
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Many studies have been conducted on the selective inhibition of human monoamine oxidase B (hMAO-B) enzyme using benzylamine-sulphonamide derivatives. Using various chemical modifications on BB-4h, which was reported previously by our team and showed a significant level of MAO-B inhibition, novel benzylamine-sulphonamide derivatives were designed, synthesised, and their MAO inhibition potentials were evaluated. Among the tested derivatives, compounds 4i and 4t achieved IC50 values of 0.041 ± 0.001 µM and 0.065 ± 0.002 µM, respectively. The mechanism of hMAO-B inhibition by compounds 4i and 4t was studied using Lineweaver–Burk plot. The nature of inhibition was also determined to be non-competitive. Cytotoxicity tests were conducted and compounds 4i and 4t were found to be non-toxic. Molecular docking studies were also carried out for compound 4i, which was found as the most potent agent, within hMAO-B catalytic site.
1. Introduction
Monoamine oxidase (MAO) is the enzyme responsible for catalysing the oxidative deamination of intracellular amines and monoamine neurotransmitters, which contributes to the regulation of the concentrations of these chemicals in the brain and in peripheral tissuesCitation1,Citation2. MAOs, which are flavin adenine dinucleotide (FAD)-containing enzymes, are localised in the outer mitochondrial membranes of glial, neuronal, and other types of cells; they are particularly abundant in the liver and the brain. MAOs have two different isoforms, MAO-A and MAO-B, with 70% homology. The genes that code for the two isoforms are linked in opposite orientation on the X chromosome, differ in the specificity of their substrates and the selectivity of their inhibitorsCitation3. For example, MAO-B is selectively inhibited by selegiline, and utilises phenylethylamine and benzylamine as substrates. On the contrary, MAO-A is selectively inhibited by clorgiline, and utilises adrenaline, noradrenaline and serotonin as substrates. However, both isoforms may also act on the same substrates such as dopamine and tyramineCitation4.
MAOs are of extensive pharmacological importance due to their roles in the metabolism of certain neurotransmitters. Selective MAO-A inhibitors are used clinically as antidepressants and anxiolytics, while MAO-B inhibitors are used to reduce the progression of Parkinson’s disease, and manage symptoms related to Alzheimer’s diseaseCitation5. Moreover, MAO-catalyzed deamination reactions produce hydrogen peroxide as a byproduct, which may typically contribute to the oxidative stress state. In this context, MAO inhibitors are thought to act as neuroprotective agents in degenerative processesCitation6,Citation7.
Parkinson's disease (PD), which affects more than 5 million people worldwide, is the second most common disease after Alzheimer's disease. Considering the loss of nigrostriatal dopaminergic cells as a pathological hallmark of PD, therapeutic strategies have been established to boost the levels of dopamine in the brainCitation8–10. Although dopamine is metabolised by both MAO isoforms, MAO-B is the more common isoform present in the basal ganglia and is therefore responsible for dopamine metabolism in this regionCitation11.
Currently, the Protein Data Bank contains more than 40 crystal structures of MAO (most of them MAO-B) in complex with different reversible and irreversible inhibitors, as observed through X-ray diffraction at refinements of 3.0–1.7 Å. Additionally, MAO-A shows a markedly different monopartite cavity (∼550 Å) compared to the bipartite cavity (290 Å) found in MAO-B. The “aromatic cage”—a hydrophobic binding pocket containing the FAD cofactor—is considered the active regionCitation4,Citation7. The FAD is covalently attached to the cysteine residue of the protein, and the 8α-thioether linkage provides this connection. It is believed that the catalytic activity of the two tyrosine residues, Tyr398 and Tyr435, found in the hMAO-B structure is due to the polarisation of the amine N pair of the substrateCitation12. Therefore, in designing a new inhibitor compound, it is desirable to have the amine group in the structure.
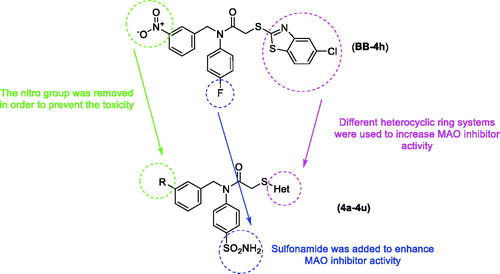
In light of the above-mentioned information, this study was conducted to develop new and potent MAO inhibitors. It has been thought that the proven MAO inhibition of benzylamine derivatives may provide MAO-B inhibitory activity due to strong interactions on the enzyme active sideCitation13–15. In our recent studyCitation2, we reported a new benzothiazole-benzylamine hybrid compound, 2-((5-chlorobenzothiazol-2-yl)thio)-N-(4-fluorophenyl)-N-(3-nitrobenzyl)acetamide (BB-4h), as shown in , with significant IC50 (2.95 ± 0.09 µM) against MAO-B. Moreover, sulphonamides and various heterocyclic ring systems have been identified as inhibitors of MAO in previous studiesCitation3,Citation16–19. Therefore, we considered the compound (BB-4h) as a lead compound, and we performed some modifications, such as removing nitro and fluoro groups, introducing a sulphonamide group, and changing heterocyclic rings in order to improve biological activity. Subsequently, 20 benzylamine derivatives containing a sulphonamide moiety and different heterocyclic ring moieties were synthesised, and their MAO inhibitory activities were evaluated in this study.
Figure 1. Design of the target compounds from compound BB-4h.
2. Experimental
2.1. Chemistry
All chemicals used in the synthesis studies were obtained from Merck Chemicals (Merck KGaA, Darmstadt, Germany) or Sigma-Aldrich Chemicals (Sigma-Aldrich Corp., St. Louis, MO). MP90 digital melting point apparatus (Mettler Toledo, Ohio) was used to determine the melting points of the resulting compounds and was presented uncorrected. A Bruker 300 MHz and 75 MHz digital FT-NMR spectrometer (Bruker Bioscience, Billerica, MA) in DMSO-d6, respectively recorded 1H NMR and 13 C NMR spectra. In the NMR spectra, splitting patterns were determined recognised as follows: s: singlet; d: doublet; t: triplet; dd: double doublet; td: triple doublet; br.s.: bronsted singlet; m: multiplet. Coupling constants (J) are reported in units of Hertz (Hz). IRAffinity-1S Fourier transform IR (FTIR) spectrometer (Shimadzu, Tokyo, Japan) was used to record the IR spectra of the compounds. Mass spectra were recorded on an LCMS-IT-TOF (Shimadzu, Kyoto, Japan) by means of ESI method. Silica gel 60 F254 by TLC (Merck KGaA, Darmstadt, Germany) was used to control the purity of the obtained compounds.
2.1.1. General procedure for the synthesis of the compounds
2.1.1.1. Synthesis of 4-(benzylideneamino)benzenesulfonamide and 4-((4-methylbenzylidene)amino)benzenesulfonamide (1a, 1b)
4-Aminobenzenesulfonamide (2.408 g, 0.014 mol) and benzaldehyde (1.427 ml, 0.014 mol) or 4-methylbenzaldehyde (1.680 g, 0.014 mol) were refluxed in EtOH (50 ml) for 48 h. Acetic acid was used as catalyst in this reaction. After completion of the reaction, the mixture was cooled, precipitated product was filtered and dried.
2.1.1.2. Synthesis of 4-(benzylamino)benzenesulfonamide and 4-((4-methylbenzyl)amino)benzenesulfonamide (2a, 2b)
4-(Benzylideneamino)benzenesulfonamide (1a) (3.328 g, 0.0128 mol) or 4-((4-methylbenzylidene)amino)benzenesulfonamide (1b) (3.507 g, 0.0128 mol) was dissolved in MeOH. Sodium borohydride was added to the reaction medium in portions of 0.5 moles. It was observed by controlling the end of the reaction with TLC that the reaction was complete when the total amount of sodium borohydride reached 1.5 moles. After completion of the reaction, the MeOH was removed by a rotavapor. The precipitated product was washed with deionised water to remove the excess of the sodium borohydride, dried, and recrystallized from EtOH.
2.1.1.3. Synthesis of N-Benzyl-2-chloro-N-(4-sulfamoylphenyl)acetamide and 2-chloro-N-(4-methylbenzyl)-N-(4-sulfamoylphenyl)acetamide (3a, 3b)
4-(Benzylamino)benzenesulfonamide (2a) (3.013 g, 0.0115 mol) or 4-((4-methylbenzyl)amino)benzenesulfonamide (2b) (3.174 g, 0.0115 mol) and triethylamine (TEA) (1.605 ml, 0.0115 mol) were dissolved in DMF (20 ml) and the reaction mixture was taken up in ice bath. The solution of chloroacetyl chloride (1.004 ml, 0.0126 mol) in DMF (10 ml) was added dropwise to the reaction mixture. After completion of the reaction, the mixture was poured into ice-water (50 ml), precipitated product was filtered, washed with deionised water, dried and recrystallized from EtOH.
2.1.1.4. General procedure for the synthesis of target compounds (4a–4u)
N-Benzyl-2-chloro-N-(4-sulfamoylphenyl)acetamide (3a) (0.305 g, 0.0009 mol) or 2-chloro-N-(4-methylbenzyl)-N-(4-sulfamoylphenyl)acetamide (3b) (0.317 g, 0.0009 mol), heterocyclic substituted thiol derivatives (0.0009 mol) and sufficient quantity of potassium carbonate (K2CO3) (0.193 g, 0.0014 mol) were reacted in acetone for 3 h. After completion of the reaction, acetone was removed under reduced pressure, the residue was washed with water, dried, and recrystallized from EtOH.
N-Benzyl-2-((1-methyl-1H-imidazol-2-yl)thio)-N-(4-sulfamoylphenyl)acetamide (4a)
Yield: 85%, M.P. = 137–139 °C, FTIR (ATR, cm−1): 3340 (N–H), 2941 (C–H), 1666 (C = O), 709, 850. 1H-NMR (300 MHz, DMSO-d6): δ = 3.55 (3H, s, –CH3), 3.81 (2H, s, –CH2–), 4.92 (2H, s, -CH2), 6.90 (1H, s, Imidazole CH), 7.17–7.31 (6H, m, Imidazole CH, Monosubstitutedbenzene), 7.39 (2H, d, J = 8.3 Hz, 1,4-Disubstitutedbenzene), 7.78 (2H, d, J = 8.3 Hz, 1,4-disubstitutedbenzene). 13 C-NMR (75 MHz, DMSO-d6): δ = 33.4, 37.9, 52.8, 123.8, 127.3, 127.7, 128.2, 128.8, 128.9, 129.1, 137.2, 139.7, 144.2, 144.5, 167.6. HRMS (m/z): [M + H]+ calcd for C19H20N4O3S2: 417.1055; found: 417.1040
N-Benzyl-2-((4-methyl-4H-1,2,4-triazol-3-yl)thio)-N-(4-sulfamoylphenyl)acetamide (4b)
Yield: 88%, M.P. = 201–203 °C, FTIR (ATR, cm−1): 3296 (N-H), 2976 (C–H), 1654 (C = O), 709, 852. 1H-NMR (300 MHz, DMSO-d6): δ = 3.55 (3H, s, –CH3), 4.01 (2H, s, –CH2–), 4.94 (2H, s, –CH2–), 7.18–7.31 (5H, m, Monosubstitutedbenzene), 7.42 (2H, s, –SO2NH2), 7.49 (2H, d, J = 8.5 Hz, 1,4-Disubstitutedbenzene), 7.81 (2H, d, J = 8.5 Hz, 1,4-Disubstitutedbenzene), 8.50 (1H, s, Triazole CH). 13 C-NMR (75 MHz, DMSO-d6): δ = 31.2, 37.8, 52.9, 127.4, 127.8, 128.3, 128.9, 129.1, 137.1, 143.8, 144.5, 146.5, 149.1, 167.0. HRMS (m/z): [M + H]+ calcd for C18H19N5O3S2: 418.1002; found: 418.1001
N-Benzyl-2-((5-methyl-1,3,4-thiadiazol-2-yl)thio)-N-(4-sulfamoylphenyl)acetamide (4c)
Yield: 82%, M.P. = 164–166 °C, FTIR (ATR, cm−1): 3334 (N-H), 3047 (C-H), 1654 (C = O), 698, 740, 856. 1H-NMR (300 MHz, DMSO-d6): δ = 2.66 (3H, s, –CH3), 4.18 (2H, s, –CH2–), 4.97 (2H, s, –CH2–), 7.21–7.32 (5H, m, Monosubstitutedbenzene), 7.42 (2H, s, –SO2NH2), 7.53 (2H, d, J = 8.5 Hz, 1,4-Disubstitutedbenzene), 7.83 (2H, d, J = 8.5 Hz, 1,4-Disubstitutedbenzene). 13 C-NMR (75 MHz, DMSO-d6): δ = 15.6, 38.3, 53.0, 127.4, 127.8, 128.3, 128.9, 129.1, 137.1, 143.9, 144.5, 164.6, 166.0, 166.6. HRMS (m/z): [M + H]+ calcd for C18H18N4O3S3: 435.0614; found: 435.0622
N-Benzyl-2-((1-methyl-1H-tetrazol-5-yl)thio)-N-(4-sulfamoylphenyl)acetamide (4d)
Yield: 86%, M.P. = 124–127 °C, FTIR (ATR, cm−1): 3305 (N-H), 2931 (C–H), 1651 (C = O), 702, 734, 848. 1H-NMR (300 MHz, DMSO-d6): δ = 3.95 (3H, s, –CH3), 4.19 (2H, s, –CH2–), 4.96 (2H, s, –CH2–), 7.20–7.32 (7H, m, Monosubstitutedbenzene, –SO2NH2), 7.51 (2H, d, J = 8.4 Hz, 1,4-Disubstitutedbenzene), 7.84 (2H, d, J = 8.4 Hz, 1,4-Disubstitutedbenzene). 13 C-NMR (75 MHz, DMSO-d6): δ = 34.1, 38.3, 53.0, 127.5, 127.8, 128.3, 128.9, 129.0, 137.0, 144.1, 144.6, 153.8, 166.5. HRMS (m/z): [M + H]+ calcd for C17H18N6O3S2: 419.0955; found: 419.0956
N-Benzyl-2-((1-phenyl-1H-tetrazol-5-yl)thio)-N-(4-sulfamoylphenyl)acetamide (4e)
Yield: 81%, M.P. = 138–140 °C, FTIR (ATR, cm−1): 3356 (N-H), 2949 (C–H), 1651 (C = O), 734, 759, 848. 1H-NMR (300 MHz, DMSO-d6): δ = 4.27 (2H, s, –CH2–), 4.95 (2H, s, –CH2–), 7.21–7.32 (5H, m, Monosubstitutedbenzene), 7.51 (2H, d, J = 8.4 Hz, 1,4-Disubstitutedbenzene), 7.67 (7H, br.s., Monosubstitutedbenzene, -SO2NH2), 7.84 (2H, d, J = 8.4 Hz, 1,4-Disubstitutedbenzene). 13 C-NMR (75 MHz, DMSO-d6): δ = 38.5, 53.0, 124.9, 124.9, 127.5, 127.8, 128.3, 128.9, 130.4, 130.6, 130.7, 131.2, 133.5, 137.0, 154.3, 166.3. HRMS (m/z): [M + H]+ calcd for C22H20N6O3S2: 481.1111; found: 481.1096
N-Benzyl-2-(pyridin-2-ylthio)-N-(4-sulfamoylphenyl)acetamide (4f)
Yield: 87%, M.P. = 106–108 °C, FTIR (ATR, cm−1): 3402 (N-H), 2929 (C-H), 1651 (C = O), 698, 763, 854. 1H-NMR (300 MHz, DMSO-d6): δ = 3.94 (2H, s, –CH2–), 4.96 (2H, s, –CH2–), 7.11 (1H, t, J = 5.9 Hz, Pyridine CH), 7.21–7.31 (6H, m, Monosubstitutedbenzene, Pyridine CH), 7.41 (2H, s, –SO2NH2), 7.52 (2H, d, J = 8.4 Hz, 1,4-Disubstitutedbenzene), 7.62 (1H, t, J = 8.5, Pyridine CH), 7.83 (2H, d, J = 8.4 Hz, 1,4-Disubstitutedbenzene), 8.36 (1H, d, J = 4.3 Hz, Pyridine CH). 13 C-NMR (75 MHz, DMSO-d6): δ = 33.4, 52.9, 120.4, 122.0, 127.4, 127.7, 128.2, 128.9, 129.1, 137.1, 137.5, 143.6, 145.2, 149.7, 157.5, 168.1. HRMS (m/z): [M + H]+ calcd for C20H19N3O3S2: 414.0941; found: 414.0950
2-(Benzoxazol-2-ylthio)-N-benzyl-N-(4-sulfamoylphenyl)acetamide (4g)
Yield: 80%, M.P. = 82–84 °C, FTIR (ATR, cm−1): 3363 (N-H), 2985 (C-H), 1651 (C = O), 704, 744, 846. 1H-NMR (300 MHz, DMSO-d6): δ = 4.25 (2H, s, –CH2–), 4.99 (2H, s, –CH2–), 7.26–7.29 (5H, m, Monosubstitutedbenzene), 7.31–7.35 (2H, m, Benzoxazole CH), 7.45 (2H, s, –SO2NH2), 7.58–7.64 (4H, m, 1,4-Disubstitutedbenzene, Benzoxazole CH), 7.88 (2H, d, J = 8.4, 1,4-Disubstitutedbenzene), 13 C-NMR (75 MHz, DMSO-d6): δ = 36.9, 53.2, 110.7, 118.7, 124.8, 125.1, 127.6, 127.8, 128.2, 128.9, 129.2, 137.1, 141.6, 144.0, 144.6, 151.7, 164.3, 166.6. HRMS (m/z): [M + H]+ calcd for C22H19N3O4S2: 454.0890; found: 454.0880
2-(Benzothiazol-2-ylthio)-N-benzyl-N-(4-sulfamoylphenyl)acetamide (4h)
Yield: 79%, M.P. = 88–90 °C, FTIR (ATR, cm−1): 3352 (N–H), 2941 (C–H), 1651 (C = O), 702, 758, 846. 1H-NMR (300 MHz, DMSO-d6): δ = 4.24 (2H, s, –CH2–), 4.99 (2H, s, –CH2–), 7.24 (5H, br.s., Monosubstitutedbenzene), 7.37 (1H, td, J1=1.0 Hz, J2=7.7 Hz, Benzothiazole CH) 7.44 (2H, s, -SO2NH2), 7.49 (1H, td, J1=1.1 Hz, J2=7.7 Hz, Benzothiazole CH), 7.62 (2H, d, J = 8.3 Hz, 1,4-Disubstitutedbenzene), 7.83 (1H, d, J = 8.0, Benzothiazole CH), 7.88 (2H, d, J = 8.4 Hz, 1,4-Disubstitutedbenzene), 8.01 (1H, d, J = 7.6 Hz, Benzothiazole CH). 13 C-NMR (75 MHz, DMSO-d6): δ = 37.2, 53.1, 121.6, 122.4, 125.0, 126.8, 127.5, 127.8, 128.3, 128.9, 129.1, 135.2, 137.2, 143.8, 144.8, 152.9, 166.3, 166.8. HRMS (m/z): [M + H]+ calcd for C22H19N3O3S3: 470.0661; found: 470.0652
N-Benzyl-2-((5-chlorobenzothiazol-2-yl)thio)-N-(4-sulfamoylphenyl)acetamide (4i)
Yield: 85%, M.P. = 114–116 °C, FTIR (ATR, cm−1): 3473 (N-H), 2995 (C–H), 1645 (C = O), 702, 732, 846. 1H-NMR (300 MHz, DMSO-d6): δ = 4.22 (2H, s, –CH2–), 4.98 (2H, s, –CH2–), 7.24 (5H, br.s., Monosubstitutedbenzene), 7.41 (1H, d, J = 1.8 Hz, Benzothiazole CH) 7.44 (2H, s, –SO2NH2), 7.62 (2H, d, J = 8.3 Hz, 1,4-Disubstitutedbenzene), 7.88–7.90 (3H, m, Benzothiazole CH, 1,4-Disubstitutedbenzene), 8.04 (1H, d, J = 8.6 Hz, Benzothiazole CH). 13 C-NMR (75 MHz, DMSO-d6): δ = 37.4, 53.1, 121.0, 123.8, 125.0, 127.6, 127.8, 128.3, 128.8, 129.2, 131.7, 134.1, 137.3, 143.9, 144.7, 153.7, 166.6, 169.2. HRMS (m/z): [M + H]+ calcd for C22H18 ClN3O3S3: 504.0272; found: 504.0250
N-Benzyl-2-((5-methoxybenzothiazol-2-yl)thio)-N-(4-sulfamoylphenyl)acetamide (4j)
Yield: 80%, M.P. = 170–172 °C, FTIR (ATR, cm−1): 3483 (N-H), 2939 (C-H), 1651 (C = O), 702, 734, 842. 1H-NMR (300 MHz, DMSO-d6): δ = 3.84 (3H, s, –OCH3), 4.23 (2H, s, –CH2–), 4.99 (2H, s, –CH2–), 7.01 (1H, dd, J1=2.5 Hz, J2=8.8 Hz, Benzothiazole CH), 7.25 (5H, br.s., Monosubstitutedbenzene), 7.33 (1H, d, J = 2.5 Hz, Benzothiazole CH) 7.43 (2H, br.s, -SO2NH2), 7.59 (2H, d, J = 8.4 Hz, 1,4-Disubstitutedbenzene), 7.86 (1H, d, J = 8.7, Benzothiazole CH), 7.88 (2H, d, J = 8.2 Hz, 1,4-Disubstitutedbenzene). 13 C-NMR (75 MHz, DMSO-d6): δ = 37.3, 53.1, 56.0, 105.0, 114.2, 122.6, 126.8, 127.5, 127.8, 128.2, 128.9, 129.1, 137.2, 143.9, 144.7, 154.2, 159.2, 166.8, 167.2. HRMS (m/z): [M + H]+ calcd for C23H21N3O4S3: 500.0767; found: 500.0761
2-((1-Methyl-1H-imidazol-2-yl)thio)-N-(4-methylbenzyl)-N-(4-sulfamoylphenyl)acetamide (4k)
Yield: 86%, M.P. = 153–155 °C, FTIR (ATR, cm−1): 3313 (N–H), 2920 (C–H), 1651 (C = O), 840. 1H-NMR (300 MHz, DMSO-d6): δ = 2.25 (3H, s, –CH3), 3.56 (3H, s, –CH3), 3.80 (2H, s, –CH2–), 4.87 (2H, s, –CH2–), 6.90 (1H, d, J = 1.1 Hz, Imidazole CH), 7.04–7.10 (4H, m, Methylbenzene), 7.21 (1H, d, J = 1.1 Hz, Imidazole CH), 7.37–7.41 (4H, m, –SO2NH2, 1,4-Disubstitutedbenzene), 7.79 (2H, d, J = 8.5 Hz, 1,4-Disubstitutedbenzene). 13 C-NMR (75 MHz, DMSO-d6): δ = 21.1, 33.4, 37.9, 52.5, 123.8, 127.3, 128.3, 128.9, 129.0, 129.44, 134.1, 136.9, 140.0, 143.6, 144.7, 167.5. HRMS (m/z): [M + H]+ calcd for C20H22N4O3S2: 431.1206; found: 431.1199
2-((4-Methyl-4H-1,2,4-triazol-3-yl)thio)-N-(4-methylbenzyl)-N-(4-sulfamoylphenyl)acetamide (4l)
Yield: 84%, M.P. = 229–231 °C, FTIR (ATR, cm−1): 3302 (N-H), 3049 (C–H), 1654 (C = O), 852. 1H-NMR (300 MHz, DMSO-d6): δ = 2.25 (3H, s, -CH3), 3.55 (3H, s, –CH3), 4.00 (2H, s, –CH2–), 4.90 (2H, s, -CH2-), 7.08 (4H, br.s., Methylbenzene), 7.35 (2H, br.s, -SO2NH2), 7.46 (2H, d, J = 8.5 Hz, 1,4-Disubstitutedbenzene), 7.81 (2H, d, J = 8.5 Hz, 1,4-Disubstitutedbenzene), 8.51 (1H, s, Triazole CH). 13 C-NMR (75 MHz, DMSO-d6): δ = 21.1, 31.2, 37.9, 52.6, 127.4, 128.3, 129.0, 129.5, 134.0, 136.9, 144.1, 144.4, 146.5, 149.1, 167.0. HRMS (m/z): [M + H]+ calcd for C19H21N5O3S2: 432.1159; found: 432.1161
2-((5-Methyl-1,3,4-thiadiazol-2-yl)thio)-N-(4-methylbenzyl)-N-(4-sulfamoylphenyl)acetamide (4m)
Yield: 79%, M.P. = 178–180 °C, FTIR (ATR, cm−1): 3288 (N-H), 3070 (C-H), 1651 (C = O), 850. 1H-NMR (300 MHz, DMSO-d6): δ = 2.25 (3H, s, –CH3), 2.66 (3H, s, –CH3), 4.16 (2H, s, –CH2–), 4.92 (2H, s, –CH2–), 7.09 (4H, br.s., Methylbenzene), 7.43 (2H, s, –SO2NH2), 7.51 (2H, d, J = 8.5 Hz, 1,4-Disubstitutedbenzene), 7.83 (2H, d, J = 8.5 Hz, 1,4-Disubstitutedbenzene). 13 C-NMR (75 MHz, DMSO-d6): δ = 15.6, 21.1, 38.3, 52.7, 127.4, 128.3, 129.1, 129.5, 134.0, 136.9, 143.9, 144.5, 164.6, 166.0, 166.6. HRMS (m/z): [M + H]+ calcd for C19H20N4O3S3: 449.0770; found: 449.0750
2-((1-Methyl-1H-tetrazol-5-yl)thio)-N-(4-methylbenzyl)-N-(4-sulfamoylphenyl)acetamide (4n)
Yield: 80%, M.P. = 154–156 °C, FTIR (ATR, cm−1): 3338 (N-H), 2947 (C–H), 1649 (C = O), 850. 1H-NMR (300 MHz, DMSO-d6): δ = 2.25 (3H, s, –CH3), 3.95 (3H, s, –CH3), 4.19 (2H, s, –CH2–), 4.91 (2H, s, –CH2–), 7.09 (4H, br.s., Methylbenzene), 7.33 (2H, br.s, –SO2NH2), 7.50 (2H, d, J = 8.4 Hz, 1,4-Disubstitutedbenzene), 7.84 (2H, d, J = 8.4 Hz, 1,4-Disubstitutedbenzene). 13 C-NMR (75 MHz, DMSO-d6): δ = 21.1, 34.1, 38.4, 52.7, 127.5, 128.3, 129.1, 129.5, 133.9, 137.0, 144.1, 144.4, 153.8, 166.4. HRMS (m/z): [M + H]+ calcd for C18H20N6O3S2: 433.1111; found: 433.1106
N-(4-Methylbenzyl)-2-((1-phenyl-1H-tetrazol-5-yl)thio)-N-(4-sulfamoylphenyl)acetamide (4o)
Yield: 83%, M.P. = 87–90 °C, FTIR (ATR, cm−1): 3294 (N-H), 2924 (C–H), 1651 (C = O), 848. 1H-NMR (300 MHz, DMSO-d6): δ = 2.25 (3H, s, –CH3), 4.27 (2H, s, –CH2–), 4.92 (2H, s, –CH2–), 7.09 (4H, br.s., Methylbenzene), 7.46 (2H, br.s., –SO2NH2), 7.53 (2H, d, J = 8.4 Hz, 1,4-Disubstitutedbenzene), 7.67 (5H, br.s., Monosubstitutedbenzene), 7.86 (2H, d, J = 8.4 Hz, 1,4-Disubstitutedbenzene). 13 C-NMR (75 MHz, DMSO-d6): δ = 21.1, 38.5, 52.7, 124.9, 127.5, 128.3, 129.2, 129.5, 130.6, 131.1, 133.5, 133.9, 137.0, 144.0, 144.3, 154.3, 166.2. HRMS (m/z): [M + H]+ calcd for C23H22N6O3S2: 495.1268; found: 495.1243
N-(4-Methylbenzyl)-2-(pyridin-2-ylthio)-N-(4-sulfamoylphenyl)acetamide (4p)
Yield: 77%, M.P. = 121–123 °C, FTIR (ATR, cm−1): 3408 (N-H), 2918 (C-H), 1645 (C = O), 848. 1H-NMR (300 MHz, DMSO-d6): δ = 2.25 (3H, s, –CH3), 3.93 (2H, s, -CH2-), 4.91 (2H, s, –CH2–), 7.09–7.13 (5H, m, Methylbenzene, Pyridine CH), 7.29 (1H, d, J = 8.1 Hz, Pyridine CH), 7.50 (2H, d, J = 8.4 Hz, 1,4-Disubstitutedbenzene), 7.62 (1H, td, J1=1.7 Hz, J2=7.7 Hz, Pyridine CH), 7.83 (2H, d, J = 8.4 Hz, 1,4-Disubstitutedbenzene). 8.36 (1H, d, J = 4.3 Hz, Pyridine CH). 13 C-NMR (75 MHz, DMSO-d6): δ = 21.1, 33.5, 52.6, 120.4, 122.0, 127.3, 128.2, 129.1, 129.4, 134.4, 136.8, 137.1, 143.8, 145.1, 149.7, 157.5, 168.0. HRMS (m/z): [M + H]+ calcd for C21H21N3O3S2: 428.1097; found: 428.1092
2-(Benzoxazol-2-ylthio)-N-(4-methylbenzyl)-N-(4-sulfamoylphenyl)acetamide (4r)
Yield: 85%, M.P. = 148–150 °C, FTIR (ATR, cm−1): 3388 (N-H), 2933 (C–H), 1666 (C = O), 856. 1H-NMR (300 MHz, DMSO-d6): δ = 2.25 (3H, s, –CH3), 4.24 (2H, s, –CH2–), 4.93 (2H, s, –CH2–), 7.07–7.11 (4H, m, Methylbenzene), 7.32–7.35 (2H, m, Benzoxazole CH), 7.55 (2H, d, J = 8.3 Hz, 1,4-Disubstitutedbenzene), 7.61–7.64 (2H, m, Benzoxazole CH), 7.88 (2H, d, J = 8.3 Hz, 1,4-Disubstitutedbenzene). 13 C-NMR (75 MHz, DMSO-d6): δ = 21.1, 37.0, 52.9, 110.7, 118.7, 124.8, 125.1, 127.5, 128.3, 129.0, 129.5, 134.1, 136.9, 141.6, 144.1, 145.0, 151.7, 164.3, 166.5. HRMS (m/z): [M + H]+ calcd for C23H21N3O4S2: 468.1046; found: 468.1030
2-(Benzothiazol-2-ylthio)-N-(4-methylbenzyl)-N-(4-sulfamoylphenyl)acetamide (4s)
Yield: 88%, M.P. = 170–172 °C, FTIR (ATR, cm−1): 3238 (N-H), 2918 (C-H), 1651 (C = O), 850. 1H-NMR (300 MHz, DMSO-d6): δ = 2.24 (3H, s, –CH3), 4.22 (2H, s, –CH2–), 4.93 (2H, s, –CH2–), 7.04–7.12 (4H, m, Methylbenzene), 7.37 (1H, td, J1=1.0 Hz, J2=7.7 Hz, Benzothiazole CH), 7.48 (1H, td, J1=1.1 Hz, J2=7.7 Hz, Benzothiazole CH), 7.56 (2H, d, J = 8.4 Hz, 1,4-Disubstitutedbenzene), 7.82 (1H, d, J = 8.0 Hz, Benzothiazole CH), 7.87 (2H, d, J = 8.4 Hz, 1,4-Disubstitutedbenzene), 8.00 (1H, d, J = 7.9 Hz, Benzothiazole CH). 13 C-NMR (75 MHz, DMSO-d6): δ = 21.1, 37.3, 52.8, 121.6, 122.3, 125.0, 126.8, 127.4, 128.2, 129.1, 129.4, 134.2, 135.2, 136.9, 144.4, 144.5, 152.9, 166.3, 166.7. HRMS (m/z): [M + H]+ calcd for C23H21N3O3S3: 484.0818; found: 484.0796
2-((5-Chlorobenzothiazol-2-yl)thio)-N-(4-methylbenzyl)-N-(4-sulfamoylphenyl)acetamide (4t)
Yield: 79%, M.P. = 109–111 °C, FTIR (ATR, cm−1): 3360 (N-H), 2927 (C-H), 1664 (C = O), 850. 1H-NMR (300 MHz, DMSO-d6): δ = 2.25 (3H, s, –CH3), 4.20 (2H, s, –CH2–), 4.94 (2H, s, –CH2–), 7.04–7.11 (4H, m, Methylbenzene), 7.40–7.43 (3H, m, -SO2NH2, Benzothiazole CH) 7.60 (2H, d, J = 8.3 Hz, 1,4-Disubstitutedbenzene), 7.85 (1H, d, J = 2.0 Hz, Benzothiazole CH), 7.90 (2H, d, J = 8.4 Hz, 1,4-Disubstitutedbenzene), 8.03 (1H, d, J = 8.6, Benzothiazole CH). 13 C-NMR (75 MHz, DMSO-d6): δ = 21.1, 37.4, 52.8, 121.1, 123.7, 125.0, 127.6, 128.3, 129.3, 129.4, 131.7, 134.1, 134.2, 136.9, 144.0, 144.7, 153.7, 166.5, 169.2. HRMS (m/z): [M + H]+ calcd for C23H20 ClN3O3S3: 518.0428; found: 518.0393
2-((5-Methoxybenzothiazol-2-yl)thio)-N-(4-methylbenzyl)-N-(4-sulfamoylphenyl)acetamide (4u)
Yield: 78%, M.P. = 82–84 °C, FTIR (ATR, cm−1): 3344 (N-H), 2933 (C-H), 1651 (C = O), 844. 1H-NMR (300 MHz, DMSO-d6): δ = 2.25 (3H, s, –CH3), 3.84 (3H, s, –OCH3), 3.84 (3H, s, –OCH3), 4.21 (2H, s, -CH2-), 4.94 (2H, s, -CH2-), 7.01 (1H, dd, J1=2.5 Hz, J2=8.8 Hz, Benzothiazole CH), 7.05–7.12 (4H, m, Methylbenzene), 7.32 (1H, d, J = 2.4 Hz, Benzothiazole CH), 7.43 (2H, br.s, –SO2NH2), 7.57 (2H, d, J = 8.4 Hz, 1,4-Disubstitutedbenzene), 7.84–7.89 (3H, m, Benzothiazole CH, 1,4-Disubstitutedbenzene). 13 C-NMR (75 MHz, DMSO-d6): δ = 21.1, 37.3, 52.8, 56.0, 105.0, 114.2, 122.6, 126.8, 127.5, 128.3, 129.2, 129.5, 134.2, 136.9, 143.8, 144.7, 154.2, 159.1, 166.7, 167.2. HRMS (m/z): [M + H]+ calcd for C24H23N3O4S3: 514.0923; found: 514.0897.
2.2. MAO-A and MAO-B inhibition assay
Ampliflu™ Red (10-Acetyl-3,7-dihydroxyphenoxazine), hMAO-A, hMAO-B, peroxidase from horseradish, tyramine hydrochloride, H2O2, clorgiline and selegiline were acquired from Sigma-Aldrich (Steinheim, Germany) and retained under the proposed conditions by supplier. A Biotek Precision XS robotic system (USA) was used for all pipetting operations. Measurements were performed with the use of BioTek-Synergy H1 microplate reader (USA) based upon the fluorescence generated (excitation, 535 nm, emission, 587 nm) over a 30 min period, in which the fluorescence increased linearly.
Enzymatic assay was performed according to recent method pronounced by our research groupCitation17,Citation20–22. Control, blank and all concentrations of obtained compounds were tested in quadruplicate and inhibition percent was calculated with following equation:
FCt2: Fluorescence of a control well measured at t2 time, FCt1: Fluorescence of a control well measured at t2 time, FIt2: Fluorescence of an inhibitor well measured at t2 time, FIt1: Fluorescence of an inhibitor well measured at t1 time,
The IC50 values were calculated using a dose-response curve achieved by plotting the percentage inhibition versus the log concentration using GraphPad ‘PRISM’ software (version 5.0). The results were showed as mean ± SD.
2.3. Enzyme kinetic studies
The same materials were used in the MAO inhibition assay. The most active compounds 4i and 4t determined according to the result of the MAO inhibition assay were experienced in three different concentrations of IC50/2, IC50 and 2(IC50) in accordance with the assay assigned in our final study.Citation17,Citation20–22. All processes were evaluated in quadruplicate. The results were analysed as Lineweaver–Burk plots by means of Microsoft Office Excel 2013. The Vmax values of the Lineweaver–Burk plots were replotted versus the inhibitor concentration, and the Ki values were determined from the x-axis intercept as −Ki.
2.4. Cytotoxicity assay
The NIH/3T3 mouse embryonic fibroblast cell line (ATCC®CRL-1658 ™, London, UK) was used for cytotoxicity assays. The incubation period of NIH/3T3 cells was based on the supplier's recommendation. NIH/3T3 cells were seeded at 1 × 104 cells into each well of 96-well plates. MTT assay was carried out in accordance with the standards previously described mannerCitation23,Citation24. The compounds were tested between 1 and 0.000316 mM concentrations. Inhibition % for each concentration was calculated according to the following formula and IC50 values were reported by plotting the% inhibition dose response curve against the compound concentrations tested.Citation23–25.
2.5. Prediction of ADME parameters and BBB permeability
Physicochemical parameters were performed with the use of QikProp 4.8 softwareCitation26 to predict pharmacokinetic profiles and BBB permeability of obtained compounds (4a–4u).
2.6. Molecular docking
A structure based in silico procedure was applied to discover the binding modes of compound 4i to hMAO-B enzyme active site. The crystal structures of hMAO-B (PDB ID: 2V5Z)Citation27, which was crystallised with safinamide, were retrieved from the Protein Data Bank server (www.pdb.org).
The structures of ligands were built using the Schrödinger MaestroCitation28 interface and then were submitted to the Protein Preparation Wizard protocol of the Schrödinger Suite 2016 Update 2Citation29. The ligands were prepared by the LigPrep 3.8Citation30 to assign the protonation states at pH 7.4 ± 1.0 and the atom types, correctly. Bond orders were assigned, and hydrogen atoms were added to the structures. The grid generation was formed using Glide 7.1Citation31. The grid box with dimensions of 20 Å × 20 Å × 20 Å was centred in the vicinity of the flavin (FAD) N5 atom on the catalytic site of the protein to cover all binding sites and neighbouring residuesCitation32–34. Flexible docking runs were performed with single precision docking mode (SP).
3. Result and discussion
3.1. Chemistry
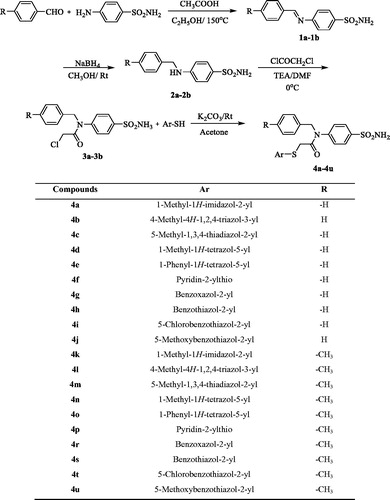
Various compounds, labelled 4a to 4u, were synthesised as outlined in Scheme 1. Initially, Schiff bases were prepared through the reaction of benzaldehyde (or 4-methylbenzaldehyde) and sulphanilamide. Then, benzylamine derivatives (2a, 2b) were obtained by a reduction reaction. Acetylation of the benzylamine derivatives (2a, 2b) gave compounds 3a and 3b. Finally, the target compounds (4a–4u) were obtained through a substitution reaction using acetylated benzylamine (3a, 3b) and corresponding heterocyclic thiols.
Scheme 1. Synthesis way of the compounds 4a–4u.
The synthesised compounds were elucidated by instrumental analyses such as infra-red spectroscopy (IR), mass spectrometry (MS), and nuclear magnetic resonance (NMR). The N-H bond of the sulphonamide group appeared as IR bands between 3238 and 3483 cm−1, and as a singlet between 7.30 and 7.60 ppm on the 1H-NMR spectrum. The presence of the carbonyl group was shown by IR bands between 1645 cm−1 and 1666 cm−1, and a 13 C-NMR peak over 160 ppm. The CH2 group bound to the nitrogen atom gave a singlet 1H-NMR peak around 4.90 ppm, and a 13 C-NMR peak over 50 ppm. The other CH2 group between the carbonyl and sulphur groups was recorded in 1H-NMR peak around 4.20 ppm and a 13 C-NMR peak between 33.4 and 38.5 ppm. The carbons of aromatic groups were observed from 105.0 to 166.7 ppm in the 13 C-NMR spectrum, and the protons of the same groups were between 6.90 and 8.51 ppm in the 1H-NMR spectrum. In mass spectroscopy, the masses were found to differ by at most 5 ppm from the expected masses.
3.2. MAO inhibition
All the obtained benzylamine-sulphonamide derivatives 4a–4u were investigated for their inhibitory activity against MAO isoforms using a previously described in vitro fluorometric method, which is based on the detection of H2O2 in a horseradish peroxidase-coupled reaction using 10-acetyl-3,7-dihydroxyphenoxazine (Amplex Red reagent)Citation17,Citation20–22.
The assay was carried out in two steps. The first step was carried out using 10−3 and 10−4 M concentrations of all synthesised compounds and reference agents, namely selegiline and clorgiline. The enzyme activity results of first step are presented in . Then, the selected compounds that displayed more than 50% inhibitory activity at 10−3 and 10−4 M concentrations were further tested, along with reference agents, at concentrations of 10−5 to 10−9 M. The IC50 values of the test compounds and reference agents are presented in .
Table 1. % Inhibition of the synthesized compounds, clorgiline and selegiline against MAO-A and MAO-B enzymes.
Table 2. IC50 values of 4b, 4d, 4f, 4i, 4t and selegiline against MAO-B.
According to the enzyme inhibition results, none of the synthesised compounds showed a significant activity against hMAO-A enzyme. All of the obtained compounds displayed selective inhibition on hMAO-B. At 1 0 −3 M concentration, all of the compounds showed more than 50% inhibitory activity. Compounds 4b, 4d, 4f, 4i and 4t could pass the second step of enzyme activity assay and the IC50 values of them were calculated by performing enzyme inhibition study at 10−5–10−9 M concentration. The most active compounds, 4i and 4t, exhibited IC50 values of 0.041 ± 0.001 µM and 0.065 ± 0.002 µM, respectively, against hMAO-B, while the reference agent, selegiline, had an IC50 of 0.037 ± 0.001 µM.
These findings from the screening of inhibitory activities against hMAO-B revealed that the compounds containing 5-chlorobenzothiazole exhibited more potent inhibitory activity than the other obtained compounds as in the previously synthesised and reported BB-4h derivative, which has a 5-chlorobenzothiazole ring. Moreover, the increased inhibitory activity of the synthesised compounds, compared to that of BB-4h, is likely due to the contribution of the sulphonamide group, which displaced the fluorine group, and the removal of the nitro group from the structure.
3.3. Kinetic studies of enzyme inhibition
Enzyme kinetics studies were performed to determine the mechanism of hMAO-B inhibition by using a procedure similar to that of the MAO inhibition assay. Compounds 4i and 4t, which were found to be the most potent agents, were included in these studies. In order to estimate the type of inhibition of these compounds, linear Lineweaver-Burk graphs were used. Substrate velocity curves in the absence and presence of compounds 4i and 4t were recorded. These compounds were prepared at concentrations of IC50/2, IC50, and 2(IC50) for enzyme kinetic studies. In each case, the initial velocity measurements were obtained at different substrate (tyramine) concentrations ranging from 20 μM to 0.625 μM. The secondary plots of slope (Km/Vmax) versus varying concentrations (0, IC50/2, IC50, and 2(IC50)) were created to calculate the Ki (intercept on the x-axis) value of these compounds. The graphical analyses of steady-state inhibition data for compounds 4i and 4t are shown in and .
Figure 2. (A) Lineweaver–Burk plots for the inhibition of hMAO-B by compound 4i. [S], substrate concentration (µM); V, reaction velocity (nmol/min/mg protein). Inhibitor concentrations are shown at the left. (B) Secondary plot for the calculation of the steady-state inhibition constant (Ki) of compound 4i. Ki was calculated as 0.036 µM.
![Figure 2. (A) Lineweaver–Burk plots for the inhibition of hMAO-B by compound 4i. [S], substrate concentration (µM); V, reaction velocity (nmol/min/mg protein). Inhibitor concentrations are shown at the left. (B) Secondary plot for the calculation of the steady-state inhibition constant (Ki) of compound 4i. Ki was calculated as 0.036 µM.](/cms/asset/f2c715c7-2978-431a-8bfa-b07d39320f6b/ienz_a_1784892_f0002_c.jpg)
Figure 3. (A) Lineweaver–Burk plots for the inhibition of hMAO-B by compound 4t. [S], substrate concentration (µM); V, reaction velocity (nmol/min/mg protein). Inhibitor concentrations are shown at the left. (B) Secondary plot for the calculation of the steady-state inhibition constant (Ki) of compound 4t. Ki was calculated as 0.055 µM.
![Figure 3. (A) Lineweaver–Burk plots for the inhibition of hMAO-B by compound 4t. [S], substrate concentration (µM); V, reaction velocity (nmol/min/mg protein). Inhibitor concentrations are shown at the left. (B) Secondary plot for the calculation of the steady-state inhibition constant (Ki) of compound 4t. Ki was calculated as 0.055 µM.](/cms/asset/e082dd96-a55d-40a7-a002-7c81cb7283d5/ienz_a_1784892_f0003_c.jpg)
The type of inhibition can be determined as either reversible or irreversible by using the Lineweaver-Burk plots. The reversible inhibition type can be classified as mixed-type, uncompetitive, competitive, or non-competitiveCitation17,Citation20–22. According to Lineweaver–Burk plots, a graph that shows parallel lines without any cross-overs is observed in the uncompetitive type of inhibition. For mixed-type inhibition, a graph with lines that do not intersect at the x-axis or the y-axis is formed. Competitive inhibition is seen if the lines intersect on the y-axis, and the slopes and x-intercepts are different. On the contrary, non-competitive inhibition has the opposite result: the plotted lines have the same x-intercept but there are diverse slopes and y-intercepts. Therefore, as shown in and , compounds 4i and 4t are reversible and non-competitive inhibitors with similar inhibition features as the substrates. Ki values for compounds 4i and 4t were calculated as 0.036 and 0.055 μM, respectively, for the inhibition of hMAO-B.
Irreversible enzymatic inhibition involves covalent interactions between the substrate and the enzyme. In contrast, there are non-covalent interactions such as hydrophobic interactions, ionic bonds, and hydrogen bonds involved in reversible inhibition. In this type of inhibition, inhibitors bind to the enzymes without forming any chemical bonds; thus, the enzyme-inhibitor complex could be separated quickly because non-covalent interactions can form rapidly and break easily. Furthermore, reversible inhibitors have a lower risk of side effects than irreversible inhibitors owing to their non-covalent binding ability. Consequently, compounds 4i and 4t, whose inhibition types were determined to be reversible and non-competitive, have pharmaceutical importance in contrast to irreversible hydrazine-type MAO inhibitors.
3.4. Cytotoxicity
Compounds 4i and 4t displayed potent hMAO-B inhibition profiles and were further tested for toxicity using the MTT assay in the NIH3T3 cell line; the IC50 values of compounds 4i and 4t are shown in . Both compounds showed an IC50 value of >1000 µM against NIH3T3 cells, which was significantly higher than their IC50 values (0.041 and 0.065 µM) against hMAO-B. Consequently, compounds 4i and 4t were found to be non-cytotoxic at their effective concentrations against hMAO-B. This result further increases the biological importance of the compounds.
Table 3. The IC50 value of the compounds 4i and 4t against NIH/3T3 cell line.
3.5. Prediction of ADME parameters and BBB permeability
Intrinsic pharmacological activity and low toxicological effects are not sufficient for a compound to become a drug nomineeCitation35. Most new drug nominees fail in clinical trials due to their reduced absorption, distribution, metabolism, and excretion (ADME) properties. These late-stage failures result in increased drug development costsCitation36. The ability to identify problematic issues early can dramatically reduce the amount of wasted time and funds, and can streamline the overall development process. Therefore, the pharmacokinetic properties of new drug candidates are very important, and it is beneficial to assess them as early as possible in the drug development processCitation37. ADME estimation can be used to focus on precursor compound optimisation thereby improving the preferred properties of a compoundCitation38. Predictions of ADME parameters of the obtained compounds (4a–4u) were performed using QikProp 4.8 softwareCitation26. The violations of Jorgensen’s “Rule of Three”Citation39 and Lipinski’s rule of fiveCitation40, which assess the ADME properties of new drug nominees, are crucial for the optimisation of a biologically active compound. The calculated ADME parameters, including molecular weight (MW), number of rotatable bonds (RB), dipole moment (DM), molecular volume (MV), number of hydrogen donors (DHB), number of hydrogen acceptors (AHB), polar surface area (PSA), octanol/water partition coefficient (log P), aqueous solubility (log S), apparent Caco-2 cell permeability (PCaco), number of likely primer metabolic reactions (PM), percent of human oral absorption (%HOA), and the violations of the rules of three (VRT) and five (VRF) are presented in . In keeping with Jorgensen’s “Rule of Three” and Lipinski’s rule of five, the obtained compounds (4a–4u) are in accordance with the set parameters as they did not cause more than one violation.
Table 4. Calculated ADME parameters of compounds 4a–4u.
Drugs that specifically target the CNS must first pass the blood–brain barrier (BBB). Although the BBB is protective in nature, the use of drug candidates with CNS effects in a clinical setting is unlikely if such drug molecules are unable to penetrate it. Therefore, this feature should be examined earlier on in the drug discovery process. Accordingly, predicting the BBB permeability of new compounds is of great significanceCitation41. Thereby, the BBB permeability of the obtained compounds (4a–4u) was also evaluated using QikProp 4.8 softwareCitation26. Brain/blood partition coefficient (logBB) and apparent MDCK cell permeability (PMDCK) were calculated for this purpose. In keeping with the software estimates, the PMDCK values of <25 and >500 nm/sec are advised as poor and great for non-active transport of compounds. In order to assess for a compound’s capacity to pass through the BBB, logBB is the other significant parameter to consider, with recommended values between −3 and +1.2. The PMDCK and logBB values of the synthesised compounds are within the advised ranges as shown in . Thus, it can be postulated that the obtained compounds are capable of crossing the BBB, which is crucial for CNS-associated drugs.
Considering the results of the ADME and BBB permeability studies, the synthesised compounds have pharmacokinetic profiles that may be appropriate for clinical use.
3.6. Molecular docking
As observed in the MAO inhibition assay studies, compounds 4i and 4t were found to be the most active derivatives in the hMAO-B enzyme inhibition series. Furthermore, compound 4i was determined to be the most potent agent with an IC50 value of 0.041 ± 0.001 µM; hence, docking studies were carried out to evaluate its inhibition capability in silico. Using the X-ray crystal structure of hMAO-B (PDB ID: 2V5Z)Citation27, docking studies were performed and the binding modes of compound 4i were assigned. Also, this compound was subjected to the molecular docking procedure with the X-ray crystal structure of hMAO-A (PDB ID: 2Z5X) to compare its binding modes on hMAO-A and hMAO-B enzymes. Unfortunately, it was determined that compound 4i did not be settle down to the active site of hMAO-A enzyme (data not shown). Thus, no important and significant interactions were observed. Actually, this evidence is consistent with in vitro enzyme inhibition assay and supports the selective effect of compound 4i and the other derivatives in the series on hMAO-B enzyme.
The docking poses of this compound are presented in . Compound 4i adequately binds to the amino acid residues lining the cavity of hMAO-B enzyme and is located very near the FAD cofactor. When analysing the docking poses of this compound, it is clear that there is a π-π interaction, formation of three hydrogen bonds, and formation of a halogen bond. Compound 4i has a sulphonamide group at the 4th position of the phenyl ring. This group is essential for polar interactions. The amino moiety of sulphonamide forms a hydrogen bond with the carbonyl of Pro102. In addition, there is another hydrogen bond between the oxygen atom of sulphonamide and the amino group of Thr201. As mentioned above, one of the main structural modifications of BB-4b, as previously reported by our research groupCitation2, is the substitution of the fluorine atom with a sulphonamide group (). All the detected interactions of the sulphonamide group prove that the structural modification of compound BB-4b is a suitable approach. The addition of the sulphonamide group made a positive contribution to the MAO enzyme inhibition profile.
Figure 4. The three-dimensional pose of compound 4i in the active region of hMAO-B (PDB ID: 2V5Z). The important residues in the active site and this compound are presented by tube model and coloured with grey and red, respectively.
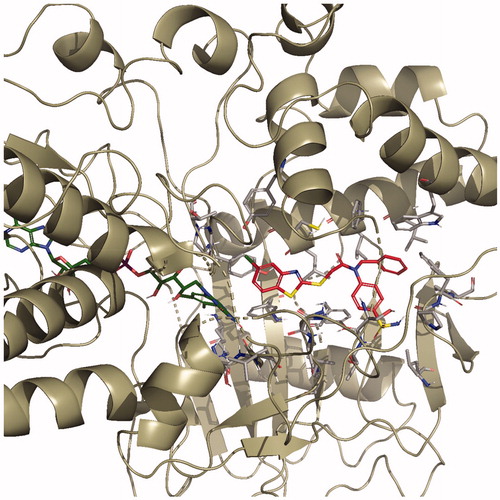
Figure 5. The three-dimensional interacting mode of compound 4i in the active region of hMAO-B. The inhibitor and the important residues in the active site of the enzyme are presented by tube model. The FAD molecule is coloured dark green with tube model.
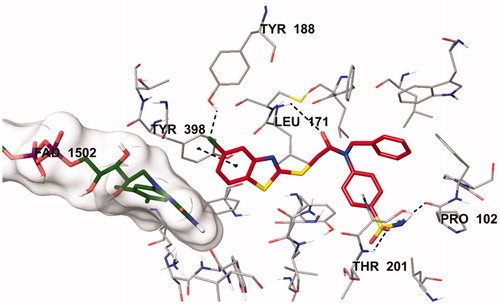
Figure 6. The van der Waals interaction of compound 4i with active region of hMAO-B. The active ligand has a lot of favourable van der Waals interactions (red and pink).
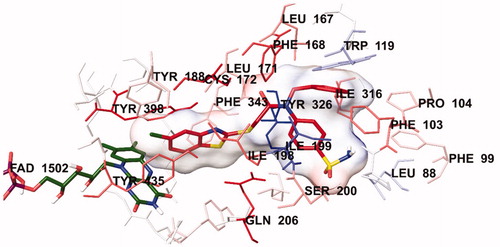
Figure 7. The electrostatic interaction of compound 4İ with active region of hMAO-B. The residues are coloured (blue, red, and pink) according to the distance from ligand by Per-Residue Interaction panel.
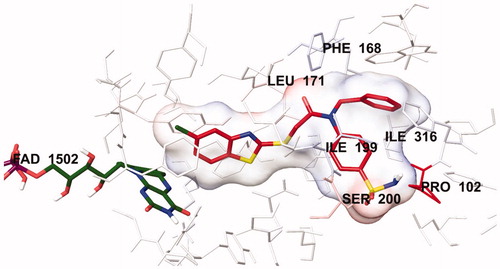
Another formation of hydrogen bond is observed between the carbonyl of the amide group in the structure and the amino group of Leu171. Compound 4i has a benzothiazole ring as a heterocyclic ring. The benzene on the benzothiazole ring interacts with the phenyl of Thr398. Interaction with the Thr398 amino acid is very important in terms of catalytic activity, and the binding of inhibitor candidates in the substrate cavity of the MAO-B enzyme. This finding indicates that compound 4i binds very effectively to the MAO-B enzyme active site.
The main structural difference between compound 4i and the other compounds in the series is that it carries a chlorine atom at the 5th position of the benzothiazole ring. It is clearly observed in that this halogen atom establishes a halogen bond with the hydrogen of the hydroxyl group of Tyr188. This additional interaction ensures that it binds more strongly to the active site. Furthermore, all these interactions explain why compound 4i exhibits a stronger inhibition profile than the other compounds.
In order to analyse the contribution of van der Waals and electrostatic interactions in binding to the enzyme active site, docking studies were performed using Glide, according to the Per-Residue Interaction panel. and present the van der Waals and electrostatic interactions of compound 4i. As shown in the figures, this compound has favourable van der Waals interactions with Leu88, Phe99, Phe103, Pro104, Tyr119, Leu167, Phe168, Leu171, Cys172, Tyr188, Ile198, Ile199, Ser200, Gln206, Ile316, Tyr326, Phe343, Tyr398 and Tyr435, which are displayed in pink and red colours as described in the user guide of GlideCitation31. Similarly, promising electrostatic contributions of compound 4i have been determined with Pro102, Phe168, Leu171, Ile199, Ser200 and Ile316 amino acids.
4. Conclusion
In conclusion, a new series of benzylamine-sulphonamide derivatives were designed, and their inhibition profile of MAO isozymes was evaluated. None of the synthesised compounds displayed a remarkable enzyme activity on hMAO-A enzyme. All of the compounds showed selectivity against hMAO-B enzyme. Among the obtained compounds, labelled 4i and 4t derivatives were found to be most active agents. Compound 4i, which contained 5-chlorobenzothiazole ring, was determined to be the most effective inhibitor candidate with an IC50 value of 0.041 ± 0.001 µM. It is thought that the 5-chlorobenzothiazole ring and sulphonamide groups were very essential for inhibiting hMAO-B enzyme by docking studies. Hence, these findings showed that the new benzylamine-sulphonamide derivatives inhibited hMAO-B enzyme and suggested that benzylamine-sulphonamide derivatives could be improved in future studies with modifications to design and gain more potent MAO enzyme inhibitor candidates.
Supplemental Material
Download PDF (3.3 MB)Disclosure statement
The authors declare no conflicts of interest.
Additional information
Funding
References
- Xu R, Xiao G, Li Y, et al. Multifunctional 5,6-dimethoxybenzo[d]isothiazol-3(2H)-one-N-alkylbenzylamine derivatives with acetylcholinesterase, monoamine oxidases and β-amyloid aggregation inhibitory activities as potential agents against Alzheimer’s disease. Bioorg Med Chem 2018;26:1885–9.
- Kaya B, Sağlık BN, Levent S, et al. Synthesis of some novel 2-substituted benzothiazole derivatives containing benzylamine moiety as monoamine oxidase inhibitory agents. J Enzyme Inhib Med Chem 2016;31:1654–61.
- Tripathi RKP, Ayyannan SR. Design, synthesis, and evaluation of 2-amino-6-nitrobenzothiazole-derived hydrazones as MAO inhibitors: role of the methylene spacer group. ChemMEdChem 2016;11:1551–67.
- Zenn RK, Abad E, Kastner J. Influence of the environment on the oxidative deamination of p-substituted benzylamines in monoamine oxidase. J Phys Chem B 2015;119:3678–86.
- Mathew B, Parambi DG, Mathew GE, et al. Emerging therapeutic potentials of dual‐acting MAO and AChE inhibitors in Alzheimer's and Parkinson's diseases. Arch Pharm 2019;352:1900177.
- Jiang T, Sun Q, Chen S. Oxidative stress: a major pathogenesis and potential therapeutic target of antioxidative agents in Parkinson's disease and Alzheimer's disease. Prog Neurobiol 2016;147:1–19.
- Mathew B, Suresh J, Mathew GE, et al. Plant secondary metabolites- potent inhibitors of monoamine oxidase isoforms. Cent Nerv Syst Agents Med Chem 2014;14:28–33.
- Fonseca A, Reis J, Silva T, et al. Coumarin versus chromone monoamine oxidase B inhibitors: quo vadis. J Med Chem 2017;60:7206–12.
- Kakkar AK, Dahiya N. Management of Parkinson's disease: current and future pharmacotherapy. Eur. J. Pharmacol 2015;750:74–81.
- Deeks ED. Safinamide: first global approval. Drugs 2015;75:705–11.
- Legoabe L, Kruger J, Petzer A, et al. Monoamine oxidase inhibition by selected anilide derivatives. Eur J Med Chem 2011;46:5162–74.
- Saddique FA, Zaib S, Jalil S, et al. Synthesis, monoamine oxidase inhibition activity and molecular docking studies of novel 4-hydroxy-N'-[benzylidene or 1-phenylethylidene]-2-H/methyl/benzyl-1,2-benzothiazine-3-carbohydrazide 1,1-dioxides. Eur J Med Chem 2018;143:1373–86.
- Lu X, Rodríguez M, Gu W, et al. Inactivation of mitochondrial monoamine oxidase B by methylthio-substituted benzylamines. Bioorg Med Chem 2003;11:4423–30.
- Upadhyay AK, Edmondson DR. Development of spin-labeled pargyline analogues as specific inhibitors of human monoamine oxidases A and B. Biochemistry 2009;48:3928–35.
- Tripathi RK, Goshain O, Ayyannan SR. Design, synthesis, in vitro MAO-B inhibitory evaluation, and computational studies of some 6-nitrobenzothiazole-derived semicarbazones. ChemMEdChem 2013;8:462–74.
- Tavari M, Malan SF, Joubert J. Design, synthesis, biological evaluation and docking studies of sulfonyl isatin derivatives as monoamine oxidase and caspase-3 inhibitors. Med Chem Commun 2016;7:1628–39.
- Ilgın S, Osmaniye D, Levent S, et al. Design and synthesis of new benzothiazole compounds as selective hMAO-B inhibitors. Molecules 2017;22:2187.
- Nam MH, Park M, Park H, et al. Indole-substituted benzothiazoles and benzoxazoles as selective and reversible MAO-B inhibitors for treatment of Parkinson's Disease. ACS Chem Neurosci 2017;8:1519–29.
- Turan-Zitouni G, Hussein W, Sağlık BN, et al. Design, synthesis and biological evaluation of novel N-pyridyl-hydrazone derivatives as potential monoamine oxidase (MAO) inhibitors. Molecules 2018;23:113.
- Can ÖD, Osmaniye D, Demir Özkay Ü, et al. MAO enzymes inhibitory activity of new benzimidazole derivatives including hydrazone and propargyl side chains. Eur J Med Chem 2017;131:92–106.
- Can NÖ, Osmaniye D, Levent S, et al. Synthesis of new hydrazone derivatives for MAO enzymes inhibitory activity. Molecules 2017;22:1381.
- Can NÖ, Osmaniye D, Levent S, et al. Design, synthesis and biological assessment of new thiazolylhydrazine derivatives as selective and reversible hMAO-A inhibitors. Eur J Med Chem 2018;144:68–81.
- Sağlık BN, Ilgın S, Özkay YS, et al. "Synthesis of new donepezil analogues and investigation of their effects on cholinesterase enzymes. Eur J Med Chem 2016;124:1026–40.
- Demir Özkay Ü, Can ÖD, Sağlık BN, et al. Design, synthesis, and AChE inhibitory activity of new benzothiazole-piperazines. Bioorg Med Chem Lett 2016;26:5387–94.
- Patel S, Ghewala N, Suthar A, et al. In-vitro cytotoxicity activity of Solanum nigrum extract against Hela cell line and Vero cell line. Int J Pharm Sci 2009;1:38–46.
- QikProp, version 4.8, Schrödinger, LLC, New York, NY; 2016.
- Claudia B, Wang J, Pisani L, et al. Structures of human monoamine oxidase B complexes with selective noncovalent inhibitors: safinamide and coumarin analogs. J Med Chem 2007;50:5848–52.
- Maestro, version 10.6, Schrödinger, LLC, New York, NY; 2016.
- Schrödinger, LLC, New York, NY; 2016.
- LigPrep, version 3.8, Schrödinger, LLC, New York, NY; 2016.
- Glide, version 7.1, Schrödinger, LLC, New York, NY; 2016.
- Toprakçı M, Yelekçi K. Docking studies on monoamine oxidase-B inhibitors: estimation of inhibition constants (K(i)) of a series of experimentally tested compounds. Bioorg Med Chem Lett 2005;15:4438–46.
- Gökhan-Kelekçi N, Şimşek ÖÖ, Ercan A, et al. Synthesis and molecular modeling of some novel hexahydroindazole derivatives as potent monoamine oxidase inhibitors. Bioorg Med Chem 2009;17:6761–72.
- Evranos-Aksöz B, Yabanoğlu-Çiftçi S, Uçar G, et al. Synthesis of some novel hydrazone and 2-pyrazoline derivatives: Monoamine oxidase inhibitory activities and docking studies. Bioorg Med Chem Lett 2014;24:3278–84.
- De Waterbeemd HV, Gifford E. ADMET in silico modelling: towards prediction paradise?. Nat Rev Drug Discov 2003;2:192–204.
- Henchoz Y, Bard B, Guillarme D, et al. Analytical tools for the physicochemical profiling of drug candidates to predict absorption/distribution. Anal Bioanal Chem 2009;394:707–29.
- Kerns EH. High throughput physicochemical profiling for drug discovery. J Pharm Sci 2001;90:1838–58.
- Kerns EH, Di L. Physicochemical profiling: overview of the screens. Drug Discov Today Technol 2004;1:343–8.
- Jorgensen WL, Duffy EM. Prediction of drug solubility from structure. Adv Drug Deliv Rev 2002;54:355–66.
- Lipinski CA, Lombardo F, Dominy BW, et al. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 2001;46:3–26.
- Dunitz JD, Taylor R. Organic fluorine hardly ever accepts hydrogen bonds. Chem-A Eur J Med Chem 1997;3:89–98.