
Abstract
As a continuation for our previous work, a novel set of N-alkylindole-isatin conjugates (7, 8a–c, 9 and 10a–e) is here designed and synthesised with the prime aim to develop more efficient isatin-based antitumor candidates. Utilising the SAR outputs from the previous study, our design here is based on appending four alkyl groups with different length (ethyl and n-propyl), bulkiness (iso-propyl) and unsaturation (allyl) on N-1 of indole motif, with subsequent conjugation with different N-unsubstituted isatin moieties to furnish the target conjugates. As planned, the adopted strategy achieved a substantial improvement in the growth inhibitory profile for the target conjugates in comparison to the reported lead VI. The best results were obtained with N-propylindole –5-methylisatin hybrid 8a which displayed broad spectrum anti-proliferative action with efficient sub-panel GI50 (MG-MID) range from 1.33 to 4.23 µM, and promising full-panel GI50 (MG-MID) equals 3.10 µM, at the NCI five-dose assay. Also, hybrid 8a was able to provoke cell cycle disturbance and apoptosis in breast T-47D cells as evidenced by the DNA flow cytometry and Annexin V-FITC/PI assays. Furthermore, hybrid 8a exhibited good inhibitory action against cell cycle regulator CDK2 protein kinase and the anti-apoptotic Bcl-2 protein (IC50= 0.85 ± 0.03 and 0.46 ± 0.02 µM, respectively). Interestingly, molecular docking for hybrid 8a in CDK2 and Bcl-2 active sites unveiled that N-propyl group is involved in significant hydrophobic interactions. Taken together, the results suggested conjugate 8a as a promising lead for further development and optimisation as an efficient antitumor drug.
1. Introduction
In the current medical era, cancer is considered as a major public health problem worldwide and one of the most leading causes of death throughout the world. So, development of more effective new drugs for management of different human malignancies is a major requirement. The expanded knowledge for the functional relationship between the different molecules which constitute the cell cycle and checkpoint pathways furnished novel promising strategies for the management of tumours.
Cyclin-dependent kinases (CDKs) are considered as crucial factors that affect diverse key transitions in cell cycle for the cancer cell, in addition to regulation of apoptosis, transcription and exocytosis, therefore therapeutic strategies based on inhibition of CDKs stand out as a promising opportunity for anticancer drug discovery and an efficient approach for management of different human malignancies. On the other hand, apoptosis, an automatic cancer cell death, was found as an important consequence of CDKs inhibition and can be assessed by cell cycle arrest at low concentration or even mitochondrial damage at high concentrationCitation1. This fact was discovered from previous studies on CDKs inhibitors as Roscovitine and Purvalanol and their effect on three different prostate cancer cell linesCitation2. In addition, the link between CDKs and apoptosis was revealed in the investigational research of the effect of Ibulocydine, a prodrug CDK inhibitor, on hepatocellular carcinomaCitation3.
Isatin moiety served as a contributor in various CDKs inhibitors having an apoptotic effect. For example, isatin dimers, such as Indirubin-3′-oxime (), revealed a potent inhibition against CDK1, CDK2, and CDK5 with IC50s = 180 nM, 500 nM and 250 nM, respectivelyCitation4. In addition to its effect against CDKs, it possessed an apoptotic effect by disruption of cell cycle phases through arresting G2/M phaseCitation5. Indirubin-3′-oxime is regarded as a lead compound. It is especially known in Chinese traditional medicine, and its other derivatives as 5-Bromoindirun and Indirubin-5-sulfonate proved to be potent CKD inhibitors against CDK2 with IC50s = 1 and 0.5 µM, respectively. Moreover, they were co-crystallized with CDK2 (2BHE and 1E9H, respectively) ()Citation6,Citation7. This dimeric scaffold is initiative for the design of other derivatives with a significant activity against CDKs as reportedCitation8,Citation9. Other isatin derivatives decorated with 3-substitution expressed potent inhibition for CDK2 as shown in SU9516 which inhibits CDK2 with IC50 = 25 nM ()Citation10. Moreover, compound (I) is another isatin derivative with hydrazino linker at position 3 (CDK2; IC50 = 60 nM) that was co-crystallized with its target and submitted to protein data bank (1VFT). Several modifications on compound (I) were performed to get compound (II) which inhibits CDK2 in sub-nanomolar range (IC50 = 0.54 nM) ()Citation11.
Figure 1. Chemical structures for certain reported isatin-based anticancer agents (I–V), and lead compound VI.
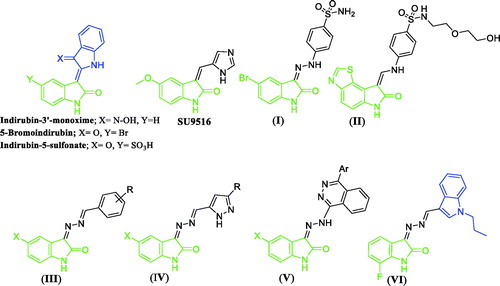
In last few years, our research team has developed diverse small molecules based on the isatin scaffold as efficient anticancer agents (structures III–VCitation12–14, ) with diverse cellular and enzymatic targets; for example triggering of apoptosis in several tumour cell linesCitation15,Citation16, inhibition of tumour-linked human carbonic anhydrase isoform IXCitation17,Citation18, and inhibition of VEGFR-2 kinaseCitation19. In 2018, we have reported the conjugation between the isatin and indole moieties via methylenehydrazono spacer (HC=N–N=) to develop new three different series of [(3-indolylmethylene)hydrazono]indolinone derivatives as anticancer agents with promising pro-apoptotic activityCitation20. As concluded from SAR analysis for this studyCitation20, hybridisation of N-propyl indole moiety with N-unsubstituted isatin moiety (compound VI, ) achieved the most effective anticancer activity.
Inspired by these findings and in connection with our previous research work, it is thought advantageous to broaden our investigations to probe new N-alkylindole-isatin conjugates 7, 8a–c, 9 and 10a–e exerting promising anticancer and pro-apoptotic actions. Utilising the SAR outputs from the previous studyCitation20, our design here is based on appending four alkyl groups with different length (ethyl 7, n-propyl 8a–c), bulkiness (isopropyl 9) and unsaturation (allyl 10a–e) on N-1 of indole motif, with subsequent conjugation with different N-unsubstituted isatin moieties to furnish the target conjugates.
All the synthesised conjugates were examined for their cytotoxicity towards a list of three different human cancer cell lines HCT-116 (Colon), A-549 (NSCLC), and MDA-MB-231 (Breast) utilising SRB assay. Furthermore, seven hybrids (8a–c, 9, 10a, 10c and 10d) were screened for their possible in vitro anticancer action in accordance with US-NCI protocol, then, 8a was furthermore chosen for testing at the five-dose assay. Subsequently, we examined the growth inhibition mechanism of hybrid 8a in relation to cell cycle regulation as well as apoptosis induction in breast T-47D cancer cells through the DNA flow cytometry and Annexin V-FITC/PI assays. Moreover, inhibitory actions of hybrid 8a against the cell cycle regulator protein CDK2 kinase and the anti-apoptotic protein Bcl-2 were explored. Finally, docking simulations were conducted in order to explore the behaviour of 8a within the active site of both CDK2 and Bcl-2, and to justify its activity.
2. Results and discussion
2.1. Chemistry
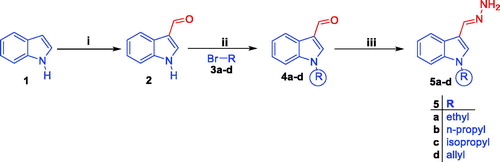
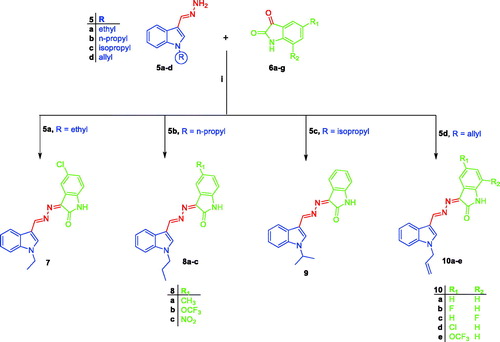
Synthetic routes herein proposed in order to get the targeted conjugates (7, 8a–c, 9 and 10a–e) have been illustrated in Schemes 1 and 2. With regard to Scheme 1, preparation of 1H-indole-3-carbaldehyde 2 was achieved through Vilsmeier formylation of indole 1 by the use of N, N-dimethylformamide (DMF) and phosphorus oxychloride (POCl3), then aldehyde 2 was undergone to N-alkylation using different alkyl halides 3a–d in DMF with the aid of sodium hydride base to get N-substituted indole-3-carbaldehyde intermediates 4a–d. The different N-substituted indole-3-carbaldehyde derivatives 4a–d were condensed with hydrazine hydrate via heating under reflux temperature in ethyl alcohol in order to produce the key intermediates N-substituted-3-(hydrazonomethyl)-1H-indoles 5a–d. In Scheme 2, the key intermediates 5a–d were reacted with different isatin derivatives 6a–g in absolute ethyl alcohol with the aid of catalytic amount of glacial acetic acid to furnish targeted [(3-indolylmethylene)hydrazono]indolinones 7, 8a–c, 9 and 10a–e.
Scheme 1. Preparation of the key intermediates 5a–d; (i) DMF, POCl3, reflux 8 h.; (ii) DMF, NaH, stirring at R.T for 24 h.; (iii) Ethyl alcohol, NH2NH2·H2O, reflux 2 h.
Scheme 2. Synthesis of target conjugates 7, 8a–c, 9 and 10a-e; (i) Ethyl alcohol, acetic acid, reflux 3 h.
Postulated structures for the newly prepared key intermediates and target compounds 7, 8a–c, 9 and 10a–e have been in full consistent with the data of both spectral and elemental analyses.
2.2. Biological evaluation
2.2.1. Antiproliferative activities towards A-549, MDA-MB-231 and HCT-116 cancer cell lines
All the newly prepared target conjugates (7, 8a–c, 9 and 10a–e) were investigated for the prospective growth inhibitory actions against three different cancer cell lines viz.; A-549, MDA-MB-231, and HCT-116, using the sulforhodamine B colorimetric (SRB) assayCitation21. Staurosporine, a clinically used antitumor drug, was co-assayed as a reference agent for the experiment. The results have been conveyed as IC50 values, and listed in .
Table 1. Anti-proliferative activity of compounds 7, 8a–c, 9 and 10a–e against human cancer A-549, MDA-MB-231 and HCT-116 cell lines. 
Investigation of the obtained IC50 values () hinted out that the tested N-alkylindole-isatin derivatives (7, 8a–c, 9 and 10a–e) were more effective against colon HCT-116 cells than NSCLC A-549 and TNBC MDA-MB-231 cancer cells, except compound 8b that displayed enhanced anti-proliferative potency against A-549 and MDA-MB-231 cell lines (IC50 = 9.2 ± 0.78 and 8.9 ± 0.61 µM, correspondingly) than HCT-116 cells (IC50 = 19.1 ± 1.34 µM), and compound 9 that exhibited slightly enhanced antiproliferative action against breast MDA-MB-231 cells (IC50 = 10.4 ± 0.80 µM) than colon HCT-116 cells (IC50 = 28.2 ± 1.73 µM).
With respect to the cytotoxic activity against colon HCT-116 cell line, hybrid 8a stood out as the most potent analogue herein reported with IC50 = 2.6 ± 0.17 µM, which is more effective than reference drug doxorubicin (IC50 = 3.7 ± 0.24 µM). Also, conjugates 8c, 10a and 10c exhibited potent anti-proliferative activity (IC50 = 6.4 ± 0.50, 7.7 ± 0.68 and 7.3 ± 0.47 µM, respectively) with about 2-fold decreased efficiency than doxorubicin. Moreover, compounds 7 and 8b possessed moderate potency with IC50 values equal 19.7 ± 1.28 and 19.1 ± 1.34 µM, respectively, whereas compounds 9, 10b, 10d and 10e elicited weak growth inhibitory activity towards HCT-116 cells (IC50 = 28.2 ± 1.73, 36.3 ± 2.35, 28.5 ± 1.59 and 37.4 ± 2.06 µM, respectively).
Exploring anti-proliferative activities towards NSCLC A-549 cells revealed that hybrid 8a was the most efficient counterpart (IC50 = 7.3 ± 0.42 µM), with about 3-fold dwindled efficiency compared to the reference compound doxorubicin (IC50 = 2.3 ± 0.17 µM) against A-549 cells. Moreover, compounds 8b, 8c and 10a exhibited efficient anti-proliferative action towards A-549 cells with IC50 values equal 9.2 ± 0.78, 12.3 ± 1.05 and 17.3 ± 1.19 µM, respectively. With regards to the cytotoxicity against the breast MDA-MB-231 cell line, the data shown in ascribed to the examined hybrids excellent to weak efficacy in inhibiting the proliferation of MDA-MB-231 cells with IC50 values ranging between 4.7 ± 0.28 and 42.9 ± 2.51 µM, except compounds 10b and 10e that failed to inhibit the growth of breast MDA-MB-231 cells up to 50 µM (IC50 = 54.0 ± 4.17, and 51.8 ± 3.77 µM, respectively). Superiorly, compound 8a emerged as the most effective derivative in the current study towards MDA-MB-231 cells with IC50 value equals 4.7 ± 0.28 µM. Besides, compounds 8b, 8c, 9 and 10a displayed good anti-proliferative activity against MDA-MB-231 cells (IC50 = 8.9 ± 0.61, 15.6 ± 0.98, 10.4 ± 0.80 and 11.7 ± 1.04 µM, respectively).
2.2.2. Nci-USA cancer cell lines screening
Seven hybrids (8a–c, 9, 10a, 10c and 10d) have been selected and screened by USA-National Cancer Institute (NCI-DTP; www.dtp.nci.nih.gov) for their possible in vitro anti-proliferative actions towards a panel of 59 human cancer cell lines representing breast, ovarian, CNS, colon, NSCLC, leukaemia, melanoma, renal and prostate cancers, according to the NCI, Bethesda, Drug Evaluation Branch protocolCitation22–24.
2.2.2.1. Preliminary single (10 µM) dose screening
Antitumor activities of here reported conjugates 8a–c, 9, 10a, 10c and 10d were first screened in an initial single dose (10 µM) screening, utilising the SRB assay to estimate cells viability and growthCitation21. The obtained results were presented as percent growth inhibition (GI %) for the examined conjugates towards the different treated tumour cell lines, as presented in .
Table 2. In vitro GI % for compounds (8a–c, 9 and 10a, c, d), at 10 μM concentration, towards the subpanel cancer cell lines
Exploring the attained GI% values (), disclosed that hybrid 8a stood out as the most efficient anti-proliferative agent in the herein reported NCI screening exhibiting mean GI % equals to 59, with broad-spectrum action towards all human cancer cell lines in all the herein examined cancer subpanels, except towards Renal cancer TK-10 cell line.
Remarkably, conjugate 8a showed excellent growth inhibition properties against Leukaemia (K-562 and SR), NSCLC (NCI-H522), Colon cancer (HT29), Ovarian cancer (OVCAR-3), Prostate cancer (PC-3), Renal cancer (RXF 393), and Breast cancer (MCF7) cell lines with percentage inhibition of 83, 89, 86, 83, 98, 89, 82 and 91%, respectively (, ). Furthermore, compound 8a exerted good activity with growth inhibition % equal to or greater than 60 towards NSCLC (HOP-92 and NCI-H460), Colon cancer (SW-620, HCT-116 and HCT-15), CNS cancer (SF-539 and U251), Melanoma (LOX IMVI, SK-MEL-2 and UACC-62), Ovarian cancer (IGROV1) and Breast cancer (MDA-MB-231, T-47D and MDA-MB-468) cell lines with inhibition percent of 63, 68, 60, 77, 64, 66, 60, 69, 65, 60, 61, 66, 61 and 75% respectivly (, ). On the other hand, the remaining herein examined hybrids 8b, 8c, 9, 10a, 10c and 10d displayed moderate to weak antitumor activities (GI % range: 4–11). It is worth stressing that hybrid 8a exerted a cytotoxic impact with GI % >100 against some tumour cell lines. Hybrid 8a was shown to be lethal towards Leukaemia (HL-60(TB), CCRF-CEM, MOLT-4 and RPMI-8226) cells with GI % values equal to 101, 123, 110 and 125, respectively, whereas, 8a exerted its lethal action towards Colon cancer HCT-116 and Melanoma MDA-MB-435 cells with GI % = 116 and 126, respectively ().
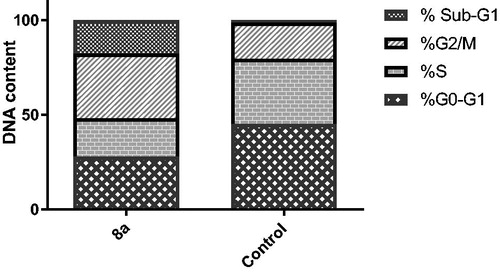
Figure 2. Impact of hybrid 8a on the cell cycle progression in breast cancer T-47D cells.
2.2.2.2. In vitro full NCI panel five dose assay
The obtained data from the preliminary single dose (10 µM) screening disclosed that hybrid 8a (NSC: D-771612/1) is the most efficient anticancer agent here in this study, displaying promising effectiveness against numerous cancer cell lines from different subpanels (, ). Subsequently, hybrid 8a was chosen to carry out further biological screening at a (0.01–100 µM) five-dose assay. Three response parameters (GI50, TGI and LC50) were evaluated for hybrid 8a towards each herein examined cancer cell line, and listed in . GI50 values reflect the level for growth inhibitory effect, whereas TGI represents cytostatic impact. In addition, LC50 parameter is considered to be the cytotoxicity parameter for the examined hybrid. Moreover, both full panel and subpanel mean graph midpoints (MG-MID), for full panel and individual subpanels cell lines, were calculated on the level of GI50 parameter, providing an average potency parameter for the tested hybrid 8a ().
Table 3. Results of NCI-USA in vitro five-dose testing for conjugate 8a (NSC: D- 795311/1).
Table 4. Median GI50 (µM) values for subpanel cancer cell lines for conjugate 8a.
As shown in , hybrid 8a displayed potent anti-proliferative action at a single-digit micromolar concentration against all herein examined human cancer cell lines with GI50 values range: 1.17–8.41 µM, except for Melanoma (UACC-257), Renal (TK-10) and Breast (HS 578 T) cell lines which possessed GI50 values equal 12.30, 11.10 and 11.30 µM, respectively (). Interestingly, hybrid 8a showed superior sub-micromolar activity towards leukaemia (MOLT-4 and SR), Ovarian cancer (OVCAR-4) and Breast cancer (T-47D) cells (GI50 = 0.68, 0.52, 0.93 and 0.41 µM, respectively).
In addition, hybrid 8a showed potent cytostatic activity at single-digit micromolar concentration (TGI range: 2.96–8.03 µM) against 22 cancer cell lines belonging to all herein examined cancer subpanels, except CNS cancer subpanel (). While hybrid 8a had no cytostatic impact (TGI > 100 µM) against leukaemia (CCRF-CEM), NSCLC (EKVX and NCI-H322M), colon cancer (HT29), CNS cancer (SNB-19), renal cancer (ACHN and TK-10), ovarian cancer (OVCAR-8), and breast cancer (HS 578 T) cells, it exhibited good to weak cytostatic activity towards the remaining cancer cell lines with TGI spanning in the interval: 10.6–96.6 µM. On the other hand, compound 8a emerged as a non-lethal agent that possesses LC50 values more than 100 µM for the most of cancer cell lines herein examined, except for Colon cancer (COLO 205, HCC-2998 and HCT-116), NSCLC (NCI-H460), Prostate cancer (PC-3), Melanoma (LOX IMVI, M14, MDA-MB-435 and SK-MEL-5), and Breast cancer (MDA-MB-468) cell lines (LC50 = 75.7, 7.46, 19.0, 9.96, 7.41, 17.2, 5.85, 39.0, 47.40 and 79.40 µM, respectively ().
With regards to the sensitivity for diverse tumour cell lines, hybrid 8a possessed relatively homologous growth inhibitory action for the whole of NCI panel, with efficient sub-panel GI50 (MG-MID) range of 1.33–4.23 µM, and promising full-panel GI50 (MG-MID) = 3.10 µM. leukaemia, prostate cancer, colon cancer, and breast cancer subpanels were the most vulnerable herein examined cancer subpanels to the impact of hybrid 8a [GI50 (MG-MID) = 1.33, 2.47, 2.85 and 3.09 µM, respectively] ().
Furthermore, dividing the full-panel MG-MID (µM) for the examined derivative by its individual subpanel MG-MID (µM) provides an index which is regarded as a measure for its selectivity. A value between three and six refers to moderate selectivity against the corresponding cancer cell line, ratio more than six indicates high selectivity, whereas if the compound does not meet any of such criteria is considered as non-selectiveCitation25. In this regards, hybrid 8a was found to be a non-selective anticancer agent that exhibits broad-spectrum potency towards all cancer subpanels herein examined at the GI50 level, with selectivity ratios spanning in the range from 0.73 to 2.33 ().
2.2.3. Cell cycle analysis
The superior sub-micromolar anti-proliferative activity of conjugate 8a on breast cancer T-47D cells (GI50 = 0.41 µM, ) prompted us to further investigate about the growth inhibitory mechanism of the target conjugates. Both regulation of cell cycle progression and apoptosis induction have been considered as significant strategies to control the proliferation of different cancer cells, accordingly, we primarily examined the growth inhibition mechanism of hybrid 8 in relation to cell cycle progression and regulation in human breast T-47D cancer cells.
The impact on cell cycle distribution was assessed by a DNA flow cytometry analysis, upon incubation of T-47D cells with conjugate 8a at its GI50 concentration (0.41 µM) for 24 h (. From the obtained results it was found that T-47D cells exposed to hybrid 8 significantly arrested at the G2/M phase of the cell cycle with an escalation in G2/M phase fraction from 19.19% (in control cells) to 34.05% (in 8a-treated T-47D cells). Furthermore, the cell population in sub-G1 phase was drastically augmented from 1.25% (in control cells) to 17.73% (in 8a-treated T-47D cells).
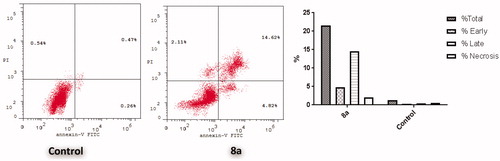
Figure 3. Effect of conjugate 8a over the AV-FITC-positive staining percentages in breast T-47D cells.
Generally the upsurge of populations in the sub-G1 phase indicates the induction of apoptotic cell death. So, a subsequent study will be conducted to reveal whether the G2/M phase cell cycle arrest afforded by conjugate 8a was accompanied by apoptosis.
2.2.4. Apoptosis assay
In an attempt to further investigate whether the antiproliferative activity for conjugate 8a is harmonious with the apoptosis induction within T-47D cells pointed out by the increased cell population in sub-G1 phase in 8a-treated T-47D cells (), Annexin V-FITC/PI dual staining analysis was used for the apoptosis assay (.
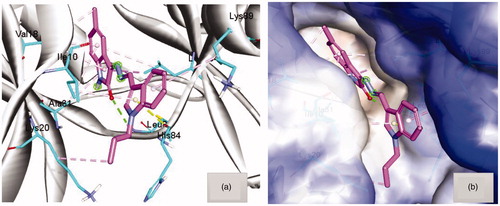
Figure 4. (a) Docking pose of hybrid 8a within the active site of CDK2 (pdb code 2BHE) showing hydrogen bond with Leu83 (green dots) and hydrophobic interaction (light purple dots). (b) Hydrophobic surface of active site of CDK2 surround compound 8a.
The outcomes of the Annexin V-FITC/PI assay suggested that treatment of T-47D cells with conjugate 8a led to an early and late cellular apoptosis, which proved through the significant increase for the apoptotic cells percentage in both the early apoptotic phase (from 0.26% to 4.82%) and the late apoptotic phase (from 0.47% to 14.62%) that signifies about 26-fold increase in total apoptosis, when compared to the untreated control (.
2.2.5. Cdk2 and Bcl-2 inhibitory activity
The promising anti-proliferative impact of conjugate 8a, in addition to its cell cycle disruption and pro-apoptotic effects, provoked a further exploration for their possible inhibitory activities against the cell cycle regulator CDK2 protein kinase and the anti-apoptotic Bcl-2 protein. The results expressed as IC50 values are presented in .
Table 5. Inhibitory activities of compound 8a against CDK2 and Bcl-2.
Results in showed that the tested hybrid 8a exhibited good inhibitory action against CDK2 and Bcl-2 with IC50 values equal 0.85 ± 0.03 and 0.46 ± 0.02 µM, respectively.
3. Molecular docking studies
The docking simulation studies were conducted to investigate the behaviour of compound 8a in the active site of both CDK2 and Bcl-2 (pdb code 2BHECitation7 and 4Citation26). From several X-ray structures for CDK2 enzyme, as mentioned before in the introduction, it was observed that the active site should be filled by planar moleculesCitation8–11. In the docking simulations 8a has taken the planar orientation, although it has a certain flexibility, with some little deviation at the indole moiety to fill the part of the corresponding hydrophobic pocket by hydrophobic contact with Lys89 as shown in (), whereas the N-propyl group interacted with the part of the terminal hydrophobic pocket by alkyl hydrophobic contact with Lys20. Moreover, the Molecular docking simulations showed an important hydrogen bond interaction with Leu83, via C=O of isatin, which is a key interaction with distance 2.5 Å. Also the docking poses of hybrid 8a showed that 5-methyl group, decorated on the benzenoid part of the isatin moiety, made an anchoring hydrophobic mixed with π interactions with Val18 and Leu134 from both sides as shown in .
On the other hand, the molecular docking studies explored the binding pattern of hybrid 8a within the active site of Bcl-2 to justify its apoptotic effect. As reported, the pro-apoptotic effect of the small molecules could be done at the molecular level by mimicking the BH3 α-helix and then binding to the hydrophobic groove of anti-apoptotic Bcl-2 proteins preventing their heterodimerization, which eventually leads to apoptosisCitation27. The attitude of α-helical BH3 domains complexed with Bcl-2 family besides the docking poses of other small molecules as Bcl-2 inhibitors were discussed as a guide for our docking studyCitation28–31.
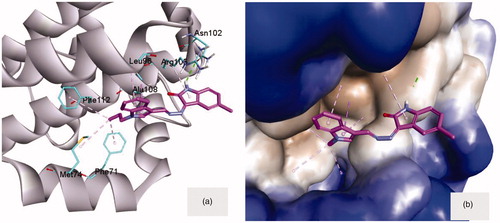
Molecular docking of compound 8a, as a small molecule inhibitor, inside the hydrophobic groove of Bcl-2 showed significant interactions at the front of this groove (). The (NH) functionality of isatin moiety can donate a hydrogen bond (2.00 Å) to the key amino acid Asn102 at the margin of the hydrophobic groove. This anchoring hydrogen bond was supported by the π-stack of isatin ring with Arg105 (). On the other side of the groove, N-propyl group attached to indole ring was embedded in the hydrophobic pocket formed of three amino acids; Phe112, Phe71 and Met74. The π-stack interaction of indole ring with Leu96 and Ala108 would act as a middle support for the whole compound inside the opening of the hydrophobic groove ().
Figure 5. (a) Compound 8a docked into the groove of Bcl-2 (4AQ3) showing the different interaction from both sides to fill this groove. (b) Compound 8a embedded inside the active site of Bcl-2. The macromolecule was surrounded by hydrophobic surface to focus on the hydrophobic contacts.
4. Experimental
4.1. Chemistry
4.1.1. General
Melting points were measured with a Stuart melting point apparatus, and are uncorrected. Nuclear magnetic resonance; 1H NMR and 13C NMR spectra were recorded using a Bruker NMR spectrometer (400/100 MHz), in deuterated dimethylsulphoxide (DMSO-d6). Chemical shifts (δH) were reported relative to TMS as an internal standard. The coupling constant values (J) were given in hertz. Chemical shifts (δC) were reported as follows: s, singlet; d, doublet; m, multiplet. HRMS spectra were performed on a Bruker MicroTOF spectrometer. Infra-red (IR) Spectra are obtained by the use of Schimadzu FT-IR 8400S spectrophotometer as KBr discs and expressed in wave number (cm−1). Compounds 2 and 4a–d were prepared as reported previouslyCitation32.
4.1.2. Synthesis of N-substituted-3-(hydrazonomethyl)-1H-indole derivatives 5a–d
To a solution of N-substituted-1H-indole-3-carbaldehyde derivatives 4a–d (4 mmol) in ethyl alcohol (25 ml), an excess of 99% hydrazine hydrate (1.25 ml, 25 mmol) was added. The reaction mixture was stirred at reflux temperature for 2 h, and then filtrated upon cooling. The obtained precipitate was washed with water several times, dried and recrystallized from isopropyl alcohol to furnish N-substituted-3-(hydrazonomethyl)-1H-indole derivatives 5a–d, respectively.
4.1.3. General procedures for preparation of target compounds 7, 8a–c, 9 and 10a–e
To a hot stirred solution of isatin derivatives 6a–g (1 mmol) in absolute ethyl alcohol (5 ml) and catalytic drops of acetic acid, the appropriate N-substituted-3-(hydrazonomethyl)-1H-indole derivative 5a–d (1 mmol) was added. The resulting mixture was stirred at reflux for 3 h, and then filtrated. The collected precipitate was dried and then recrystallized from isopropyl alcohol to afford target hybrids 7, 8a–c, 9 and 10a–e, respectively.
Full characterisation for the key intermediates (5a, 5c and 5d) as well as the target conjugates (7, 8a–c, 9 and 10a–e) have been presented in the Supporting Information.
4.2. Biological evaluation
Experimental procedures for biological evaluations for the herein reported conjugates (7, 8a-c, 9 and 10a-e) were provided in the Supplementary material (Supplementary materials can be found at www.mdpi.com/xxx/s1).
4.2.1. Antiproliferative action against A-549, MDAMB-231 and HCT-116 cancer cell lines
The three examined cancer cell lines (non-small cell lung A-549, Breast MDA-MB-231 and colon HCT-116 cells) were obtained from American Type Culture Collection (ATCC). Examination of the cytotoxic activity for the target conjugates was carried out in accordance with the SRB colorimetric assay protocolCitation21, as described previouslyCitation33.
4.2.2. Nci-60 cancer cell lines screening
The NCI-USA anticancer screening was carried out following the NCI, Bethesda, Drug Evaluation Branch protocolCitation22–24, utilising the SRB protein assayCitation21, as reported earlierCitation34,Citation35.
4.2.3. Cell cycle analysis
Flow cytometric analysis (FACS) was carried out to examine the cell cycle distributions in breast T-47D cancer cells upon treatment with conjugate 8a, by BD FACS Calibur flow cytometer, as described earlierCitation36.
4.2.4. Apoptosis assay
Phosphatidylserine externalisation effect of conjugate 8a over breast T-47D cancer cell line was investigated using Annexin-V-FITC Apoptosis Detection Kit by flow cytometry, as described earlierCitation37.
4.2.5. Cdk2 and bcl-2 inhibitory activity
These assays were carried out as reported earlierCitation38,Citation39.
4.3. Molecular docking studies
The target proteins, CDK2 (2BHE) and BCl-2 (4AQ3), were downloaded from protein databank and prepared by the default protocol in Discovery Studio 4. All other steps including preparation of small molecules, determination of the active site and docking protocol were followed as reportedCitation40.
5. Conclusions
To conclude, a novel series of N-alkylindole-isatin conjugates (7, 8a–c, 9 and 10a-e) was designed and synthesised, utilising the SAR outputs from the previous study. The design is based on appending four alkyl groups with different length (ethyl, n-propyl), bulkiness (isopropyl) and unsaturation (allyl) on N-1 of indole motif, with subsequent conjugation with different N-unsubstituted isatin moieties to furnish the target conjugates. As planned, the adopted strategy achieved a substantial improvement in the growth inhibitory profile for the target conjugates in comparison to the reported lead VI. All conjugates were screened for their potential cytotoxicity towards a panel of three different human cancer cell lines HCT-116 (Colon), A-549 (NSCLC), and MDA-MB-231 (Breast) utilising SRB assay. Conjugate 8a, superiorly, displayed the best anti-proliferative activity against the three examined cell lines (IC50 = 7.3 ± 0.42, 4.7 ± 0.28 and 2.6 ± 0.17 µM, respectively). Furthermore, 8a displayed broad spectrum anti-proliferative action with efficient subpanel GI50 (MG-MID) range of 1.33–4.23 µM, and promising full panel GI50 (MG-MID) = 3.10 µM, at the NCI five-dose assay. In particular, hybrid 8a showed superior sub-micromolar activity towards leukaemia (MOLT-4 and SR), ovarian cancer (OVCAR-4) and breast cancer (T-47D) cell lines (GI50 = 0.68, 0.52, 0.93 and 0.41 µM, respectively). Conjugate 8a was able to provoke cell cycle disturbance and apoptosis in breast T-47D cells as evidenced by the DNA flow cytometry and Annexin V-FITC/PI assays. Moreover, hybrid 8a exhibited good inhibitory action against cell cycle regulator CDK2 protein kinase and the anti-apoptotic Bcl-2 protein (IC50 = 0.85 ± 0.03 and 0.46 ± 0.02 µM, respectively). The molecular docking for hybrid 8a in CDK2 and Bcl-2 active sites unveiled that N-propyl group is involved in significant hydrophobic interactions. Taken together, the results suggested conjugate 8a as a promising lead for further development and optimisation as an efficient antitumor drug.
Supplemental Material
Download PDF (2.3 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Related Research Data
References
- Schwartz GK. CDK inhibitors: cell cycle arrest versus apoptosis. Cell Cycle 2002;1:113–4.
- Damla Arisan E, Obakan P, Coker-Gurkan A, et al. CDK inhibitors induce mitochondria-mediated apoptosis through the activation of polyamine catabolic pathway in LNCaP, DU145 and PC3 prostate cancer cells. Curr Pharm Des 2014;20:180–8.
- Cho S-J, Kim Y-J, Surh Y-J, et al. Ibulocydine is a novel prodrug Cdk inhibitor that effectively induces apoptosis in hepatocellular carcinoma cells. J Biol Chem 2011;286:19662–71.
- Damiens E, Baratte B, Marie D, et al. Anti-mitotic properties of indirubin-3'-monoxime, a CDK/GSK-3 inhibitor: induction of endoreplication following prophase arrest. Oncogene 2001;20:3786–97.
- Perabo FGE, Frössler C, Landwehrs G, et al. Indirubin-3′-monoxime, a CDK inhibitor induces growth inhibition and apoptosis-independent up-regulation of survivin in transitional cell cancer. Anticancer Res 2006;26:2129–35.
- Davies TG, Tunnah P, Meijer L, et al. Inhibitor binding to active and inactive CDK2: the crystal structure of CDK2-cyclin A/indirubin-5-sulphonate. Structure 2001;9:389–97.
- Jautelat R, Brumby T, Schäfer M, et al. From the insoluble dye indirubin towards highly active, soluble CDK2-inhibitors. ChemBioChem 2005;6:531–40.
- Yan L, Lai F, Chen X, Xiao Z. Discovery of novel indirubin-3'-monoxime derivatives as potent inhibitors against CDK2 and CDK9 . Bioorg Med Chem Lett 2015;25:2447–51.
- Ahn M-Y, Kim T-H, Kwon S-M, et al. 5-Nitro-5'-hydroxy-indirubin-3'-oxime (AGM130), an indirubin-3'-oxime derivative, inhibits tumor growth by inducing apoptosis against non-small cell lung cancer in vitro and in vivo. Eur J Pharm Sci 2015;79:122–31.
- Wood DJ, Korolchuk S, Tatum NJ, et al. Differences in the conformational energy landscape of CDK1 and CDK2 suggest a mechanism for achieving selective CDK inhibition. Cell Chem Biol 2019;26:121–30.
- Bramson HN, Corona J, Davis ST, et al. Oxindole-based inhibitors of cyclin-dependent kinase 2 (CDK2): design, synthesis, enzymatic activities, and X-ray crystallographic analysis. J Med Chem 2001;44:4339–58.
- Attia MI, Eldehna WM, Afifi SA, et al. New hydrazonoindolin-2-ones: Synthesis, exploration of the possible anti-proliferative mechanism of action and encapsulation into PLGA microspheres. PLoS One 2017;12:e0181241.
- Eldehna WM, Al-Wabli RI, Almutairi MS, et al. Synthesis and biological evaluation of certain hydrazonoindolin-2-one derivatives as new potent anti-proliferative agents. J Enzyme Inhib Med Chem 2018;33:867–78.
- Abdel-Aziz HA, Eldehna WM, Keeton AB, et al. Isatin-benzoazine molecular hybrids as potential antiproliferative agents: Synthesis and in vitro pharmacological profiling. Drug Des Devel Ther 2017;11:2333–46.
- Eldehna WM, Almahli H, Al-Ansary GH, et al. Synthesis and in vitro antiproliferative activity of some novel isatins conjugated with quinazoline/phthalazine hydrazines against triple-negative breast cancer MDA-MB-231 cells as apoptosis-inducing agents. J Enzym Inhib Med Chem 2017;32:600–13.
- El-Naggar M, Eldehna WM, Almahli H, et al. Novel thiazolidinone/thiazolo [3, 2-a] benzimidazolone-isatin conjugates as apoptotic anti-proliferative agents towards breast cancer: one-pot synthesis and in vitro biological evaluation. Molecules 2018;23:1420.
- Eldehna WM, Nocentini A, Al-Rashood ST, et al. Tumor-associated carbonic anhydrase isoform IX and XII inhibitory properties of certain isatin-bearing sulfonamides endowed with in vitro anticancer activity towards colon cancer. Bioorg Med Chem 2018;81:425–32.
- Eldehna WM, Abo-Ashour MF, Nocentini A, et al. Enhancement of the tail hydrophobic interactions within the carbonic anhydrase IX active site via structural extension: design and synthesis of novel N-substituted isatins-SLC-0111 hybrids as carbonic anhydrase inhibitors and antitumor agents. Eur J Med Chem 2019;162:147–60.
- Eldehna WM, Fares M, Ibrahim HS, et al. Indoline ureas as potential anti-hepatocellular carcinoma agents targeting VEGFR-2: synthesis, in vitro biological evaluation and molecular docking. Eur J Med Chem 2015;100:89–97.
- Eldehna WM, Abo-Ashour MF, Ibrahim HS, et al. Novel [(3-indolylmethylene) hydrazono] indolin-2-ones as apoptotic anti-proliferative agents: design, synthesis and in vitro biological evaluation. J Enzym Inhib Med Chem 2018;33:686–700.
- Skehan P, Storeng R, Scudiero D, et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst 1990;82:1107–12.
- Monks A, Scudiero D, Skehan P, et al. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J Natl Cancer Inst 1991;83:757–66.
- Boyd MR. Cancer. In: Teicher BA, ed. Drug discovery and development: anticancer drug development guide: preclinical screening, clinical trials and approval, 2nd ed. Totowa, NJ: Humana Press; 2014:41–62.
- Boyd MR, Paull KD. Some practical considerations and applications of the National Cancer Institute in vitro anticancer drug discovery screen. Drug Dev Res 1995;34:91–109.
- Ibrahim HS, Eldehna WM, Fallacara AL, et al. One-pot synthesis of spiro(indoline-3,4'-pyrazolo[3,4-b]pyridine)-5'-carbonitriles as p53-MDM2 interaction inhibitors. Future Med Chem 2018;10:2771–89.
- Perez HL, Banfi P, Bertrand J, et al. Schmidt, Identification of a phenylacylsulfonamide series of dual Bcl-2/Bcl-xL antagonists. Bioorg Med Chem Lett 2012;22:3946–50.
- Delgado-Soler L, Pinto M, Tanaka-Gil K, Rubio-Martinez J. Molecular determinants of Bim(BH3) peptide binding to pro-survival proteins. J Chem Inf Model 2012;52:2107–18.
- Czabotar PE, Lee EF, van Delft MF, et al. Structural insights into the degradation of Mcl-1 induced by BH3 domains. Proc. Natl. Acad. Sci. U.S.A 2007;104:6217–22.
- Porter J, Payne A, de Candole B, et al. Whitcombe, tetrahydroisoquinoline amide substituted phenyl pyrazoles as selective Bcl-2 inhibitors. Bioorg Med Chem Lett 2009;19:230–3.
- Enyedy IJ, Ling Y, Nacro K, et al. Discovery of small-molecule inhibitors of Bcl-2 through structure-based computer screening. J Med Chem 2001;44:4313–24.
- Oltersdorf T, Elmore SW, Shoemaker AR, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005;435:677–81.
- Abo-Ashour MF, Eldehna WM, George RF, et al. Novel indole-thiazolidinone conjugates: design, synthesis and whole-cell phenotypic evaluation as a novel class of antimicrobial agents. Eur J Med Chem 2018;160:49–60.
- Eldehna WM, El Kerdawy AM, Al-Ansary GHA, et al. Type IIA–Type IIB protein tyrosine kinase inhibitors hybridization as an efficient approach for potent multikinase inhibitor development: Design, synthesis, anti-proliferative activity, multikinase inhibitory activity and molecular modeling of novel indolinone-based ureides and amides. Eur J Med Chem 2019;163:37–53.
- Abou-Seri SM, Eldehna WM, Ali MM, El Ella DA. 1-Piperazinylphthalazines as potential VEGFR-2 inhibitors and anticancer agents: Synthesis and in vitro biological evaluation. Eur J Med Chem 2016;107:165–79.
- Eldehna WM, Hassan GS, Al-Rashood ST, et al. Synthesis and in vitro anticancer activity of certain novel 1-(2-methyl-6-arylpyridin-3-yl)-3-phenylureas as apoptosis-inducing agents. J Enzyme Inhib Med Chem 2019;34:322–32.
- Sabt A, Abdelhafez OM, El-Haggar RS, et al. Novel coumarin-6-sulfonamides as apoptotic anti-proliferative agents: Synthesis, in vitro biological evaluation, and QSAR studies. J Enzyme Inhib Med Chem 2018;33:1095–107.
- Eldehna WM, El-Naggar DH, Hamed AR, et al. One-pot three-component synthesis of novel spirooxindoles with potential cytotoxic activity against triple-negative breast cancer MDA-MB-231 cells. J Enzyme Inhib Med Chem 2018;33:309–18.
- Said MA, Eldehna WM, Nocentini A, et al. Sulfonamide-based ring-fused analogues for CAN508 as novel carbonic anhydrase inhibitors endowed with antitumor activity: design, synthesis, and in vitro biological evaluation. Eur J Med Chem 2020;189:112019.
- Abo-Ashour MF, Eldehna WM, Nocentini A, et al. 3-Hydrazinoisatin-based benzenesulfonamides as novel carbonic anhydrase inhibitors endowed with anticancer activity: Synthesis, in vitro biological evaluation and in silico insights. Eur J Med Chem 2019;184:111768.
- Ibrahim HS, Albakri ME, Mahmoud WR, et al. Synthesis and biological evaluation of some novel thiobenzimidazole derivatives as anti-renal cancer agents through inhibition of c-MET kinase. Bioorg Chem 2019;85:337–48.