
Abstract
Trypanothione synthetase (TryS) produces N1,N8-bis(glutathionyl)spermidine (or trypanothione) at the expense of ATP. Trypanothione is a metabolite unique and essential for survival and drug-resistance of trypanosomatid parasites. In this study, we report the mechanistic and biological characterisation of optimised N5-substituted paullone analogues with anti-TryS activity. Several of the new derivatives retained submicromolar IC50 against leishmanial TryS. The binding mode to TryS of the most potent paullones has been revealed by means of kinetic, biophysical and molecular modelling approaches. A subset of analogues showed an improved potency (EC50 0.5–10 µM) and selectivity (20–35) against the clinically relevant stage of Leishmania braziliensis (mucocutaneous leishmaniasis) and L. infantum (visceral leishmaniasis). For a selected derivative, the mode of action involved intracellular depletion of trypanothione. Our findings shed light on the molecular interaction of TryS with rationally designed inhibitors and disclose a new set of compounds with on-target activity against different Leishmania species.
Introduction
The genus Trypanosoma and Leishmania encompasses parasitic protozoa that are the aetiological agents of several highly disabling and/or fatal diseases for humans and livestock (e.g. African sleeping sickness, Chagas’ disease, the different forms of leishmaniasis, Nagana and Surra disease). These parasites have a unusual thiol redox system that depends on the low molecular weight thiol trypanothione [N1,N8-bis(glutathionyl)spermidine, T(SH)2], which is absent in mammalsCitation1. From the pharmacological point of view, trypanosomatids present another attractive peculiarity; they lack genes for glutathione reductase and thioredoxin reductaseCitation2. Being devoid of backup pathways to maintain redox homeostasis, T(SH)2 is pivotal for parasite viability and virulence by providing reducing equivalents to cope with oxidative stress and sustain DNA synthesis and repairCitation3. Indeed, most of the drugs used to treat trypanosomiasis and leishmaniasis are known to directly (e.g. efluornithine, antimonials, arsenates) or indirectly (e.g. benznidazole, nifurtimox and amphotericin) interfere with T(SH)2 metabolismCitation4–7.
The biosynthesis of T(SH)2 is achieved in two consecutive steps each involving the ligation of a glutathione (GSH) molecule by its glycine carboxyl group to the free N1 and N8 amine groups of spermidine (SP). Both reactions are catalysed by the C-terminal ligase domain of trypanothione synthetase (TryS; EC 6.3.1.9), at the expense of ATP. The indispensability of TryS for the infective form of Trypanosoma brucei (Tb)Citation8,Citation9, Leishmania infantum (Li)Citation10 and, more recently, Trypanosoma cruzi (Tc)Citation11 has been confirmed by means of reverse genetic or chemical inhibition approaches. The high conservation of the trypanothione system among trypanosomatidsCitation2,Citation3 suggests that TryS is essential for this protozoan lineage. The development of TryS inhibitors is rather limited compared to work done on the metabolically-related enzyme trypanothione reductaseCitation12. Overall, the TryS inhibitors can be grouped in substrate or transition state analoguesCitation13,Citation14, natural derivativesCitation15,Citation16 and other synthetic compoundsCitation17–19 ().
Figure 1. Trypanothione synthetase inhibitors. The structure and biological activities (IC50 against TryS or Ki, EC50 against trypanosomatids and selectivity index) are shown for representatives singletons or scaffolds reported to inhibit TryS. The data shown in the upper box were reported inCitation14 for phosphinopeptides, inCitation19 for oxabicyclo nonanone, inCitation15 for conessine, and inCitation16 for cynaropicrin. Data shown in the middle box was reported inCitation17 for the phenyl-indazole derivative and inCitation18 for the phenyl-tetrazole and -thiazole derivatives. Lower panel, the 7,12-dihydroindolo[3,2-d][1]benzazepin-6(5H)-ones core scaffold, referred as paullone, is shown in red. Incorporation of a Cl and Br atom at position 3 and 9, respectively, give rise to chlorokenpaullone. The asterisk denotes data reported in this work whereas information for FS-554 {9-trifluoromethylpaullone with N-[2-(methylamino)ethyl]acetamide side chain} and MOL2008 {3-chlorokenpaullone with N-[2-(methylamino)ethyl]acetamide side chain} was reported elsewhereCitation10,Citation20. Compound 20 is a 3-chlorokenpaullone with 2-oxo-2-piperazinoethyl side chain as hydrochloride.
![Figure 1. Trypanothione synthetase inhibitors. The structure and biological activities (IC50 against TryS or Ki, EC50 against trypanosomatids and selectivity index) are shown for representatives singletons or scaffolds reported to inhibit TryS. The data shown in the upper box were reported inCitation14 for phosphinopeptides, inCitation19 for oxabicyclo nonanone, inCitation15 for conessine, and inCitation16 for cynaropicrin. Data shown in the middle box was reported inCitation17 for the phenyl-indazole derivative and inCitation18 for the phenyl-tetrazole and -thiazole derivatives. Lower panel, the 7,12-dihydroindolo[3,2-d][1]benzazepin-6(5H)-ones core scaffold, referred as paullone, is shown in red. Incorporation of a Cl and Br atom at position 3 and 9, respectively, give rise to chlorokenpaullone. The asterisk denotes data reported in this work whereas information for FS-554 {9-trifluoromethylpaullone with N-[2-(methylamino)ethyl]acetamide side chain} and MOL2008 {3-chlorokenpaullone with N-[2-(methylamino)ethyl]acetamide side chain} was reported elsewhereCitation10,Citation20. Compound 20 is a 3-chlorokenpaullone with 2-oxo-2-piperazinoethyl side chain as hydrochloride.](/cms/asset/1b403c68-673c-4062-8d62-0e171ff3c443/ienz_a_1780227_f0001_c.jpg)
In the search of novel scaffolds targeting TryS inhibition, we recently reported the identification of a subset of N5-substituted paullones {7,12-dihydroindolo[3,2-d][1]benzazepin-6(5H)-ones} ()Citation10,Citation20,Citation21. Substitutions at position N5 of paullones render them almost inactive against kinasesCitation21,Citation22 while the nature of the substituent conferred species-selectivity towards TryS. Indeed, the inclusion of a N-(2-methylamino)ethyl)acetamide group on 9-trifluoromethyl paullone (FS-554) or 3-chlorokenpaullones (MOL2008) () produced the most potent inhibitors of LiTryS so far reported with an IC50 value of 0.35 µM and 0.15 µM, respectivelyCitation10,Citation20. In contrast, less polar substitutions (e.g. 3-chlorokenpaullones with N-benzylacetamide substituent at N5) rendered better and moderate inhibitors of TryS from T. cruzi (IC50 ∼10 µM) or T. brucei TryS (IC50 ∼30 µM)Citation21, suggesting species-specific differences at the enzyme binding site interacting with the N5-group of paullones. The most active inhibitors of TcTryS (IC50 ∼10 µM) allowed the chemical validation of the enzyme and of T(SH)2 as a key metabolite for T. cruzi proliferation and drug-resistanceCitation11. Similarly, FS-554Citation10 and MOL2008Citation20 displayed on-target effect against the extracellular form of L. infantum. However, both compounds displayed a high cytotoxicity towards murine macrophages (selectivity index, SI <1)Citation20, which clearly indicates that they have additional molecular targets on mammalian cells.
With the aim to improve the biological selectivity of N5-acetamide substituted paullones and get insights towards mechanistic aspects of the TryS-inhibition mode, 36 novel derivatives of 3-chlorokenpaullones were synthesised and tested in vitro against TryS from different trypanosomatids, and Leishmania species associated with visceral and (muco)cutaneous diseases in humans.
Materials and methods
Reagents
Unless otherwise stated all chemical reagents were of analytical grade and purchased from Sigma-Aldrich (Chicago, IL) and Acros Organics. Cell culture medium, antibiotics and foetal bovine serum (FBS) were from Gibco® or InvitrogenTM (Carlsbad, CA). T(SH)2 and mono(glutathionyl)spermidine (GSP) were kindly provided by Dr Luise Krauth-Siegel (Heidelberg University, Heidelberg, Germany).
Synthetic chemistry
Melting points (mp) or decomposition points (dec) were determined in an open-glass capillary on an electric variable heater (Barnstead Electrothermal IA 9100, Electrothermal, Staffordshire, Great Britain) and were not corrected. KBr pellets were prepared for the recording of IR-spectra with a Thermo Nicolet FT-IR 200 (Thermo Nicolet, Madison, WI). DMSO-d6 solutions of compounds were prepared to record the 1H NMR and 13 C NMR-spectra on a Bruker Avance DRX-400 and a Bruker Avance II-600 (Bruker, Billerica, MA; NMR Laboratories of the Chemical Institutes of the Technische Universität Braunschweig). Chemical shifts were related to tetramethylsilane (δ = 0 ppm). The CE Instruments FlashEA®1112 Elemental Analyser (Thermo Quest, San Jose, CA) was used to perform elementary analyse. The MAT95XL spectrometer (Thermo Finnigan MAT, Bremen, Germany; Department of Mass Spectrometry of the Chemical Institutes of the Technische Universität Braunschweig) was used to perform the EI mass spectrometry and the LTQ-Orbitrap Velos (Thermo Fisher SCIENTIFIC, Bremen, Germany; Department of Mass Spectrometry of the Chemical Institutes of the Technische Universität Braunschweig) was used to perform the ESI mass spectrometry. The Mass spectra were recorded on a double-focus sector field mass spectrometer Finnigan-MAT 90 (EI) or on a linear ion trap coupled with orbitrap mass analyser (ESI). Accurate measurements were conducted according to the peakmatch method using perfluorokerosene as an internal mass reference (EI) or were recorded by direct infusion in methanol with tetradecyltrimethylammonium bromide (final concentration 50 µg/mL) (ESI); (EI)-MS: ionisation energy 70 eV; (ESI)-MS: positive modus, 2.3–2.8 V. TLC: Polygram® Sil G/UV254, 40 × 80 mm (Machery-Nagel, Düren, Germany), visualisation by UV-illumination at 254 nm. The purity of 95% was ensured with HPLC under following conditions, devices and settings: LaChrome Elite® (isocratic elution): pump L-2130, column oven L-2300, autosampler L-2200, diode array detector L-2450, OrganizerBox L-2000, column Merck LiChroCART® 125–4, LiChrospher® 100, RP-18, 5 µM, (Merck/Hitachi, Darmstadt, Germany); LaChrom Elite® (gradient elution): pump L-2130, autosampler L-2200, UV detector L-2400, OrganizerBox L-2000, column Merck LiChroCART® 125–4, LiChrospher® 100, RP-18, 5 µM, (Merck/Hitachi, Darmstadt, Germany); isocratic eluent: acetonitrile/buffer mixture or acetonitrile/water mixture; gradient elution (method A): concentration acetonitrile 0–2 min: 10%, 2–12 min: 10% → 90% (linear), 12–20 min: 90%; gradient elution (method B): concentration acetonitrile 0–10 min: 10% → 70%, 10–10.5 min: 70% → 90%, 10.5–22 min: 90%; elution rate: 1000 mL/min; detection wavelength: 254 nm and 280 nm (isocratic), 254 nm (gradient); overall run time: 15 min at 40 °C (isocratic); tM (DMSO) (dead time related to DMSO); tM+S (total retention time); wavelength maxima were extracted from spectra generated by diode array detector during HPLC analyses; preparation of buffer pH 2.7: To a solution of 20.0 mL triethylamine and 242 mg sodium hydroxide in 980 mL water, sulphuric acid was added drop wise to reach pH 2.7.
THF was pre-dried over potassium hydroxide and subsequently distilled from CaH2. CH2Cl2 was dried by distillation from CaH2.
3-Chlorokenpaullone (1) was synthesised in a four-step synthetic sequence according to a published methodCitation23. 2-(Piperidin-1-yl)ethan-1-amine and 2-morpholinoethan-1-amine were synthesised according to a method previously reportedCitation24.
Tested substituted paullones were synthesised according to the procedures described in Supplementary Information.
Expression and purification of recombinant TryS
TryS expression and purification were performed according to protocols previously reportedCitation20, except for the size exclusion chromatography. Elution fractions from the metal-affinity chromatography (∼60 mg for TbTryS and LiTryS and ∼16 mg for TcTryS) were loaded onto a HiLoad® 16/60 Superdex®200 pg column (GE Healthcare, Piscataway, NJ) pre-equilibrated with 100 mM HEPES pH 7.4, 10 mM MgSO4, 0.5 mM EDTA, 1 mM DTT and 150 mM NaCl. The chromatography was performed at room temperature (20–25 °C) using an Äkta-FPLC device (GE Healthcare, Piscataway, NJ) and a flow rate of 0.75 mL/min. The fractions containing recombinant TryS were pooled, concentrated to ≥1 mg protein/mL using a 10 kDa cut-off Amicon filter (Millipore, Billerica, CA). Glycerol was added at a final concentration of 40% (v/v) and samples stored at −20 °C. Enzyme purity and activity were assessed by SDS-PAGE (12%) under reducing conditions and by the end-point TryS assay (see below “TryS activity assay”), respectively.
Protein concentration was determined using the bicinchoninic acid assay with bovine serum albumin as standard. The protocols described above yielded 4–8 mg TcTryS and 25–30 mg TbTryS or LiTryS per litre of culture medium with all enzyme preparations presenting ≥95% purity and homogeneous specific activity.
TryS activity assay
TryS activity was determined by the end-point assayCitation20, which is described below. For all TryS, ATP was used at 150 µM and SP fixed at 2 mM, whereas GSH was adjusted to 0.57, 0.25 and 0.05 mM for TcTryS, LiTryS and TbTryS, respectively, to avoid substrate inhibition or to approach physiological concentrations. A master mix (MM) solution containing all the substrates at 1.25-fold their end concentration in assay was prepared in the screening reaction buffer (5 mM DTT, 10 mM MgSO4, 0.5 mM EDTA, 100 mM HEPES pH 7.4). Ninety-six wells microtitre plates were loaded with 5 µL/well of the test compounds, DMSO (reaction control) or TryS-specific inhibitor (inhibition control), and 40 µL of MM. The reactions were started by adding 5 µL of TryS and stopped after 15 min with 200 µL BIOMOL GREENTM reagent (Enzo Life Sciences, Farmingdale, NY). The plates were incubated 20 min at room temperature and then A650 nm was measured with a MultiScan EX plate reader (Thermo Fisher Scientific, Waltham, MA). Blanks were prepared for each condition by adding 5 µL of the screening reaction buffer instead of enzyme. The interference of the compounds with the colorimetric reaction was evaluated in a sample containing different concentrations of the compound and 20 µM K2HPO4 dissolved in MM. The activity values were corrected accordingly. All determinations were performed at least by quadruplicates.
Kinetic assays for LiTryS
Assays were conducted at 28 °C, using 96-well plates and a total reaction volume of 50 µL, 5 µL of test compound or DMSO and 40 µL of MM. The reactions were started by adding 5 µL enzyme and stopped after 15 min with 200 µL BIOMOL GREENTM reagent. The colorimetric reaction was allowed to develop for 20 min and then A650 nm was measured with a MultiScan FC plate reader (Thermo Fisher Scientific, Waltham, MA).
Before running the kinetic studies, the enzyme concentration was adjusted in order to obtain a linear progression during the first 15 min of reaction under optimal substrates concentration for LiTryS (13 mM SP or 250 µM GSP, 250 µM GSH and 200 µM ATP; data not shown), which corresponded to an enzyme activity of 7.2 × 10−6 µmol/min mL with SP or 2.1 × 10−5 µmol/min mL with GSP.
Compound 20 and MOL2008 were tested at concentrations 1, 2.5, 5, 7.5 and 10 times their IC50. Due to substrate inhibition by GSHCitation20, the assays were performed at sub-KM [GSH] (from 100 to 3 µM), whereas SP and ATP were added at 13 mM (∼9.1-fold the KM value) and 200 µM (∼4.6-fold the KM value), respectively. When ATP was the variable substrate, the concentrations tested ranged from 200 to 6 µM, whereas SP and GSH were fixed to 13 mM and 250 µM (∼1.5-fold the KM value), respectively. For SP, the concentrations were varied from 13 to 0.41 mM, ATP and GSH were fixed to 200 µM and 250 µM, respectively. For GSP, the concentrations tested were from 250 to 7.8 µM, while ATP and GSH were fixed to 200 µM and 250 µM, respectively.
All tests were performed in quadruplicates for each concentration of compound evaluated. Blanks were prepared for each condition by adding 5 µL of screening reaction buffer instead of enzyme.
The assays were run manually and yielded an intra-assay coefficient variation ≤2.5%. Lineweaver–Burk plots (1/[Abs] versus 1/[substrate]) were prepared and analysed using the OriginPro 8 software (SAS Inc., Cary, NC). An R-square (R2) ≥0.96 was used as criteria to eliminate outlier’ values and for linear fitting of plots.
Apparent Ki for MOL2008 against LiTryS in the presence of SP and GSP was determined assuming a pure competitive inhibitionCitation25 and employing the equation apparent KM/KM = [Inhibitor].Ki−1 + 1.
Isothermal titration calorimetry assay
ITC experiments were conducted by titrating 60 µM MOL2008 with LiTryS in its apo- or GSH-bound forms at concentrations of 429 µM and 615 µM, respectively, or by titrating 60 µM MOL2008 on ADP-bound LiTryS at 6.7 µM and ADP/GSH-bound LiTryS at 6.2 µM. The first step consisted in buffer exchange of LiTryS in titration buffer [97.5 mM HEPES pH 7.4, 0.49 mM EDTA, 9.75 mM MgSO4 and 2.5% (v/v) DMSO] in the absence (apo-form) or presence of substrate/product: 2 mM GSH (5 mM DTT) or 2 mM GSH/2 mM ADP (5 mM DTT). For apo- and GSH-bound LiTryS, an extensive dialysis using a SnakeSkin™ Dialysis Tubing, 3.5 K MWCO, 16 mm (Thermo Fisher Scientific, Waltham, MA) was performed for 10 h at 4 °C. For GSH/ADP-bound LiTryS, buffer exchange was performed at 4 °C by diafiltration on a 10 kDa cut-off Amicon filter (Millipore, Billerica, CA) and repeated cycles of centrifugation-dilution at 3500 g (RT1 Thermo-Sorvall centrifuge). Prior to ITC assay, TryS activity was assayed to estimate the fraction of active enzyme.
Table 1. Inhibition of trypanothione synthetase (TryS) by N5-substituted, 3-chlorokenpaullones.
To reduce buffer composition mismatch for both interaction solutions, MOL2008 was dissolved (final concentration of 60 µM) in the dialysed buffer from the protein sample by sonication at room temperature for 5 min in an Ultrasonic bath (Branson Model B200), and further incubated 1 h at 37 °C. All samples were degassed immediately before ITC.
The ITC experiments were done at 28 °C in a MicroCal VP-ITC equipment (Malvern Instruments, Malvern, UK) by collecting data from twenty-seven injections of 10 µL (at 0.5 µL/s) spaced by periods of 500 s, to achieve an accurate post injection baseline.
Data analysis was carried out by automated baseline adjustment and heat integration using NITPIC softwareCitation26,Citation27. Integrated heat dataset was fitted to an A + B<–>AB heterodimer association model with SEDPHATCitation28 and plotted with GUSSICitation29 software.
Molecular simulations
Docking calculations were performed with AutoDock Vina 1.1.2Citation30. Input PDBQT files for the receptors and ligands were prepared with AutoDockTools 1.5.6Citation31. All torsions of the ligand were set as fully rotatable applying to the receptors a partially flexible treatment, allowing the following side chains to rotate: for Escherichia coli mono(glutathionyl)-spermidine synthetase (EcGspS): Trp226, Leu227, Tyr237, Asn241, Gln243, Val244, Ile245, Asn246, Glu341, Trp349, Glu391, Tyr394, Asp610 and Ile611; and for LmTryS: Trp234, Leu235, Phe245, Phe249, Val263, Ser264, Glu355, Trp363, Glu405, Glu407, Tyr410, Asp623 and Phe626. We retained a total of 15 different binding poses per ligand, setting an energy range of 5 kcal.mol−1. The best binding scoring was of 11 kcal.mol−1 corresponding to 1 in LmTryS. Subsequent docking scores are presented as percentages of this reference value. The search space was defined by a grid with X, Y and Z dimensions of 20 × 22 × 36 angstroms. The grid was centred so that the binding pockets corresponding to GSH and GSP could be included in the search space.
The loop comprising Glu248 to Val263 of LmTryS was modelled with ModLoopCitation32. Three possible conformations were obtained by changing the starting and ending position of the loop segment provided to the server.
Multiple sequences analysis
The sequences of L. major, L. infantum, Crithidia fasciculata, T. cruzi and T. brucei brucei TryS proteins (accession identification numbers: CAC83968.1, CAM69145.1, AAC39132.1, AAO00722.1 and CAC87573.1, respectively) were aligned with Clustal OmegaCitation33 tool from the server of the European Bioinformatic Institute (http://www.ebi.ac.uk/Tools/msa/clustalo).
Cell culture and cytotoxicity assays
Mouse macrophages from the cell line J774 (ATCC® TIB67TM) were cultivated in vented-cap culture flasks (Corning®) containing Dulbecco’s Modified Eagle Medium supplemented with 10% (v/v) FBS, 10 U/mL penicillin and 10 µg/mL streptomycin. The cell culture was incubated in a humidified 5% CO2/95% air atmosphere at 37 °C. Two hundred µL of a cell suspension containing 6 × 104 cells/mL in fresh culture medium was added per well in a 96-well culture plate (Corning®). Next day, cells were incubated for 24 h with the selected paullones or the control drug Amphotericin B (Fungizone, sodium deoxycholate form, Gibco®, Carlsbad, CA) added at different concentrations (from 100 to 0.001 µM, and 200 µM for 1). Thereafter, cytotoxicity was evaluated using a colorimetric assay (WST-1 reagent from Roche, Basel, Switzerland) and an EL 800 microplate reader (Biotek®). The following equation was used to estimate cytotoxicity: cytotoxicity (%) = [(A450 nm for cells treated with compound X − A450 nm for DMSO-treated cells)/(A450 nm for non-treated cells − A450 nm for DMSO-treated cells)] × 100.
Leishmania infantum promastigotes (strain MHOM MA67I7MAP263, wild type: WT or single TryS-knock out cell line: sKO)Citation10 and L. braziliensis promastigotes (strain MHOM/BR/01/BA788) were cultured in plug seal-cap culture-flask (Corning®) at 28 °C in Roswell Park Memorial Institute 1640 Medium containing GlutaMAX™, 25 mM HEPES sodium salt (pH 7.4) and at 26 °C in Schneider’s medium, respectively. Both culture media were supplemented with 10% (v/v) FBS, 10 U/mL penicillin and 10 µg/mL streptomycin.
Macrophages seeded in a 24-well plate containing glass cover-slips [1 × 105 cells/mL × well in Dulbecco’s Modified Eagle Medium with 1% (v/v) FBS] were infected with 2 × 106 stationary-phase promastigotes from L. braziliensis or L. infantum (macrophage:Leishmania ratio of 1:20) for 24 h (2 h at room temperature and 22 h at 35 °C for L. braziliensis or at 37 °C for L. infantum). Thereafter, the plates were washed with phosphate buffered saline (pH 7.0) to remove non-internalized parasites and then compounds were added at different concentrations (5 or 10 µM for primary screening or 10–0.01 µM for EC50 determination). After 24 h incubation at 37 °C and 5% CO2, the wells were washed three times with phosphate buffered saline (pH 7.0), and the cells attached to the coverslips were fixed (EtOH 100%, 10 min) and incubated with fluoroshieldTM-DAPI mounting medium to stain nucleic acids. Fluorescence and bright field images from a total of 100 to 200 macrophages distributed in 4 or 5 microscope fields were acquired with an Olympus IX-81 inverted fluorescence microscope equipped with an ORCA, Hamamatsu camera and the µManager free softwareCitation34. The percentage of infected cells by compound X at concentration Y was calculated as follow: [number of infected cells treated with compound X at concentration Y/number of infected cells treated with DMSO 1% (v/v)] × 100%. For all compounds, the end concentration of the vehicle (DMSO) in the assay was 1% (v/v). The EC50 for Amphotericin B was determined from drug-response curves for concentrations spanning from 0.27 to 0.01 µM and following the protocol described above. The negative control corresponded to cells treated with 1% (v/v) DMSO. Each condition was tested at least in duplicates.
All data corresponding to cytotoxicity or infection assays were analysed using GraphPad Prism 6 (GraphPad Software, La Jolla, CA). The EC50 values were determined from concentration–response curves (up to seven experimental concentrations of compounds were tested in at least duplicate) fitted to a four-parameter sigmoid equation or extrapolated from non-linear fitting plots. All errors are expressed as one SD, estimated as σ n–1.
Determination of intracellular low molecular mass thiols
The method is a modified version of that previously described by Fairlamb et al.Citation35. Leishmania infantum promastigotes from the WT and sKO cell lines were seeded at 5 × 105 cells/mL and cultured for 24 h as described above in the presence of 7 µM 20 or DMSO 1% (v/v). Thereafter, cells were harvested by centrifugation (2000 g for 10 min at room temperature) and low molecular weight thiols labelled and extracted as described below.
A 50% confluent culture of murine macrophages (cell line J774) was infected with L. infantum promastigotes (WT and sKO), from a 24 h stationary-phase culture at a ratio 1:40 (1 × 106 macrophages:4 × 107 parasites), during 24 h (2 h at room temperature and 22 h at 37 °C). Next, the cell monolayer was washed with phosphate buffer saline (pH 7.0) to remove extracellular parasites, and then treated or not with 1 µM 20 for 24 h. Cells were scraped and harvested by centrifugation (2500 g for 10 min at room temperature). Non-infected macrophages were included as control.
All samples (promastigotes or infected macrophages) were washed twice with phosphate buffered saline (pH 7.0) and resuspended at a density ≥ 1.0 × 107 cells/0.1 mL of 40 mM HEPES pH 8.0, treated during 1 h at room temperature with NaBH4 70 mM (stock 1 M in distilled water) and then added of monobromobimane up to 2 mM (stock 100 mM in ethanol). After incubation at 70 °C for 3 min, the samples were cooled on ice, then added of 100 µL 4 M methanesulfonic acid and incubated overnight at 4 °C. The supernatant containing the derivatized low molecular mass thiols was centrifuged at 13,000 g for 40 min at 0 °C. Labelled thiols were separated using solvent A: 0.25% camphorsulfonic acid pH 2.6 and solvent B: 25% 1-propanol in solvent A, and the following conditions: 100 µL of the sample, corresponding to ≥ 0.5 × 107 cells, was injected onto a C18, 5 µm, 4.6 × 150 mm column connected to an HPLC (both from Agilent, Palo Alto, CA) pre-equilibrated in 12% solvent B followed by a linear gradient from 12 to 50% solvent B over 36 min at a flow rate of 1 mL/min. The column was calibrated with derivatized standards of GSH and T(SH)2 in the concentration range of 2.5 to 75 nmol. For the different samples, the relative level of these molecular weight thiols was determined by integrating the corresponding HPLC peak areas and, when corresponding, subtracting the GSH content contributed by non-infected, non-treated macrophages. For promastigotes, the values for each thiol were expressed as % relative to non-treated WT cells, whereas for samples from infected macrophages, the values were expressed as fold-change normalised against non-treated samples. At least duplicate assays were performed for each condition and the data were analysed using the GraphPad Prism 6 programme (GraphPad Software, La Jolla, CA), applying an unpaired t-test for statistical comparison.
Results
TryS-based screening of N5-substituted paullones
Previous reports show that 3-chlorokenpaullones containing substitutions at position N5 are species-specific TryS inhibitorsCitation10,Citation20,Citation21. However, the contribution of the paullone scaffold to enzyme inhibition was unknown. The core scaffold 3-chlorokenpaullone (1) tested at 30 µM proved inactive against Trys from L. infantum () as well as from T. brucei and T. cruzi (Table S1). In contrast, a paullone with a small hydroxyethyl group at position N5 (2) displayed low (10–30% inhibition) to moderate (IC50 ∼30 µM) activity against at least one of the TryS. This result suggests that the paullone core alone is not a major determinant of TryS inhibition.
Given the relevance of this finding, a set of derivatives with different substituents consisting of acetic acid and its ester derivatives, acetamides, N-(2-aminoethyl)acetamides or N-(4-aminobutyl)acetamide (N-acetyl putrescine) were synthesised and tested against TryS. Overall, the new derivatives proved more active against the leishmanial (32 out of 36 with IC50 ≤30 µM, ) than towards the trypanosomal TryS (5 out of 36 with IC50 ≤30 µM, Table S1). Based on these results, structure-activity relationship analysis was performed only for LiTryS.
Comparison of the four linkers shows that an acetic acid side chain is detrimental for activity (3 inactive at 30 µM) whereas the switch to a primary acetamide renders an active derivative (7, IC50 = 5.1 µM). Compared to the simple primary acetamide (7), paullones with longer linkers and a terminal amine showed a 10-fold increased inhibitory activity (27, IC50 = 0.3 µM and 35, IC50 = 0.4 µM).
Modification of the carboxylic acid of 3 to an acetic acid ester with methyl (4, IC50 = 3.0 µM), tert-butyl (6, IC50 = 2.4 µM) or, to a minor extent, with ethyl (5, 36% inhibition at 30 µM) substituents proved beneficial for anti-LiTryS activity. This indicates that the negative charge of the acetate substituent may interfere with paullone binding to the active site.
For paullones containing an acetamide linker, high activities (IC50 = 0.5–0.8 µM) were observed for derivatives with small N-alkyl substituents. A secondary amide (N-methylacetamide, 9) as well as tertiary amides (N,N-dimethylacetamide, 10 and N,N-diethylacetamide, 12) proved active, which implies that an H-bond donor at this position is not essential for inhibitor-enzyme interaction. The primary acetamide (7), the acetohydrazide (8) or amino-substituted paullone (11) and the analogues containing terminal hydroxyl functions (13, 14) or the bulkier tert-butylacetamide group (15), retained low µM activity (IC50 = 1.5–5.2 µM). In contrast, the activity was significantly impaired for analogues containing terminal heterocyclic (i.e. 1,3,4-thiadiazole, 4,5-dihydro-1,3,thiazole, 1,3-oxazole, IC50 = 20–30 µM for 16, 17, 18, respectively) or phenyl (IC50 ∼30 µM for 19) rings.
Compounds where the nitrogen atom of the acetamide linker is part of a heterocycle, such as the piperazine (20, IC50 = 0.3 µM) or the 4-methylpiperazine (21, IC50 = 0.4 µM) derivatives, presented submicromolar activity. In contrast, addition of a bulky BOC group (22, IC50 = 30 µM) or pyrimidine (23, IC50 = 12.5 µM) to the piperazine moiety affected negatively the activity. A low activity was also observed for a derivative containing a piperidine substituent (24, 39% LiTryS inhibition at 30 µM). On the other hand, exchange of the piperidine for a smaller pyrrolidine group, impacted positively enzyme inhibition (25, IC50 = 0.5 µM). An intermediate activity was observed for the derivative containing a morpholine moiety (26, IC50 = 2.8 µM).
These data suggest that the enzyme binding pocket for the N5-substituent of paullones allows the accommodation of an acetamide linker with either smaller and apolar groups (e.g. methyl: 9 and 10, ethyl: 11, pyrrolidine: 25) or larger (a)polar groups (e.g. piperazine: 20 or 4-methylpiperazine: 21). Overall, compounds bearing tertiary amides (N,N-dimethyl: 10 and N,N-diethyl: 12) were more potent than those with secondary (9, 11, 13–19) or primary (7) amides. In addition, the apolar nature of relatively smaller substituents appears to be important because derivatives containing groups with H-bond donors or acceptors (e.g. a terminal primary alcohol, 1,3,4-thiadiazole, 4,5-dihydro-1,3-thiazole or 1,3-oxazole: 13, 14, 16, 17 and 18, all with µM IC50) were less active than those lacking them (e.g. N-methyl, N-ethyl, N,N-dimethyl, N,N-diethyl and pyrrolidine: 9, 11, 10, 12 and 25, all with nM IC50).
MOL2008 contains a side chain consisting of an N-[2-(methylamino)ethyl]acetamide and, so far, is the most active representative within this compound family (IC50 = 0.15 µM). Modification of the terminal amine did not prove critical for inhibitory activity, because analogues with unsubstituted terminal primary amine (27, IC50 = 0.3 µM) as well as the tertiary amine (N-[2-(diethylamino)ethyl]acetamide, 28, IC50 = 0.5 µM) showed IC50 values within the same order of magnitude as MOL2008. However, amine protection with a sterically demanding BOC group and the concomitant reduction of its basic properties decreased by almost two orders of magnitude compound potency (29, 37% LiTryS inhibition at 30 µM) when compared to the unprotected amine derivative (27). Compounds where the nitrogen atom of the ethylendiamine moiety is part of a 6-membered heterocycle (i.e. piperidine: 31, IC50 = 0.8 µM and piperazine: 32, IC50 = 0.6 µM) presented submicromolar activity.
Furthermore, paullones substituted with an ethylmorpholine moiety (30, IC50 of 3.3 µM) or a modified piperazine (4-methylpiperazine, 33, IC50 = 1.1 µM and 4-BOC-piperazine, 34, IC50 ∼30 µM) presented comparatively low anti-LiTryS activity.
Finally, a paullone with a long and flexible alkyl linker, an N-acetyl putrescine, retained nanomolar inhibitory activity against LiTryS (35, IC50 = 0.4 µM), which is decreased almost 100-fold upon protection of the terminal amine with a BOC group (36, IC50 ∼30 µM).
With respect to the trypanosomal TryS (Table S1), few compounds that were moderately active towards LiTryS showed a similar activity against TcTryS (i.e. 34, IC50 = 20 µM; 36, IC50 = 25 µM; 16, IC50 = 30 µM) or TbTryS (19, IC50 = 30 µM). Few highly active compounds against LiTryS with an IC50 in the nanomolar range showed modest activity against TbTryS (12 and 20 both with IC50 ∼30 µM). Despite these examples, the low inhibitory activity of most paullone analogues against trypanosomal TryS precluded the elaboration of a structure-activity relationship analysis. On the other hand, the fact that many of the compounds reported as (highly) active against LiTryS and not or poorly active against Tb- and TcTryS highlights remarkable species-specific differences at the TryS´s binding pocket for the N5-substituted moiety of paullones.
Kinetic inhibition and binding thermodynamics of N5-substituted paullones to LiTryS
The mechanism that characterise the inhibition of LiTryS by N5-substituted paullones was studied for two of the most active analogues that harbour structurally unrelated substituents: N-[2-(methylamino)ethyl]acetamide (MOL2008; ) and an acetyl piperazine (20; S1 Figure). For both paullones, the kinetic analysis reveals a parallel pattern of the double reciprocal plots for ATP and GSH that is characterised by a decrease in both substrates’ KM and Vmax at increasing concentrations of inhibitor ( and Figure S1(A,B)). Such behaviour is compatible with an uncompetitive inhibition of LiTryS. This type of inhibition is preceded by the formation of an enzyme-substrate complex (TryS-ATP, TryS-GSH and/or TryS-ATP/GSH) prior to binding of the inhibitor to the active site. In contrast, for both polyamine-based substrates, namely SP and GSP, the compounds yielded double reciprocal plots with convergent lines; where increasing concentrations of inhibitor correlated with increasing apparent KM values for these substrates and no changes on Vmax ( and Figure S1(C)). This behaviour corresponds to a competitive inhibition mechanism. Although we have previously communicated that MOL2008 competes with SP binding to LiTrySCitation20, our new data shows that the binding of N5-substituted paullones extents to the second GSH-binding site of the enzyme independently whether the substituent is linear or a cycle. In fact, the Ki for MOL2008 with GSP as competing substrate (Ki = 59 nM) was nearly two-fold higher than with SP (Ki = 33 nM), which suggests that GSP competes more efficiently than SP against paullone binding.
Figure 2. Mode of Leishmania infantum TryS (LiTryS) inhibition by MOL2008. Lineweaver–Burk reciprocal plots for the LiTryS activity measured at different inhibitor (black square: 0 µM, red circle: 0.15 µM, blue triangle: 0.375 µM, violet star: 0.75 µM, green inverted triangle: 1.12 µM and magenta inclined triangle: 1.5 µM) and varying substrate concentrations while maintaining fixed the concentration of the co-substrates (13 mM for SP, 200 μM for ATP and 100 μM GSH). The enzyme velocities were measured using an end-point assay (see the section Materials and methods for details) and the reciprocal plots are shown for the varying substrate: (A) GSH, (B) ATP, (C) SP and (D) GSP [mono(glutathionyl)spermidine].
![Figure 2. Mode of Leishmania infantum TryS (LiTryS) inhibition by MOL2008. Lineweaver–Burk reciprocal plots for the LiTryS activity measured at different inhibitor (black square: 0 µM, red circle: 0.15 µM, blue triangle: 0.375 µM, violet star: 0.75 µM, green inverted triangle: 1.12 µM and magenta inclined triangle: 1.5 µM) and varying substrate concentrations while maintaining fixed the concentration of the co-substrates (13 mM for SP, 200 μM for ATP and 100 μM GSH). The enzyme velocities were measured using an end-point assay (see the section Materials and methods for details) and the reciprocal plots are shown for the varying substrate: (A) GSH, (B) ATP, (C) SP and (D) GSP [mono(glutathionyl)spermidine].](/cms/asset/6900522f-0349-4a80-b1f4-0174539db527/ienz_a_1780227_f0002_c.jpg)
To get an insight into the thermodynamics of the inhibitor–enzyme interaction, ITC assays were performed with MOL2008, as model paullone, and LiTryS alone or in the presence of the substrate GSH and/or the product ADP. SP and GSP were excluded from the titration experiments because of the competitive nature exhibited by MOL2008 towards polyamine substrates. Titration in the presence of ATP alone or with GSH could not be performed due to enzyme-catalysed ATP hydrolysis, an exothermic reaction that overlaps the heat released by the interaction enzyme–inhibitor (not shown). ADP was considered an adequate surrogate of ATP because, as shown previously, it remains bound to the nucleotide-binding site of the enzyme (Ki 90 µM)Citation20.
ITC data was fitted to a 1:1 interaction model, which is compatible with the binding of a single molecule of paullone per TryS subunitCitation20. For apo-LiTryS, the dissociation constant (KD) for the LiTryS-MOL2008 complex was estimated in 6.5 µM with ΔH and ΔS values of −13.3 kcal.mol−1 and −20.6 cal.mol−1.K−1, respectively (). In the presence of GSH (2 mM), the affinity for MOL2008 was 3-fold higher (KD = 1.9 µM; ) than that obtained for the apo-form, which suggests a marginal increase in the affinity for the inhibitor. Accordingly, the ΔH (−17.0 kcal.mol−1) and ΔS (−30.4 cal.mol−1.K−1) values decreased maintaining a similar thermodynamic signature (enthalpy driven association). Next, titration performed in the presence of the non-hydrolyzable product ADP (2 mM) yielded a KD for MOL2008 = 52 nM with a similar thermodynamic signature (ΔH −13.2 kcal.mol−1 and ΔS −10.5 cal.mol−1.K−1, ). Interestingly, the TryS/ADP complex showed a KD for the inhibitor 37- and 125-fold higher than that estimated for the TryS/GSH complex and apo-TryS, respectively. This clearly indicates a superior affinity of the compound for the nucleotide-bound enzyme. Finally, titration of the enzyme with MOL2008 in the presence of both ADP (2 mM) and GSH (2 mM) yielded a KD = 417 nM with both enthalpic and entropic contributions (less dependent on the former than in the other cases, ΔH −7.9 kcal.mol−1, with a favourable entropic component, ΔS = 2.9 cal.mol−1.K−1; ). The affinity is 4.5-fold higher than that estimated for the TryS-GSH complex and 8-fold lower than that exhibited by the TryS/ADP complex. This shows that, to some extent, high GSH concentrations interfere with inhibitor binding to the TryS/nucleotide complex. Such behaviour agrees well with the kinetic analysis performed with GSP as variable substrate, which suggests that binding of N5-substituted paullones extents to the second GSH-binding site of TryS. At saturating concentrations of substrate (e.g. 2 mM GSH for ITC), which exert enzyme inhibitionCitation20, this site will likely be partially occupied by GSH and, hence, competing for paullone binding.
Figure 3. Isothermal titration calorimetry of Leishmania infantum TryS (LiTryS) with MOL2008. Top plots: baseline subtracted thermogram showing differential heating power (DP) versus time. Bottom Plots: integrated and normalised heats of injections versus molar ratio fitted to a heterodimer association model with the corresponding residuals plots. (A) LiTryS (429 μM) versus MOL2008 (60 μM; non-competent fraction of compound = 0.218). (B) LiTryS (615 μM), glutathione (GSH, 2 mM), DTT (5 mM) versus MOL2008 (60 μM; non-competent fraction of compound = 0.329). (C) MOL2008 (60 μM) versus LiTryS (6.7 μM; non-competent fraction of enzyme = 0.459), ADP (2 mM). (D) MOL2008 (60 μM) versus LiTryS (6.2 μM; non-competent fraction of enzyme = 0.386), ADP (2 mM), GSH (2 mM) and DTT (5 mM).
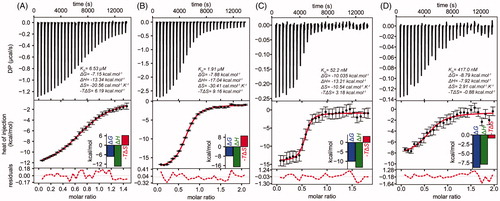
In agreement with the differences in KD values, comparison of the Gibbs free energy indicates that paullone binding to LiTryS is thermodynamically more favoured in the following order: TryS/ADP (ΔG −10.0 kcal.mol−1) ≫ TryS/GSH/ADP (ΔG −8.8 kcal.mol−1) ≫ TryS/GSH (ΔG −7.9 kcal.mol−1) > apo-TryS (ΔG −7.2 kcal.mol−1). The comparatively less negative T*ΔS values observed for the titration performed in the presence of ADP (−3.2) and ADP/GSH (0.88) indicate that these ligands may induce conformational changes in the enzyme that favour inhibitor binding. The positive ΔS and the less negative ΔH for the titration of the TryS/ADP/GSH complex are compatible with water displacement from the inhibitor binding-site and the establishment of hydrophobic interactions between TryS and MOL2008. Although to a significantly minor extent, such phenomenon is also observed for the complex TryS/ADP/MOL2008.
The kinetic and thermodynamic studies strongly suggest that the binding site of MOL2008 and 20, at least partially, overlaps with that of SP and GSP. GspS is an enzyme capable to catalyse the formation of GSP but not of T(SH)2 due to steric effects that preclude binding of GSP to the enzyme’s active siteCitation36. Thus, we reasoned that if occupation of the GSP-binding site by N5-substituted paullones is relevant for TryS inhibition, then GspS should not be inhibited by these compounds. Confirming our hypothesis, GspS from the Kinetoplastid Crithidia fasciculata proved refractory to MOL2008, which caused a minor inhibition (20%) of the enzyme at the highest concentration tested of 300 µM (see Supplementary Information for details about protein preparation and enzymatic assay). This concentration is 2000-fold higher than that needed to produce 50% inhibition of LiTryS ().
In summary, binding of N5-substituted paullones to LiTryS: (a) is enhanced if an ATP- and/or GSH-enzyme complex is pre-formed, (b) competes with SP- and GSP-binding, (c) involves occupancy of the second GSH-binding site, and (d) interferes with catalysis.
Binding model and selectivity of N5-substituted paullones for non-trypanosomal TryS
To get structural insights into the binding mode of N5-substituted paullones with TryS, molecular models and target sequences were analysed.
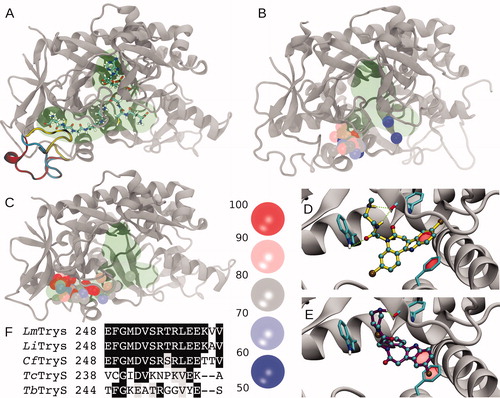
In TryS, the polyamine binding pocket is an open cleft bounded by flexible loops that allow binding of polyamines of different length and GSPCitation37,Citation38. One important element of this region is the loop comprising amino acids Glu248 to Val263 (hereafter loop248-263), which was not solved in the X-ray structure of LmTrySCitation37 and has been proposed to facilitate GSP bindingCitation3,Citation39. Molecular modelling suggests a large conformational plasticity for loop248-263. Conformations ranging from completely solvent exposed to compactly folded can be modelled onto the protein scaffold without obvious steric impediments (). For E. coli GspS (EcGspS), the segment equivalent to loop248-263 is five residues shorter and visible from the electronic densityCitation36, which suggests it is a less dynamic elementCitation39. Next, comparative docking experiments on both protein structures with paullones that do (i.e. MOL2008 and 20) or do not inhibit (1) LiTryS were performed. Docking onto EcGspS reveals that the preferred binding sites for paullones lie within a region corresponding to the SP region and in close proximity to loop248-263, while only low energy solutions are located towards the GSH region (). Repeating the same calculations on LmTryS () yielded higher docking affinities (compare ) with docking solutions shifted towards the GSP region. Despite some degree of dispersion in the docking poses, all of them are located mainly in the GSP region. A hallmark of the best-ranked solutions was the presence of π-stacking interactions between the paullones indole ring and Phe249, alternating hydrogen bonding between the secondary amines from the N5-substituents with the Phe626 backbone oxygen, or the acetamide oxygen and the Trp363 indolic NH (). While Trp363 is highly conserved in Kinetoplastid´s TryS and EcGspS, Phe249 is replaced by an Asparagine, Leucine or Cysteine in EcGspS, CfGspSCitation40 and TcTryS (), respectively. This provides additional support for the striking refractoriness of the last two enzymes to inhibition by the model paullone MOL2008 (see previous section and Table S1). Furthermore, multiple sequence analysis of the segment corresponding to loop248-263 reveals that this region is two residues shorter and poorly conserved in trypanosomal TryS with respect to the highly conserved signature of leishmanial and crithidial TryS (13 out of 16 residues are strictly conserved; ). Adding value to a potential contribution of residues from this loop to paullone’s TryS species-specificity, CfTryS was highly sensitive to MOL2008 with an IC50 of 30 nMCitation41. This contrasted with the 40–60% inhibition of trypanosomal TryS achieved by 30 µM MOL2008 (Table S1). The putative conformational diversity generated by the flexibility of the loop248-263 hampers the identification of candidate residues for the interactions with paullones. However, the data presented here support an important role of this loopy region in determining the selective binding of N5-substituted paullones to TryS.
Figure 4. Molecular modelling and docking of selected compounds. (A) Cartoon representation of Leishmania major Trypanothione synthetase (LmTryS) bound to ADP (top), GSH (right) and GSP (left). The green shading represents the volume occupied by the ligands. Three different possible conformations of the loop248-263 (not solved in the X-ray structure of LmTryS , PDB id: 2VPS) were modeled and are represented by yellow, cyan and red ribbons. (B) Superimposition of the docking solutions for compounds 1, 20 and MOL2008 on the structure of EcGspS (PDB id: 2IOA). The volume of the ligands ADP and a phosphinate inhibitor solved in the X-ray structure are shown in green. These ligands were removed prior to docking calculations. Spheres represent the position of the N5 nitrogen in each docking pose. The binding affinities are indicated by colour from red to blue as percentage of the absolute best docking solution. (C) Top: superimposition of the docking solutions for the three paullones on the X-ray structure of LmTryS. Docking solutions are coloured as in B. The docking was performed in absence of the loop248-263. (D) and (E) Inset of the best docking solutions for MOL2008 and 20, respectively. Hydrogen bonds are depicted in green dashed lines and π-stacking interactions are represented with a translucent red surface. (F) Multiple sequence analysis of the segment corresponding to loop248-263 in TryS from different trypanosomatids coloured by conservation (black and grey background denote aminoacid conservation and homology, respectively, in at least 4 out 5 sequences). From the top to the bottom: TryS from L. major (LmTryS), L. infantum (LiTryS), Crithidia fasciculata (CfTryS), Trypanosoma cruzi (TcTryS) and Trypanosoma brucei (TbTryS).
Phenotypic-based screenings
In South America, the incidence of (muco)cutaneous leishmaniasis is at least two orders of magnitude higher than that of visceral leishmaniasisCitation42. Furthermore, L. braziliensis is the species responsible for the vast majority of the reported cases of (muco)cutaneous leishmaniasis in this regionCitation42. Therefore, the anti-proliferative activity of a subset of paullones was preliminary tested against the clinically relevant form (i.e., intracellular amastigote) of L. braziliensis. For each set of paullones harbouring different N5-linkers and substituents, the most active ones against LiTryS were selected for the screening assay (). The final concentration of the compounds in the bioassay was 10 µM or, for those causing detachment of the macrophage monolayer, 5 µM. Paullones containing at N5-position a tert-butyl acetate group (6) or an N-alkyl acetamide linker (9, 10, 12 and 15) proved poorly active in preventing the infection of macrophages with L. braziliensis (72–107% infection). Similar to the behaviour observed against LiTryS, compounds with an acetamide linker and bearing tertiary amides (N,N-dimethyl: 10 and N,N-diethyl: 12) displayed, although minor, anti-leishmanial activity compared to the inactive mono-substituted derivatives 9 and 15. Notably, compounds for which the nitrogen atom of the acetamide linker is part of a heterocycle (20, 21, 25 and 26) were more effective in inhibiting macrophage infection (15–68% infection). Also derivatives with an N-(2-aminoethyl)acetamide side chain lacking (27) or containing terminal alkyl groups (MOL2008 and 28) displayed similar anti-L. braziliensis activity (38–68% infection). From the two paullones with an ethylacetamide linker, only the piperidine (49% infection at 5 µM) but not the piperazine derivative showed anti-leishmanial activity. Interestingly, the piperazine group incorporated to a shorter acetyl linker (20) resulted in an analogue with the highest activity (15% infection at 5 µM). In contrast to the paullones with a short alkyl linker (N-(2-aminoethyl)acetamide; MOL2008, 27 and 28), a derivative harbouring a longer N5-extension and a terminal amine (i.e. N-(4-aminobutyl)acetamide, 35) was inactive at 10 µM.
Table 2. Biological activity of N5-substituted, 3-chlorokenpaullones.
Based on the analysis of host cell viability, paullones can be grouped according to the following degree of cytotoxicity: CC50 >200 µM (1), CC50 ≥100 µM (10, 12, 15, 27, 32 and 35), CC50 ≤100 µM (6, 9, 21 and 25) and CC50 ≤ 10 µM (20, 26, MOL2008, 28 and 31). Thus, except for 35, the incorporation of an N5-substitution to the core scaffold (1) conferred cytotoxicity to paullones. On the other hand, paullones showing high activity against the intracellular stage of L. braziliensis were also the most cytotoxic towards macrophages (CC50 ≤10 µM for 20, 26, MOL2008, 28 and 31).
The EC50 against L. braziliensis amastigotes was determined for paullones showing capacity to lower the infection rate at values ≤65%. Compound 20 represented the most active derivative with a submicromolar EC50 (value of 0.2–0.4 µM) and SI ≥ 21, whereas the potency of the remaining compounds ranged from 1.6 to 7 µM and their SI from 2 to 6. Interestingly, the switch from a terminal secondary (i.e. 20) to a tertiary amine (N-methylpiperazine moiety, 21: EC50 of 7 µM and SI of 5) affected negatively the potency and selectivity. Overall, the derivatives consisting of an acetyl linker substituted with heterocyclic rings (20, 21 and 26) appeared more selective than those containing a N-(2-aminoethyl)acetamide linker substituted with further alkyl groups (MOL2008 and 28) or a heterocyclic ring (31). To assess a potential wide spectrum activity of the new paullones, the compounds with SI >2 towards L. braziliensis were tested against a species responsible for visceral leishmaniasis. All three derivatives based on paullones with an acetyl linker substituted with heterocyclic rings (20, 21 and 26) were more active and selective (EC50 = 0.5–1 µM and SI = 9–35) against L. infantum amastigotes than MOL2008 (EC50 = ∼10 µM and SI = 1).
In summary, the incorporation of cyclic substituents at the N5-position yielded analogues with higher and more selective anti-leishmanial activity than those containing linear substituents.
On-target effect of compound 20
For paullones containing a N-[2-(methylamino)ethyl]acetamide substituent at position N5, their on-target effect against the promastigote stage of L. infantum was demonstrated previouslyCitation10,Citation20. Here, we extended the study to 20, a derivative with an acetylpiperazine substituent. Intracellular thiols were measured in L. infantum promastigotes () and intracellular amastigotes () from the wild-type and TryS-single KO cell lines exposed for 24 h to the compound’ EC50 determined for each parasite stage.
Figure 5. On-target effect of compound 20 in promastigotes and amastigotes of Leishmania infantum. (A) L. infantum promastigotes from the wildtype (WT) or TryS single knockout (sKO) cell line were incubated for 24 h in the absence (white bars) or presence of 7 µM 20 (black bars). The content of free glutathione (GSH) and trypanothione [T(SH)2] is expressed as % relative to samples from non-treated WT parasites. * and **, denote p values < 0.025 and = 0.0052, respectively (two-tailed t-test). (B) Murine macrophages (cell line J774) infected with L. infantum amastigotes from the WT and sKO cell lines were incubated for 24 h in the absence (white bars) or presence of 1 µM 20 (black bars). The results are expressed as relative fold-change in GSH and T(SH)2 content with respect to the corresponding non-treated control from each sample. ***, denotes a p values ≤ 0.001 (two-tailed t-test).
![Figure 5. On-target effect of compound 20 in promastigotes and amastigotes of Leishmania infantum. (A) L. infantum promastigotes from the wildtype (WT) or TryS single knockout (sKO) cell line were incubated for 24 h in the absence (white bars) or presence of 7 µM 20 (black bars). The content of free glutathione (GSH) and trypanothione [T(SH)2] is expressed as % relative to samples from non-treated WT parasites. * and **, denote p values < 0.025 and = 0.0052, respectively (two-tailed t-test). (B) Murine macrophages (cell line J774) infected with L. infantum amastigotes from the WT and sKO cell lines were incubated for 24 h in the absence (white bars) or presence of 1 µM 20 (black bars). The results are expressed as relative fold-change in GSH and T(SH)2 content with respect to the corresponding non-treated control from each sample. ***, denotes a p values ≤ 0.001 (two-tailed t-test).](/cms/asset/6721465b-51f9-4300-8ce7-1701514d1e3c/ienz_a_1780227_f0005_b.jpg)
For non-treated promastigotes, and in agreement with the reduced expression of TryS from the single encoding alleleCitation10, the content of T(SH)2 in sKO cells was about one quarter (24%) of that present in WT cells (). Although to a minor extent, sKO cells also showed a lower GSH content (63%) compared to WT cells. Treatment with 20 (7 µM) produced a significant depletion of T(SH)2 in WT (88% decrease) and in sKO cells, where it became undetectable [detection limit of 0.5 nmol T(SH)2/100 million cells]. Overall, the metabolic changes induced by 20, namely depletion of GSH and T(SH)2, resemble those caused by MOL2008 in L. infantum promastigotesCitation20.
For amastigotes, and compared to non-treated controls, 20 led to a significant accumulation of GSH in WT (16-fold) and sKO (10-fold) parasites, respectively (). This suggests a differential metabolic regulation of GSH in non-infective and infective parasite stages. In contrast, the intracellular content of T(SH)2 was depleted about 40% in WT and 82% in sKO amastigotes with respect to the corresponding non-treated controls. This resembles the metabolic phenotype of bloodstream African trypanosomes with downregulated expression of TryS8.
In summary, the metabolic changes exerted by 20 in both life cycle stages of L. infantum are compatible with TryS inhibition.
Discussion
Historically, paullones belong to a series of molecules reported as inhibitors of mammalian CMGC superfamily protein kinasesCitation43–46. Considering that paullones interfere with the activity of several kinases that regulate multiple biochemical and biological processes in cellsCitation47–49, the development of selective inhibition against specific targets represents a challenge. In this regard, structural analysis revealed that hydrogen bonding to the lactam nitrogen at position 5 (N5) of an alsterpaullone is necessary for inhibitor´ binding to the ATP binding site of GSK-3βCitation22,Citation50. This led to the assumption that N5-substituted paullones will lose affinity for kinases, a hypothesis that was confirmed with a subset of 3-chlorokenpaullones with N-benzylacetamide substituent at N5 showing potent and selective anti-T. b. brucei activity and lack of inhibition of mammalian kinases at 10 µM (e.g. compound 3a)Citation21. Further evidence that attainment of selectivity is possible within the paullone scaffold was provided by the inclusion of a N-[2-(methylamino)ethyl]acetamide side chain on position 5 of 9-trifluoromethyl paullone (FS-554) or 3-chlorokenpaullones (MOL2008), which yielded potent and selective inhibitors of L. infantum TryS (IC50 = 0.35 µM and 0.15 µM, respectively)Citation10,Citation20. The almost 100-fold lower activity of these compounds against trypanosomal TryS posed the question about the molecular determinants of such selectivity. On the other hand, the poor biological discriminatory activity of these paullones suggests that modification of the N5-position, although detrimental for kinase inhibition, may confer them new molecular targets in the host cell.
Thus, in an attempt to extend and improve the selectivity of paullones towards trypanosomal TryS and the clinically relevant stages of different trypanosomatid species, novel N5-substituted analogues of 3-chlorokenpaullones were synthesised and characterised. From the 36 new derivatives, 12 of them showed submicromolar activity against LiTryS. However, none of the new compounds surpassed the potency of MOL2008 against LiTryS neither extended the selectivity towards trypanosomal TryS. In fact, the IC50 for the best inhibitors of T. cruzi and T. brucei TryS were yet two orders of magnitude higher than those obtained against LiTryS. This likely suggests steric restrictions at the binding site of paullones to trypanosomal TryS that probably extent beyond the nature of the N5-substituent. Strategies to overcome this limitation may include the simplification of the paullone scaffold to reduce compound size while keeping a variety of the linkers tested here and in other analoguesCitation21 that, as shown here, appear important for anchoring the paullone to the polyamine binding site.
The inhibition mechanism of N5-substituted paullones against LiTryS has been elucidated for the first time. Two compounds harbouring structurally unrelated linkers at N5 position showed uncompetitive inhibition for the nucleotide ligand and GSH, and competitive for SP or GSP. The ATP-uncompetitive inhibition of TryS by kenpaullones seems atypical, because it has been well documented that related analogues behave as ATP-competitive inhibitors of CDK1/cyclin BCitation43 and GSK-3βCitation50. However, our mechanistic observations are fully compatible with the molecular and binding energy models that support anchoring of the N5-substituent to the polyamine pocket of TryS whereas the paullone ring occupies, partially, the polyamine binding site and the second GSH binding site. In this binding conformation, the paullone scaffold sits away from the nucleotide and first GSH binding sites, and, hence, does not interfere or compete with their binding to TryS. Also, biophysical characterisation of the interaction of TryS with the inhibitor and different physiological ligands yielded data in line with the inhibition kinetics and binding model. Compared to the apo-form, the affinity of the enzyme for the inhibitor increased in the presence of ADP alone (125-folds) or in combination with GSH (37-folds). In comparison, titration performed at high concentration of GSH alone (2 mM) increased only marginally (3-folds) the affinity of the enzyme for the paullone. This behaviour may have two possible explanations that are not mutually exclusive: (i) the conformational changes induced by substrate binding, which subsequently favour paullone interaction with TryS, are more prominent for the nucleotide than for the thiol substrate, and (ii) at the saturating concentration of GSH used in the ITC assay, this ligand may compete with paullone for binding to the second GSH site within the GSP-binding pocket.
The relevance of the second GSH-pocket for paullone binding to TryS was further evidenced by the refractoriness of the related enzyme GspS, which lacks this site, to inhibition by the model paullone MOL2008. Furthermore, within this region, we have identified a non-structured element (loop248-263) that may contribute to the selectivity displayed by paullones for leishmanial and crithidial TryS. This element is two amino acids shorter in trypanosomal TryS and has low amino acid sequence conservation with respect to the highly conserved motif present in leishmanial and crithidial TryS. Analysis of point mutants of this region may shed light onto the specific role of individual residues for loop conformation and paullone interaction. This information would be highly valuable for the rational design of compounds with broad selectivity for TryS.
In regard to the biological performance of the new derivatives, and compared to the reference molecule MOL2008, several of them showed higher potency (from 2.5- to 28-fold) and selectivity (from 5- to 35-fold) against pathogenic Leishmania that causes mucocutaneous (L. braziliensis) and visceral leishmaniasis (L. infantum). The derivatives containing a N5-acetamide substituent with a terminal heterocycle (i.e. piperazine: 20, 4-methylpiperazine: 21 and morpholine: 26) are the most representative examples from this successful optimisation strategy. This suggests, that in comparison to derivatives containing linear alkyl substituents, the heterocyclic ring at N5-position is detrimental for the interaction of paullones with host´s molecular targets. Moreover, the on-target effect of 20 was confirmed in both life cycle stages of different L. infantum cell lines.
As shown here, N5-substituted paullones are uncompetitive inhibitors of TryS with respect to ATP and GSH. Uncompetitive and mechanistic-based inhibitors are the most wanted, albeit rarely found, hits in screening campaigns since their design usually requires several rounds of hit optimisation. In an in vivo context, uncompetitive inhibition is not overcome, but enhanced, by substrate accumulation because the enzyme/ligand equilibrium is shifted to the formation of the enzyme–substrate complex for which the uncompetitive inhibitor has a higher affinityCitation51. Probably, the higher potency of MOL2008 and 20 towards intracellular amastigotes than against extracellular promastigotes is in part exacerbated by the significant accumulation of GSH.
The results presented here should contribute to a better understanding of TryS molecular features and interaction with inhibitors, while providing a rationale platform for the design of improved inhibitors against this top drug target candidate. Future studies will address the pharmacological potential of the most potent and selective N5-substituted paullones in a murine model of cutaneous leishmaniasis.
Supplemental Material
Download PDF (381 KB)Supplemental Material
Download PDF (500.3 KB)Acknowledgements
The authors thank Dr. Sergio Guerrero (Universidad Nacional del Litoral, Santa Fe, Argentina), Dr. Ana Tomas, Dr Helena Castro (Instituto de Biologia Molecular e Celular, Universidade do Porto, Porto, Portugal), Dr. Alan Fairlamb (Dundee University, Dundee, Scotland) and Dr. Luise Krauth-Siegel (Heidelberg University, Germany) for providing expression plasmids for different TryS, CfGspS and L. infantum cell lines. The Recombinant Protein Unit from the Institut Pasteur de Montevideo is acknowledged for providing facilities and assistance during protein purification. Dr. Lucía Piacenza, Dr. Rafael Radi and Damian Estrada from the CEINBIO (Centro de Investigaciones Biomédicas, Universidad de la República, Uruguay) are gratefully acknowledged for providing facilities and assistance for the HPLC analysis. We acknowledge support by German Research Foundation and the Open Access Publication Funds of the Technische Universität Braunschweig.
Disclosure statement
The authors report no conflict of interest.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Fairlamb AH, Blackburn P, Ulrich P, et al. Trypanothione: a novel bis(glutathionyl)spermidine cofactor for glutathione reductase in trypanosomatids. Science 1985;227:1485–7.
- Krauth-Siegel RL, Comini M. Redox control in trypanosomatids, parasitic protozoa with trypanothione-based thiol metabolism. Biochim Biophys Acta 2008;1780:1236–48.
- Manta B, Bonilla M, Fiestas L, et al. Polyamine-based thiols in trypanosomatids: evolution, protein structural adaptations, and biological functions. Antioxid Redox Signal 2018;28:463–86.
- Croft SL, Sundar S, Fairlamb AH. Drug resistance in leishmaniasis. Clin Microbiol Rev 2006;19:111–26.
- Franco J, Scarone L, Comini MA. Drugs and drug resistance in African and American trypanosomiasis. Annu Rep Med Chem 2018;51:97–133.
- Ghosh AK, Saini S, Das S, et al. Glucose-6-phosphate dehydrogenase and Trypanothione reductase interaction protects Leishmania donovani from metalloid mediated oxidative stress. Free Radic Biol Med 2017;106:10–23.
- Pountain AW, Barrett MP. Untargeted metabolomics to understand the basis of phenotypic differences in amphotericin B-resistant Leishmania parasites. Wellcome Open Res 2019;4:176.
- Comini M, Guerrero SA, Haile S, et al. Validation of Trypanosoma brucei trypanothione synthetase as drug target. Free Radic Biol Med 2004;36:1289–302.
- Wyllie S, Oza SL, Patterson S, et al. Dissecting the essentiality of the bifunctional trypanothione synthetase-amidase in Trypanosoma brucei using chemical and genetic methods. Mol Microbiol 2009;74:529–40.
- Sousa AF, Gomes-Alves AG, Benítez D, et al. Genetic and chemical analyses reveal that trypanothione synthetase but not glutathionylspermidine synthetase is essential for Leishmania infantum . Free Radic Biol Med 2014;73:229–38.
- Mesías AC, Sasoni N, Arias DG, et al. Trypanothione synthetase confers growth, survival advantage and resistance to anti-protozoal drugs in Trypanosoma cruzi. Free Radic Biol Med 2019;130:23–34.
- Leroux AE, Krauth-Siegel RL. Thiol redox biology of trypanosomatids and potential targets for chemotherapy. Mol Biochem Parasitol 2016;206:67–74.
- D’Silva C, Daunes S, Rock P, et al. Structure-activity study on the in vitro antiprotozoal activity of glutathione derivatives. J Med Chem 2000;43:2072–8.
- Oza SL, Chen S, Wyllie S, et al. ATP-dependent ligases in trypanothione biosynthesis-kinetics of catalysis and inhibition by phosphinic acid pseudopeptides. Febs J 2008;275:5408–21.
- Saudagar P, Dubey VK. Cloning, expression, characterization and inhibition studies on trypanothione synthetase, a drug target enzyme, from Leishmania donovani. Biol Chem 2011;392:1113–22.
- Zimmermann S, Oufir M, Leroux A, et al. Cynaropicrin targets the trypanothione redox system in Trypanosoma brucei. Bioorg Med Chem 2013;21:7202–9.
- Torrie LS, Wyllie S, Spinks D, et al. Chemical validation of trypanothione synthetase a potential drug target for human trypanosomiasis. J Biol Chem 2009;284:36137–45.
- Spinks D, Torrie LS, Thompson S, et al. Design, synthesis and biological evaluation of Trypanosoma brucei trypanothione synthetase inhibitors. *ChemMedChem 2012;7:95–106.
- Saudagar P, Saha P, Saikia AK, Dubey VK. Molecular mechanism underlying antileishmanial effect of oxabicyclo[3.3.1]nonanones: inhibition of key redox enzymes of the pathogen. Eur J Pharm Biopharm 2013;85:569–77.
- Benítez D, Medeiros A, Fiestas L, et al. Identification of novel chemical scaffolds inhibiting trypanothione synthetase from pathogenic trypanosomatids. PLoS Negl Trop Dis 2016;10:e0004617.
- Orban OC, Korn RS, Benítez D, et al. 5-Substituted 3-chlorokenpaullone derivatives are potent inhibitors of Trypanosoma brucei bloodstream forms. Bioorg Med Chem 2016;24:3790–800.
- Kunick C, Lauenroth K, Wieking K, et al. Evaluation and comparison of 3D-QSAR CoMSIA models for CDK1, CDK5, and GSK-3 inhibition by paullones. J Med Chem 2004;47:22–36.
- Orban OCF, Korn RS, Unger L, et al. Chlorokenpaullone. Molbank 2015;2015:M856.
- Jo MN, Seo HJ, Kim Y, et al. Novel T-type calcium channel blockers: dioxoquinazoline carboxamide derivatives. Bioorg Med Chem 2007;15:365–73.
- Ascenzi P, Ascenzi MG, Amiconi G. Enzyme competitive inhibition. Graphical determination of Ki and presentation of data in comparative studies. Biochem Educ 1987;15:134–5.
- Keller S, Vargas C, Zhao H, et al. High-precision isothermal titration calorimetry with automated peak-shape analysis. Anal Chem 2012;84:5066–73.
- Scheuermann T, Brautigam C. High-precision, automated integration of multiple isothermal titration calorimetric thermograms: new features of NITPIC. Methods 2015;76:87–98.
- Houtman JC, Brown PH, Bowden B, et al. Studying multisite binary and ternary protein interactions by global analysis of isothermal titration calorimetry data in SEDPHAT: application to adaptor protein complexes in cell signaling. Protein Sci 2007;16:30–42.
- Brautigam CA. Calculations and publication-quality illustrations for analytical ultracentrifugation data. Meth Enzymol 2015;562:109–34.
- Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010;31:455–61.
- Morris GM, Huey R, Lindstrom W, et al. Autodock4 and AutoDockTools4: automated docking with selective receptor flexiblity. J Comput Chem 2009;30:2785–91.
- Fiser A, Sali A. ModLoop: automated modeling of loops in protein structures. Bioinformatics 2003;19:2500–1.
- Sievers F, Wilm A, Dineen D, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 2011;7:539.
- Edelstein AD, Tsuchida MA, Amodaj N, et al. Advanced methods of microscope control using µManager software. J Biol Methods 2014;1:e11.
- Fairlamb AH, Henderson GB, Bacchi CJ, Cerami A. In vivo effects of difluoromethylornithine on trypanothione and polyamine levels in bloodstream forms of Trypanosoma brucei. Mol Biochem Parasitol 1987;24:185–91.
- Pai CH, Chiang BY, Ko TP, et al. Dual binding sites for translocation catalysis by Escherichia coli glutathionylspermidine synthetase. Embo J 2006;25:5970–82.
- Oza SL, Tetaud E, Ariyanayagam MR, et al. A single enzyme catalyses formation of trypanothione from glutathione and spermidine in Trypanosoma cruzi. J Biol Chem 2002;277:35853–61.
- Fyfe PK, Oza SL, Fairlamb AH, Hunter WN. Leishmania trypanothione synthetase-amidase structure reveals a basis for regulation of conflicting synthetic and hydrolytic activities. J Biol Chem 2008;283:17672–80.
- Koch O, Cappel D, Nocker M, et al. Molecular dynamics reveal binding mode of glutathionylspermidine by trypanothione synthetase. PLoS One 2013;8:e56788.
- Comini M, Menge U, Wissing J, Flohé L. Trypanothione synthesis in Crithidia revisited. J Biol Chem 2005;280:6850–60.
- Jäger T, Koch O, Flohé L, Selzer PM, (Eds.). Trypanosomatid diseases: molecular routes to drug discovery (Drug Discovery in Infectious Diseases). Oxford, UK: Wiley-Blackwell; 2013.*
- Burza S, Croft SL, Boelaert M. Leishmaniasis. Lancet 2018;392:951–70.
- Schultz C, Link A, Leost M, et al. Paullones, a series of cyclin-dependent kinase inhibitors: synthesis, evaluation of CDK1/cyclin B inhibition, and in vitro antitumor activity. J Med Chem 1999;42:2909–19.
- Zaharevitz DW, Gussio R, Leost M, et al. Discovery and initial characterization of the paullones, a novel class of small-molecule inhibitors of cyclin-dependent kinases. Cancer Res 1999;59:2566–9.
- Bain J, Plater L, Elliott M, et al. The selectivity of protein kinase inhibitors: a further update. Biochem J 2007;408:297–315.
- Egert-Schmidt AM, Dreher J, Dunkel U, et al. Identification of 2-anilino-9-methoxy-5,7-dihydro-6H-pyrimido[5,4-d][1]benzazepin-6-ones as dual PLK1/VEGF-R2 kinase inhibitor chemotypes by structure-based lead generation. J Med Chem 2010;53:2433–42.
- Lyssiotis CA, Foreman RK, Staerk J, et al. Reprogramming of murine fibroblasts to induced pluripotent stem cells with chemical complementation of Klf4. Proc Natl Acad Sci USA 2009;106:8912–7.
- Stukenbrock H, Mussmann R, Geese M, et al. 9-Cyano-1-azapaullone (cazpaullone), a glycogen synthase kinase-3 (GSK-3) inhibitor activating pancreatic beta cell protection and replication. J Med Chem 2008;51:2196–207.
- Tolle N, Kunick C. Paullones as inhibitors of protein kinases. Curr Top Med Chem 2011;11(11):1320–32.
- Bertrand JA, Thieffine S, Vulpetti A, et al. Structural characterization of the GSK-3beta active site using selective and non-selective ATP-mimetic inhibitors. J Mol Biol 2003;333:393–407.
- Cornish-Bowden A. Why is uncompetitive inhibition so rare? A possible explanation, with implications for the design of drugs and pesticides. FEBS Lett 1986;203:3–6.