
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
A series of novel calix[4]azacrown substituted sulphonamide Schiff bases was synthesised by the reaction of calix[4]azacrown aldehydes with different substituted primary and secondary sulphonamides. The obtained novel compounds were investigated as inhibitors of six human (h) isoforms of carbonic anhydrases (CA, EC 4.2.1.1). Their antioxidant profile was assayed by various bioanalytical methods. The calix[4]azacrown substituted sulphonamide Schiff bases were also investigated as inhibitors of acetylcholinesterase (AChE), butyrylcholinesterase (BChE) and tyrosinase enzymes, associated with several diseases such as Alzheimer, Parkinson, and pigmentation disorders. The new sulphonamides showed low to moderate inhibition against hCAs, AChE, BChE, and tyrosinase enzymes. However, some of them possessed relevant antioxidant activity, comparable with standard antioxidants used in the study.
1. Introduction
Carbonic anhydrases are metalloenzymes present in Archaea, prokaryotes, and eukaryotes, and catalyse a physiologically very simple but relevant reaction, i.e. the interconversion of CO2 to HCO3− and protons via a ping-pong mechanismCitation1–6. In humans, 16 different isoforms have been described from eight genetically distinct CA families (α-, β-, γ-, δ-, ζ-, η-, θ-, and ι-CAs)Citation1–6. The isozymes have different subcellular localisation, catalytic activity, and inhibitory properties in body fluid and tissues. Since these isoforms play an important role in acid–base regulation, gluconeogenesis and other biosynthetic reactions, electrolyte secretion, bone resorption/calcification, and tumorigenicity, their inhibition/activation may be exploited in several diseases, including glaucoma, obesity, neuropathic pain, arthritis, Alzheimers` disease and more recently cancerCitation1–6.
Acetylcholinesterase (AChE; EC 3.1.1.7), which hydrolyses the neurotransmitter acetylcholine, is found in high concentrations over the peripheral and central nervous systems but also in other tissuesCitation7,Citation8. The imbalance in AChE activity can cause various types of neurodegenerative pathologies such as Alzheimer’s disease (AD) and Parkinson’s disease (PD). Butyrylcholinesterase (BChE) is a non-specific cholinesterase enzyme that hydrolyses many different choline-based estersCitation9,Citation10. Thus, AChE and BChE inhibition have been documented as an interesting approach in the palliative treatment of ADCitation11,Citation12.
Several major classes of macrocyclic derivatives such as the crown ethers, cyclodextrins, and calixarenes are important third-generation derivatives in supramolecular chemistryCitation13,Citation14. Among them, calixarenes may have biomedical applications thanks to their exceptional structural properties, which may be exploited for designing antitumor, antiviral, antimicrobial, anti-thrombotic, and antifungal derivativesCitation15–17. From this perspective, synthesising new derivatised calixarenes as antioxidant and enzyme inhibitors, and exploring the molecular mechanisms underlying their effect are ongoing new fields.
In continuation of our recent interest in developing new metalloenzymes inhibitorsCitation18–21, in this study, we report the first calix[4]azacrown substituted sulphonamide Schiff bases (to the best of our knowledge) acting as an antioxidant and metalloenzyme inhibitors (such as carbonic anhydrase, acetylcholinesterase, butyrylcholinesterase, and tyrosinase inhibitors).
2. Materials and methods
2.1. General
All materials were purchased from commercial suppliers and used without further purification. All reactions were conducted under an atmosphere of nitrogen unless noted otherwise. Anhydrous solvents were distilled over appropriate drying agents before use. 1H NMR spectra were recorded on a Varian 400 MHz spectrometer in DMSO-d6 solution with the internal solvent signal peak at 2.50 ppm. 13C NMR was recorded at 100 MHz spectrometer in DMSO-d6 solution and referenced to the internal solvent signal at 39.5 ppm. Proton NMR data are reported as follows: chemical shift (ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartette, dd = doublet of doublets, dt = triplet of doublets, m = multiplet, brs. = broad singlet), and coupling constants (Hz). Infra-red spectra were measured using a Bruker Spectrometer transform-infra-red (FT-IR) spectrometer. The progress of the reactions was monitored by thin layer chromatography. For the detection of the spots, the plates were exposed to UV light (254 and 365 nm).
2.2. Synthesis procedure for preparation of sulphonamides
2.2.1. Synthesis of SA-1
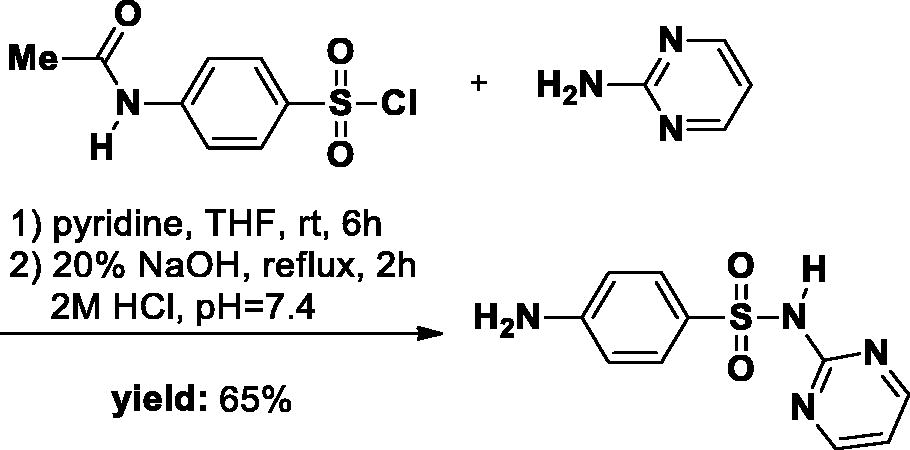
The SA-1 was synthesised as described in literatureCitation22. Briefly, pyrimidin-2-amine (1.14 g, 12 mmol) was added to a 50 ml round-bottom flask and dissolved in a solution of THF (20 ml) and pyridine (1 ml). p-Acetamidobenzenesulfonyl chloride (2.81 g, 12 mmol) (6 g, 2.81 mmol) was then added to the reaction solution. After completion of the reaction followed by TLC, the THF was removed in the evaporator. Subsequently, 30 ml 0.5 M HCl was added and a large amount of precipitate was formed. The precipitate was filtrated and then washed with water and used directly in the next step. The Intermediate was dissolved in a 20% NaOH solution and the reaction mixture was refluxed for 2 h. After the reaction was completed, the mixture was quenched to approximately pH 7.4 with 2 M HCl, resulting in a large number of white solids. The precipitate was obtained and washed with water, then dried overnight in a desiccator (white solid, yield 65%).
2.2.2. Synthesis of SA-2
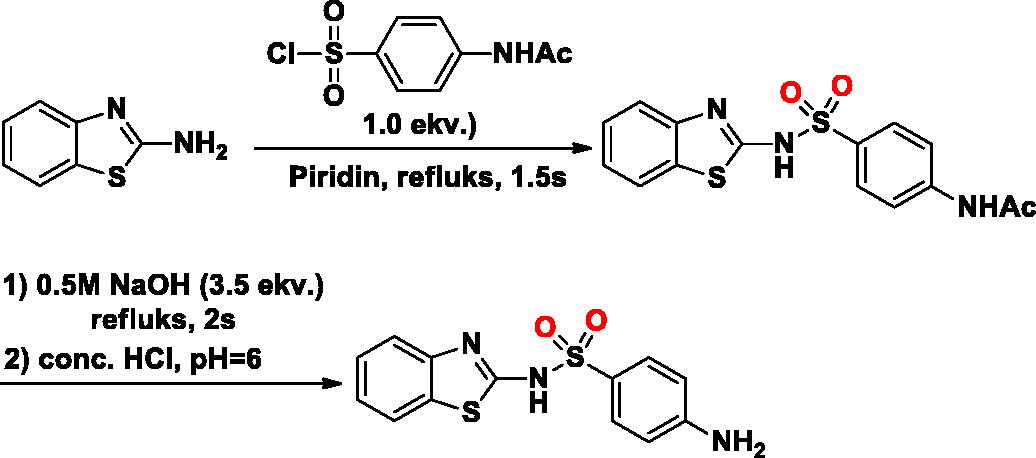
The SA-2 was synthesised as described in literatureCitation23. Briefly, in a 50 ml round-bottom flask, to a stirred solution of benzo[d]thiazol-2-amine (1.0 g, 6.66 mmol) in dry pyridine (6 ml) was added 4-acetamidobenzenesulfonyl chloride (1.55 g, 6.66 mmol). The reaction mixture was refluksed for 2 h. The reaction mixture was poured onto acidic crushed ice and filtered. The crude solid was washed with water and directly used in the next step. The intermediate was dissolved in 1 M NaOH solution (3.5 ekv. of the intermediate) and the reaction mixture was refluksed for 2 h before being cooled down to room temperature. Then the reaction solution was quenched with 2 M HCl to approximately pH 6, resulting in a large number of white solids. To obtain the desired product, the crude product was filtered, washed with water, and then dried in the desiccator (white solid, yield 68%).
2.2.3. Synthesis of SA-3
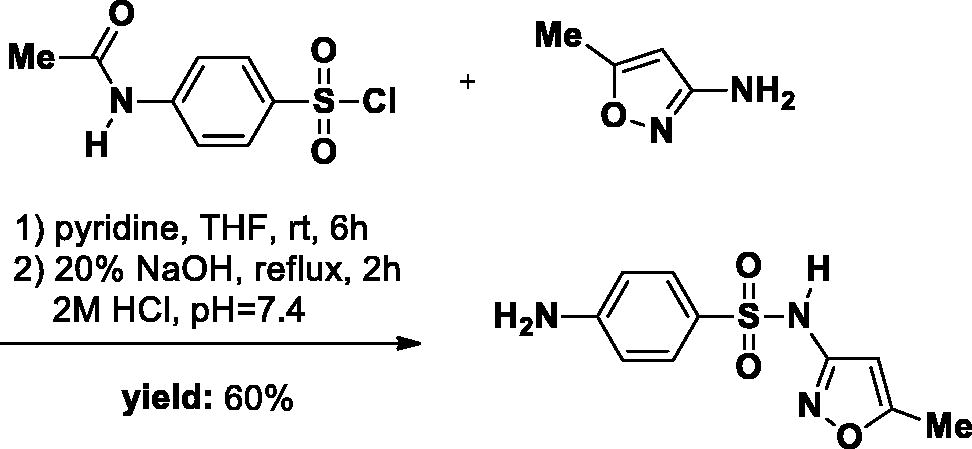
The SA-3 was synthesised as described in literatureCitation24. Briefly, 3-amino-5-methylisoxazole (1.18 g, 12 mmol) was added to a 50 ml round-bottom flask and dissolved in a solution of THF (20 ml) and pyridine (1 ml). p-Acetamidobenzensulfonyl chloride (6 g, 2.81 mmol) was then added to the reaction solution. The reaction mixture was stirred at room temperature for 12 h. The reaction was followed by TLC. After completion of the reaction, the THF was removed in the evaporator. Subsequently, 30 ml 0.5 M HCl was added to the crude mixture and a large amount of precipitate was formed. The precipitate was filtrated, washed with water, and directly used in the next step. The Intermediate was dissolved in a 20% NaOH solution and the reaction mixture was refluxed for 2 h. After the reaction was completed, the mixture was quenched to approximately pH 7.4 with 2 M HCl, resulting in a large number of white solids. The precipitate was obtained and washed with water, then dried overnight in a desiccator (white solid, yield 60%).
2.2.4. Synthesis of SA-4 and SA-5
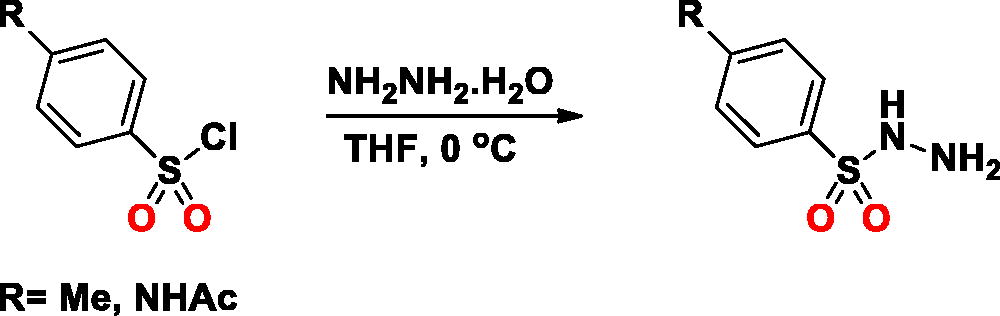
General Procedure: The SA-5 and SA-6 were synthesised as described in literatureCitation25. Briefly, a solution of an arylsulfonyl chloride (10 mmol) in tetrahydrofuran (40 ml) was added hydrazine monohydrate (1.25 ml, 2.5 equiv.) dropwise at 0 °C. The reaction mixture was stirred vigorously for 30 min at 0 °C. Upon completion, the reaction mixture was added ethyl acetate (50 ml) and extracted with saturated brine (3 × 50 ml). The organic layer was separated, dried over anhydrous Na2SO4, filtrated, concentrated in an evaporator and added to hexane (10 ml) over 5 min. The precipitate was filtered, collected, and dried in vacuum.
2.3. General procedure for the synthesis of compounds CX (1–6)
In a 25-ml round-bottomed flask equipped with a magnetic stirrer, sulphonamide derivative (0.4 mmol) was added to the solution of calix[4]arene-aldehyde (0.2 mmol, 146.6 mg) in a mixture of 10 ml CHCl3/MeOH (1:1). The resulting mixture heated to reflux overnight. After the reaction was complete, the solvent was removed. The crude mixture was dissolved with 2 ml of methylene chloride. Upon the addition of hexane to the solution, the target product was precipitated. Then, the product was filtered off and dried under vacuum at 40 °C. The obtained final pure compounds CX(1–6) were fully characterised by 1H-NMR and 13C-NMR techniques.
CX-1: A white solid, yield 717%. 1H NMR (400 MHz, DMSO-d6) δ; 9.19 (s, 2H, CONH), 8.34 (s, 2H, CH = N), 7.88 (d, J = 8.5 Hz, 4H, ArH) , 7.57 (d, J = 8.5 Hz, 4H, ArH) , 7.55 (s, 4H, calix-ArH), 7.11 (s, 4H, calix-ArH), 4.51 (s, 4H, OCH2), 4.20 (d, J = 13.5 Hz, 4H, ArCH2Ar), 3.69 (d, J = 13.5 Hz, 4H, ArCH2Ar), 3.49 (m, 4H, CH2CH2), 1.10 (s, 18H, tBu); 13C NMR (100 MHz, DMSO-d6) δ 166.9, 161.4, 153.2, 150.0, 141.9, 140.3, 131.7, 130.3, 129.6, 129.0, 128.3, 127.6, 126.8, 125.8, 74.1, 43.6, 38.6, 34.4, 32.5; Anal. Calcd for C64H66N8O12S2 (1041.24): C, 64.60; H, 5.81; N, 8.07; S, 6.16. Found: C, 64.83; H, 5.80; N, 8.07; S, 6.14.
CX-2: A yellow solid, yield 70%. 1H NMR (400 MHz, DMSO-d6) δ 9.31 (s, 2H, CONH), 8.30 (s, 2H, CH = N), 7.92 (d, J = 8.5 Hz, 4H, ArH), 7.60 (d, J = 8.5 Hz, 4H, ArH), 7.56 (s, 4H, calix-ArH), 7.15 (s, 4H, calix-ArH), 6.07 (s, 2H, Het-ArH), 4.50 (s, 4H, OCH2), 4.21 (d, J = 13.5 Hz, 4H, ArCH2Ar), 3.68 (d, J = 13.5 Hz, 4H, ArCH2Ar), 3.50 (m, 4H, CH2CH2), 2.25 (s, 6H, Me), 1.12 (s, 18H, tBu); 13C NMR (100 MHz, DMSO-d6) δ 168.8, 167.7, 160.5, 152.2, 151.1, 150.4, 143.3, 141.9, 132.4, 130.1,129.7, 129.2, 128.7, 127.1, 126.3, 124.6, 95.6, 74.7, 42.0, 38.7, 34.4, 32.0, 12.9; Anal. Calcd for C64H66N8O12S2 (1197.38): C, 63.88; H, 5.53; N, 9.31; S, 5.33. Found: C, 63.97; H, 5.50; N, 9.29; S, 5.35.
CX-3: A yellow solid, yield 72%. 1H NMR (400 MHz, DMSO-d6) δ 9.42 (s, 2H, CONH), 8.45 (d, J = 8.8 Hz, 4H, Het-ArH), 8.33 (s, 2H, CH = N), 7.96 (d, J = 8.8 Hz, 4H, ArH), 7.60 (d, J = 8.8 Hz, 4H, ArH), 7.58 (s, 4H, calix-ArH), 7.15 (s, 4H, calix-ArH), 6.97 (t, J = 4.8 Hz, 2H, ArH), 4.50 (s, 4H, OCH2), 4.21 (d, J = 13.4 Hz, 4H, ArCH2Ar), 3.70 (d, J = 13.4 Hz, 4H, ArCH2Ar), 3.50 (m, 4H, CH2CH2), 1.12 (s, 18H, tBu); 13C NMR (100 MHz, DMSO-d6) δ 167.4, 162.6, 159.7, 158.0, 157.4, 157.1, 155.9, 144.5, 142.2, 138.8,130.1, 129.9, 129.2, 128.7, 128.3, 127.7, 126.0, 75.6, 44.0, 38.2, 34.2, 31.1; Anal. Calcd for C64H64N10O10S2 (1197.38): C, 64.20; H, 5.39; N, 11.70; S, 5.36. Found: C, 64.42; H, 5.41; N, 11.74; S, 5.37.
CX-4: A yellow solid, yield 70%. 1H NMR (400 MHz, DMSO-d6) δ 9.41 (bs, 2H, CONH), 8.31 (s, 2H, CH = N), 7.92 (d, J = 8.5 Hz, 4H, ArH), 7.85–7.70 (m, 4H, Het-ArH), 7.57 (s, 4H, calix-ArH), 7.55–7.48 (m, 8H, ArH), 7.14 (s, 4H, calix-ArH), 4.50 (s, 4H, OCH2) 4.21 (d, J = 13.5 Hz, 4H, ArCH2Ar), 3.69 (d, J = 13.5 Hz, 4H, ArCH2Ar), 3.49 (m, 4H, CH2CH2), 1.12 (s, 18H, tBu); 13C NMR (100 MHz, DMSO-d6) δ 169.6, 166.1, 161.9, 152.6, 152.0, 151.4, 147.3, 144.2, 132.5, 130.9, 130.0, 129.4, 128.7, 128.2, 127.9, 127,4, 127.4, 127.0, 126.8, 126.2, 125.4, 75.9, 44.0, 38.5, 34.6, 31.0; Anal. Calcd for C70H66N8O10S4 (1307.58): C, 64.30; H, 5.09; N, 8.57; S, 9.81. Found: C, 64.49; H, 5.09; N, 8.59; S, 9.79.
CX-5: As a yellow solid, yield 90%. 1H NMR (400 MHz, DMSO-d6) δ 8.92 (s, 2H, CONH), 8.31 (s, 2H, CH = N), 7.72 (d, J = 8.5 Hz, 4H, ArH), 7.45 (s, 4H, calix-ArH), 7.34 (d, J = 8.5 Hz, 4H, ArH), 7.15 (s, 4H, calix-ArH), 2.30 (s, 6H, Me), 4.46 (s, 4H, OCH2), 4.10 (d, J = 13.2 Hz, 4H, ArCH2Ar), 3.58 (d, J = 13.2 Hz, 4H, ArCH2Ar), 3.47 (m, 4H, CH2CH2), 2.30 (s, 6H, Me), 1.09 (s, 18H, tBu); 13C NMR (100 MHz, DMSO-d6) δ 167.6, 153.1, 151.9, 148.2, 143.7, 141.4, 133.6, 131.8, 130.0, 129.5, 129.1, 128.2, 127.3, 125.6, 75.5, 44.3, 38.2, 34.5, 30.9, 21.8; Anal. Calcd for C58H64N6O10S2 (1069.29): C, 65.15; H, 6.03; N, 7.86; S, 6.00. Found: C, 65.39; H, 6.02; N, 7.86; S, 5.59.
CX-6: As a yellow solid. (203 mg, yield 88%); 1H NMR (400 MHz, DMSO-d6) δ 8.89 (s, 2H, CONH), 8.30 (s, 2H, CH = N), 7.78 (d, J = 9.0 Hz, 4H, ArH), 7.73 (d, J = 9.0 Hz, 4H, ArH), 7.42 (s, 4H, calix-ArH), 7.11 (s, 4H, calix-ArH), 2.30 (s, 6H, Me), 4.46 (s, 4H, OCH2), 4.10 (d, J = 13.1 Hz, 4H, ArCH2Ar), 3.57 (d, J = 13.1 Hz, 4H, ArCH2Ar), 3.47 (m, 4H, CH2CH2), 2.03 (s, 6H, Me), 1.06 (s, 18H, tBu); 13C NMR (100 MHz, DMSO-d6) δ 168.2, 167.6, 156.9, 152.4, 147.9, 143.0, 141.2, 132.2, 131.0, 129.7, 129.2, 128.4, 127.5, 126.4, 124.9, 75.2, 44.1, 38.0, 34.0, 30.1, 23.8; Anal. Calcd for C60H66N8O12S2 (1155.34): C, 62.37; H, 5.76; N, 9.70; S, 5.55. Found: C, 62.59; H, 5.76; N, 9.73; S, 5.54.
2.4. CA inhibition assay
An SX.18 MV-R Applied Photophysics (Oxford, UK) stopped-flow instrument has been used to assay the inhibition of various CA isozymesCitation26. Phenol Red (at a concentration of 0.2 mM) has been used as an indicator, working at the absorbance maximum of 557 nm, with 10 mM Hepes (pH 7.4) as a buffer, 0.1 M Na2SO4 or NaClO4 (for maintaining constant the ionic strength; these anions are not inhibitory in the used concentration), following the CA-catalyzed CO2 hydration reaction for a period of 5–10 s. Saturated CO2 solutions in water at 25 °C were used as substrate. Stock solutions of inhibitors were prepared at a concentration of 10 mM (in DMSO-water 1:1, v/v) and dilutions up to 0.01 nM done with the assay buffer mentioned above. At least 7 different inhibitor concentrations have been used for measuring the inhibition constant. Inhibitor and enzyme solutions were pre-incubated together for 10 min at room temperature before assay, to allow for the formation of the E-I complex. Triplicate experiments were done for each inhibitor concentration, and the value reported throughout the paper is the mean of such results. The inhibition constants were obtained by nonlinear least-squares methods using the Cheng-Prusoff equation, as reported earlier, and represent the mean from at least three different determinations1Citation27–32. All CA isozymes used here were recombinant proteins obtained as reported earlier by our group.
2.5. Determination of antioxidant, anticholinesterase and tyrosinase activity of calix[4]arene sulphonamides CX(1–6)
2.5.1. Dpph radical scavenging assay
The DPPH (2,2-diphenyl-1-picrylhydrazyl) radical scavenging activity of the synthesised compounds was determined by a spectrophotometric method based on the reduction of an ethanol solution of DPPHCitation33,Citation34. 2, 5, 10, 20 µL of 1 mM stock solution of each compound was completed to 40 µL with the DMSO and mixed with 160 µL of 0.1 mM of DPPH free radical solution. The mixture was led to stand for 30 min in the dark and the absorbance was then measured at 517 nm against a blank. Inhibition of free radical, DPPH, in percent (I %), was calculated according to the formula:
where Acontrol is the absorbance of the control reaction (containing all reagents except for the tested compounds), and Asample is the absorbance of the test compounds. Tests were carried out in triplicate. BHA and BHT were used as positive control.
2.5.2. Abts cation radical decolorisation assay
The percent inhibition of decolorisation of ABTS [2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid)] cation radical is obtained as a function of time and concentration and evaluated by comparison with the BHT and BHA compounds used as standardCitation35,Citation36. The tested compounds at different concentrations are added to each well and 160 µL of 7 mM ABTS solution is added. After 6 min at room temperature, the absorbances were measured at 734 nm. ABTS cation radical decolorisation activities were determined by using the equation below:
where A is the absorbance. Tests were carried out in triplicate. BHA and BHT were used as positive control.
2.5.3. Metal chelating activity
The chelating ability of synthesised compounds was examined according to the method of Dinis et al.Citation37. The tested compounds at different concentrations were added to each well and 4 µL of 2 mM ferrous (II) chloride was added. Then 8 µL of 5 mM ferrozine was added and the reaction was started. After 10 min at room temperature, the absorbance was measured at 562 nm against a blank. The results were expressed as a percentage of inhibition of the ferrozine-Fe2+ complex formation. The percentage inhibition of the ferrozine -Fe2+ complex formation was calculated using the formula given below:
where A is the absorbance. Tests were carried out in triplicate. EDTA was used as a positive control.
2.5.4. Anticholinesterase assay
The inhibitory effect of novel calix[4]azacrown substituted sulphonamide Schiff bases CX(1–6) on AChE and BChE activities was determined according to the slightly modified spectrophotometric method of Ellman et al.Citation38. All compounds were dissolved in DMSO to prepare stock solutions at 4 mM concentration. Aliquots of 150 µL of 100 mM sodium phosphate buffer (pH 8.0), 10 µL of sample solution and 20 µL AChE (or BChE) solution were mixed and incubated for 15 min at 25 °C, and DTNB [5,5′-Dithio-bis(2-nitro-benzoic)acid] (10 µL) is added. The reaction was then initiated by the addition of acetylthiocholine iodide (or butyrylthiocholine iodide) (10 µL). The final concentration of the tested compounds’ solution was 200 µM.
where A is the absorbance. Tests were carried out in triplicate. Galantamine was used as positive control.
2.5.5. Anti-tyrosinase activity
Anti-tyrosinase activity of the compounds was performed according to the method designed by Hearing and JimenezCitation39. Firstly, the inhibition of diphenolase function of the compounds was evaluated and L-DOPA was used as substrate. Tyrosinase from mushroom (E.C. 1.14.18.1) (30 U, 28 nM) was dissolved in Na-phosphate buffer (pH = 6.8, 50 nM) and the compounds were added to the solution for pre-incubation at room temperature for ten minutes. After incubation, 0.5 mM L-DOPA was added to the mixture and the change in absorbance was measured at 475 nm at 37 °C. For positive control, kojic acid was used. The following formula was used to calculate the percentage of all enzyme inhibitions:
A: Absorbance.
2.6. Statistical analysis
The results of the antioxidant, anticholinesterase, and tyrosinase activity assays are expressed as the mean ± SD of three parallel measurements. The statistical significance was estimated using a Student’s t-test, where p values < 0.05 were considered significant.
3. Results and discussion
3.1. Chemistry
To develop novel and effective enzyme inhibitors and antioxidant agents based on calixarenes, we used the calix[4](aza)crown dialdehyde as a scaffold to design a series of new derivatives bearing different sulphonamide moieties. The sulphonamide-substituted calix[4]zacrown derivatives CX(1–6) were obtained in four steps (Scheme 1). The required starting compound p-tert-butylcalix[4]arene was synthesised according to literature procedureCitation16 and then the calix[4]arene diester was obtained in satisfactory yield by reflux with bromomethyl acetate in the presence of K2CO3 in CH3CNCitation16. The diester derivative of calixarene was reacted with ethylenediamine in CH3OH/CHCl3 (1:1) at room temperature, being converted to p-tert-butycalix[4](aza-)crown derivative which was isolated by recrystallisation from the methanolCitation16. The desired p-tert-butycalix[4](aza)crown dialdehyde derivative was prepared by treating p-tert-butycalix[4](aza)crown with hexamethylenetetraamine with refluxing trifluoroacetic acidCitation16. In the last step, the sulphonamide derivatives (SA-1–5) were reacted with calix(aza)crown dialdehyde in absolute ethanol to synthesise the novel calix(aza)crown substituted sulphonamide derivatives CX(1–6). All the synthesised compounds were fully characterised by using spectroscopic techniques (see the experimental part for details).
Scheme 1. General synthetic route for the synthesis of the calix[4]arene substituted sulphonamide Schiff base derivatives CX(1–6).
![Scheme 1. General synthetic route for the synthesis of the calix[4]arene substituted sulphonamide Schiff base derivatives CX(1–6).](/cms/asset/04ad8029-3bbf-4a51-ac40-f568ca7022e0/ienz_a_1765166_sch0001_c.jpg)
3.2. CA inhibition studies
The newly synthesised calix[4]azocrown substituted sulphonamide Schiff bases CX(1–6) were assessed as inhibitors of six physiologically relevant CA isoforms, the cytosolic hCA I, hCA II, and hCA VII, membrane-bound hCA IV, and the transmembrane tumor-associated hCA IX and hCA XII, by a stopped flow CO2 hydrase assayCitation26. The clinically used sulphonamide acetazolamide (AAZ) was used as a positive control. The compounds, in general, showed low inhibition potency against the tested CA isoforms. Specifically, all the secondary sulphonamides exhibited inhibition constants >100 µM activity against the tested CA isoforms, except the isoform hCA IX, for which some of the compounds (CX-2, CX-3 and CX-6) showed moderate activity with KIs of 67.6, 46.0, and 64.6 µM, respectively. On the other hand, the primary sulphonamide substituted derivative (CX-1) showed medium potency activity against the tumor-associated isoforms hCA IX and hCA XII with KIs in the range of 0.15 to 0.27 µM, respectively, also inhibiting in the micromolar ramge the other isoforms ().
Table 1. In vitro hCA I, hCA II, hCA IV, hCA VII, hCA IX, and hCA XII inhibition data with calix[4]azacrown substituted sulphonamide Schiff base derivatives CX(1–6) investigated here, and standard sulphonamide inhibitor Acetazolamide (AAZ) by a stopped flow CO2 hydrase assayCitation26.
3.3. Antioxidant activity
The antioxidant capacities of the newly synthesised compounds CX(1–6) were demonstrated by using three different methods, namely, DPPH free radical scavenging, ABTS cation radical scavenging, and metal chelating methods. All of the compounds showed antioxidant activities in a dose-dependent manner and shown in , and the IC50 values were compared with the standards BHA, BHT, and EDTA. The three compounds (CX-1, CX-2, and CX-3) showed no activity against DPPH free radical assay with IC50 values of >1000 μM, but CX-5 and CX-6 had an activity comparable with standards, having IC50 values of 16.79 ± 0.85 and 9.02 ± 0.05 μM, respectively. Interestingly, these two compounds (CX-5 and CX-6) were also sensitive to ABTS radical scavenging activity with IC50 values of 9.79 ± 0.09 and 7.74 ± 0.04 μM, respectively. On the other hand, none of the tested compounds showed any metal chelating activity.
Table 2. The antioxidant activity of calix[4]azacrown substituted sulphonamide Schiff base derivatives CX(1–6) and controls BHA, BHT, and EDTA.
3.4. Acetylcholinesterase, butyrylcholinesterase, and tyrosinase activity
The calix[4]azacrown substituted sulphonamide Schiff bases CX(1–6) were also evaluated for their anti-cholinesterase (AChE and BChE) and anti-tyrosinase activities. None of the compounds from the series showed any inhibition potency against AChE and BChE enzymes, except for compounds CX-6, which showed moderate activity against BChE with % inhibition value of 35.41 ± 0.90. The tyrosinase activity of the compounds was also moderate and close the each other, with % inhibition values in the range of 16.48 ± 0.21 to 35.52 ± 0.82, except compound CX-5, which showed no activity against tyrosinase ().
Table 3. Anti-cholinesterase and anti-tyrosinase activity of calix[4]azacrown substituted sulphonamide Schiff base derivatives CX(1–6) and controls galantamine and kojik acid.
4. Conclusion
In the current work, we report a novel series of six calix[4]azacrown substituted sulphonamide Schiff bases which were synthesised by the reaction of calix[4]arene dialdehydes with different substituted primary and secondary sulphonamide derivatives. The newly synthesised novel compounds were investigated as antioxidant and metabolic enzyme inhibitors namely, carbonic anhydrase, acetylcholinesterase, butyrylcholinesterase, and tyrosinase enzymes. The results revealed that these calix[4]azacrown based sulphonamides CX(1–6) show, in general, low to moderate metabolic enzyme inhibition against hCAs, AChE, BChE, and tyrosinase enzymes. More specifically, only primary sulphonamide substituted compound (CX-1) showed moderate activity against six different isoforms of carbonic anhydrases with Ki values ranging from 0.15 to 5.55 µM. On the other hand, some of the synthesised compounds showed great antioxidant activity comparable with standards used in the study, such as CX-6 (IC50 of 9.02 ± 0.05 μM) for DPPH radical scavenging assay and CX-5 and CX-6 (IC50 9.79 ± 0.09 and 7.74 ± 0.04 μM, respectively) for ABTS radical decolarisation assay.
Acknowledgements
The authors thank the Research Foundation of Selcuk University for financial support of this work.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- (a) Supuran CT. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin Drug Discov 2017;12:61–88. (b) Capasso C, Supuran CT. An overview of the alpha-beta- and gamma-carbonic anhydrases from bacteria: can bacterial carbonic anhydrases shed new light on evolution of bacteria? J Enzyme Inhib Med Chem 2015;30:325–32. (c) Supuran CT. Carbon-versus sulphur-based zinc binding groups for carbonic anhydrase inhibitors? J Enzyme Inhib Med Chem 2018;33:485–95.
- (a) Supuran CT, Capasso C. The eta-class carbonic anhydrases as drug targets for antimalarial agents. Expert Opin Ther Targets 2015;19:551–63. (b) Del Prete S, De Luca V, De Simone G, et al. Cloning, expression and purification of the complete domain of the g-carbonic anhydrase from Plasmodium falciparum. J Enzyme Inhib Med Chem 2016;31:54–9. (c) Vullo D, Del Prete S, Fisher GM, et al. Sulfonamide inhibition studies of theg-class carbonic anhydrase from the malaria pathogen Plasmodium falciparum. Bioorg Med Chem 2015;23:526–31.
- (a) Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31:345–60. (b) Supuran CT. Structure and function of carbonic anhydrases. Biochem J 2016;473:2023–32. (c) Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81. (d) Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77. (e) Supuran CT, Vullo D, Manole G, et al. Designing of novel carbonic anhydrase inhibitors and activators. Curr Med Chem Cardiovasc Hematol Agents 2004;2:49–68.
- (a) Akocak S, Ilies MA. Next-generation primary sulfonamide carbonic anhydrase inhibitors. In: Supuran CT, Cappasso C, eds. Targeting carbonic anhydrases. London: Future Science; 2014:35–51. (b) Akocak S, Alam MR, Shabana AM, et al. PEGylated bis-sulfonamide carbonic anhydrase inhibitors can efficiently control the growth of several carbonic anhydrase IX-expressing carcinomas. J Med Chem 2016;59:5077–88. (c) Shabana AM, Mondal UK, Alam R, et al. pH-Senstive multiligand gold nanoplatform targeting carbonic anhydrase IX enhances the delivery of Doxorubicin to hypoxic tumor spheroids and overcomes the hypoxia-induced chemoresistance. ACS Appl Mater Interfaces 2018;10:17792–808. (d) Akocak S, Supuran CT. Activation of α-, β-, γ-, δ-, ζ-, and η- class of carbonic anhydrases with amines and amino acids: a review. J Enzyme Inhib Med Chem 2019;34:1652–9.
- (a) Supuran CT. Exploring the multiple binding modes of inhibitors to carbonic anhydrases for novel drug discovery. Expert Opin Drug Discov 2020. (b) Nocentini A, Supuran CT. Advances in the structural annotation of human carbonic anhydrases and impact on future drug discovery. Expert Opin Drug Discov 2019; 14:1175–97. (c) Berrino E, Supuran CT. Novel approaches for designing drugs that interfere with pH regulation. Expert Opin Drug Discov 2019;14:231–48.
- (a) Gul HI, Yamali C, Yesilyurt F, et al. Microwave-assisted synthesis and bioevaluation of new sulfonamides. J Enzyme Inhib Med Chem 2017;32:369–74. (b) Supuran CT. Carbonic anhydrases and metabolism. Metabolites 2018;8:25. (c) Guzel-Akdemir O, Akdemir A, Karali N, et al. Discovery of novel isatin-based sulfonamides with potent and selective inhibition of the tumor-associated carbonic anhydrase isoforms IX and XII. Org Biomol Chem 2015;13:6493–9. (d) Alterio V, Di Fiore A, D’Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012; 112:4421–68. (e) da Silva Cardoso V, Vermelho AB, Ricci Junior E, et al. Antileishmanial activity of sulphonamide nanoemulsions targeting the β-carbonic anhydrase from Leishmania species. J Enzyme Inhib Med Chem 2018;33:850–7. (f) Supuran CT, Capasso C. An overview of the bacterial carbonic anhydrases. Metabolites 2017;7:56.
- (a) Bag S, Tulsan R, Sood A, et al. Sulfonamides as multifunctional agents for Alzheimer’s disease. Bioorg Med Chem 2015;25:626–30. (b) Gocer H, Akincioglu A, Goksu S, et al. Carbonic anhydrase and acetylcholinesterase inhibitory effects of carbamates and sulfamoylcarbamates. J Enzyme Inhib Med Chem 2014;30:316–20. (c) Supuran CT. Carbonic anhydrase activators. Future Med Chem 2018;10:561–73.
- (a) Supuran CT. Carbonic anhydrase inhibitors and their potential in a range of therapeutic areas. Expert Opin Ther Pat 2018;28:709–12. (b) Gulcin I, Scozzafava A, Supuran CT, et al. The effect of caffeic acid phenethyl ester (CAPE) on metabolic enzymes including acetylcholinesterase, butyrylcholinesterase, glutathione S-transferase, lactoperoxidase, and carbonic anhydrase isoenzymes I, II, IX, and XII. J Enzyme Inhib Med Chem 2016;31:1095–101. (c) Supuran CT. Applications of carbonic anhydrases inhibitors in renal and central nervous system diseases. Expert Opin Ther Pat 2018;28:713–21.
- (a) Rishton GM, Retz DM, Tempest PA, et al. Fencyhylamine sulfonamide inhibitors of amyloid beta peptide production by the gamma-secretase proteolytic pathway: potential small-molecule therapeutic agents for the treatment of Alzheimer’s disease. J Med Chem 2000;43:2297–9. (b) Gocer H, Topal F, Topal M, et al. Acetylcholinesterase and carbonic anhydrase isoenzymes I and II inhibition profiles of taxifolin. J Enzyme Inhib Med Chem 2016;31:441–7. (c) Provensi G, Carta F, Nocentini A, et al. A new kid on the block? Carbonic anhydrases as possible new targets in Alzheimer’s disease. Int J Mol Sci 2019;20:4724.
- (a) Wang J, Gu BJ, Masters CL, Wang YJ. A systemic view of Alzheimer disease-insights from amyloid-b metabolism beyond the brain. Nat Rev Neurol 2017;13:612–23. (b) Lolak N, Boga M, Tuneg M, et al. Sulphonamides incorporating 1,3,5-triazine structural motifs show antioxidant, acetylcholinesterase, butyrylcholinesterase, and tyrosinase inhibitory profile. J Enzyme Inhib Med Chem 2020;35:424–31.
- (a)Yılmaz S, Akbaba Y, Özgeriş B, et al. Synthesis and inhibitory properties of some carbamates on carbonic anhydrase and acetylcholine esterase. J Enzyme Inhib Med Chem 2016;31:1484–91. (b) Ozgun DO, Yamali C, Gul HI, et al. Inhibitory effects of isatin Mannich bases on carbonic anhydrases, acetylcholinesterase, and butyrylcholinesterase. J Enzyme Inhib Med Chem 2016;31:1498–501. (c) Tars K, Vullo D, Kazaks A, et al. Sulfocoumarins (1,2-benzoxathiine-2,2-dioxides): a class of potent and isoform-selective inhibitors of tumor-associated carbonic anhydrases. J Med Chem 2013;56:293–300. (d) Nocentini A, Supuran CT, Winum JY. Benzoxaborole compounds for therapeutic uses: a patent review (2010–2018). Expert Opin Ther Pat 2018;28:493–504.
- (a) Ozgeris B, Goksu S, Kose LP, et al. Acetylcholinesterase and carbonic anhydrase inhibitory properties of novel urea and sulfamide derivatives incorporating dopaminergic 2-aminotetralin scaffolds. Bioorg Med Chem 2016;24:2318–29. (b) Akıncıoğlu A, Akıncıoğlu H, Gülçin İ, et al. Discovery of potent carbonic anhydrase and acetylcholine esterase inhibitors: novel sulfamoyl carbamates and sulfamides derived from acetophenones. Bioorg Med Chem 2015;23:3592–602.
- (a) Yilmaz M, Sayin S. Calixarenes in organo and biomimetic catalysis. In: Neri P, Sessler J, Wang MX, eds. Calixarenes and beyond. Cham: Springer; 2016:719–42. (b) Bhatti AA, Oguz M, Yilmaz M. New water soluble p-sulphonatocalix [4] arene chemosensor appended with rhodamine for selective detection of Hg2+ ion. J. Mol 2020;1203:127436.
- (a) Yilmaz M, Karanastasis AA, Chatziathanasiadou M, et al. Inclusion of quercetin in gold nanoparticles decorated with supramolecular hosts amplifies its tumor targeting properties. ACS Appl. Bio Mater 2019;2:2715–25. (b) Oguz M, Bhatti AA, Dogan B, et al. Formation of the inclusion complex of water soluble fluorescent calix [4] arene and naringenin: solubility, cytotoxic effect and molecular modeling studies. J Biomol Struct Dyn 2019.
- (a) Oguz M, Gul A, Karakurt S, Yilmaz M. Synthesis and evaluation of the antitumor activity of Calix [4] arene L-proline derivatives. Bioorg Chem 2020;94:103207. (b) Karakurt S, Kellici TF, Mavromoustakos T, et al. Calixarenes in lipase biocatalysis and cancer therapy. Curr Org Chem 2016;20:1043–57. (c) Bardi B, Tosi I, Faroldi F, et al. A calixarene-based fluorescent ratiometric temperature probe. Chem Commun (Camb) 2019;55:8098–101.
- (a) Gasparello J, Manicardi A, Casnati A, et al. Efficient cell penetration and delivery of peptide nucleic acids by an argininocalix[4]arene. Sci Rep 2019;9:3036. (b) Bayrakci M, Kursunlu AN, Güler E, Ertul Ş. A new calix [4] azacrown ether based boradiazaindacene (Bodipy): selective fluorescence changes towards trivalent lanthanide ions. Dyes Pigm 2013;99:269–74. (c) Sestito SE, Facchini FA, Morbioli I, et al. Amphiphilic guanidinocalixarenes inhibit lipopolysaccharide (LPS)- and lectin-stimulated toll-like receptor 4 (TLR4) signaling. J Med Chem 2017;60:4882–92.
- (a) Rouge P, Pires VS, Gaboriau F, et al. Antiproliferative effect on HepaRG cell cultures of new calix[4]arenes. J Enzyme Inhib Med Chem 2010;25:216–27. (b) Pires VS, Gaboriau F, Guillon J, et al. Modulation of cell proliferation in rat liver cell cultures by new calix[4]arenes. J Enzyme Inhib Med Chem 2006;21:261–70. (c) Giuliani M, Morbioli I, Sansone F, Casnati A. Moulding calixarenes for biomacromolecule targeting. Chem Commun (Camb) 2015;51:14140–59.
- (a) Akocak S, Lolak N, Nocentini A, et al. Synthesis and biological evaluation of novel aromatic and heterocyclic bis-sulfonamide Schiff bases as carbonic anhydrase I, II, VII and IX inhibitors. Bioorg Med Chem 2017;25:3093–7. (b) Akocak S, Lolak N, Bua S, et al. Synthesis and biological evaluation of novel N,N′-diaryl cyanoguanidines acting as potent and selective carbonic anhydrase II inhibitors. Bioorg Chem 2018;77:245–51. (c) De Simone G, Supuran CT. (In)organic anions as carbonic anhydrase inhibitors. J Inorg Biochem 2012;111:117–29.
- (a) Supuran CT, Altamimi ASA, Carta F. Carbonic anhydrase inhibition and the management of glaucoma: a literature and patent review 2013-2019. Expert Opin Ther Pat 2019;29:781–92. (b) Supuran CT. The management of glaucoma and macular degeneration. Expert Opin Ther Pat 2019;29:745–7. (c) Supuran CT, Alterio V, Di Fiore A, et al. Inhibition of carbonic anhydrase IX targets primary tumors, metastases, and cancer stem cells: three for the price of one. Med Res Rev 2018;38:1799–836. (d) Supuran CT. Carbonic anhydrase inhibitors as emerging agents for the treatment and imaging of hypoxic tumors. Expert Opin Investig Drugs 2018;27:963–70.
- (a) Lolak N, Akocak S, Bua S, Supuran CT. Design, synthesis and biological evaluation of ureido benzenesulfonamides incorporating 1,3,5-triazine moieties as potent carbonic anhydrase IX inhibitors. Bioorg Chem 2019;82:117–22. (b) Lolak N, Akocak S, Bua S, et al. Discovery of new ureido benzenesulfonamides incorporating 1,3,5-triazine moieties as carbonic anhydrase I, II, IX and XII inhibitors. Bioorg Med Chem 2019;27:1588–94. (c) Lolak N, Akocak S, Bua S, et al. Design and synthesis of novel 1,3-diaryltriazene-substituted sulfonamides as potent and selective carbonic anhydrase II inhibitors. Bioorg Chem 2018;77:542–7. (d) Nocentini A, Supuran CT. Carbonic anhydrase inhibitors as antitumor/antimetastatic agents: a patent review (2008-2018). Expert Opin Ther Pat 2018;28:729–40.
- Durgun M, Türkeş C, Işık M, et al. Synthesis, characterization, biological evaluation, and in silico studies of novel series sulfonamide derivatives. J Enzyme Inhib Med Chem 2020;35:950–62.
- Wang L, Li L, Zhou ZH, et al. Structure-based virtual screening and optimization of modulators targeting Hsp90-Cdc37 interaction. Eur J Med Chem 2017;136:63.
- Patel NB, Agravat SN. Synthesis and microbial studies of new pyridine derivatives III. Chin J Chem 2007;25:1363–69.
- Sunduru N, Salin O, Gylfe Å, Elofsson M. Design, synthesis and evaluation of novel polypharmacological antichlamydial agents. Eur J Med Chem 2015;101:595–603.
- Shinde PS, Patil NT. Copper-catalyzed sulfonyl radical-enabled regioselective cyclization of 1, 6-enynes. Eur J Org Chem 2017;2017:3512–15.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73.
- (a) Draghici B, Vullo D, Akocak S, et al. Ethylene bis-imidazoles are highly potent and selective activators for isozymes VA and VII of carbonic anhydrase, with a potential nootropic effect. Chem Commun 2014;50:5980–3. (b) Akocak S, Lolak N, Vullo D, et al. Synthesis and biological evaluation of histamine Schiff bases as carbonic anhydrase I, II, IV, VII, and IX activators. J Enzym Inhib Med Ch 2017;32:1305–12. (c) Buzás GM, Supuran CT. The history and rationale of using carbonic anhydrase inhibitors in the treatment of peptic ulcers. In Memoriam Ioan Puşcaş (1932-2015). J Enzyme Inhib Med Chem 2016;31:527–533. d) Supuran CT. Bacterial carbonic anhydrases as drug targets: toward novel antibiotics? Front Pharmacol 2011;2:34.(e) Nishimori I, Onishi S, Takeuchi H, Supuran CT. The alpha and beta classes carbonic anhydrases from Helicobacter pylori as novel drug targets. Curr Pharm Des 2008;14:622–630.
- (a) Şentürk M, Gülçin İ, Beydemir Ş, et al. In vitro inhibition of human carbonic anhydrase I and II isozymes with natural phenolic compounds. Chem Biol Drug Des 2011;77:494–499. (b) Temperini C, Scozzafava A, Vullo D, Supuran CT. Carbonic anhydrase activators. Activation of isoforms I, II, IV, VA, VII, and XIV with L- and D-phenylalanine and crystallographic analysis of their adducts with isozyme II: stereospecific recognition within the active site of an enzyme and its consequences for the drug design. J Med Chem 2006;49:3019–3027. (c) Nocentini A, Bonardi A, Gratteri P, et al. Steroids interfere with human carbonic anhydrase activity by using alternative binding mechanisms. J Enzyme Inhib Med Chem 2018;33:1453–59.
- (a) Rotondi G, Guglielmi P, Carradori S, et al. Design, synthesis and biological activity of selective hCAs inhibitors based on 2-(benzylsulfinyl)benzoic acid scaffold. J Enzyme Inhib Med Chem 2019;34:1400–13. (b) Supuran CT. Carbonic anhydrase inhibition and the management of hypoxic tumors. Metabolites 2017;7:E48. (c) Métayer B, Angeli A, Mingot A, et al. Fluoroenesulphonamides: N-sulphonylurea isosteres showing nanomolar selective cancer-related transmembrane human carbonic anhydrase inhibition. J Enzyme Inhib Med Chem 2018;33:804–808. (d) Casey JR, Morgan PE, Vullo D, et al. Carbonic anhydrase inhibitors. Design of selective, membrane-impermeant inhibitors targeting the human tumor-associated isozyme IX. J Med Chem 2004;47:2337–47.
- (a) Krall N, Pretto F, Decurtins W, et al. A small-molecule drug conjugate for the treatment of carbonic anhydrase IX expressing tumors. Angew Chem Int Ed Engl 2014;53:4231–35. (b) Rehman SU, Chohan ZH, Gulnaz F, Supuran CT. In-vitro antibacterial, antifungal and cytotoxic activities of some coumarins and their metal complexes. J Enzyme Inhib Med Chem 2005;20:333–40. (c) Clare BW, Supuran CT. Carbonic anhydrase activators. 3: structure-activity correlations for a series of isozyme II activators. J Pharm Sci 1994;83:768–73. (d) Ammazzalorso A, Carradori S, Angeli A, et al. Fibrate-based N-acylsulphonamides targeting carbonic anhydrases: synthesis, biochemical evaluation, and docking studies. J Enzyme Inhib Med Chem 2019;34:1051–61. (e) Chohan ZH, Munawar A, Supuran CT. Transition metal ion complexes of Schiff-bases. Synthesis, characterization and antibacterial properties. Met Based Drugs 2001;8:137–43. (f) Zimmerman SA, Ferry JG, Supuran CT. Inhibition of the archaeal β-class (Cab) and γ-class (Cam) carbonic anhydrases. Curr Top Med Chem 2007;7:901–908.
- (a) Köhler K, Hillebrecht A, Schulze Wischeler J, et al. Saccharin inhibits carbonic anhydrases: possible explanation for its unpleasant metallic aftertaste. Angew Chem Int Ed Engl 2007;46:7697–99. (b) Ozensoy Guler O, Capasso C, Supuran CT. A magnificent enzyme superfamily: carbonic anhydrases, their purification and characterization. J Enzyme Inhib Med Chem 2016;31:689–94. (c) De Simone G, Langella E, Esposito D, et al. Insights into the binding mode of sulphamates and sulphamides to hCA II: crystallographic studies and binding free energy calculations. J Enzyme Inhib Med Chem 2017;32:1002–11. (d) Alper Türkoğlu E, Şentürk M, Supuran CT, Ekinci D. Carbonic anhydrase inhibitory properties of some uracil derivatives. J Enzyme Inhib Med Chem 2017;32:74–7. (e) Alterio V, Esposito D, Monti SM, et al. Crystal structure of the human carbonic anhydrase II adduct with 1-(4-sulfamoylphenyl-ethyl)-2,4,6-triphenylpyridinium perchlorate, a membrane-impermeant, isoform selective inhibitor. J Enzyme Inhib Med Chem 2018;33:151–57.
- (a) Akocak S, Lolak N, Bua S, et al. a-Carbonic anhydrases are strongly activated by spinaceamine derivatives. Bioorg Med Chem 2019;27:800–4. (b) Akocak S, Lolak N, Bua S, et al. Activation of human a-carbonic anhydrase isoforms I, II, IV and VII with bis-histamine Schiff bases and bis-spinaceamine substituted derivatives. J Enzyme Inhib Med Chem 2019;34:1193–8.
- Blois MS. Antioxidant determinations by the use of a stable free radical. Nature 1958;181:1199–1200.
- Akocak S, Boga M, Lolak N, et al. Design, synthesis and biological evaluation of 1,3-diaryltriazene-substituted sulfonamides as antioxidant, acetylcholinesterase, and butyrylcholinesterase inhibitors. J Turk Chem Soc Sect A: Chem 2019;6:63–70.
- Pellegrini RRN, Proteggente A, Pannala A, et al. Antioxidant activity applying and improved ABTS radical cation decolorization assay. Free Rad Bio Med 1999;26:1231–37.
- Akocak S, Lolak N, Tuneg M, Boga M. Antioxidant, acetylcholinesterase and butyrylcholinesterase inhibition profiles of histamine Schiff bases. J Turk Chem Soc Sect A: Chem 2019;6:157–64.
- Dinis TCP, Madeira VMC, Almeida LM. Action of phenolic derivatives (acetoaminophen, salycilate and 5-aminosalycilate) as inhibitors of membrane lipid preoxidation and as preoxyl radical scavengers. Arc Biochem Biophy 1994;315:161–9.
- Ellman GL, Courtney KD, Andres V, Feather-Stone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 1961;7:88–95.
- Hearing VJ, Jiménez M. Mammalian tyrosinase – the critical regulatory control point in melanocyte pigmentation. Int J Biochem 1987;19:1141–47.