
Abstract
Oxazolidinone hydroxamic acid derivatives were synthesised and evaluated for inhibitory activity against leukotriene (LT) biosynthesis in three in vitro cell-based test systems and on direct inhibition of recombinant human 5-lipoxygenase (5-LO). Thirteen of the 19 compounds synthesised were considered active ((50% inhibitory concentration (IC50) ≤ 10 µM in two or more test systems)). Increasing alkyl chain length on the hydroxamic acid moiety enhanced activity and morpholinyl-containing derivatives were more active than N-acetyl-piperizinyl derivatives. The IC50 values in cell-based assay systems were comparable to those obtained by direct inhibition of 5-LO activity, confirming that the compounds are direct inhibitors of 5-LO. Particularly, compounds PH-249 and PH-251 had outstanding potencies (IC50 < 1 µM), comparable to that of the prototype 5-LO inhibitor, zileuton. Pronounced in vivo activity was demonstrated in zymosan-induced peritonitis in mice. These novel oxazolidinone hydroxamic acid derivatives are, therefore, potent 5-LO inhibitors with potential application as anti-allergic and anti-inflammatory agents.
1. Introduction
The biosynthesis of bioactive leukotrienes (LTs) from arachidonic acid (AA) is catalysed by the crucial enzyme 5-Lipoxygenase (5-LO) with the help of 5-LO-activating protein (FLAP), the accessory protein that presents AA to 5-LOCitation1–4. LTs are pro-inflammatory mediators that are implicated in a variety of human inflammatory and allergic diseases, including, asthma, allergic rhinitis, cardiovascular diseases (e.g. atherosclerosis and myocardial infarction), arthritis, inflammatory bowel diseases and certain forms of cancerCitation3,Citation5–8. Since 5-LO and its isoforms have been implicated in the pathophysiology and progression of several human diseases, it is therefore identified as a viable therapeutic target. Therefore, discovery and development of selective inhibitors of 5-LO for therapeutic intervention have been subjects of active research, which are presented in patents and scientific publicationsCitation3,Citation7,Citation9,Citation10.
Generally, 5-LO inhibitors can be classified into 4 main types based on their mechanism of action: (i) substrate or competitive analogues, (ii) inhibitors of 5-LO activating protein (FLAP), (iii) redox-active inhibitors that could interfere with the free radical chemistry, (iv) iron-chelating (Fe3+ ion) inhibitors – bind the putative iron at the active siteCitation11. Several compounds based on these four different types have been synthesised, isolated as natural products and investigated as potent selective inhibitors of 5-LO with promising therapeutic usefulnessCitation3,Citation7. However, only few 5-LO inhibitors progress to clinical trials due to insufficient bioavailability, pharmacokinetics and/or toxicity related problemsCitation12.
The scientific literature contains several reports on the discovery and synthesis of iron-complexing hydroxamates and hydroxyureas as 5-LO inhibitors with therapeutic potentials. The N-hydroxy-arachidonamides (1a–c, ) and the arylhydroxamic acids (2a, ) containing a terminal N-hydroxyl group (designated as “type A hydroxamic acids”) were the most potent inhibitors of 5-LO in vitro in their series but suffered rapid metabolic hydrolysis in vivoCitation13,Citation14. However, further studies identified the arylhydroxamic acids of structure (2b, ) having terminal small N-acyl groups like acetyl, which are designated as “type B hydroxamic acids” containing reversed substitution pattern. These “type B hydroxamic acids” are potent 5-LO inhibitors that are orally active and less prone to rapid in vivo metabolic hydrolysis. In addition, they showed higher plasma bioavailability, longer duration, and are more potent orally active inhibitors of leukotriene biosynthesis compared with the type A hydroxamic acidsCitation15. Further intensive investigations led to the discovery of Zileuton, (±)1–(1-(benzo[b] thiophen-2-yl)-ethyl)-1-hydroxyurea (3a, ), a hydroxyurea derivative of the Fe3+-chelating type inhibitor as the only 5-LO inhibitor currently in clinical use. Zileuton is commercially available as a racemate (R and S enantiomers), with both enantiomers exhibiting in vitro 5-LO inhibitory activity. However, it is plagued with significant drawbacks such as liver toxicity, weak potency and short half-lifeCitation7, thus requiring higher frequency of administration (an extended-release dosage form has been introduced) accompanied by liver enzyme test. Based on these disadvantages, the need for intensive research to discover newer and potentially more effective 5-LO inhibitors with favourable pharmacokinetic, pharmacodynamic and safety profiles for treating related human diseases is highly imperative. In this light, the N-hydroxy urea atreleuton (VA-2291, 3b, ) is currently in clinical trials for the treatment of cardiovascular diseases and vascular inflammationCitation7, while the orally active hydroxamate containing a sulphonamide linker (4, ) has been reported as a 5-LO inhibitor with potential use in 5-LO mediated cancersCitation3.
Figure 1. Chemical structures of 5-lipoxygenase inhibitors and antibacterial agents.
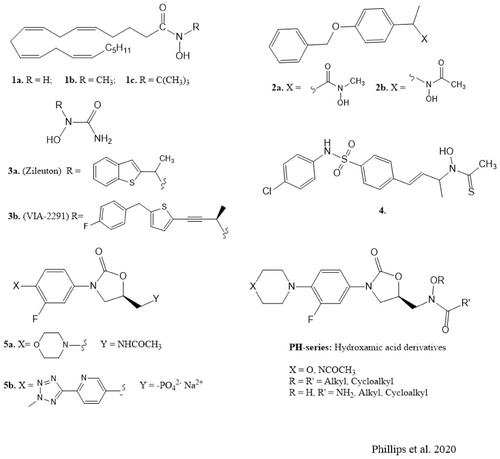
The oxazolidinone scaffold is an important pharmacophore in some clinically used antibacterial, psychoactive and anticoagulant agentsCitation16–20. In addition, structural modifications around the oxazolidinone 5-membered ring have resulted in novel compounds with antiepileptic, anticancer and antithyroid activitiesCitation19,Citation21–23. Based on the clinical success of drug molecules containing oxazolidinone scaffold, namely the antibacterial drugs linezolid (5a, ) and tedizolid phosphate (5b, ) and the well-documented pharmacological properties of the hydroxamic acid derivatives we decided to investigate a series of novel oxazolidinones containing type B hydroxamic acid functionality as inhibitors of 5-LO.
We hereby report the synthesis and structure-activity relationships of novel oxazolidinone hydroxamic acid (PH-series intermediates and PH-series, ) derivatives with potent in vitro 5-LO inhibitory activity based on the structural modifications around the oxazolidinone heterocycle as a scaffold for drug discovery.
2. Results and discussion
2.1. Chemistry
The oxazolidinone hydroxamic acid derivatives (PH-series intermediates and PH-series, ) evaluated in this study were prepared as outlined in the chemical reaction sequence in Scheme 1 according to previously reported experimental methods with some modificationsCitation24,Citation25. Starting from the commercially available starting materials morpholine 6, piperazine 7 and 3,4-difluoronitrobenzene 8, the key intermediate hydroxylamine derivatives 15 and 16 (TFA salt) were prepared in seven and ten chemical reaction steps, respectively. The nucleophilic acylation reactions of the hydroxylamine derivatives 15 and 16 (TFA salt) with different acyl anhydrides or acid chlorides yielded the respective 5-(N-alkanoxy-N-alkanamide) methyl oxazolidinone intermediate derivatives, PH-series intermediates. The final hydroxamic acid derivatives PH-series were obtained from base-promoted hydrolysis of the 5-N-alkanoxy-N-alkanamide oxazolidinones, PH-series intermediates. Ten of the compounds reported in this study were previously reported by our laboratory and were shown to be devoid of significant antibacterial or monoamine oxidases inhibitory activitiesCitation24,Citation25. Six of the previously reported compounds are morpholinyl derivatives (PH-23, PH-199, PH-204, PH-205, PH-206, and PH-211), and others are N-acetyl piperazinyl derivatives (PH-201, PH-212, PH-208, and PH-213). While the novel oxazolidinone hydroxamic acid derivatives, namely PH-237, PH-239, PH-241, PH-244, PH-245, PH-246, PH-247, PH-249, and PH-251) and their PH-series intermediates were prepared and reported for the first time in the present study. These novel compounds were fully characterised by appropriate spectroscopic and analytical methods as described in the experimental section. The characteristic signals for the N-hydroxamic acid N–OH appeared between 9.90 and 10.20 ppm, which is exchangeable with D2O and as broadband around 3171–3467 cm−1, in the nuclear magnetic resonance and infra-red spectra, respectively. The calculated log of the partition coefficient (Clog P), which is an indication of the lipophilicity of the compounds was estimated using the PerkinElmer ChemBioDraw Ultra 19.0 Computer Software and was reported as Clog P values in .
Scheme 1. Synthetic route for the oxazolidinone hydroxamic acid derivatives. (i) DCM/TFA/0 °C r.t.; (ii) DCM/TEA/acetic anhydride/0 °C r.t.; (iii) DMF/NaH/tert-Butyl-N-(tertbutoxycarbonyl)carbamate/0 °C r.t. or 0–60 °C.; (iv) DCM/TFA/0 °C r.t.; (v) DCM/TEA/RCOCl or (RCO)2O/0 °C r.t.; (vi) MeOH/THF/NaOH/0 °C.
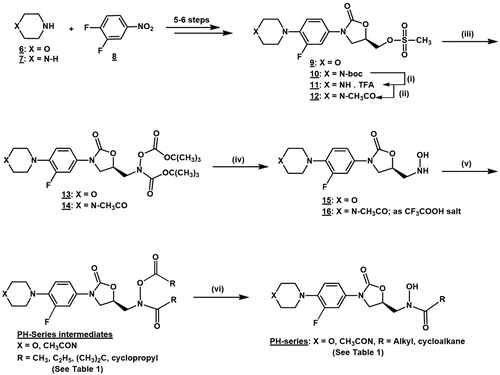
Table 1. Clog P values and in vitro inhibitory activity of oxazolidinone hydroxamates.
2.2. Pharmacology
Nineteen synthesised oxazolidinones containing the hydroxamic acid functionality were evaluated in three different cell-based in vitro assay systems for inhibition against the release of LTs, and by direct inhibition of 5-LO activity in a cell-free assay, using zileuton as a reference drug. The cell-based test systems included inhibition of 5-LO-dependent generation of LTB4 from activated human whole blood, inhibition of 5-LO product LTC4 from isolated human monocytes and inhibition of 5-LO product LTC4 from allergen/Immunoglobulin E (IgE)-activated bone marrow-derived mouse mast cells (BMMC). Furthermore, in order to confirm that the in vitro inhibitory activity of the compounds was not due to the direct toxic effects on the cells, the effect of the compounds on the viability of isolated human monocytes after 3 h and 24 h treatment was performed. Finally, in vivo anti-inflammatory studies were performed using the zymosan-induced peritoneal inflammation model in mice, which is a well-known model in which the LTs are known to play a critical roleCitation26.
The inhibitory activity data for the tested compounds obtained in three cell-based in vitro test systems - LTB4 release from human whole blood, LTC4 release from isolated human monocytes and LTC4 release from IgE/antigen-activated mouse mast cells, are shown in . Similar data on the direct inhibitory effect on the enzymatic activity of recombinant human 5-LO, together with the calculated Clog P (log of partition coefficient) values, which is an indication of the lipophilicity of each compound, are also shown in . In each case, the 50% inhibitory concentration (IC50) (95% confidence interval) values of the compounds were compared with those of the reference drug, and the only clinically available 5-LO inhibitor, zileuton.
Of the 19 compounds tested, 13 were found to have good (IC50 < 10 µM) to excellent (IC50 < 1 µM) inhibitory activity in at least two of the test systems, while six compounds (PH-211, PH-239, PH-241, PH-247, PH-249, and PH-251) were active in all four test systems. On human whole blood and isolated human monocytes, compound PH-249 (IC50 = 0.7 µM and 0.9 µM, respectively), containing the heptanoyl moiety on the hydroxamate nitrogen, had potencies that were similar to those of zileuton (IC50 = 0.7 µM and 0.5 µM, respectively, ), whereas on mast cells that were activated by an allergic mechanism, compound PH-251, containing the octanoyl moiety on the hydroxamate nitrogen, was the most active (IC50 = 0.2 µM). In this later test system, compound PH-251 was slightly more potent than zileuton (IC50 = 0.4 µM). In the cell-free system, both PH-249 and PH-251 were also the most active (IC50 = 1.9 µM and 1.6 µM, respectively, ).
Structure-activity relationship revealed that the inhibitory activities of the compounds were highly dependent on the substitution pattern around the phenyloxazolidinone moiety, whereby the morpholinyl-containing derivatives were more active compared with the N-acetyl-piperizinyl-containing derivatives. Also, the N–OH hydroxamate-containing oxazolidinones were significantly more active than their respective precursors, with the exception of the N-(hexanoyloxy)hexanamide derivative PH-246, which was also quite active in cell-based systems (IC50 values of 1.5 µM, 7.1 µM and 3.3 µM, for whole blood, isolated monocytes and mast cells, respectively, ). The reason for this discrepancy is not immediately clear given that the compound contains an N-hexanoyloxy functionality which might make it liable to hydrolysis by plasma esterases. Moreover, it is possible that the observed activity in these systems may reflect that of its hydrolytic metabolite, PH-247. However, since PH-246 is the only 5-N-alkanyloxy-N-alkanamide derivative with reasonable activity compared to that of PH-247, this may suggest that the N-hexanoyloxy moiety is more readily hydrolysed by hydrolases than the other N-alkanoyloxy groups. Hence, PH-246 may be serve as a pro-drug for the release of PH-247 in vivo. In addition, the absence of cellular esterase activity may explain why PH-246 is very weak in inhibiting the activity of isolated 5-LO enzyme in cell-free experiments. Further studies are planned to investigate the stability of these compounds in vitro at different pH conditions and in the presence of hydrolases in plasma.
The finding that most of the active compounds were generally more active in human whole blood than on isolated human monocytes is very interesting, given that in drug discovery studies, compound instability in plasma is a well-recognised problem. One possible explanation could be that the compounds are more active on granulocytes than monocytes or that a plasma factor enhances their entry into cells. These possibilities are currently being investigated.
It was further observed that all the N-acetyl-piperizinyl containing oxazolidinone derivatives tested showed either weak activity (IC50 values in the ranges of 10.0–30 µM) or were essentially inactive (IC50 values > 30 µM, ). This demonstrates that the presence of the morpholine heterocycle is essential for activity in these compounds.
Further analysis also revealed that among the hydroxamates, activity generally increased with the length of the straight-chain hydrocarbon moiety on the hydroxamate nitrogen, until the heptanoyl group. The increase in chain length to the octanoyl group resulted in a slight decrease in potency. On the other hand, cyclic hydrocarbon-containing analogues (cyclobutyl and cyclopentyl), were generally less potent, with the exception of the cyclopropyl derivative, PH-211. In the morpholine containing hydroxamic acids, IC50 values seem to correlate with the Clog P values (indicative of the lipophilicity of the compounds), thus suggesting that the optimal Clog P values are in the range of 3.3598 for PH-249 and 3.8888 for PH-251, ().
Data from studies with an isolated 5-LO enzyme () showed that the inhibitory effect of the compounds paralleled their effects in cell-based assays. For example, compounds PH-249 and PH-251, which were the most active in inhibiting 5-LO activity in the cell-free system were also the most active in inhibiting LT biosynthesis in cell-based systems, whereas the inactive analogue, PH-237 (containing the isovaleryloxy and isovaleryl moieties on the hydroxamate nitrogen), was inactive in both systems. These results indicate that the mechanism of inhibition of LT biosynthesis by the active compounds is by direct inhibition of 5-LO, and that their potencies are comparable to that of zileuton. In addition, one of the most active compounds, PH-251 demonstrated an outstanding potency in inhibiting LT biosynthesis in mouse mast cells (IC50 = 0.2 µM), which was slightly better than that of zileuton (IC50 = 0.4 µM, ). Since mast cell activation through IgE/allergen interaction is the basis of all allergic diseasesCitation27, compounds like PH-251 have a particularly high potential of being developed into useful drugs for the treatment of allergic diseases, including asthma.
From these results, it can be concluded that these novel oxazolidinones, especially the morpholinyl-containing derivatives, are potent inhibitors of 5-LO that affect cells from both humans and mice. Their action is also independent of the mode of cell activation, as they equally affect cells activated by lipopolysaccharide (LPS)/N-formyl methyl-leucyl-phenylalanine (FMLP), calcium ionophore and antigen-antibody immune complex. These findings are very important in that they show that the active compounds have the potential of being useful, not only in allergic diseases, such as asthma, allergic rhinitis, atopic dermatitis, but also in many other inflammatory diseases in which LTs, induced by a variety of stimuli, are known to be involvedCitation6,Citation7.
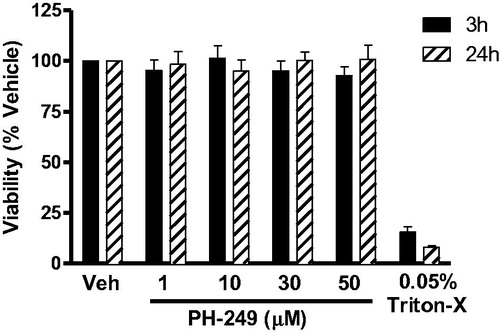
In order to exclude the possibility that the in vitro inhibitory activity of the compounds, was a result of direct toxicity of the compound on the cells, we studied the effect of the most active compound (PH-249) on the viability of isolated human monocytes after 3 h and 24 h treatment. As shown in , at concentrations up to 50 μM (a concentration far beyond that required for 100% inhibition of LT release), no significant effect on cell viability was detected, whether cells were cultured with the drug for 3 h or 24 h. This shows that the compounds are not cytotoxic.
Figure 2. Effect of compound PH-249 on the viability of isolated human monocytes. Cells were exposed to the test compound or triton-X, as positive control, for 3 h or 24 h, and viability was assessed by the MTT method, n = 3.
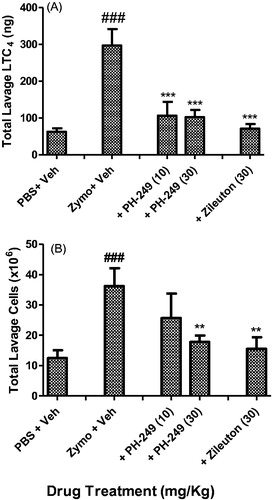
To determine if the compounds were active in vivo, the representative compound PH-249 was also tested in the zymosan-induced peritoneal inflammation, a well-known model in which LT is known to play a critical roleCitation26. As shown in , intra-peritoneal administration of zymosan (plus vehicle) resulted in a highly significant increase in the cellular influx and LTC4 content of the recovered peritoneal lavage fluid after 3 h. Pre-treatment with 10 or 30 mg/kg of PH-249 resulted in highly significant inhibition of total LTC4 content of the lavage fluid (p < 0.001), , but for cellular infiltration, only the inhibition by the higher dose reached statistical significance, p < 0.01, (). At 30 mg/kg, zileuton achieved a similar effect with respect to both parameters. These results show that the representative compound PH-249 containing the heptanoyl moiety has in vivo biological activity that is comparable to that of the reference drug, zileuton and thus suggests that these novel oxazolidinone hydroxamic acid derivatives have potential anti-inflammatory actions in vivo.
Figure 3. In vivo inhibitory effect of compound PH-249 on zymosan-induced peritoneal inflammation in mice. Animals were pre-treated subcutaneously with PH-249, zileuton or vehicle, 30 min before induction of peritoneal inflammation with intraperitoneal injection of 0.2 ml activated zymosan (2 mg/ml). Peritoneal lavage fluids obtained after 2 h were analysed for total LTC4 content (A) and total cellular content (B). Values are means ± sem, n = 6–7. ###p < 0.001 with respect to PBS plus vehicle; ***p < 0.001; **p < 0.01, with respect to zymosan plus vehicle.
3. Conclusion
In conclusion, the synthesised novel oxazolidinone hydroxamic acid derivatives were screened in four in vitro test systems for inhibitory activity against the release of LTs or direct inhibition of 5-LO enzyme activity. Thirteen of the compounds had good or excellent activity in at least two test systems, while six compounds were active in all four systems. The most active compounds had activities that were comparable to those of zileuton. One compound, PH-251, was particularly effective on mast cell LT release induced by allergen/IgE interaction (IC50 = 0.2 µM). Structure-activity relationship studies revealed that oxazolidinone derivatives containing the (N-(OH)COR) hydroxamate functionality with relatively longer straight-chain alkyl groups (-R) along with the morpholine heterocycle demonstrated the best activity. The optimal alkyl chain length appeared to be C = 6 (PH-249, R = hexyl), as extending the length to C = 7 (PH-251, R = heptyl) resulted in either a slight loss or no change in activity, except for mast cells in which a further increase in activity was observed. Results also suggest that the active compounds are non-toxic and possess strong in vivo anti-inflammatory activity. Hence, they have the potential for development as drugs for the treatment of allergic and inflammatory diseases.
4. Experimental
4.1. Synthesis
4.1.1. Materials and methods
The starting materials 3,4-difluoronitrobenzene, morpholine, piperazine, n-butyllithium, sodium hydride, and other common reagents and solvents used for the synthesis of the oxazolidinones, including, dichloromethane (DCM), diethyl ether, dimethylformamide (DMF), ethyl acetate, methanol, tetrahydrofuran (THF) were purchased from Merck (formerly Sigma-Aldrich) Germany. Among the previously reported compounds, six are morpholinyl derivatives (PH-23, PH-199, PH-204, PH-205, PH-206, and PH-211), and the others are N-acetyl piperazinyl derivatives (PH-201, PH-212, PH-208 and PH-213). These were previously reported from our laboratory and synthesised according to literature methodsCitation24,Citation25. The remaining nine oxazolidinone hydroxamic acid derivatives, namely PH-237, PH-239, PH-241, PH-244, PH-245, PH-246, PH-247, PH-249 and PH-251 and their respective PH-series intermediates are reported for the first time (). Purification of compounds was performed on silica gel column chromatography using silica gel (Kieselgel 60, 70–230 mesh; Aldrich-Sigma, Germany) and thin-layer chromatography (TLC) was conducted on 0.25 mm pre-coated silica gel plates (60F254, Merck, Darmstadt, Germany). Solid products were recrystallised from a suitable solvent and or solvent mixtures. Melting points were determined on a Stuart Scientific melting point apparatus (SMP1, Stuart, Stone, UK) and were uncorrected. Further structural elucidation was performed using 1H- and 13 C-NMR (decoupled experiments) spectra in DMSO-d6 using solvent peaks as reference signals and were recorded on Bruker DPX 400 MHz and Bruker Avance II 600 NMR spectrometers. Two-dimensional NMR spectra experiments, namely 2 D COSY (COrrelated SpectroscopY) and 2 D HSQC (Heteronuclear Signal Quantum Coherence) experiments were also performed on representative compounds (PH-242 (containing the cyclobutylcarbonyloxy and the cyclobutylcarbonyl moieties on the hydroxamate nitrogen), PH-245 (containing the cyclobutylcarbonyl group on the hydroxamate nitrogen), PH-246 (containing hexanoyloxy and the hexanoyl moieties on the hydroxamate nitrogen), PH-247 (containing the hexanoyl moiety on the hydroxamate nitrogen) to assist in proton and carbon assignments. Chemical shifts and coupling constants (J, Hz) of protons and carbons are reported in parts per million (ppm) downfield and up-field from solvent DMSO-d6 (δ = 2.5; 39.7) peak as reference. Mass spectra data were recorded on a Thermo Scientific DFS Gas Chromatography/Mass Spectrometer (DFS GC-MS, Thermo Fisher Scientific, Bremen, Germany) and Waters QToF/Mass Spectrometer (LC-MS/MS ESI, Waters Corporation, Milford, MA). Infra-red (IR) spectra were recorded on JASCO FT-IR-6300 (JASCO, Tokyo, Japan) Spectrometer. Elemental analyses were performed on an Elementar Vario Micro Cube CHN Analyser apparatus (Elementar, Langenselbold, Germany), and analyses indicated by the symbols of the elements (CNH) were within ±0.4% of the theoretical values. Elemental analyses (CHN) were used to confirm the purity of all newly synthesised compounds (>95%), Analyses were performed by the General Facility-Science (GF-S), Faculty of Science, Kuwait University, Kuwait. The structures of the oxazolidinones and their Clog P values were sketched and estimated, respectively using the PerkinElmer ChemBioDraw Ultra 19.0 Software.
4.1.2. (R)-tert-butyl (tert-butoxycarbonyl)oxy ((3-3-fluoro-4-morpholinophenyl)-2-oxooxazolidin-5-yl) methyl)carbamate (13)
An ice-cooled (0 °C) solution of tert-butyl N-(tert-butoxycarbonyloxy) carbamate (4.97 g, 21.31 mmol) in anhydrous DMF (30 ml) under nitrogen was treated portion-wise with 60% sodium hydride in mineral oil (770 mg, 22.75 mmol) and stirred for 30 min. The reaction mixture was treated with drop-wise addition of a solution of [N-3-(3-fluoro-4-morpholinylphenyl)-2-oxo-5-oxazolidinyl] methyl methanesulfonate (6.00 g, 16.02 mmol) in DMF (70 ml). The reaction mixture was stirred to room temperature for 60 h, quenched by addition of water (200 ml) and extracted with ethyl acetate (3 × 100 ml). The ethyl acetate (EtOAc) extract was diluted with hexane (60 ml) and washed with water, brine, dried (Na2SO4), filtered and concentrated to give the crude as a brownish oil. Silica gel column chromatography eluted with EtOAc-Hexane 2:1 gave the title compound 13Citation24,Citation25 as a pale-yellow viscous oil (7.0 g, yield 85%), solidifies upon cooling in the fridge to a yellow solid after a long period of time. 1H-NMR (DMSO-d6, 600 MHZ): δ 7.51 (dd, 1H, J = 15.0 Hz, 2.5 Hz, phenyl H), 7.19 (dd, 1H, J = 8.8 Hz, 2.5 Hz, phenyl H), 7.09 (t, 1H J = 9.6 Hz, phenyl H), 4.87 (m, 1H, oxazolidinone H), 4.15 (t, 1H, J = 4.6 Hz, oxazolidinone H), 3.97 (m, 1H, oxazolidinone H), 3.82 (m, 1H, methylene H), 3.72–3.76 (br. t, 5H, morpholine H and methylene H), 2.97 (br. t, 4H, morpholine H), 1.41–1.48 (br,18H). MS 511.2 (M+)Citation24,Citation25
4.1.3. (R)-3-(3-fluoro-4-morpholinophenyl)-5-((hydroxyamino)methyl)oxazolidin-2-one (15)
Compound 13 (14.69 g, 28.71 mmol) was dissolved in anhydrous DCM (25 ml) and cooled to 0 °C in an ice-bath. This solution was treated with rapid dropwise addition of trifluoroacetic acid (20 ml) and the reaction mixture was stirred overnight. The reaction mixture was concentrated to give a gummy residue, which was treated with a 10% potassium carbonate solution in water (100 ml) to give a basic solution. The resulting gelatinous precipitate was collected by filtration to give an off-white solid (8.40 g), the filtrate was extracted with DCM and the DCM layer was dried (Na2SO4) and concentrated to give a second crop (0.240 g). The titled compound 15 was obtained as an off-white solid (8.64 g, yield 94%)Citation24,Citation25, mp 134–137 °C. This product was used for further reactions without further purification. 1H-NMR (DMSO-d6, 400 MHZ): δ 7.50 (dd, 1H, J = 15.1 Hz, 2.5 Hz, phenyl H), 7.45 (s, N–OH, 1H), 7.20 (dd, 1H, J = 2.1 Hz, 8.7 Hz, phenyl H), 7.06 (t, 1H, J = 9.6 Hz, phenyl H), 6.02 (br. s, 1H, N–H), 4.70–4.83 (m, 1H, oxazolidinone H), 4.09 (t, 1H, J = 8.9 Hz, oxazolidinone H), 3.82 (dd, 1H, J = 6.8 Hz, 8.9 Hz, oxazolidinone H), 3.73 (br. t, 4H, J = 4.6 Hz, morpholine H), 2.97–3.10 (br. m, 6H, CH2N(OH)H, partially overlaps with the morpholine H triplet signal at 2.96, J = 4.6 Hz), 13 C-NMR (DMSO-d6, 600 MHZ): δ 165.28, 155.35, 154.21, 153.74, 135.42, 135.37, 133.64, 133.57, 119.19, 119.17, 114.04, 114.02, 106.69, 106.52, 70.66, 70.04, 66.11, 56.31, 50.68, 48.12. MS 311.1 (M+)Citation24,Citation25.
4.1.4. General procedure for the synthesis of the (R)-N-((3-(3-fluoro-4-morpholinophenyl)-2-oxoxazolidin-5-yl)methyl)-N-hydroxyalkanamide (PH-series)
A solution of the N-hydroxylamine derivative, compound 15 (1.0 eq.) in anhydrous DCM or CH3CN (30–50 ml) was treated with triethylamine (6.0 eq.) and dropwise addition of the respective acid anhydride or acid chloride (3.0 eq.) at 0 °C and stirred to room temperature overnight. The reaction mixture was diluted with a 10% solution of potassium carbonate (20 ml) and the DCM layer was separated and washed with water, brine, dried (Na2SO4), filtered and concentrated to give the crude product. Purification by normal phase silica gel column chromatography and/or recrystallised using suitable organic solvent or mixtures to give the intermediate products N-alkanoxy-N-((3-3-fluoro-4-morpholinophenyl)-2-oxooxazolidin-5-yl) methyl)alkanamide PH-series intermediates (PH-237, PH-240, PH-242, PH-243, PH-246, PH-248 and PH-250). A solution of the PH-series intermediates (1 eq.) in methanol:THF (4:1, v/v) was stirred at 0 °C and treated with a 1.0 eq. NaOH solution in water (∼20 ml). The reaction mixture was stirred for 1 h 10 min and neutralised to pH of ∼7 by addition of a solution of 1.0 eq. HCl in water (30 ml). The reaction mixture was concentrated to remove THF and methanol, and the aqueous residue was saturated with NaCl and extracted with DCM (2 × 30 ml) and the organic layer was separated, dried (Na2SO4), filtered and concentrated to give the crude product. Purification was performed either by silica gel column chromatography and/or recrystallisation using suitable organic solvent or mixtures to give the (R)-N-((3-(3-fluoro-4-morpholinophenyl)-2-oxoxazolidin-5-yl)methyl)-N-hydroxy-alkanamide derivatives (PH-239, PH-241, PH-244, PH-245, PH-247, PH-249, and PH-251) as the final products.
4.1.4.1. (R)-N-((3-(3-fluoro-4-morpholinophenyl)-2-oxooxazolidin-5-yl)methyl)-N-hydroxy-3-methylbutanamide (PH-239)
The intermediate compound PH-237, was prepared via the general procedure from compound 15 (1.50 g, 4.82 mmol), isovaleric anhydride (2.83 ml, 14.46 mmol), triethylamine (4.05 ml; 28.41 mmol) in anhydrous DCM (30 ml) to give crude product. Purification by silica gel column chromatography (EtOAc-Hexane, 1:2 to 1:1) gave the intermediate (R)-N-((3-(3-fluoro-4-morpholinophenyl)-2-oxooxazolidin-5-yl)methyl)-3-methyl-N-((3-methylbutanoyl)oxy)butanamide PH-237, as a white solid 0.815 g, yield 35%, mp 89–90 °C. IR (KBr, cm−1): ν 2960, 2871, 2822, 1797, 1759, 1672, 1520, 1471, 1445, 1404, 1329, 1289, 1169, 1136, 1119, 1065. 1H-NMR (CDCl3, 600 MHZ): δ 7.46 (dd, 1H, J = 2.6 Hz, 14.3 Hz, phenyl H), 7.11 (m, 1H, phenyl H), 6.94 (t, 1H, J = 9.1 Hz, phenyl H), 4.80–4.84 (br. m, 1H, oxazolidinone H), 4.10 (d, 2H, J = 4.6 Hz, methylene CH2), 4.05 (t, 1H, J = 8.9 Hz, oxazolidinone H), 3.88–3.91 (m, 5H, morpholine H and oxazolidinone H), 3.07 (t, 4H, J = 4.7 Hz, morpholine H), 2.39 (d, 2H, J = 7.1 Hz, NOCOCH2CH(CH3)2), 2.19–2.51 (m, 1H, NOCOCH2CH(CH3)2), 2.06–2.12 (m, 3H, NCOCH2CH (CH3)2 and NCOCH2CH (CH3)2), 1.05 (dd, 6H, J = 1.4 Hz, 6.6 Hz, NOCOCH2CH(CH3)2), 0.93 (dd, 6H, J = 6.1 Hz, 13.0 Hz, NCOCH2CH(CH3)2). 13 C-NMR (DMSO-d6, 600 MHZ): δ 155.33 (d, J = 243.20 Hz), 153.70, 135.55 (d, J = 8.76 Hz), 133.32 (d, J = 10.80 Hz), 119.22 (d, J = 4.16 Hz), 114.13, (d, J = 2.80 Hz), 106.67 (d, J = 25.88 Hz), 70.10, 66.10, 50.66, 50.30, 47.15, 24.95, 22.11. MS 479.4 (M+). Anal. Calcd. for C24H34FN3O6: C: 60.11, H: 7.15, N: 8.76; found C: 59.71, H: 7.41, N: 8.55. A solution of the intermediate PH-237 (0.80 g, 1.67 mmol) in MeOH:THF (28:7 ml) was treated with NaOH solution (133 mg in 20 ml water). Purification by recrystallisation (EtOAc-hexane 2:1) gave the titled compound PH-239 as a white solid (328 mg, yield, 55%), mp 122–124.5 °C. IR (KBr, cm−1): ν 3384, 3196, 2955, 2869, 1752, 1729, 1633, 1610, 1521, 1447, 1429, 1333, 1272, 1233, 1173, 1119, 1068. 1H-NMR (DMSO-d6, 600 MHZ): δ 9.93 (s, 1H, N–OH, exchangeable with D2O), 7.48 (dd, 1H, J = 2.5 Hz, 14.9 Hz, phenyl H), 7.17 (dd, 1H, J = 2.3 Hz, 8.7 Hz, phenyl H), 7.06 (t, 1H, J = 8.4 Hz, phenyl H), 4.85–4.90 (br. m, 1H, oxazolidinone H), 4.13 (t, 1H, J = 8.9 Hz, oxazolidinone H), 4.05 (dd, 1H, J = 6.5 Hz, 13.7 Hz, oxazolidinone H), 3.72–3.76 (m, 5H, morpholine H and methylene H), 3.67 (dd, 1H, J = 4.5 Hz, 14.6 Hz, methylene H), 2.96 (t, 4H, J = 4.6 Hz, morpholine H), 2.25–2.30 (m, 2H, NCOCH2CH(CH3)2), 1.99–2.04 (m, 1H, NCOCH2CH(CH3)2), 0.88 (t, 6H, J = 5.9 Hz, NCOCH2CH(CH3)2). 13 C-NMR (DMSO-d6, 600 MHZ): δ 173.22, 154.55 (d, J = 243.45 Hz), 153.95, 135.53 (d, J = 8.77 Hz), 133.43 (d, J = 10.83 Hz), 119.24 (d, J = 4.11 Hz), 114.11 (d, J = 2.79 Hz), 106.66 (d, J = 26.24 Hz), 69.87, 66.13, 50.69, 50.27, 47.48, 24.42, 22.48. MS ES+ (m/z): 396.1874 (M+ + H), MS (m/z): 395.2 (M+). Anal. Calcd. for C19H26FN3O5: C: 57.71, H: 6.63, N: 10.63; found C: 57.28, H: 6.94, N: 10.32.
4.1.4.2. (R)-N-((3-(3-fluoro-4-morpholinophenyl)-2-oxooxazolidin-5-yl)methyl)-N-hydroxypentanamide (PH-241)
The intermediate compound 0PH-240, was prepared via the general procedure from compound 15 (2.20 g, 7.07 mmol), valeric anhydride (4.18 ml, 21.20 mmol), triethylamine (5.94 ml; 42.40 mmol) in anhydrous DCM (30 ml) to give crude product. Purification by silica gel column chromatography (EtOAc-Hexane, 1:2 to 1:1) gave the intermediate (R)-N-((3-(3-fluoro-4-morpholinophenyl)-2-oxooxazolidin-5-yl)methyl)-N-(pentanoyloxy)pentamide PH-240 as a white solid 1.10 g, yield 33%, mp 75–77.5 °C. IR (KBr, cm−1): ν 2963, 2930, 2857, 1797, 1739, 1683, 1627, 1572, 1518, 1447, 1409, 1327, 1236, 1214, 1138, 1120, 1057. 1H-NMR (DMSO-d6, 600 MHZ): δ 7.48 (dd, 1H, J = 2.5 Hz, 14.9 Hz, phenyl H), 7.18 (dd, 1H, J = 2.2 Hz, 8.8 Hz, phenyl H), 7.07 (t, 1H, J = 9.4 Hz, phenyl H), 4.84–4.88 (br. m, 1H, oxazolidinone H), 4.10–4.19 (br. t, 2H, oxazolidinone H, overlaps with oxazolidinone H triplet, at 4.11 ppm, J = 9.0 Hz), 3.84–3.92 (br., 1H, methylene H), 3.74 (t, 5H, J = 4.6 Hz, morpholine H and methylene H), 2.96 (t, 4H, J = 4.6 Hz, morpholine H), 2.50 (br., 2H, methylene –CH2– overlapping with DMSO signal), 2.11–2.27 (br., 2H, methylene H), 1.56–1.61 (m, 2H, methylene H), 1.40–1.50 (br., 2H, methylene –CH2–), 1.31–1.37 (m, 2H, methylene H) 1.20–1.29 (br., 2H, methylene H), 0.88 (t, 3H, J = 7.4 Hz, methyl H), 0.82 (br., t, 6H, J = 6.5 Hz, methyl H). 13 C-NMR (DMSO-d6, 600 MHZ): δ 154.56 (d, J = 243.26 Hz), 153.75, 135.56 (d, J = 8.72 Hz), 133.36 (d, J = 10.62 Hz), 119.24 (d, J = 4.16 Hz), 114.13 (d, J = 2.78 Hz), 106.66 (d, J = 25.88 Hz), 70.16, 66.13, 50.69, 50.67, 47.15, 30.85, 30.75, 25.97, 21.55, 21.49, 13.62, 13.49. MS ES+ (m/z): 480.2300 (M+ + H), MS (m/z): 479.3 (M+). Anal. Calcd. for C24H34FN3O6: C: 60.11; H: 7.15; N: 8.76; found C: 60.06; H, 6.87; N, 8.74. A solution of the intermediate PH-240 (0.900 g, 1.88 mmol) in MeOH:THF (28:7 ml) was treated with NaOH solution (150 mg in 20 ml water). Purification by recrystallisation (EtOAc-hexane 2:1) gave the titled compound PH-241 as an off-white solid 700 mg, yield 94%, mp 121–122.5 °C. IR (KBr, cm−1): ν 3187, 2959, 2931, 2860, 1743, 1720, 1626, 1522, 1447, 1425, 1331, 1271, 1234, 1196, 1115, 1071. 1H-NMR (DMSO-d6, 600 MHZ): δ 9.94 (s, 1H, N–OH, exchangeable with D2O), 7.48 (dd, 1H, J = 2.5 Hz, 15.0 Hz, phenyl H), 7.17 (dd, 1H, J = 2.3 Hz, 8.9 Hz, phenyl H), 7.06 (t, 1H, J = 9.4 Hz, phenyl H), 4.85–4.89 (br. m, 1H, oxazolidinone H), 4.12 (t, 1H, J = 8.9 Hz, oxazolidinone H), 4.04 (dd, 1H, J = 6.6 Hz, 15.6 Hz, oxazolidinone H), 3.72–3.76 (m, 5H, morpholine H and methylene H), 3.67 (dd, 1H, J = 4.6 Hz, 14.9 Hz, methylene H), 2.96 (t, 4H, J = 4.7 Hz, morpholine H), 2.36–2.39 (m, 2H, NCOCH2CH2CH2CH3), 1.44–1.48 (m, 2H, NCOCH2CH2CH2CH3), 1.24–1.30 (m, 2H, NCOCH2CH2CH2CH3), 0.85 (t, 2H, J = 7.4 Hz, NCOCH2CH2CH2CH3). 13 C-NMR (DMSO-d6, 600 MHZ): δ 173.93, 154.53 (d, J = 244.06 Hz), 153.90, 135.49 (d, J = 8.78 Hz), 133.40 (d, J = 10.79 Hz), 119.20 (d, J = 4.06 Hz), 114.10 (d, J = 2.84 Hz), 106.64 (d, J = 26.00 Hz), 69.87, 66.09, 50.66, 50.64, 50.30, 47.45, 31.22, 26.21, 21.81, 13.68. MS ES+ (m/z): 396.2037 (M+ + H), MS (m/z): 395.2 (M+). Anal. Calcd. for C19H26FN3O5: C: 57.71, H: 6.63, N: 10.63; found C: 58.10, H: 6.61, N: 10.39.
4.1.4.3. (R)-N-((3-(3-fluoro-4-morpholinophenyl)-2-oxooxazolidin-5-yl)methyl)-N-hydroxycyclobutanecarboxamide (PH-245)
The intermediate compound PH-242 was prepared via the general procedure from compound 15 (2.20 g, 7.07 mmol), cyclobutanecarbonylchloride (2.42 ml, 21.20 mmol), triethylamine (5.94 ml; 42.40 mmol) in anhydrous DCM (30 ml) to give a brown-yellowish gummy crude product. Purification by silica gel column chromatography (EtOAc-Hexane, 1:2 to 1:1) gave the intermediate (R)-N-((cyclobutanecarbonyl)oxy)-N-((3-(3-fluoro-4-morpholino phenyl) −2-oxooxazolidin-5-yl)methyl)cyclobutanecarboxamide PH-242 as a white solid 0.487 g, yield 15%, mp 122–124 °C. IR (KBr, cm−1): ν 2963, 2857, 1788, 1741, 1678, 1517, 1445, 1410, 1329, 1261, 1096, 1021. 1H-NMR (DMSO-d6, 600 MHZ): δ 7.48 (dd, 1H, J = 2.4 Hz, 14.9 Hz, phenyl H), 7.17 (dd, 1H, J = 2.3 Hz, 8.8 Hz, phenyl H), 7.07 (t, 1H, J = 9.3 Hz, phenyl H), 4.83–4.87 (br. m, 1H, oxazolidinone H), 4.09–4.15 (br. t, 2H, oxazolidinone H, overlaps with oxazolidinone H, triplet, at 4.10 ppm, J = 9.0 Hz), 3.87–3.90 (br., 1H, methylene H), 3.73–3.75 (m, 5H, morpholine H and methylene H), 3.32–3.35 (br., 1H, cyclobutane H), 3.09–3.19 (br., 1H, cyclobutane H), 2.96 (t, 4H, J = 4.7 Hz, morpholine H), 1.17–2.34 (m, 12H, cyclobutane H). 13 C-NMR (DMSO-d6, 600 MHZ): δ 154.54 (d, J = 243.26 Hz), 153.75, 135.57 (d, J = 8.72 Hz), 133.34 (d, J = 10.75 Hz), 119.23 (d, J = 4.13 Hz), 114.16, 106.69 (d, J = 26.26 Hz), 70.20, 66.12, 50.68, 50.67, 47.15, 34.94, 24.43, 24.32, 24.21, 24.11, 17.99, 17.44. MS ES+ (m/z): 498.2177 (M+ + Na), MS (m/z): 475.2 (M+). A solution of the intermediate PH-242 (0.487 g, 1.02 mmol) in MeOH:THF (28:7 ml) was treated with NaOH solution (84 mg in 20 ml water). Purification by recrystallisation (EtOAc-hexane 2:1) gave the titled compound PH-245 as off-white solid 248 mg, yield 92%, mp 132–133.5 °C. IR (KBr, cm−1): ν 3224, 2953, 2858, 1740, 1722, 1629, 1523, 1476, 1425, 1331, 1233, 1198, 1114, 1081. 1H-NMR (DMSO-d6, 600 MHZ): δ 9.80 (s, 1H, N–OH, exchangeable with D2O), 7.49 (dd, 1H, J = 2.5 Hz, 14.9 Hz, phenyl H), 7.18 (dd, 1H, J = 2.3 Hz, 8.9 Hz, phenyl H), 7.06 (t, 1H, J = 9.4 Hz, phenyl H), 4.84–4.89 (br. m, 1H, oxazolidinone H), 4.12 (t, 1H, J = 8.9 Hz, oxazolidinone H), 4.02 (dd, 1H, J = 6.8 Hz, 14.6 Hz, oxazolidinone H), 3.73–3.76 (m, 5H, morpholine H and methylene H), 3.66 (dd, 1H, J = 4.5 Hz, 14.6 Hz, methylene H), 3.46–3.52 (m, 1H, cyclobutane H), 2.96 (t, 4H, J = 4.7 Hz, morpholine H), 1.75–1.93 (m, 6H, cyclobutane H). 13 C-NMR (DMSO-d6, 600 MHZ): δ 175.29, 155.55 (d, J = 243.92 Hz), 153.95, 135.52 (d, J = 8.77 Hz), 133.42 (d, J = 10.69 Hz), 119.23 (d, J = 3.77 Hz), 114.11 (d, J = 2.89 Hz), 106.66 (d, J = 26.03 Hz), 69.80, 66.13, 50.69, 50.67, 50.56, 47.50, 35.72, 24.39, 17.73. MS ES+ (m/z): 394.1899 (M+ + H), MS (m/z): 393.2 (M+). Anal. Calcd. for C19H24FN3O5: C: 58.01, H: 6.15, N: 10.68; found C: 57.61, H: 6.02, N: 10.42.
4.1.4.4. (R)-N-((3-(3-fluoro-4-morpholinophenyl)-2-oxooxazolidin-5-yl)methyl)-N-hydroxycyclopentanecarboxamide (PH-244)
The intermediate compound PH-243 was prepared via the general procedure from compound 15 (2.20 g, 7.07 mmol), cyclopentanecarbonylchloride (2.58 ml, 21.20 mmol), triethylamine (5.94 ml; 42.40 mmol) in anhydrous DCM (30 ml) to give a brown-yellowish gummy crude product. Purification by silica gel column chromatography (EtOAc-Hexane, 1:2 to 1:1) gave the intermediate (R)-N-((cyclopentanecarbonyl)oxy)-N-((3-(3-fluoro-4-morpholino phenyl)-2-oxooxazolidin-5-yl)methyl)cyclopentanecarboxamide PH-243 as a white solid 1.20 g, yield 34%, recrystallised from EtOAc-Et2O, mp 114.5–116 °C. IR (KBr, cm−1): ν 2960, 2853, 1779, 1754, 1671, 1515, 1449, 1415, 1327, 1225, 1193, 1117, 1087. 1H-NMR (DMSO-d6, 600 MHZ): δ 7.48 (dd, 1H, J = 2.3 Hz, 14.9 Hz, phenyl H), 7.17 (dd, 1H, J = 2.3 Hz, 8.9 Hz, phenyl H), 7.07 (t, 1H, J = 9.4 Hz, phenyl H), 4.83–4.88 (br. m, 1H, oxazolidinone H), 4.09–4.16 (br. t, 2H, oxazolidinone H, overlaps with oxazolidinone H triplet, at 4.12 ppm, J = 9.0 Hz), 3.84–3.95 (br., 1H, methylene H), 3.74 (t, 5H, J = 4.6 Hz, morpholine H and methylene H), 2.96 (t, 5H, J = 4.6 Hz, morpholine H and cyclopentane H), 2.64–2.79 (br., 1H, cyclopentane H), 1.44–1.96 (m, 16H, cyclopentane H). 13 C-NMR (DMSO-d6, 600 MHZ): δ 154.52 (d, J = 243.39 Hz), 153.71, 135.36 (d, J = 8.85 Hz), 133.33 (d, J = 10.77 Hz), 119.22 (d, J = 4.16 Hz), 114.10 (d, J = 2.37 Hz), 106.64 (d, J = 26.22 Hz), 70.24, 66.10, 50.66, 50.65, 47.15, 29.49, 29.22, 29.08, 29.05, 25.53, 25.26. MS ES+ (m/z): 504.2700 (M+ + H), MS (m/z): 503.2 (M+). Anal. Calcd. for C26H34FN3O6: C: 62.01; H: 6.81; N: 8.34; found C: 62.25; H: 6.64; N: 8.08. A solution of the intermediate PH-243 (0.900 g, 1.79 mmol) in MeOH:THF (28:7 ml) was treated with NaOH solution (143 mg in 20 ml water). Purification by recrystallisation (EtOAc-hexane 2:1) gave the titled compound PH-244 as off-white solid 578 mg, yield 76%, recrystallised from EtOAc-Et2O; mp 164.5–166.5 °C. IR (KBr, cm−1): ν 3301, 2964, 2869, 1752, 1655, 1640, 1520, 1477, 1444, 1398, 1328, 1239, 1132, 1107. 1H-NMR (DMSO-d6, 600 MHZ): δ 9.90 (s, 1H, N–OH, exchangeable with D2O), 7.49 (dd, 1H, J = 2.5 Hz, 15.00 Hz, phenyl H), 7.18 (dd, 1H, J = 2.3 Hz, 8.8 Hz, phenyl H), 7.06 (t, 1H, J = 9.4 Hz, phenyl H), 4.84–4.89 (br. m, 1H, oxazolidinone H), 4.13 (t, 1H, J = 8.9 Hz, oxazolidinone H), 4.02 (dd, 1H, J = 6.5 Hz, 14.2 Hz, oxazolidinone H), 3.73–3.76 (m, 5H, morpholine H and methylene H), 3.68 (dd, 1H, J = 4.2 Hz, 14.6 Hz, methylene H), 3.10–3.20 (m, 1H, cyclopentane H), 2.96 (t, 4H, J = 4.6 Hz, morpholine H), 1.45–1.80 (m, 8H, cyclopentane H). 13 C-NMR (DMSO-d6, 600 MHZ): δ 176.77, 154.55 (d, J = 243.43 Hz), 153.96, 135.51 (d, J = 8.75 Hz), 133.43 (d, J = 10.72 Hz), 119.23 (d, J = 4.18 Hz), 114.10 (d, J = 2.87 Hz), 106.64 (d, J = 26.14 Hz), 69.90, 66.13, 50.69, 50.68, 50.58, 47.48, 29.27, 29.20, 25.58, 25.56. MS ES+ (m/z): 408.2039 (M+ + H), MS (m/z): 407.2 (M+). Anal. Calcd. for C20H26FN3O5: C, 58.96; H, 6.43; N, 10.31; found C, 58.57; H, 6.31; N, 10.01.
4.1.4.5. (R)-N-((3-(3-fluoro-4-morpholinophenyl)-2-oxooxazolidin-5-yl) methyl)-N-hydroxyhexanamide (PH-247)
The intermediate compound PH-246 was prepared via the general procedure from compound 15 (6.0 g, 19.27 mmol), hexanoyl chloride (8.08 ml, 57.82 mmol), triethylamine (16.20 ml; 115.64 mmol) in anhydrous DCM (90 ml) to give crude product. Purification by silica gel column chromatography (EtOAc-Hexane, 1.3:2) gave the intermediate (R)-N-((3-(3-flouro-4-morpholino phenyl)methyl)-N-(hexanoyloxy)-hexanamide PH-246 as a white solid 4.50 g, yield 46%, mp 80–82.8 °C. IR (KBr, cm−1): ν 2958, 2930, 2856, 1794, 1740, 1685, 1517, 1446, 1408, 1329, 1237, 1216, 1140, 1119, 1063. 1H-NMR (DMSO-d6, 600 MHZ): δ 7.48 (dd, 1H, J = 2.5 Hz, 14.9 Hz, phenyl H), 7.18 (dd, 1H, J = 2.3 Hz, 8.8 Hz, phenyl H), 7.06 (t, 1H, J = 9.3 Hz, phenyl H), 4.84–4.88 (br. m, 1H, oxazolidinone H), 4.10–4.18 (br. t, 2H, oxazolidinone H, overlaps with oxazolidinone H triplet at 4.11, J = 9.0 Hz), 3.84–3.94 (br., 1H, methylene H), 3.74 (t, 5H, J = 4.6 Hz, morpholine H and methylene H), 2.96 (t, 4H, J = 4.6 Hz, morpholine H), 2.50 (br., 2H, methylene –CH2– overlapping with DMSO signal), 2.10–2.28 (br., 2H, methylene H), 1.16–1.64 (m, 12H, methylene H), 0.82–0.88 (m, 6H, two methyl H). 13 C-NMR (DMSO-d6, 600 MHZ): δ 154.49 (d, J = 246.37 Hz), 153.67, 135.50 (d, J = 8.78 Hz), 133.27 (d, J = 10.43 Hz), 119.17 (d, J = 4.13 Hz), 114.10 (d, J = 2.71 Hz), 106.64 (d, J = 26.06 Hz), 70.09, 66.06, 50.62, 50.61, 47.11, 31.07, 30.94, 30.53, 30.40, 23.50, 21.68, 21.57, 13.64, 13.60. MS ES+ (m/z): 508.3000 (M+ + H), MS (m/z): 507.3 (M+). Anal. Calcd. for C26H38FN3O6: C: 61.52; H: 7.55; N: 8.28; found C: 61.44; H, 7.53; N, 7.95. A solution of the intermediate compound PH-246 (4.50 g, 8.87 mmol) in MeOH:THF (84:21 ml) was treated with NaOH solution (709 mg in 20 ml water). Purification by recrystallisation (EtOAc-hexane 2:1) gave the titled compound PH-247 as an off-white solid 3.23 g, yield 89%, mp 118.5–120.5 °C. IR (KBr, cm−1): ν 3187, 2957, 2930, 2858, 1745, 1719, 1626, 1524, 1475, 1426, 1332, 1270, 1258, 1234, 1196, 1114, 1073. 1H-NMR (DMSO-d6, 600 MHZ): δ 9.92 (s, 1H, N–OH, exchangeable with D2O), 7.48 (dd, 1H, J = 2.5 Hz, 15.0 Hz, phenyl H), 7.17 (dd, 1H, J = 2.2 Hz, 8.8 Hz, phenyl H), 7.06 (t, 1H, J = 9.4 Hz, phenyl H), 4.85–4.89 (br. m, 1H, oxazolidinone H), 4.12 (t, 1H, J = 8.9 Hz, oxazolidinone H), 4.04 (dd, 1H, J = 6.7 Hz, 14.7 Hz, oxazolidinone H), 3.73–3.76 (m, 5H, morpholine H and methylene H), 3.67 (dd, 1H, J = 4.3 Hz, 14.8 Hz, methylene H), 2.96 (t, 4H, J = 4.7 Hz, morpholine H), 2.35–2.38 (m, 2H, NCOCH2CH2CH2CH2CH3), 1.45–1.49 (m, 2H, NCOCH2CH2CH2CH2CH3), 1.22–1.30 (m, 4H, NCOCH2CH2CH2CH2CH3), 0.85 (t, 3H, J = 7.0 Hz, NCOCH2CH2CH2CH2CH3). 13 C-NMR (DMSO-d6, 600 MHZ): δ 173.90, 154.51 (d, J = 244.10 Hz), 153.88, 135.46 (d, J = 8.79 Hz), 133.38 (d, J = 10.70 Hz), 119.17 (d, J = 4.18 Hz), 114.07 (d, J = 2.64 Hz), 106.62 (d, J = 26.24 Hz), 69.86, 66.07, 50.64, 50.62, 50.26, 47.42, 31.46, 30.88, 23.70, 21.80, 13.71. MS ES+ (m/z): 410.2000 (M+ + H), MS (m/z): 409.2 (M+). Anal. Calcd. for C20H28FN3O5: C: 58.67; H: 6.89; N: 10.26; found C: 58.32, H: 6.87, N: 10.61.
4.1.4.6. (R)-N-((3-(3-fluoro-4-morpholinophenyl)-2-oxooxazolidin-5-yl) methyl)-N-hydroxyheptanamide (PH-249)
The intermediate compound PH-248 was prepared via the general procedure from compound 15 (6.00 g, 19.27 mmol), heptanoyl chloride (8.95 ml, 57.81 mmol), triethylamine (16.21 ml; 115.62 mmol) in anhydrous DCM (30 ml) to give crude product. Purification by silica gel column chromatography (EtOAc-Hexane, 3:4) gave the intermediate (R)-N-(3-(3-fluoro-4-morpholinophenyl)-2-oxooxazolidin-5-yl) methyl)-N-(heptanoyloxy) heptanamide PH-248 as a white solid 6.82 g, yield 66%, mp 68–70.5 °C. IR (KBr, cm−1): ν 2960, 2929, 2855, 1794, 1740, 1685, 1519, 1446, 1410, 1329, 1236, 1217, 1140, 1119, 1066. 1H-NMR (DMSO-d6, 600 MHZ): δ 7.48 (dd, 1H, J = 2.5 Hz, 14.7 Hz, phenyl H), 7.17 (dd, 1H, J = 2.2 Hz, 8.8 Hz, phenyl H), 7.06 (t, 1H, J = 9.3 Hz, phenyl H), 4.84–4.88 (br. m, 1H, oxazolidinone H), 4.10–4.18 (br. t, 2H, oxazolidinone H, overlaps with oxazolidinone H triplet at 4.11, J = 9.0 Hz), 3.84–3.94 (br., 1H, methylene H), 3.73 (t, 5H, J = 4.6 Hz, morpholine H and methylene H), 2.96 (t, 4H, J = 4.6 Hz, morpholine H), 2.50 (br., 2H, methylene –CH2– overlapping with DMSO signal), 2.10–2.26 (br., 2H, methylene H), 1.15–1.64 (m, 16H, methylene H), 0.86–0.87 (m, 6H, two methyl H). 13 C-NMR (DMSO-d6, 600 MHZ): δ 154.53 (d, J = 244.26 Hz), 153.72, 135.54 (d, J = 8.77 Hz), 133.31 (d, J = 10.73 Hz), 119.19 (d, J = 4.01 Hz), 114.11 (d, J = 2.43 Hz), 106.66 (d, J = 26.01 Hz), 70.13, 66.10, 50.66, 50.65, 47.13, 31.03, 30.87, 30.74, 28.04, 27.94, 23.83, 21.87, 21.83, 13.79. MS ES+ (m/z): 535.3498 (M+ + H), 538.3384 (M+ + Na), MS (m/z): 535.4 (M+). Anal. Calcd. for C28H42FN3O6: C: 62.78; H: 7.90; N: 7.84; found C: 62.72; H, 7.90; N, 7.51. A solution of the intermediate PH-248 (6.28 g, 11.72 mmol) in MeOH:THF (84:21 ml) was treated with NaOH solution (928 mg in 20 ml water). Purification by recrystallisation (EtOAc-hexane 2:1) gave the titled compound PH-249 as an off-white solid 4.30 g, yield 87%, mp 123–125 °C. IR (KBr, cm−1): ν 3188, 2957, 2923, 2855, 1743, 1719, 1626, 1525, 1473, 1426, 1332, 1271, 1235, 1196, 1115, 1073. 1H-NMR (DMSO-d6, 600 MHZ): δ 9.92 (s, 1H, N–OH, exchangeable with D2O), 7.48 (dd, 1H, J = 2.5 Hz, 14.9 Hz, phenyl H), 7.18 (dd, 1H, J = 2.2 Hz, 8.8 Hz, phenyl H), 7.06 (t, 1H, J = 9.3 Hz, phenyl H), 4.85–4.89 (br. m, 1H, oxazolidinone H), 4.12 (t, 1H, J = 8.9 Hz, oxazolidinone H), 4.04 (dd, 1H, J = 6.7 Hz, 14.7 Hz, oxazolidinone H), 3.73–3.75 (m, 5H, morpholine H and methylene H), 3.67 (dd, 1H, J = 4.2 Hz, 14.9 Hz, methylene H), 2.96 (t, 4H, J = 4.7 Hz, morpholine H), 2.35–2.38 (m, 2H, NCOCH2CH2CH2CH2CH2CH3), 1.44–1.49 (m, 2H, NCOCH2CH2CH2CH2CH2CH3), 1.21–1.27 (m, 6H, NCOCH2CH2CH2CH2CH2CH3), 0.85 (t, 3H, J = 7.0 Hz, NCOCH2CH2CH2CH2CH2CH3). 13 C-NMR (DMSO-d6, 600 MHZ): δ 173.93, 154.53 (d, J = 244.01 Hz), 153.92, 135.49 (d, J = 8.73 Hz), 133.41 (d, J = 10.27 Hz), 119.19 (d, J = 4.15 Hz), 114.07 (d, J = 2.80 Hz), 106.63 (d, J = 26.27 Hz), 69.90, 66.10, 50.67, 50.65, 50.27, 47.43, 31.54, 31.02, 28.36, 24.02, 21.91, 13.85. MS ES+ (m/z): 424.2521 (M+ + H), MS (m/z): 423.3 (M+). Anal. Calcd. for C21H30FN3O5: C: 59.56; H: 7.14; N: 9.92; found C: 59.58, H: 7.52, N: 10.05.
4.1.4.7. (R)-N-((3-(3-fluoro-4-morpholinophenyl)-2-oxooxazolidin-5-yl) methyl)-N-hydroxyoctanamide (PH-251)
The intermediate compound PH-250 was prepared via the general procedure from compound 15 (2.43 g, 7.80 mmol), octanoyl chloride (3.27 ml, 23.40 mmol), triethylamine (6.60 ml; 46.80 mmol) in anhydrous DCM (35 ml) to give crude product. Purification by silica gel column chromatography (EtOAc-hexane, 1:2; 3:4) gave the intermediate(R)-N-((3-(-Fluoro-4-morpholinophenyl)-2-oxooxazolidin-5-yl)methyl)-N-(octanoyloxy)octanamide PH-250 as a white solid 2.20 g, yield 50%, mp 49–53 °C. IR (KBr, cm−1): ν 2954, 2925, 2853, 1790, 1750, 1673, 1520, 1442, 1417, 1377, 1328, 1272, 1225, 1193, 1120, 1075. 1H-NMR (DMSO-d6, 600 MHZ): δ 7.48 (dd, 1H, J = 2.5 Hz, 14.9 Hz, phenyl H), 7.17 (dd, 1H, J = 2.3 Hz, 8.8 Hz, phenyl H), 7.06 (t, 1H, J = 9.3 Hz, phenyl H), 4.84–4.88 (br. m, 1H, oxazolidinone H), 4.09–4.18 (br. t, 2H, oxazolidinone H, overlaps with oxazolidinone H triplet at 4.11, J = 9.1 Hz), 3.84–3.94 (br., 1H, methylene H), 3.73 (t, 5H, J = 4.6 Hz, morpholine H and methylene H), 2.96 (t, 4H, J = 4.6 Hz, morpholine H), 2.50 (br., 2H, methylene –CH2– overlapping with DMSO signal), 2.10–2.24 (br., 2H, methylene H), 1.20–1.64 (m, 20H, methylene H), 0.83–0.87 (m, 6H, two methyl H). 13 C-NMR (DMSO-d6, 600 MHZ): δ 154.53 (d, J = 244.26 Hz), 153.72, 135.56 (d, J = 8.77 Hz), 133.31 (d, J = 10.72 Hz), 119.18 (d, J = 4.01 Hz), 114.08 (d, J = 2.43 Hz), 106.66 (d, J = 26.01 Hz), 70.14, 66.10, 50.66, 50.65, 47.11, 31.07, 31.03, 28.34, 28.33, 28.25, 28.23, 23.89, 21.97, 13.86. MS ES+ (m/z): 564.3386 (M+ + H), MS (m/z): 563.3 (M+). Anal. Calcd. for C30H46FN3O6: C: 63.92; H: 8.23; N: 7.45; found C: 64.01; H, 8.26; N, 7.43. A solution of the intermediate PH-250 (2.15 g, 3.81 mmol) in MeOH:THF (56:14 ml) was treated with NaOH solution (305 mg in 20 ml water). Purification by recrystallisation (EtOAc-hexane 2:1) gave the titled compound PH-251 as an off-white solid 1.30 g, yield 78%, mp 119–122 °C. IR (KBr, cm−1): ν 3184, 2956, 2923, 2854, 1743, 1720, 1626, 1524, 1471, 1446, 1427, 1332, 1272, 1235, 1197, 1115, 1075. 1H-NMR (DMSO-d6, 600 MHZ): δ 9.93 (s, 1H, N–OH, exchangeable with D2O), 7.48 (dd, 1H, J = 2.5 Hz, 14.9 Hz, phenyl H), 7.17 (dd, 1H, J = 2.2 Hz, 8.8 Hz, phenyl H), 7.06 (t, 1H, J = 9.4 Hz, phenyl H), 4.85–4.89 (br. m, 1H, oxazolidinone H), 4.12 (t, 1H, J = 8.9 Hz, oxazolidinone H), 4.04 (dd, 1H, J = 6.7 Hz, 14.7 Hz, oxazolidinone H), 3.73–3.75 (m, 5H, morpholine H and methylene H), 3.67 (dd, 1H, J = 4.2 Hz, 14.9 Hz, methylene H), 2.96 (t, 4H, J = 4.6 Hz, morpholine H), 2.34–2.38 (m, 2H, NCOCH2CH2CH2CH2CH2CH2CH3), 1.44–1.50 (m, 2H, NCOCH2CH2CH2CH2CH2CH2CH3), 1.18–1.30 (m, 8H, NCOCH2CH2CH2CH2CH2CH2CH3), 0.85 (t, 3H, J = 7.0 Hz, NCOCH2CH2CH2CH2CH2CH2CH3). 13 C-NMR (DMSO-d6, 600 MHZ): δ 173.96, 154.56 (d, J = 244.06 Hz), 153.95, 135.51 (d, J = 8.78 Hz), 133.43 (d, J = 10.79 Hz), 119.21 (d, J = 4.06 Hz), 114.08 (d, J = 2.84 Hz), 106.64 (d, J = 26.00 Hz), 69.94, 66.13, 50.69, 50.68, 50.26, 47.44, 31.57, 31.14, 28.70, 28.49, 24.10, 22.04, 13.93.
MS ES+ (m/z): Calcd for C22H32FN3O5 + H: 438.2404, found: 438.2402 (M+ + H), Calcd. for C22H32FN3O5 + Na: 460.2224, found: 460.2214 (M+ + Na), MS (m/z): 437.2 (M+). Anal. Calcd. for C22H32FN3O5: C: 60.40; H: 7.37; N: 9.60; found C: 60.54; H, 7.10; N, 9.43.
4.2. Biological activity
4.2.1. General
The solvents, drugs and other reagents, including, polyethylene glycol (PEG), dimethylsulphoxide (DMSO), N-formyl methyl-leucyl-phenylalanine (FMLP), zileuton, zymosan, mouse anti-dinitrophenyl (DNP) IgE, dinitrophenyl-conjugated bovine serum albumin (DNP-BSA), foetal bovine serum (FBS), heparin, calcium ionophore A23187, LPS, DTT and glutathione peroxidase (GPx) were obtained from Sigma-Aldrich (St. Louis, MO). All compounds for in vitro experiments were solubilised in DMSO and diluted down in PBS. The final concentration of the solvent did not exceed 0.05%. For in vivo experiments, the compounds were made up in drug vehicle (4% DMSO/67.2% PEG/28.8% PBS).
4.2.2. In vitro assay for the inhibition of 5-LO product LTB4 from human whole blood
All compounds were evaluated for inhibitory activity against the 5-LO-dependent generation of LTB4 from activated human whole blood. With ethical approval from the Health Sciences Centre Ethical Committee of Kuwait University, heparinised fresh human blood samples from apparently healthy individuals of both sexes were obtained from the Kuwait Central Blood Bank. All donors gave informed consent and the work was carried out in accordance with the “Declaration of Helsinki” for experiments involving human subjects. Aliquots of 185 µl of whole blood were dispensed into each well of a 96-well culture plate containing 5 µl of the priming agent, LPS (1 µg/ml final concentration). After a 15 min incubation at 37 °C, 5 µl of the test compounds (0.001–30 µM) or the reference drug, zileuton (as positive control) or the vehicle (0.05% DMSO), was added. After further incubation for 15 min at 37 °C, 5 µl of FMLP (1 µM final concentration) was added to stimulate LT production. The reaction was stopped 15 min later, and the supernatants were recovered by centrifugation and stored at −40 °C pending analysis of LTB4 content.
4.2.3. In vitro assay for the inhibition of 5-LO product LTC4 from isolated human monocytes
Mononuclear cells were first isolated from heparinised blood using the Ficoll-Hypaque gradient centrifugation method. Monocytes were subsequently purified by adherence to plastic according to standard protocols. The purity of the adherent monocytes (CD14+) was routinely confirmed by flow cytometry to be >95% and viability (by trypan blue exclusion method) was routinely >97%. Adherent monocytes were then washed and incubated in 190 µl culture medium RPMI-1640 supplemented with 100 U/ml penicillin, 100 µg/ml streptomycin and 10% heat-inactivated foetal bovine serum (Sigma-Aldrich, St Louis, MO). Cells were then incubated with the various test compounds (0.001–30 μM) or vehicle (0.03% DMSO) or zileuton (positive control) for 15 min before being stimulated with 5 µl of the calcium ionophore A23187 at a final concentration of 2.5 µM. After further incubation for 15 min, the culture supernatants were recovered by centrifugation and stored at −40 °C pending LTC4 determination.
4.2.4. In vitro assay for the inhibition of 5-LO product LTC4 from allergen/IgE-activated bone marrow-derived mouse mast cells (BMMC)
Bone marrow-derived mast cells (BMMC) were generated from pathogen-free 5 to 7-weeks-old male Balb/c mice according to the method of Davis et al.Citation28. Essentially, bone marrow cells were obtained by flushing the femoral bone marrow and the recovered cells cultured in RPMI 1640 medium supplemented with 10% FBS, 100 U/ml penicillin, and 100 µg/ml streptomycin, 25 mM HEPES, 1.0 mM sodium pyruvate, 0.1 mM nonessential amino acids, 0.0035% 2-mercaptoethanol, and 30 ng/ml mouse recombinant IL-3, with culture medium, replaced every 2 days. Cells were used after 4–8 weeks of culture, by which time at least 97% of the cells would have differentiated into mast cells, as routinely obtained in our laboratory.
The generated BMMCs were seeded at 5 × 104 cells/well in a 96-well flat-bottom culture plate and passively sensitised overnight with 0.5 µg/ml anti-DNP monoclonal IgE antibody. The cells were then washed twice to remove any unbound antibody and subsequently resuspended in reaction buffer (135 mM NaCl, 5 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 5.6 mM glucose, 0.05% BSA and 20 mM HEPES, pH 7.4). They were then pre-incubated with the test compounds (0.001 − 30 μM) or the solvent (0.05% DMSO) for 15 min before being stimulated with the specific antigen, DNP-BSA (10–30 ng/ml). After 30 min incubation at 37 °C, the amount of LTC4 released into the supernatant was determined by ELISA as described below.
4.2.5. Assay of released leukotrienes
Appropriately diluted supernatants were assayed for the released products – LTs (LTB4 and LTC4), by the enzyme immunoassay (EIA) method using assay kits supplied by R&D Systems (Minneapolis, MN) and following the manufacturer’s instructions.
4.2.6. Evaluation of the direct inhibition of 5-LO activity in a cell-free assay
The assay was based on the oxidation of the dye 2′,7′-dichlorodihydrofluorescein diacetate (H2DCF-DA) to a highly fluorescent product by 5-LO enzymatic productsCitation29. H2DCF-DA (60 µM) was first pre-cleaved by incubating with 450 mU/ml recombinant human 5-LO enzyme in Tris buffer (containing 50 mM Tris, pH = 7.5, 2 mM EDTA, 2 mM CaCl2, 1 mM DTT and 0.6 U/ml glutathione peroxidase) for 10 min at room temperature. Then, to each well of a black 96-well plate was added 25 µl of the above enzyme/dye solution, followed by 25 µl of the test compounds or zileuton (0.001–30 µM) or drug vehicle, in duplicates. After 10 min incubation at room temperature, the reaction was started with the addition of 50 µl of substrate solution (Tris buffer containing 20 µM ATP and 20 µM arachidonic acid (AA). After a further 20 min, the reaction was terminated with 100 µl acetonitrile. Fluorescence was read at 500 nm excitation and 520 nm emission, with NovostarR microplate reader (BMG Labtech, Offenburg, Germany). Appropriate controls, including 100 µM nordihydroguaiaretic acid (NDGA), were included to isolate only the 5-LO-attributable, NDGA-inhibitable RFU values.
4.2.7. In vitro toxicity testing
Adherent human monocytes, prepared as described above were cultured with various concentrations of the test compounds or vehicle, or with 0.05% Triton-X as a positive control for 3 h or 24 h. At the end of the culture, cell viability was determined using the standard MTT assay method. Briefly, 15 μL of the MTT solution (5 mg/ml) was added to each well and incubated at 37 °C for 4 h. After removing the supernatant, 200 μL of DMSO was added to dissolve the crystals. Absorbance at 570 nm was then measured in a microplate reader. Viability was expressed as a percentage of the vehicle-treated cells.
4.2.8. Evaluation of in vivo activity in zymosan-induced peritoneal inflammation model in mice
Female Balb/c mice 6 to 8-weeks-old obtained from the Animal Resources Centre of the Health Sciences Centre, Kuwait University, were used. They were maintained under temperature-controlled conditions with an artificial 12-h light/dark cycle and allowed standard chow and water ad libitum. The study was carried out in compliance with the Regulations for the Use of Laboratory Animals in the Health Sciences Centre, Kuwait University, and complied with the National Institute of Health guide for the care and use of laboratory animals (NIH Publication # 8023, revised 1978).
The zymosan-induced peritonitis model - a recognised LT-mediated inflammatory reactionCitation26, was used. Five groups of mice, 7 per group, were treated subcutaneously with either PH-249 (the most active compound on human cells) at doses of 10 or 30 mg/kg or drug vehicle alone or zileuton for comparison. After 30 min, all groups were injected intraperitoneally with 0.2 ml of activated zymosan (2 mg/ml) except the control group that received 0.2 ml PBS. After 2 h, all animals were killed, and the peritoneal exudate collected by washing the cavity with 3 ml of heparinised (10 IU/ml) PBS. Cells in the exudate were recovered by centrifugation and counted in a haemocytometer while the supernatant volume was recorded and then stored frozen at −78 °C until used for the determination of LTC4 by ELISA as detailed above.
4.2.9. Statistical analysis
All data were analysed using GraphPad Prism software (GraphPad Software, San Diego, CA). The 50% inhibition concentration (IC50) values were determined from the concentration-response curves by non-linear regression analysis (normalised variable). Differences between experimental groups were first analysed by one-way ANOVA, followed by Bonferroni’s post-hoc test. A p-value of less than 0.05 was taken as statistically significant.
Ethical approval statement
Experiments involving living animals and their care were performed in strict compliance with the Regulations for the Use of Laboratory Animals in the Health Sciences Centre, Kuwait University, and complied with the National Institute of Health guide for the care and use of laboratory animals (NIH Publication # 8023, revised 1978). The protocol was approved by the Health Sciences Centre Animal Experimentation Ethical Committee of Kuwait University. All efforts were made to minimise the animal’s suffering and to reduce the number of animals used.
All experiments involving human blood were conducted with ethical approval from the Health Sciences Centre Ethical Committee of Kuwait University and carried out in accordance with the “Declaration of Helsinki” for experiments involving human subjects. All donors gave informed consent.
Supplemental Material
Download PDF (8.5 MB)Acknowledgements
The authors acknowledge the HSC Animal Resources Centre for assistance with research equipment and animals. The authors are also grateful to Ms. Elizabeth Philip, for her excellent technical assistance.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Ferguson AD, McKeever BM, Xu S, et al. Crystal structure of inhibitor-bound human 5-lipoxygenase-activating protein. Science 2007;317:510–2.
- Radmark O, Werz O, Steinhilber D, et al. 5-Lipoxygenase: regulation of expression and enzyme activity. Trends Biochem Sci 2007;32:332–41.
- Hofmann B, Steinhilber D. 5-Lipoxygenase inhibitors: a review of recent patents (2010-2012). Expert Opin Ther Pat 2013;23:895–909.
- Radmark O, Werz O, Steinhilber D, Samuelsson B. 5-Lipoxygenase, a key enzyme for leukotriene biosynthesis in health and disease. Biochim Biophys Acta 2015;1851:331–9.
- Montuschi P. Leukotrienes, antileukotrienes and asthma. Mini Rev Med Chem 2008;8:647–56.
- Riccioni G, Back M. Leukotrienes as modifiers of preclinical atherosclerosis? The Sci World J 2012;2012:490968.
- Steinhilber D, Hofmann B. Recent advances in the search for novel 5-lipoxygenase inhibitors. Basic Clin Pharmacol Toxicol 2014;114:70–7.
- Sarveswaran S, Chakraborty D, Chitale D, et al. Inhibition of 5-lipoxygenase selectively triggers disruption of c-Myc signaling in prostate cancer cells. J Biol Chem 2015;290:4994–5006.
- Pergola C, Werz O. 5-Lipoxygenase inhibitors: a review of recent developments and patents. Expert Opin Ther Pat 2010;20:355–75.
- Boudreau LH, Lassalle-Claux G, Cormier M, et al. New hydroxycinnamic acid esters as novel 5-lipoxygenase inhibitors that affect leukotriene biosynthesis. Mediators Inflamm 2017;2017:6904634.
- Boschi D, Giorgis M, Cena C, et al. Multitarget drugs: synthesis and preliminary pharmacological characterization of zileuton analogues endowed with dual 5-LO inhibitor and NO-dependent activities. ChemMedChem 2010;5:1444–9.
- Stewart AO, Bhatia PA, Martin JG, et al. Structure-activity relationships of N-hydroxyurea 5-lipoxygenase inhibitors. J Med Chem 1997;40:1955–68.
- Corey EJ, Cashman JR, Kantner SS, Wright SW. Rationally designed, potent competitive inhibitors of leukotriene biosynthesis. J Am Chem Soc 1984;106:1503–4.
- Summers JB, Gunn BP, Mazdiyasni H, et al. In vivo characterization of hydroxamic acid inhibitors of 5-lipoxygenase. J Med Chem 1987;30:2121–6.
- Summers JB, Gunn BP, Martin JG, et al. Structure-activity analysis of a class of orally active hydroxamic acid inhibitors of leukotriene biosynthesis. J Med Chem 1988;31:1960–4.
- Reck F, Zhou F, Girardot M, et al. Identification of 4-substituted 1,2,3-triazoles as novel oxazolidinone antibacterial agents with reduced activity against monoamine oxidase A. J Med Chem 2005;48:499–506.
- Bolasco A, Carradori S, Fioravanti R. Focusing on new monoamine oxidase inhibitors. Expert Opin Ther Pat 2010;20:909–39.
- Leach KL, Brickner SJ, Noe MC, Miller PF. Linezolid, the first oxazolidinone antibacterial agent. Ann N Y Acad Sci 2011;1222:49–54.
- Pandit N, Singla RK, Shrivastava B. Current updates on oxazolidinone and its significance. Int J Med Chem 2012;2012:159285.
- Phillips OA, Sharaf LH. Oxazolidinone antimicrobials: a patent review (2012-2015). Expert Opin Ther Pat 2016;26:591–605.
- Kombian SB, Phillips OA. In vitro electrophysiological investigations of the acute effects of linezolid and novel oxazolidinones on central nervous system neurons. Neuroscience 2011;180:53–63.
- Hedaya OM, Mathew PM, Mohamed FH, et al Antiproliferative activity of a series of 5-(1H-1,2,3-triazolyl) methyl- and 5-acetamidomethyl-oxazolidinone derivatives. Mol Med Rep 2016;13:3311–8.
- Qaddoumi MG, Phillips OA, Kombian SB. A novel oxazolidinone derivative PH192 demonstrates anticonvulsant activity in vivo in rats and mice. Eur J Pharm Sci 2019;130:21–6.
- Phillips OA, Udo EE, Ali AA, Al-Hassawi N. Synthesis and antibacterial activity of 5-substituted oxazolidinones. Bioorg Med Chem 2003;11:35–41.
- Phillips OA, D’Silva R, Bahta TO, et al. Synthesis and biological evaluation of novel 5-(hydroxamic acid)methyl oxazolidinone derivatives. Eur J Med Chem 2015;106:120–31.
- Hanke T, Dehm F, Liening S, et al Aminothiazole-featured pirinixic acid derivatives as dual 5-lipoxygenase and microsomal prostaglandin E2 synthase-1 inhibitors with improved potency and efficiency in vivo. J Med Chem 2013;56:9031–44.
- Adcock IM, Caramori G, Chung KF. New targets for drug development in asthma. Lancet 2008;372:1073–87.
- Davis BJ, Flanagan BF, Gilfillan AM, et al. Nitric oxide inhibits IgE-dependent cytokine production and Fos and Jun activation in mast cells. J Immunol 2004;173:6914–20.
- Pufahl RA, Kasten TP, Hills R, et al. Development of a fluorescence-based enzyme assay of human 5-lipoxygenase. Anal Biochem 2007;364:204–12.