.19 (s, 1H), 8.04 (t, J = 6.0 Hz, 1H), 7.62–7.56 (m, 4H), 7.50 (d, J = 8.1 Hz, 2H), 7.33 (d, J = 8.1 Hz, 2H), 3.93 (d, J = 5.7 Hz, 2H), 1.95 (s, 3H). MS (EI) m/z 371.1 [M-H]−.
2.1.1.8. N-(4-(N-(3-(Trifluoromethyl)benzyl)sulfamoyl)phenyl)acetamide (3h)
White solid. Yield 88%. Mp 154–156 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.19 (s, 1H), 8.05 (t, J = 6.3 Hz, 1H), 7.61–7.54 (m, 4H), 7.46–7.35 (m, 4H), 3.95 (d, J = 6.3 Hz, 2H), 1.95 (s, 3H). MS (EI) m/z 371.1 [M-H]−.
2.1.1.9. N-(4-(N-Phenethylsulfamoyl)phenyl)acetamide (3i)
White solid. Yield 85%. Mp 130–132 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.19 (s, 1H), 7.59 (q, J = 8.5 Hz, 4H), 7.46 (t, J = 5.9 Hz, 1H), 7.16–7.00 (m, 5H), 2.82–2.75 (m, 2H), 2.52 (t, J = 7.5 Hz, 2H), 1.95 (s, 3H). MS (EI) m/z 317.2 [M-H]−.
2.1.2. General preparation of compounds 4a-f
To a solution of compounds 3 (0.68 mmol) in ethanol (15 ml) was added hydrochloric acid (1 ml)Citation39. Then the mixture was stirred at 70 °C for 12 h. After the reaction was completed, the solvent was evaporated under reduced pressure. Water was added and the pH was adjusted to 7–8 with saturated NaHCO3 solution. The aqueous phase was extracted with EtOAc (3 × 30 ml). The combined organic layers were washed with water, brine, and dried. The solvent was removed in vacuo and the crude product was recrystallization to afford target compounds 4a-f.
2.1.2.1. 4-Amino-N-phenylbenzenesulfonamide (4a)
Light yellow solid. Yield 89%. Mp 188–190 °C. 1H NMR (300 MHz, DMSO-d6) δ 9.81 (s, 1H), 7.34 (d, J = 8.6 Hz, 2H), 7.15 (t, J = 7.8 Hz, 2H), 7.01 (d, J = 7.7 Hz, 2H), 6.94 (d, J = 7.3 Hz, 1H), 6.48 (d, J = 8.7 Hz, 2H), 5.92 (s, 2H). MS (EI) m/z 247.1 [M-H]−.
2.1.2.2. 4-Amino-N-(p-tolyl)benzenesulfonamide (4b)
White solid. Yield 89%. Mp 188–190 °C. 1H NMR (300 MHz, DMSO-d6) δ 9.55 (s, 1H), 7.21 (d, J = 8.5 Hz, 2H), 6.83 (dd, J = 8.0, 8.3 Hz, 4H), 6.37 (d, J = 8.6 Hz, 2H), 5.82 (s, 2H), 2.03 (s, 3H). MS (EI) m/z 261.1 [M-H]−.
2.1.2.3. 4-Amino-N-benzylbenzenesulfonamide (4c)
White solid. Yield 93%. Mp 116–118 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.53 (t, J = 6.1 Hz, 1H), 7.32 (d, J = 8.6 Hz, 2H), 7.09–7.18 (m, 5H), 6.49 (d, J = 8.6 Hz, 2H), 3.73 (d, J = 5.9 Hz, 2H). MS (EI) m/z 261.1 [M-H]−.
2.1.2.4. 4-Amino-N-(4-chlorobenzyl)benzenesulfonamide (4d)
White solid. Yield 95%. Mp 172–174 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.66 (t, J = 6.2 Hz, 1H), 7.21–7.40 (m, 6H), 6.56 (d, J = 8.4 Hz, 2H), 5.91 (s, 2H), 3.83 (d, J = 6.2 Hz, 2H). MS (EI) m/z 295.1 [M-H]−.
2.1.2.5. 4-Amino-N-(3-chlorobenzyl)benzenesulfonamide (4e)
White solid. Yield 90%. Mp 119–121 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.70 (s, 1H), 7.42 (d, J = 8.6 Hz, 2H), 7.36–7.11 (m, 4H), 6.60 (d, J = 8.4 Hz, 2H), 5.92 (s, 2H), 3.90 (d, J = 5.0 Hz, 2H). MS (EI) m/z 295.1 [M-H]−.
2.1.2.6. 4-Amino-N-phenethylbenzenesulfonamide (4f)
White solid. Yield 90%. Mp 138–140 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.37 (d, J = 8.4 Hz, 2H), 7.14–7.25 (m, 5H), 6.56 (d, J = 8.5 Hz, 2H), 2.82 (m, 2H), 2.60 (t, J = 7.4 Hz, 2H). MS (EI) m/z 275.1 [M-H]−.
2.1.3. General preparation of compounds 5a-m
To a solution of compounds 4 (0.55 mmol) in DCM (15 ml) was added TEA (1.1 mmol). Then acryloyl chloride (0.61 mmol) was added dropwise to the mixture at 0 °C for 0.5 h. The reaction was stirred at rt overnight. After the reaction was completed, the water was added to quench the reaction. The mixture was extracted with DCM to afford the crude product. The crude residue was recrystallization to afford target compounds 5a-m.
2.1.3.1. N-(4-(N-Benzylsulfamoyl)phenyl)acrylamide (5a)
White solid. Yield 75%. Mp 116–118 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.50 (s, 1H), 8.02 (t, J = 6.2 Hz, 1H), 7.81 (d, J = 8.7 Hz, 2H), 7.72 (d, J = 8.7 Hz, 2H), 7.27–7.16 (m, 5H), 6.48–6.39 (m, 1H), 6.28 (d, J = 15.6 Hz, 1H), 5.79 (d, J = 9.9 Hz, 1H), 3.92 (d, J = 6.0 Hz, 2H). MS (EI) m/z 315.1 [M-H]−.
2.1.3.2. N-(4-(N-(4-Methoxybenzyl)sulfamoyl)phenyl)acrylamide (5b)
White solid. Yield 81%. Mp 156–158 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.38 (s, 1H), 7.82 (t, J = 6.0 Hz, 1H), 7.70 (d, J = 8.7 Hz, 2H), 7.61 (d, J = 8.7 Hz, 2H), 7.00 (d, J = 8.4 Hz, 4H), 6.69 (d, J = 8.7 Hz, 2H), 6.38–6.29 (m, 1H), 6.18 (d, J = 17.1 Hz, 1H), 5.69 (d, J = 9.9 Hz, 1H), 3.75 (d, J = 6.0 Hz, 2H), 3.57 (s, 3H). MS (EI) m/z 345.2 [M-H]−.
2.1.3.3. N-(4-(N-(4-Chlorobenzyl)sulfamoyl)phenyl)acrylamide (5c)
White solid. Yield 80%. Mp 190–192 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.40 (s, 1H), 7.98 (t, J = 6.3 Hz, 1H), 7.70 (d, J = 8.4 Hz, 2H), 7.61 (d, J = 8.4 Hz, 2H), 7.17 (dd, J = 8.4, 8.1 Hz, 4H), 6.37–6.28 (m, 1H), 6.18 (d, J = 16.5 Hz, 1H), 5.69 (d, J = 2.7 Hz, 1H), 3.82 (d, J = 6.0 Hz, 2H). MS (EI) m/z 349.1 [M-H]−.
2.1.3.4. N-(4-(N-(3-Chlorobenzyl)sulfamoyl)phenyl)acrylamide (5d)
White solid. Yield 51%. Mp 157–159 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.58 (s, 1H), 8.15 (s, 1H), 7.83 (dd, J = 9.0, 3.2 Hz, 2H), 7.78–7.65 (m, 2H), 7.47–7.06 (m, 4H), 6.44 (dd, J = 9.8, 3.4 Hz, 1H), 6.37–6.22 (m, 1H), 5.88 –5.74 (m, 1H), 3.99 (d, J = 3.3 Hz, 2H). 13C NMR (126 MHz, DMSO) δ 164.08, 142.99, 140.90, 135.28, 133.36, 131.94, 130.53, 128.38, 128.13, 127.79, 127.47, 126.65, 119.53, 45.86. MS (EI) m/z 349.1 [M-H]−.
2.1.3.5. N-(4-(N-Phenethylsulfamoyl)phenyl)acrylamide (5e)
White solid. Yield 75%. Mp 154–156 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.39 (s, 1H), 7.70 (d, J = 8.1 Hz, 2H), 7.61 (d, J = 7.5 Hz, 2H), 7.49 (s, 1H), 7.09 (m, 4H), 6.37–6.28 (m, 1H), 6.17 (d, J = 15.6 Hz, 1H), 5.69 (d, J = 8.7 Hz, 1H), 2.80 (s, 1H), 2.53 (s, 1H). MS (EI) m/z 353.1 [M + Na]+.
2.1.3.6. N-(4-(N-(4-(Trifluoromethyl)benzyl)sulfamoyl)phenyl)acrylamide (5f)
White solid. Yield 80%. Mp 198–200 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.38 (s, 1H), 8.07 (s, 1H), 7.69 (d, J = 7.8 Hz, 2H), 7.61 (d, J = 8.1 Hz, 2H), 7.50 (d, J = 7.2 Hz, 4H), 7.33 (d, J = 7.2 Hz, 2H), 6.37–6.15 (m, 1H), 6.18 (d, J = 16.5 Hz, 1H), 5.69 (d, J = 9.9 Hz, 1H), 3.94 (d, J = 4.5 Hz, 2H). MS (EI) m/z 407.1 [M + Na]+.
2.1.3.7. N-(4-(N-(3-(Trifluoromethyl)benzyl)sulfamoyl)phenyl)acrylamide (5g)
White solid. Yield 77%. Mp 152–154 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.55 (s, 1H), 8.19 (t, J = 6.2 Hz, 1H), 7.79 (d, J = 8.7 Hz, 2H), 7.69 (d, J = 8.7 Hz, 2H), 7.55–7.44 (m, 4H), 6.49–6.40 (m, 1H), 6.27 (d, J = 16.8 Hz, 1H), 5.78 (d, J = 3.9 Hz, 1H), 4.06 (d, J = 6.0 Hz, 2H). MS (EI) m/z 383.1 [M-H]−.
2.1.3.8. N-(4-(N-(4-Fluorobenzyl)sulfamoyl)phenyl)acrylamide (5h)
White solid. Yield 82%. Mp 178–180 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.40 (s, 1H), 7.95 (t, J = 6.0 Hz, 1H), 7.70 (d, J = 8.7 Hz, 2H), 7.61 (d, J = 8.4 Hz, 2H), 7.14 (t, J = 6.8 Hz, 1H), 6.97 (t, J = 8.6 Hz, 2H), 6.37–6.28 (m, 1H), 6.18 (d, J = 17.1 Hz, 1H), 5.70 (d, J = 9.9 Hz, 1H), 3.82 (d, J = 6.0 Hz, 2H). MS (EI) m/z 357.1 [M + Na]+.
2.1.3.9. N-(4-(N-(3-Fluorobenzyl)sulfamoyl)phenyl)acrylamide (5i)
White solid. Yield 80%. Mp 152–154 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.39 (s, 1H), 8.01 (t, J = 6.3 Hz, 1H), 7.70 (d, J = 8.4 Hz, 2H), 7.61 (d, J = 5.7 Hz, 2H), 7.22–7.15 (m, 1H), 6.95–6.89 (m, 3H), 6.37–6.28 (m, 1H), 6.18 (d, J = 16.8 Hz, 1H), 5.69 (d, J = 9.9 Hz, 1H), 3.87 (d, J = 6.3 Hz, 2H). MS (EI) m/z 357.1 [M + Na]+.
2.1.3.10. N-(4-(N-(2-Chlorobenzyl)sulfamoyl)phenyl)acrylamide (5j)
White solid. Yield 78%. Mp 178–180 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.41 (s, 1H), 8.00 (s, 1H), 7.71 (d, J = 7.5 Hz, 2H), 7.64 (d, J = 8.7 Hz, 2H), 7.28–7.15 (m, 4H), 6.39–6.29 (m, 1H), 6.18 (d, J = 17.4 Hz, 1H), 5.69 (d, J = 2.7 Hz, 1H), 3.91 (d, J = 4.5 Hz, 2H). MS (EI) m/z 349.1 [M-H]−.
2.1.3.11. N-(4-(N-(4-Bromobenzyl)sulfamoyl)phenyl)acrylamide (5k)
White solid. Yield 83%. Mp 194–196 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.44 (s, 1H), 7.98 (t, J = 6.2 Hz, 1H), 7.71 (d, J = 8.4 Hz, 2H), 7.61 (d, J = 8.7 Hz, 2H), 7.34 (d, J = 7.8 Hz, 2H), 7.07 (d, J = 7.8 Hz, 2H), 6.38–6.29 (m, 1H), 6.17 (d, J = 15.9 Hz, 1H), 5.69 (d, J = 10.2 Hz, 1H), 3.80 (d, J = 6.3 Hz, 2H). MS (EI) m/z 393.1, 395.1 [M-H]−.
2.1.3.12. N-(4-(N-(3-Bromobenzyl)sulfamoyl)phenyl)acrylamide (5l)
White solid. Yield 81%. Mp 156–158 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.40 (s, 1H), 8.01 (t, J = 6.0 Hz, 1H), 7.70 (d, J = 8.4 Hz, 2H), 7.60 (d, J = 8.7 Hz, 2H), 7.26 (s, 2H), 7.11 (d, J = 3.9 Hz, 2H), 6.38–6.29 (m, 1H), 6.18 (d, J = 15.9 Hz, 1H), 5.69 (d, J = 10.2 Hz, 1H), 3.86 (d, J = 6.0 Hz, 2H). MS (EI) m/z 393.1, 395.1 [M-H]−.
2.1.3.13. N-(4-(N-(3,4-Dichlorobenzyl)sulfamoyl)phenyl)acrylamide (5m)
White solid. Yield 82%. Mp 148–150 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.41 (s, 1H), 8.05 (s, 1H), 7.65 (d, J = 29.4 Hz, 4H), 7.34 (d, J = 30.0 Hz, 2H), 7.11 (s, 1H), 6.38–6.15 (m, 2H), 5.70 (s, 1H), 3.82 (d, J = 6.0 Hz, 2H). MS (EI) m/z 383.1 [M-H]−.
2.1.4. General preparation of N-phenylacrylamide 6
To a solution of aniline (0.043 mol) in DCM (50 ml) was added TEA (0.086 mol). Then acryloyl chloride (0.047 mol) was added dropwise to the mixture at 0 °C for 0.5 h. The reaction was allowed to stir at rt overnight. After the reaction was completed, the water was added to quench the reaction. The mixture was extracted with DCM to afford the crude product that was purified by flash column chromatography on silica gel to afford N-phenylacrylamide 6 (white solid, Yield 72.8%)Citation40. 1H NMR (300 MHz, Chloroform-d) δ 10.14 (s, 1H), 7.67 (d, J = 8.1 Hz, 2H), 7.32 (t, J = 7.8 Hz, 2H), 7.07 (t, J = 7.2 Hz, 1H), 6.44 (dd, J = 16.8, 9.9 Hz, 1H), 6.26 (dd, J = 16.8, 1.5 Hz, 1H), 5.75 (dd, J = 9.9, 1.5 Hz, 1H).
2.1.5. General preparation of 4-acrylamidobenzenesulfonyl chloride 7
N-phenylacrylamide (0.031 mol) was added slowly to sulphuryl chloride (15 ml) at 0 °C for more than 0.5 h. The reaction was stirred at 70 °C for 1 h. After the reaction was completed, the mixture was poured into ice water slowly to quench the reaction. The mixture was stirred at rt for 1 h and the solid was filtered-off and washed with water (2 × 50 ml). The crude solid was dissolved in CH2Cl2 (50 ml), the organic layer was washed with brine, and the solvent was evaporated under reduced pressure to afford 4-acrylamidobenzenesulfonyl chloride as off-white solid (Yield 72.8%).
2.1.6. General preparation of compounds 8a-j
To a solution of substituted aniline or heterocyclic amine (0.98 mmol) in DCM (15 ml) was added pyridine (1.22 mmol). A solution of 4-acrylamidobenzenesulfonyl chloride (0.81 mmol) in DCM (10 ml) was added dropwise to the mixture at 0 °C. The reaction was stirred at rt for 4 h. The solvent was evaporated under reduced pressure and the crude product was recrystallization to afford target compounds 8a-j.
2.1.6.1. N-(4-(N-Phenylsulfamoyl)phenyl)acrylamide (8a)
White solid. Yield 72%. Mp 212–213 °C. 1H NMR (300 MHz, Chloroform-d) δ 10.50 (s, 1H), 10.19 (s, 1H), 7.74 (dd, J = 23.7, 8.7 Hz, 4H), 7.22 (t, J = 7.8 Hz, 2H), 7.09–6.98 (m, 3H), 6.42 (dd, J = 16.8, 9.9 Hz, 1H), 6.28 (d, J = 15.3 Hz, 1H), 5.81 (d, J = 10.2 Hz, 1H). MS (EI) m/z 301.1 [M-H]−.
2.1.6.2. N-(4-(N-(p-Tolyl)sulfamoyl)phenyl)acrylamide (8b)
White solid. Yield 79%. Mp 230–232 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.46 (s, 1H), 9.98 (s, 1H), 7.75 (d, J = 8.7 Hz, 2H), 7.65 (d, J = 8.7 Hz, 2H), 6.96 (dd, J = 19.5, 8.4 Hz, 4H), 6.40 (dd, J = 17.1, 9.9 Hz, 1H), 6.26 (d, J = 17.1 Hz, 1H), 5.82–5.70 (m, 1H), 2.15 (s, 3H). MS (EI) m/z 315.1 [M-H]−.
2.1.6.3. N-(4-(N-(4-Methoxyphenyl)sulfamoyl)phenyl)acrylamide (8c)
White solid. Yield 80%. Mp 234–235 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.46 (s, 1H), 9.77 (s, 1H), 7.75 (d, J = 8.7 Hz, 2H), 7.60 (d, J = 8.7 Hz, 2H), 6.94 (d, J = 9.0 Hz, 2H), 6.77 (d, J = 8.7 Hz, 2H), 6.41 (dd, J = 17.1, 9.9 Hz, 1H), 6.26 (d, J = 15.3 Hz, 1H), 5.78 (d, J = 9.6 Hz, 1H), 3.64 (s, 3H). MS (EI) m/z 331.1 [M-H]−.
2.1.6.4. N-(4-(N-(3-Methoxyphenyl)sulfamoyl)phenyl)acrylamide (8d)
White solid. Yield 77%. Mp 197–199 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.38 (s, 1H), 10.09 (s, 1H), 7.63 (dd, J = 21.3, 8.7 Hz, 4H), 6.98 (t, J = 8.4 Hz, 1H), 6.53–6.51 (m, 2H), 6.45 (d, J = 8.4 Hz, 1H), 6.29 (dd, J = 16.8, 9.9 Hz, 1H), 6.15 (d, J = 16.8 Hz, 1H), 5.67 (d, J = 9.9 Hz, 1H), 3.52 (s, 3H). MS (EI) m/z 331.1 [M-H]−.
2.1.6.5. N-(4-(N-(3-Fluorophenyl)sulfamoyl)phenyl)acrylamide (8e)
White solid. Yield 81%. Mp 212–215 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.39–10.37 (m, 2H), 7.68 (d, J = 8.7 Hz, 2H), 7.60 (d, J = 8.7 Hz, 2H), 7.12 (t, J = 8.1 Hz, 1H), 6.97–6.91 (m, 3H), 6.29 (dd, J = 16.8, 9.6 Hz, 1H), 6.15 (d, J = 15.9 Hz, 1H), 5.68 (d, J = 9.3 Hz, 1H). MS (EI) m/z 319.1 [M-H]−.
2.1.6.6. N-(4-(N-(3-Chlorophenyl)sulfamoyl)phenyl)acrylamide (8f)
White solid. Yield 83%. Mp 217–219 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.39 (d, J = 8.1 Hz, 2H), 7.69 (d, J = 9.0 Hz, 2H), 7.61 (d, J = 9.0 Hz, 2H), 7.12 (t, J = 7.8 Hz, 1H), 6.97–6.92 (m, 3H), 6.30 (dd, J = 17.1, 10.2 Hz, 1H), 6.16 (d, J = 16.8 Hz, 1H), 5.70–5.62 (m, 1H). MS (EI) m/z 335.1 [M-H]−.
2.1.6.7. N-(4-(N-(3-Chloro-4-fluorophenyl)sulfamoyl)phenyl)acrylamide (8g)
White solid. Yield 70%. Mp 224–226 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.42 (s, 1H), 7.66 (d, J = 8.7 Hz, 2H), 7.56 (d, J = 8.4 Hz, 2H), 7.16 (t, J = 9.1 Hz, 1H), 7.05 (d, J = 4.5 Hz, 1H), 6.93–6.89 (m, 1H), 6.28 (dd, J = 16.5, 9.6 Hz, 1H), 6.15 (d, J = 16.8 Hz, 1H), 5.68 (d, J = 9.6 Hz, 2H). 13C NMR (126 MHz, DMSO) δ 164.19, 154.52 (d, JC–F = 244.44 Hz, 1C), 143.66, 135.54, 133.30, 128.44, 128.38, 122.42, 121.28, 121.22, 120.13 (d, JC–F = 6.3 Hz, 1 C), 119.60, 117.90 (d, JC–F = 22.68 Hz, 1C). MS (EI) m/z 353.1 [M-H]−.
2.1.6.8. N-(4-(N-(Pyrimidin-2-yl)sulfamoyl)phenyl)acrylamide (8h)
White solid. Yield 70%. Mp 229–230 °C. 1H NMR (300 MHz, DMSO-d6) δ 11.73 (s, 1H), 10.52 (s, 1H), 8.50 (d, J = 4.9 Hz, 2H), 7.95 (d, J = 9.0 Hz, 2H), 7.93 (d, J = 8.7 Hz, 2H), 7.05 (t, J = 4.9 Hz, 1H), 6.45 (dd, J = 17.0, 9.9 Hz, 1H), 6.30 (dd, J = 16.9, 2.1 Hz, 1H), 5.82 (dd, J = 9.8, 2.0 Hz, 1H). MS (EI) m/z 327.1 [M + Na]+.
2.1.6.9. N-(4-(N-(3,4-Dimethylisoxazol-5-yl)sulfamoyl)phenyl)acrylamide (8i)
White solid. Yield 81%. Mp 206–208 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.96 (s, 1H), 10.61 (s, 1H), 7.89–7.86 (d, J = 9.0 Hz, 2H), 7.74–7.71 (d, J = 9.0 Hz, 2H), 6.51–6.42 (m, 1H), 6.34–6.29 (d, J = 15.0 Hz, 1H), 5.85–5.82 (d, J = 9.0 Hz, 1H), 2.08 (s, 3H), 1.63 (s, 3H). MS (EI) m/z 320.1 [M-H]−.
2.1.6.10. N-(4-(N-(5-Methylisoxazol-3-yl)sulfamoyl)phenyl)acrylamide (8j)
White solid. Yield 85%. Mp 232–233 °C. 1H-NMR (300 MHz, DMSO-d6) δ 11.35 (s, 1H), 10.56 (s, 1H), 7.87–7.80 (dd, J = 12.0, 9.0 Hz, 4H), 6.49–6.40 (m, 1H), 6.33–6.28 (d, J = 15.0 Hz, 1H), 6.13 (s, 1H), 5.85–5.81 (dd, J = 9.0, 3.0 Hz, 1H), 2.29 (s, 3H). MS (EI) m/z 306.1 [M-H]−.
2.2. Biological assays
The main bioassays performed in the study, including HeLa cell-based IDO1/kynurenine assay, Western blot analysis, T cell proliferation and Treg cell differentiation experiments, were carried out as described previouslyCitation32.
2.2.1. Cellular thermal shift assay
HeLa cells were seeded in 6-well culture plates at a density of 2 × 105 per well. On the next day, human IFN-γ (100 ng/mL) were added and incubated for 24 h, and then the cells were treated with 5d for 2 h. At the end of incubation, cells collected and subjected to CETSA assay. Briefly, incubated cells were equally divided into 10 parts, each part was heated for 3 min under different temperature (43, 46, 49, 52, 55, 58, 61, 64, 67, and 70 °C), and then the heated cells were kept at −80 °C for 12 h, then at room temperature for 5 min, and the process repeated one more time. After that, cell lysates were extracted by centrifugation at 20,000 × g, 20 min. Levels of IDO1 protein were assessed by Western blot analysis.
2.2.2. In vivo antitumour activity assay
BALB/c mice (6–8 weeks old, 18–22 g) were supplied by Model Animal Research Centre of Nanjing University (Nanjing, China). Mice were maintained under standard specific-pathogen-free (SPF) conditions (21 ± 2 °C and 12-h light: dark cycle). Animal welfare and experimental procedures were carried out strictly in accordance with the Guide for the Care and Use of Laboratory Animals (The Ministry of Science and Technology of China, 2006) and the related ethical regulations of our university. All efforts were made to minimise animals’ suffering and to reduce the number of animals used. Mouse colon cancer cells (CT26) and melanoma cells (B16F1) were cultured and collected by centrifugation (1000 rpm, 5 min) and washed twice with ice-cold PBS. Then cells were diluted to 1 × 107/ml and 1 × 106 CT26/B16F1 cells (in 0.1 ml PBS) were injected subcutaneously into the right flanks of mice. All mice formed tumours three days after injection. Then mice were randomly distributed into five groups (n = 10/8) according to tumour volumes. 5d (3.125, 6.5, 12.5 mg/kg) were administered (i.g) to each group respectively every day. 5-FU (25 mg/kg) and Cis-platinum (1 mg/kg) were administered (i.p) every three days. Tumour length and tumour width were measured with a Vernier calliper every three days. Tumour volumes were measured and calculated using the equation volume = a × b2/2, where “a” is the maximal width and “b” is maximal orthogonal width. After the administration were completed, mice were weighed, euthanized, and tumours were removed and the weight were taken.
Eight-week-old male BALB/c nude mice, weighing 18–22 g, were supplied by Model Animal Research Centre of Nanjing University (Nanjing, China). 1 × 107/ml and 1 × 106 CT26 cells (in 0.1 ml PBS) were injected subcutaneously into the right flanks of mice. After CT26 xenograft BALB/c nude mouse model was established, all mice were randomly divided into three groups (n = 8). 5d (12.5 mg/kg) were administered (i.g) every day and 5-FU (25 mg/kg) awere administered (i.p) every three days. The following experimental procedures were similar to the described above.
2.2.3. Immunohistochemistry analysis
For immunohistochemistry staining, the sections were deparaffinized, rehydrated, and washed in 1% PBS-Tween 20, and then treated with 2% hydrogen peroxide, blocked with 3% goat serum (Life Technology, 16210–064) and incubated for 2 h at room temperature with specific primary antibodies. Then the slides were incubated with streptavidin-HRP (Shanghai Gene Company, GK500705) for 40 min, then stained with DAB (Shanghai Gene Company, GK500705) substrate and counter-stained with haematoxylin. Images were acquired by microscopy (Olympus IX51).
2.3. Molecular modelling
In order to consider the flexibility of both ligand and protein, the induced fit docking (IFD) protocol in Schrödinger was employed. In IFD calculations, the ligands were first docked into the rigid receptor using softened energy function in Glide. By default, a maximum 20 poses per ligand were retained. Then, the protein degrees of freedom for each complex were sampled and the protein-ligand complexes were minimised. The protein structure in each pose now reflected an induced fit to the ligand structure and conformation. The best protein-ligand complex was then identified based on the predicted binding affinities of the docked ligand. Here, the residues within 5 Å of each of the 20 ligand poses were subjected to a conformational search and energy minimizations, and the residues outside this range were fixed. Finally, the minimised ligand was rigorously redocked into the induced-fit protein structure using Glide XP scoring mode, and metal constraints can be applied to both Glide docking stages in IFD protocol. The choice of the best-docked structure for each ligand was made using a model energy score that combines the energy grid score, the binding affinity predicted by GlideScore, and the internal strain energy for the model potential used to direct the conformational-search algorithm.
3. Results and discussion
3.1. Chemistry
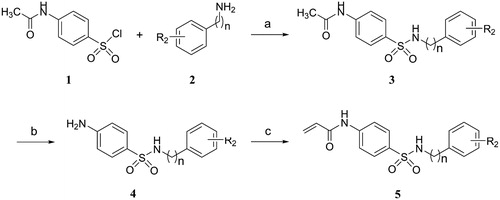
The synthetic pathways for the target compounds were shown in Scheme 1 and 2. N-acetyl secondary sulphonamides 3 were prepared by the condensation reaction of substituted 4-acetamidobenzenesulfonyl chloride 1 with various amines 2. Deacetylation of compounds 3 with concentrated HCl in ethanol gave amino secondary sulphonamides hydrochloride salts, and followed by neutralisation with saturated NaHCO3 solution to afford the corresponding amino secondary sulphonamides 4. Finally, the compounds 5 were prepared via the condensation reaction of compounds 4 with acryloyl chloride (Scheme 1).
Scheme 1. Reagents and conditions: (a) TEA, DCM, 0 °C, rt; (b) HCl, ethanol, 70 °C; (c) Acryloyl chloride, TEA, DCM, 0 °C, rt.
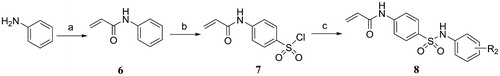
Alternatively, N-phenylacrylamide 6 was synthesised by condensation of aniline with acryloyl chloride in the presence of triethylamine, and followed by chlorosulfonation with chlorosulfonic acid to give 4-acetamidobenzenesulfonyl chloride 7. Subsequently, the 4-acetamidobenzenesulfonyl chloride was treated with substituted aniline in the presence of pyridine to form the target compounds 8 (Scheme 2).
Scheme 2. Reagents and conditions: (a) Acryloyl chloride, TEA, DCM, 0 °C, rt; (b) HSO3Cl, 70 °C, 1 h; (c) Substituted aniline, pyridine, DCM, 0 °C, rt.
3.2. Effect of secondary sulphonamides on IDO1 activity
It is well known that IDO1 is broadly expressed in human cancers and is induced by pro-inflammatory cytokines, such as interferon gamma (IFN-γ) and tumour necrosis factor alpha (TNF-α)Citation41. Here we utilised a human cervical cancer cell line HeLa that endogenously expresses IDO1 upon IFN-γ treatment to evaluate the IDO1 inhibitory activity of the synthesised compounds by measuring kynurenine secreted into the media. This cell-based assay, as a rapid and simple method for determination IDO1/kynurenine, is closely related to the potency of the enzyme assay and has been widely used to identify various IDO1 inhibitorsCitation19,Citation23,Citation29,Citation42. In addition, cytotoxic compounds can reduce cell viability, which will decrease the IDO1 levels and then lower the conversion of tryptophan to kynurenine. Thus, we also employed this cellular assay to detect false positive IDO1 inhibitors and cytotoxic compounds by measuring HeLa cell viability. Clinical candidate navoximod was used as an assay control. The results were shown in and .
Table 1. IDO1 inhibitory activity of the target compounds 3a-i, 4a-f, 5a-e and 8a-b.
Table 2. IDO1 inhibitory activity of the target compounds 5f-m and 8c-j.
Our group had already synthesised a series of N-acetyl secondary sulphonamides as the analogs of compound BS-1 and evaluated their inhibitory activityCitation43. To our surprise, although most of these compounds had weak inhibitory activity against IDO1, N-(4-(N-(4-methoxybenzyl)sulfamoyl)phenyl)acetamide (HeLa IC50: 5.88 µM) showed good inhibitory activity, and its potency was remarkably increased by 8.4-fold as compared with the hit BS-1. Inspired by N-(4-(N-(4-methoxybenzyl)sulfamoyl)phenyl)acetamide, we further synthesised the analogues 3a-i bearing an electron-donor group on the benzyl group, but they displayed only weak activity. Subsequently, we tried to remove the acetyl group of N-acetyl secondary sulphonamides, and found that all the obtained aniline compounds 4a-f had no improvement in activity. These results indicated that a free amino group on the phenyl ring with strong electron-donating ability appeared to be unfavourable for the inhibitory activity of these compounds. To this end, we tried to introduce an acryloyl group instead of the acetyl group to the amine group. As expected, compounds 5a-e bearing an acetyl group showed moderate to good inhibitory activity. Especially, compound 5d (HeLa IC50: 6.26 µM) exhibited significantly stronger activity than the hit compound BS-1 (HeLa IC50: 49.42 µM) and its corresponding acetyl compound 4e (HeLa IC50 > 50 µM).
We then further optimised the inhibitory activity of N-acryloyl secondary sulphonamides by modifying the substituents, including n, R2 and R3, while retaining the acryloyl group at R1. As shown in , most of N-acryloyl secondary sulphonamides displayed good inhibitory activity against IDO1. Among them, benzylamine derivative 5l and aniline derivative 8g exhibited potent activity with IC50 values of 6.64 µM and 3.76 µM, which were significantly increased by 7.4-fold and 13.1-fold as compared with the hit BS-1, respectively. It is worth noting that the compounds 5d, 5g, 5l, and 8d with a substituent at 3-position on aniline or benzylamine had higher activity than those compounds 5c, 5f, 5k and 8c with a substituent at 4-position (5c vs 5d, 5f vs 5g, 5k vs 5l, 8c vs 8d). However, compound 5i (IC50: 39.53 µM) with a fluoro substituent at 3-position on aniline showed lower inhibitory activity compared with the corresponding compound 5h with a fluorine atom at 4-position on aniline (IC50: 20.54 µM), which showed similar inhibitory activity with compound 5a without substituent on aniline (IC50: 22.36 µM), likely due to the similar size of the fluorine and hydrogen atoms, Interestingly, compound 8g containing a m-chlorine and a p-fluorine atoms showed the strongest potency (IC50: 3.76 µM), while compound 5m with m,p-di-chlorine atoms had only moderate activity (IC50: 37.27 µM). It might be duo to the large volume of the 4-substituent on aniline or benzylamine that cause the bad clashes with the neighbouring residues in active site of IDO1. In addition, we also tried to replace benzene or benzyl rings with aromatic heterocycles (8 h-8j), including pyrimidine and isoxazole, but they showed only weak activity.
Taken together, compounds 5d, 5l and 8g showed good inhibitory activity in the HeLa cell-based IDO1/kynurenine assay. Furthermore, the viability of HeLa cells was evaluated by the MTT method at the end of the assay. The readouts showed that most of the synthesised compounds did not affect cell viability under the experimental conditions (data not shown), indicating that IDO1 inhibitory activity of these compounds is not mediated by their cytotoxicity.
3.3. Effect of 5d, 5 l, 8f and 8g on IDO1 protein expression
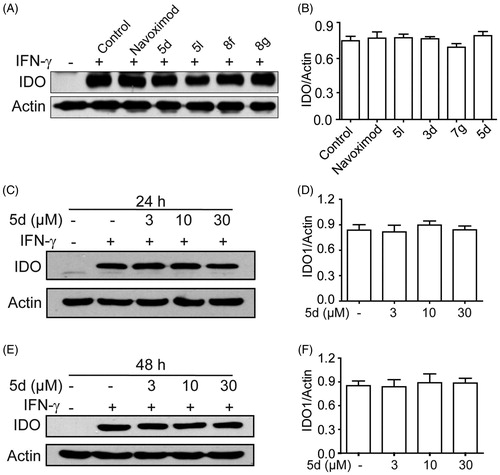
To exclude the possibility that these secondary sulphonamides inhibit IDO1 activity by down-regulating IDO1 expression, we utilised Western blot to measure the expression of IDO1 protein in HeLa cells treated with different compounds. As depicted in , compared with the blank group, the expression of IDO1 protein was significantly induced in IFN-γ–treated HeLa cells. Evidently, all tested compounds 5d, 5l, 8f and 8g did not influence IDO1 expression as compared with the control group. In addition, we further confirmed that treatment of HeLa cells with different concentrations of compound 5d (3, 10, 30 µM) for 24 h or 48 h also did not affect IDO1 expression. The results demonstrated that these compounds blocked the kynurenine pathway by affecting IDO1 enzyme activity rather than its expression and/or HeLa cell viability.
Figure 3. Compounds 3d, 5d, 5l and 8g did not affect the expression of IDO1 protein. (A) HeLa cells were treated with IFN-γ (100 ng/mL) for 24 h, then incubated with or without test compounds at their IC50 concentrations for 24 h, and expression of IDO was analysed by Western blot using an anti-IDO antibody. (B) The greyscale scanning statistical results of A. (C) HeLa cells were incubated with compound 5d (3, 10, 30 μM) for 24 h. (D) The greyscale scanning statistical results of C. (E) HeLa cells were incubated with compound 5d (3, 10, 30 μM) for 48 h. (F) The greyscale scanning statistical results of E. Data are presented as means ± SEM (n = 3).
3.4. Compounds 5d and 8g could effectively restore T cell proliferation
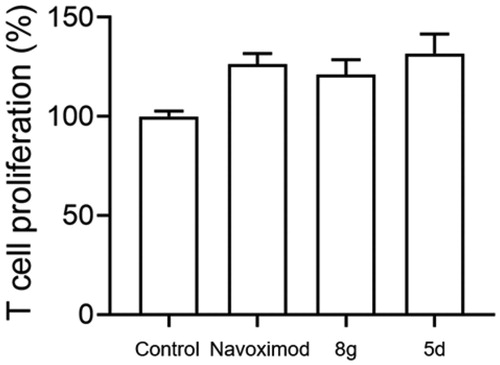
T lymphocytes are exceedingly sensitive to low tryptophan levels. Overactivation of the kynurenine pathway by high IDO1 expression leads to local tryptophan depletion, which can suppress T cell proliferation by activating the GCN2 kinase pathway and eventually result in T cell anergyCitation44. Here, to determine whether secondary sulphonamides could rescue IDO1-mediated blockade of T cell proliferation, we used a co-culture of IDO1-expressing B16F1 melanoma cells with naïve CD4+ T cells to mimic the tumour microenvironment. Given that melanoma is a highly immunogenic tumour and IDO1 constitutively expresses in melanoma cellsCitation40, B16F1 murine melanoma cell lines that express IDO1 were then used to co-culture with T cells. Naïve CD4+ T cells were isolated from spleens of C57BL/6 mice. As shown in , compounds 5d, 8g and navoximod displayed a significant augmentation on T cell activity stimulated with B16F1 cells. Especially, compound 5d showed better enhancement of T cell proliferation than navoximod. These findings demonstrated that compounds 5d and 8g could reverse the suppression of T lymphocytes caused by IDO1.
Figure 4. Compounds 5d and 8 g could promote the proliferation of T cells. Based on the B16F1-T cell co-culture system, use MTT assay to test T cells proliferation. The Navoximod and compounds were added to the system at the concentration of 1.5 and 18 μM. Data represent mean ± SEM (n = 3).
3.5. Compounds 5d and 8g could effectively inhibit FoxP3+ Treg cell differentiation
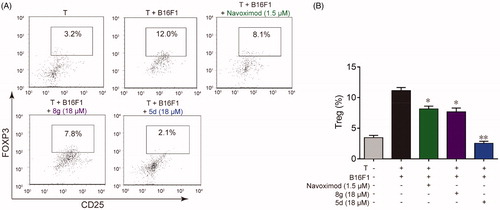
Besides causing local tryptophan depletion, high IDO1 expression can also accumulate metabolites of kynurenine pathway. The elevated metabolites can not only induce effector T cell death directly through their cytotoxicity, but also promote the differentiation of naïve CD4+ T cells into CD4+ FoxP3+ Treg cells by activating the AHR pathwayCitation10. As a highly immunosuppressive subset of CD4+ T cells, FoxP3+ Treg cells are critical to the maintenance of immune homeostasis and are involved in tumour immune escape, thereby contributing to tumour development and progressionCitation45. Therefore, we also utilised co-culture of B16F1 cells with naïve CD4+ T cells to test whether the synthesised compounds could suppress IDO1 mediated Treg cell differentiation. As illustrated in , when the naïve CD4+ T cells were co-cultured with B16F1 cells under Con A stimulation for 48 h, the number of the FoxP3+ Treg cells increased approximately 4 times compared with naïve T cells cultured alone, indicating that IDO1-expressing B16F1 cells could significantly stimulate naïve T cell differentiation into FoxP3+ Treg cells. However, treatment with 5d, 8g and navoximod could significantly reduce FoxP3+ Treg cell differentiation. Especially, compound 5d displayed the best effect in this test, and its FoxP3+ Treg cell number returned to the initial level. These data evidenced that compounds 5d and 8g could effectively suppress the differentiation of naïve CD4+ T cell into FoxP3+ Treg cells, which would help reverse IDO1-mediated immune suppression.
Figure 5. Compounds 5d and 8 g inhibited Treg cell differentiation in the cell co-culture system. (A) After co-cultured with B16F1 cells, T cells were collected. Surface staining was performed with CD4+ FTTC and CD25+ PE for 30 min at 4 °C. The cells were fixed and permeabilized with fixation buffer and permeabilization wash buffer. The intracellular staining was performed with FoxP3+ APC for 20 min. The cells were then analysed by flow cytometry analysis. (B) Statistical analysis of result A. Data are represented as ± SEM (n = 3). *p < 0.05, **p < 0.01 vs T + B16F1.
3.6. Compound 5d could bind to IDO1 protein in HeLa cells
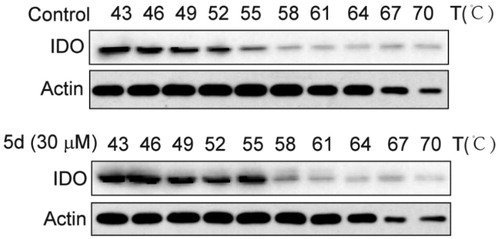
The cellular thermal shift assay (CETSA) is usually used to identify compounds and their potential targets. CESTA can detect the target engagement of drugs in a cellular context based on the biophysical principle of ligand-induced thermal stabilisation of target proteinsCitation46. To this end, we further employed the CETSA assay to detect whether compound 5d could directly bind to the target protein IDO1 in HeLa cells. As presented in , treatment with 5d could improve the thermal stability of IDO1 protein in HeLa cells as compared with the vehicle control, and still maintained the protein stability at 55 °C. These results suggested that compound 5d could enter HeLa cells and then directly bind to IDO1 protein.
Figure 6. Compound 5d could bind to IDO1 protein in HeLa cells. HeLa cells were treated with human IFN-γ (100 ng/mL) for 24 h, and then treated with 5d for 2 h. The cells were collected and subjected to CETSA assay. The levels of IDO1 protein were assessed by Western blot analysis.
3.7. Compound 5d could effectively inhibit colon carcinoma growth in immunocompetent mice, but not in immunodeficient mice
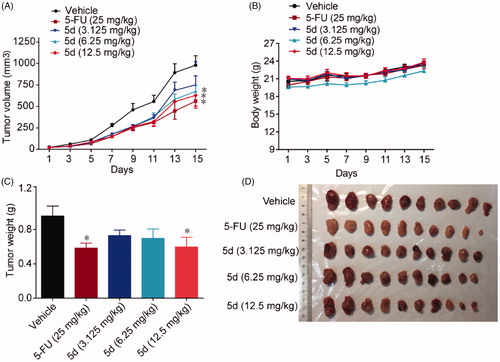
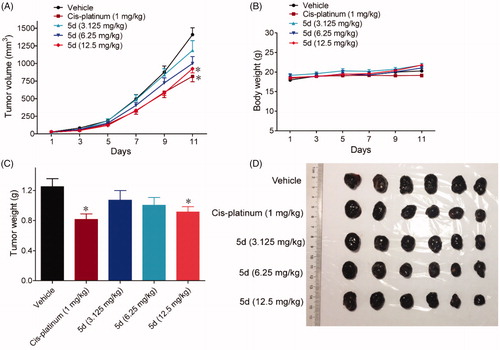
Given that it was the most prominent inhibitor in vitro, we chose compound 5d for further pharmacodynamic evaluation in vivo. Bowel cancer is the third leading cause of cancer in the world, thus we first evaluated the efficacy of compound 5d on the growth of murine CT26 colon carcinoma xenograft model in immunocompetent BALB/c mice. Fluorouracil (5-FU) is one of the most commonly used drugs to different cancers including bowel cancer, and was used as the positive control. The CT26 tumour-bearing mice were treated with 5d (3.125, 6.25, 12.5 mg/kg, i.g, every day) or 5-FU (25 mg/kg, i.p, every other day). Tumour volumes were measured every two days for 14 days. As outlined in , treatment of 5d significantly suppressed the growth of tumour in a dose-dependent manner as compared with the vehicle group. Especially, 5d reduced tumour volume by 35.9% and average tumour weight by 37.5% at the dose of 12.5 mg/kg, which were comparable to that of 5-FU. Moreover, no significant change in body weight of the mice was observed in administration groups compared with the vehicle group during the 14-day treatment.
Figure 7. Compound 5d suppressed colon cancer growth in immunocompetent mice. 1 × 106 CT26 cells were transplanted subcutaneously into the armpit of the BALB/c mice. Three days after transplantation, mice were randomly allocated to vehicle control or treatment groups (n = 10). Drugs were administered on days 1–14. Tumour volume (A) and mice body weight (B) were recorded. After sacrifice, solid tumours were separated and weighed (C, D). Data represent mean ± SEM, n = 10, *p < 0.05, **p < 0.01 vs vehicle.
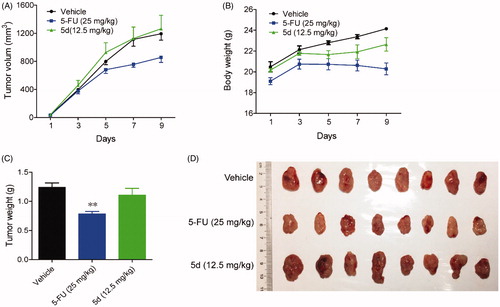
Then, in order to clarify whether the in vivo antitumour efficacy of compound 5d was related to the mouse immune system, we repeated the above experiment in immunodeficient nude mice. Briefly, BALB/c nude mice bearing established CT26 colon carcinoma xenograft were treated with compound 5d and 5-FU for 10 days. As introduced in , administration of 5d did not block tumour growth as compared with the vehicle group, while classical cytotoxics 5-FU significantly suppressed tumour growth. Taken together, these results implied that the in vivo antitumour efficacy of compound 5d was achieved by activating mouse immune system rather than its cytotoxicity.
Figure 8. Compound 5d did not suppress the growth of CT26 tumour in nude mice. 1 × 106 CT26 cells were transplanted subcutaneously into the armpit of immunodeficient BALB/c nude mice. Three days after transplantation, mice were randomly allocated to vehicle control or treatment groups (n = 8). Drugs were administered on days 1–9. (A) Tumour volume and mouse body weight (B) were recorded. After sacrifice, solid tumours were separated and weighed (C, D). Data represent mean ± SEM, n = 8, *p < 0.05, **p < 0.01 vs vehicle.
3.8. Compound 5d could inhibit the growth of tumour cells by curbing proliferation and inducing apoptosis in CT26 tumour tissues
In order to further observe morphological changes in tumour tissues after administration of compound 5d, the tumour tissue sections obtained from the BALB/c mice bearing CT26 colorectal tumours were stained with haematoxylin-eosin (H&E) to highlight different tissue structures, such as the nucleus and cytoplasm. As illustrated in , the number of tumour cells in 5-FU-treated group was significantly reduced as compared with the vehicle group. Similarly, administration of 5d decreased the number of tumour cells, as well as led to the morphological changes including cell shrinkage and chromatin condensation in a dose-dependent manner. In addition, it was clearly observed from the enlarged local image that there are more blood vessels in the compound 5d group, and their morphology appears more normal, suggesting that compound 5d might contribute to tumour vascular normalisation, thereby promoting antitumour effect of the immune systemCitation47.
Figure 9. Compound 5d dose-dependently reduced tumour cells and induced apoptosis in mice. The tumour tissues were infused in formaldehyde solution for paraffin section. (A) H&E staining of tumour tissue. (B) Expression of PCNA. (C) TUNEL staining of tumour sections. Scale bar: 50 μm.
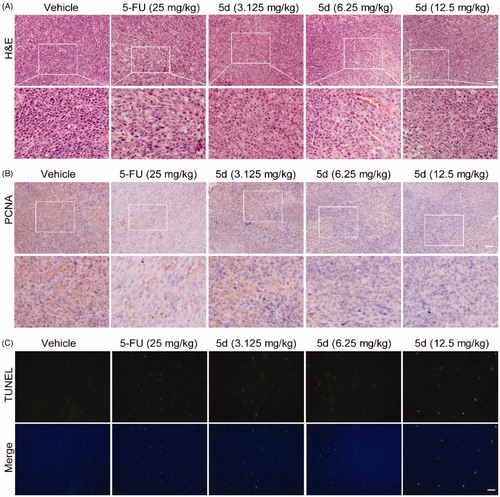
Then, to understand the mechanism of tumour cell death, we detected the level of proliferating cell nuclear antigen (PCNA) protein and tumour cell apoptosis by immunohistochemistry (IHC) and terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) experiments, respectively. PCNA is known as a molecular marker for assessing cell proliferation status, and TUNEL assay is the most widely used method to identify apoptotic cells. The results of IHC analysis showed that the expression of PCNA protein in the 5-FU group was significantly reduced as compared with the vehicle group. The similar results were observed in the 5d treatment group and showed a dose-response relationship (). In addition, the results of TUNEL assay showed that the tumour cells had less apoptosis in the vehicle group, while the apoptosis increased in the 5-FU group. Likewise, treatment with 5d also significantly induced the tumour cell apoptosis in a dose-dependent manner (). These data revealed that compound 5d displayed in vivo antitumour efficacy by inhibiting tumour cell proliferation and inducing apoptosis.
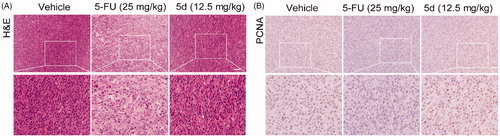
To verify the above conclusions, we further performed H&E and IHC staining on the nude mouse CT26 tumour tissues. As depicted in , the tumour cells in the 5d-treated group remained intact morphology and were arranged in an orderly manner as that in the vehicle-treated group, while the results of 5-FU treatment were just the opposite. Moreover, the results of TUNEL assay showed that administration of 5d had no influence on the expression of PCNA (). These findings further supported the above conclusions.
Figure 10. Compound 5d could not suppress the proliferation of tumour cells in immunodeficient mice. Tumour sections were infused in formaldehyde solution for immunohistochemistry. (A) H&E staining of tumour tissue. (B) Expression of PCNA. Scale bar: 50 μm.
3.9. Compound 5d facilitated immune system rejuvenation in CT26 tumour-bearing mice
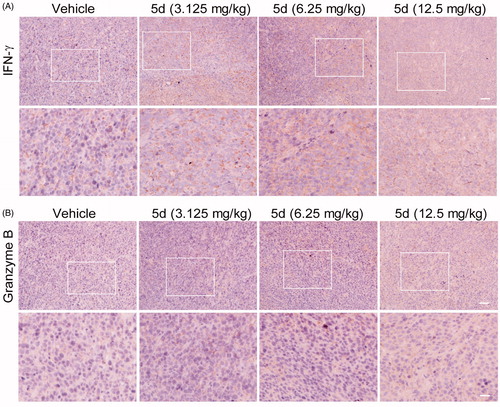
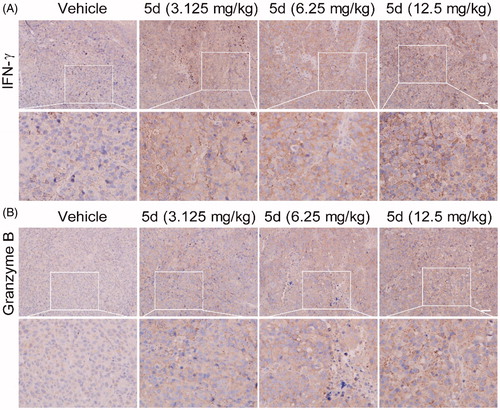
To investigate the effect of compound 5d on the immune function in CT26 tumour-bearing mice, we carried out the IHC experiment to detect the expression of IFN-γ and granzyme B in the tumour tissues. IFN-γ is a key cytokine secreted primarily by activated T cells and NK cells, which is essential for both innate and adaptive immunity. Therefore, IFN-γ is an important sign of rejuvenation of the immune system. Moreover, cytotoxic T lymphocytes (CTLs) is the key effector cells of the adaptive antitumour immune response. Granzyme B is the primary mediator of apoptosis by CTLs and NK cells. It can initiate the processing of caspases and apoptosis when it is released from activated CTLsCitation48. As detailed in , compared with the vehicle, administration of compound 5d increased the expression of IFN-γ in a dose-dependent manner in the CT26 tumour tissues, suggesting that 5d treatment could facilitate T cell activation and the rejuvenation of the immune system in the tumour microenvironment. Similarly, all treatments of 5d elevated granzyme B levels, which was consistent with the results of the TUNEL assay described above.
Figure 11. Compound 5d up-regulated the expression of IFN-γ and granzyme B. Tumour sections were infused in formaldehyde solution for immune stain. (A) Expression of IFN-γ. (B) Expression of Granzyme B. Scale bar: 50 μm.
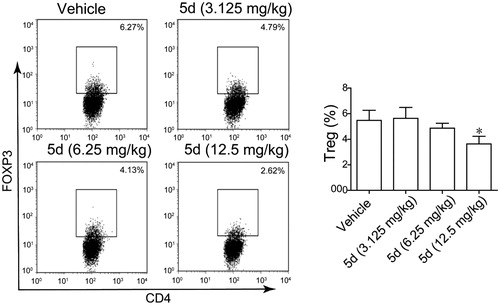
Furthermore, we also detected the content of FoxP3+ Treg cells in the CT26 tumour-bearing mouse spleens by fluorescence activated cell sorting (FACS) assay (). The results showed that 5d treatment decreased the number of FoxP3+ Treg cells in a dose-dependent manner. Especially, the amount in the high dose group was reduced 2.4-fold compared with the vehicle group. Taken together, these results revealed that the compound 5d could promote T cell activation, decrease FoxP3+ Treg cell population and heighten the function of CTLs.
Figure 12. Compound 5d decreased Treg cell differentiation in the CT26 tumour-bearing mouse. (A) Decreased differentiation of Treg cells in the spleen of tumour-bearing mice treated by compounds. (B) Statistical analysis of result A. Data represent mean ± SEM, *p < 0.05 vs vehicle.
3.10. Compound 5d could effectively suppress melanoma growth in immunocompetent mice
In addition, we also utilised a B16F1 melanoma mouse xenograft model to observe the efficacy of compound 5d on the other tumours. Malignant melanoma is a highly invasive tumour and its immunotherapy has been receiving attention in recent years. As mentioned above, B16F1 is a murine melanoma cell line that expresses IDO1. The mouse xenograft model was established by subcutaneously implanting B16F1 melanoma cells into BALB/c mice and treated with 5d (3.125, 6.25, 12.5 mg/kg, i.g, every day) or the positive control Cis-platinum (1 mg/kg, i.p, every other day). Tumour volumes were measured every two days for 11 days. As shown in , treatment with 5d significantly suppressed the growth of tumour in a dose-dependent manner both in tumour volume and in tumour weight as compared with the vehicle group without significant body weight loss. Especially at high dose, the average volume of tumour was reduced by 34.3% after 5d treatment, which were comparable to Cis-platinum.
Figure 13. Compound 5d suppressed melanoma growth in mice. 1 × 106 melanoma cells were transplanted subcutaneously into the armpit of the BALB/c mice. Three days after transplantation, mice were randomly allocated to vehicle control or treatment groups (n = 6). Drugs were administered on days 1–11. (A) Tumour volume and mouse body weight (B) were recorded. After sacrifice, solid tumours were separated and weighed (C, D). Data represent mean ± SEM, n = 6, *p < 0.05, **p < 0.01 vs vehicle.
Analogously, we also performed H&E, IHC and TUNEL experiments on the B16F1 melanoma tissues ( and ). These results indicated that compound 5d could effectively contribute to the morphological changes of B16F1melanoma cells, reduce the number of tumour cells by inhibiting proliferation and inducing apoptosis, and facilitate tumour vascular normalisation. Importantly, compound 5d also promoted T cell activation by up-regulating IFN-γ expression, and elevated CTLs function by stimulating granzyme B secretion. These results reaffirm that compound 5d could inhibit tumour growth by activating the mouse immune system.
Figure 14. Compound 5d dose-dependently reduced tumour cells and induced apoptosis in mice. Tumour sections were infused in formaldehyde solution for immunohistochemistry. (A) H&E staining of tumour tissue. (B) Expression of PCNA. (C) TUNEL staining of tumour sections. Scale bar: 50 μm.
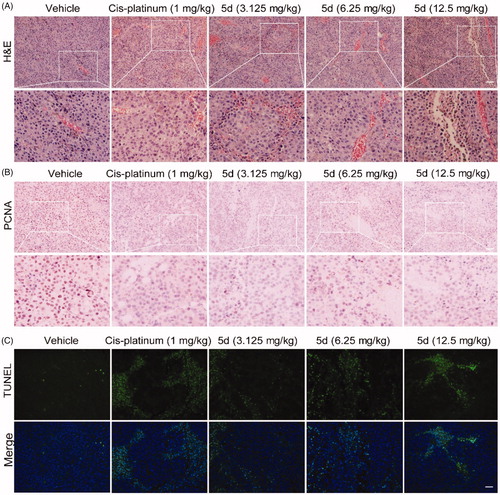
Figure 15. Compound 5d promoted T cells activation while reduced Foxp3+ Treg cell number in tumour tissue. Tumour sections were infused in formaldehyde solution for immune stain. (A) Expression of IFN-γ. (B) Expression of Granzyme B. Scale bar: 50 μm.
3.11. Molecular modelling studies
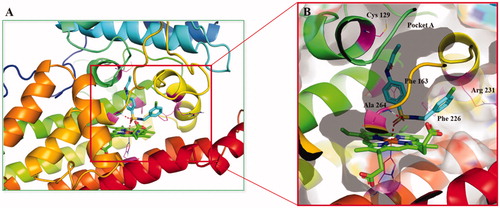
Based on their outstanding performance in biological activity, we performed computational studies to elucidate the interactions of compounds 5d with IDO1. Since the high flexibility of the side chain and backbone of IDO1 protein, the classical semi-flexible docking method often poses a problem for IDO1 docking, whose algorithm relies on a rigid protein structure. Thus, the induced fit docking (IFD) method was utilised to overcome the difficulty mentioned above. The cocrystallization of Amg-1 in complex with human IDO1 protein (PDB ID: 4PK5) was employed as the template in the following docking calculations because of its relatively high resolution and more structural information at pocket B. As shown in , The benzsulfamide moiety situated deeply in pocket A and their benzene ring was interacted with Phe163 through π-π interaction, while the benzylamine moiety extended towards pocket B formed by Phe163, Phe226, Arg231, Leu234 and Ile354. Specifically, one oxygen atom of the sulphonyl group bond directly to the haem iron of IDO1 while the other oxygen atom formed a hydrogen bond with the main chain NH group of Ala264. Acryloyl groups located at the top of pocket A, and ethenyl moiety of acryloyl group extended towards a small hydrophobic subpocket formed by Met88, Leu124, Cys129, Leu234 and Gly262. This computational study finds that the occupation of the small hydrophobic subpocket in pocket A may a key factor affecting IDO1 inhibitory activity. These data could be useful for further optimisation.
Figure 16. (A) Proposed binding mode of compound 5d docked into the crystal structure of human IDO1 (PDB: 4PK5) by IFD methods. (B) The interactions between compound 5d and IDO1. Polar interactions were presented by red dotted line.
4. Conclusions
In this study, the hit compound BS-1 was found as an IDO1 inhibitor in-house compound library. After structural optimisation, a series of sulphonamide derivatives were designed, synthesised, and evaluated in the HeLa cell-based IDO1/kynurenine assay, resulting in the identification of secondary sulphonamides as novel IDO1 inhibitors. Among them, the compounds 5d, 5l and 8g displayed the strongest inhibitory effect, and their activities were significantly improved over the hit compound BS-1. The in vitro results showed that these compounds could bind to IDO1 protein in HeLa cells without affecting the cell viability and the IDO1 protein expression under experimental conditions, indicating that these compounds blocked the kynurenine pathway by influencing IDO1 enzyme activity rather than IDO1 expression and/or cell viability. In addition, these compounds could promote the T cell proliferation and suppress the differentiation of naïve CD4+ T cell into FoxP3+ Treg cells, suggesting that these sulphonamides could reverse IDO1-mediated immunosuppression.
Based on its excellent performance in vitro, compound 5d was selected for further evaluating the pharmacodynamic effects in three types of tumour-bearing mouse models, including two immunocompetent mouse xenografts and one immunodeficient mouse xenograft. These in vivo experiments demonstrated that treatment with 5d could effectively inhibit the growth of murine CT26 coloreatic carcinoma xenograft tumour in a dose-dependent manner in immunocompetent mice, but not in immunodeficient mice, which implied that the in vivo efficacy of compound 5d was closely associated with the mouse immune system. Similarly, 5d also significantly blocked the growth of B16F1 melanoma in immunocompetent mice. Functionally, biochemical experiment and pathological analysis were utilised to detect these tumour tissues. The results from two immunocompetent mouse models indicated that compound 5d could induce the morphological changes of both CT26 and B16F1 tumour cells, decrease the number of tumour cells by suppressing proliferation and inducing apoptosis. Importantly, compound 5d could contribute to T cell activation by stimulating IFN-γ expression, improve CTLs function by inducing granzyme B secretion, and inhibit FoxP3+ Treg cell differentiation, which would help facilitate the rejuvenation of the immune system. These promising results motivate us to further develop this new type of IDO1 inhibitors.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Lizée G, Overwijk WW, Radvanyi L, et al. Harnessing the power of the immune system to target cancer. Annu Rev Med 2013;64:71–90.
- Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 2015;27:450–61.
- Schadendorf D, Hodi FS, Robert C, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in Unresectable or Metastatic Melanoma. J Clin Oncol 2015;33:1889–94.
- Szostak B, Machaj F, Rosik J, et al. CTLA4 antagonists in phase I and phase II clinical trials, current status and future perspectives for cancer therapy. Expert Opin Investig Drugs 2019;28:149–59.
- Alsaab HO, Sau S, Alzhrani R, et al. PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: mechanism, combinations, and clinical outcome. Front Pharmacol 2017;8:561
- Majzner RG, Mackall CL. Clinical lessons learned from the first leg of the CAR T cell journey. Nat Med 2019;25:1341–55.
- Kreamer KM. Immune checkpoint blockade: a new paradigm in treating advanced cancer. J Adv Pract Oncol 2014;5:418–31.
- Cervenka I, Agudelo LZ, Ruas JL. Kynurenines: tryptophan’s metabolites in exercise, inflammation, and mental health. Science 2017;357:eaaf9794.
- Munn DH, Mellor AL. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol 2013;34:137–43.
- Mándi Y, Vécsei L. The kynurenine system and immunoregulation. J Neural Transm (Vienna) 2012;119:197–209.
- Munn DH, Sharma MD, Baban B, et al. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity 2005;22:633–42.
- Frumento G, Rotondo R, Tonetti M, et al. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J Exp Med 2002;196:459–68.
- Mezrich JD, Fechner JH, Zhang X, et al. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol 2010;185:3190–8.
- Okamoto A, Nikaido T, Ochiai K, et al. Indoleamine 2,3-dioxygenase serves as a marker of poor prognosis in gene expression profiles of serous ovarian cancer cells. Clin Cancer Res 2005;11:6030–9.
- Ino K, Yoshida N, Kajiyama H, et al. Indoleamine 2,3-dioxygenase is a novel prognostic indicator for endometrial cancer. Br J Cancer 2006;95:1555–61.
- Pan K, Wang H, Chen MS, et al. Expression and prognosis role of indoleamine 2,3-dioxygenase in hepatocellular carcinoma. J Cancer Res Clin Oncol 2008;134:1247–53.
- Folgiero V, Goffredo BM, Filippini P, et al. Indoleamine 2,3-dioxygenase 1 (IDO1) activity in leukemia blasts correlates with poor outcome in childhood acute myeloid leukemia. Oncotarget 2014;5:2052–64.
- Speeckaert R, Vermaelen K, van Geel N, et al. Indoleamine 2,3-dioxygenase, a new prognostic marker in sentinel lymph nodes of melanoma patients. Eur J Cancer 2012;48:2004–11.
- Liu X, Shin N, Koblish HK, et al. Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood 2010;115:3520–30.
- Holmgaard RB, Zamarin D, Munn DH, et al. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J Exp Med 2013;210:1389–402.
- Tu W, Yang F, Xu G, et al. Discovery of imidazoisoindole derivatives as highly potent and orally active indoleamine-2,3-dioxygenase inhibitors. ACS Med Chem Lett 2019;10:949–53.
- Gomes B, Driessens G, Bartlett D, et al. Characterization of the selective indoleamine 2,3-dioxygenase-1 (IDO1) catalytic inhibitor EOS200271/PF-06840003 supports IDO1 as a critical resistance mechanism to PD-(L)1 blockade therapy. Mol Cancer Ther 2018;17:2530–42.
- Yue EW, Douty B, Wayland B, et al. Discovery of potent competitive inhibitors of indoleamine 2,3-dioxygenase with in vivo pharmacodynamic activity and efficacy in a mouse melanoma model. J Med Chem 2009;52:7364–7.
- Markwalder JA, Seitz SP, Blat Y, et al. Identification and optimization of a novel series of indoleamine 2,3-dioxygenase inhibitors. Bioorg Med Chem Lett 2017;27:582–5.
- Yue EW, Sparks R, Polam P, et al. INCB24360 (Epacadostat), a highly potent and selective indoleamine-2,3-dioxygenase 1 (IDO1) inhibitor for immuno-oncology. ACS Med Chem Lett 2017;8:486–91.
- Williams DK, Markwalder JA, Balog AJ, et al. Development of a series of novel o-phenylenediamine-based indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors. Bioorg Med Chem Lett 2018;28:732–6.
- Kumar S, Waldo JP, Jaipuri FA, et al. Discovery of clinical candidate (1R,4r)-4-((R)-2-((S)-6-fluoro-5H-imidazo[5,1-a]isoindol-5-yl)-1-hydroxyethyl)cyclohexan-1-ol (Navoximod), a potent and selective inhibitor of indoleamine 2,3-dioxygenase 1. J Med Chem 2019;62:6705–33.
- Brastianos HC, Vottero E, Patrick BO, et al. Exiguamine A, an indoleamine-2,3-dioxygenase (IDO) inhibitor isolated from the marine sponge Neopetrosia exigua. J Am Chem Soc 2006;128:16046–7.
- Yang S, Li X, Hu F, et al. Discovery of tryptanthrin derivatives as potent inhibitors of indoleamine 2,3-dioxygenase with therapeutic activity in Lewis lung cancer (LLC) tumor-bearing mice. J Med Chem 2013;56:8321–31.
- Platten M, Nollen EAA, Röhrig UF, et al. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat Rev Drug Discov 2019;18:379–401.
- Zou Y, Wang F, Wang Y, et al. Systematic study of imidazoles inhibiting IDO1 via the integration of molecular mechanics and quantum mechanics calculations. Eur J Med Chem 2017;131:152–70.
- Zou Y, Wang Y, Wang F, et al. Discovery of potent IDO1 inhibitors derived from tryptophan using scaffold-hopping and structure-based design approaches. Eur J Med Chem 2017;138:199–211.
- Zou Y, Wang F, Wang Y, et al. Discovery of imidazoleisoindole derivatives as potent IDO1 inhibitors: design, synthesis, biological evaluation and computational studies. Eur J Med Chem 2017;140:293–304.
- Zou Y, Hu Y, Ge S, et al. Effective virtual screening strategy toward heme-containing proteins: identification of novel IDO1 inhibitors. Eur J Med Chem 2019;184:111750
- Gaspari P, Banerjee T, Malachowski WP, et al. Structure-activity study of brassinin derivatives as indoleamine 2,3-dioxygenase inhibitors. J Med Chem 2006;49:684–92.
- Gulçin T, Taslimi P. Sulfonamide inhibitors: a patent review 2013-present. Expert Opin Ther Pat 2018;28:541–9.
- Penning TD, Talley JJ, Bertenshaw SR, et al. Synthesis and biological evaluation of the 1,5-diarylpyrazole class of cyclooxygenase-2 inhibitors: identification of 4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide (SC-58635, Celecoxib). J Med Chem 1997;40:1347–65.
- Cheng MF, Hung MS, Song JS, et al. Discovery and structure-activity relationships of phenyl benzenesulfonylhydrazides as novel indoleamine 2,3-dioxygenase inhibitors. Bioorg Med Chem Lett 2014;24:3403–6.
- Wang X, Ahn YM, Lentscher AG, et al. Design, synthesis, and evaluation of substituted nicotinamide adenine dinucleotide (NAD+) synthetase inhibitors as potential antitubercular agents. Bioorg Med Chem Lett 2017;27:4426–30.
- Uyttenhove C, Pilotte L, Théate I, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med 2003;9:1269–74.
- Negmeldin AT, Knoff JR, Pflum M. The structural requirements of histone deacetylase inhibitors: C4-modified SAHA analogs display dual HDAC6/HDAC8 selectivity. Eur J Med Chem 2018;143:1790–806.
- Lin SY, Yeh TK, Kuo CC, et al. Phenyl benzenesulfonylhydrazides exhibit selective indoleamine 2,3-dioxygenase inhibition with potent in vivo pharmacodynamic activity and Antitumor Efficacy. J Med Chem 2016;59:419–30.
- Jin S, Wang F, Zou Y, et al. Design, synthesis and biological evaluation of phenylsulfonamide-based IDO1 inhibitors. J China Pharm Univ 2018;49:34–8.
- Afonina IS, Cullen SP, Martin SJ. Cytotoxic and non-cytotoxic roles of the CTL/NK protease granzyme B. Immunol Rev 2010;235:105–16.
- Togashi Y, Shitara K, Nishikawa H. Regulatory T cells in cancer immunosuppression – implications for anticancer therapy. Nat Rev Clin Oncol 2019;16:356–71.
- Jafari R, Almqvist H, Axelsson H, et al. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat Protoc 2014;9:2100–22.
- Xu Q, Gu J, Lv Y, et al. Angiogenesis for tumour vascular normalization of Endostar on hepatoma 22 tumour-bearing mice is involved in the immune response. Oncol Lett 2018;15:3437–46.
- Munn DH, Shafizadeh E, Attwood JT, et al. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med 1999;189:1363–72.