
Abstract
Herein, two new series of N-substituted indole-based analogues were rationally designed, synthesized via microwave heating technology, and evaluated as noteworthy MAO-B potential inhibitors. Compared to the reported indazole-based hits VI and VII, compounds 4b and 4e exhibited higher inhibitory activities over MAO-B with IC50 values of 1.65 and 0.78 µM, respectively. When compared to the modest selectivity index of rasagiline (II, a well-known MAO-B inhibitor, SI > 50), both 4b and 4e also showed better selectivity indices (SI > 60 and 120, respectively). A further kinetic evaluation of the most potent derivative (4e) displayed a competitive mode of inhibition (inhibition constant (Ki)/MAO-B = 94.52 nM). Reasonable explanations of the elicited biological activities were presented via SAR study and molecular docking simulation. Accordingly, the remarkable MAO-B inhibitory activity of 4e (N-(1-(3-fluorobenzoyl)-1H-indol-5-yl)pyrazine-2-carboxamide), with its selectivity and competitive inhibition, advocates its potential role as a promising lead worthy of further optimization.
1. Introduction
Parkinson’s disease (PD), a neurodegenerative disorder with no currently available cure, is characterised by the impairment of motor function caused by the death of dopaminergic neurons in the substantia nigra pars compacta. Thus, the available therapies for ameliorating Parkinson’s symptoms mostly target dopamine depletion in the brainCitation1–6. Monoamine oxidase B (MAO-B), which is localized on the outer membrane of mitochondria (mainly in the liver and brain), catalyzes the oxidative deamination of monoamine neurotransmitters such as dopamine which results in hydrogen peroxide (H2O2) production leading to oxidative stress and neuronal cell deathCitation7–9. Recently, MAO-B has been reported to be highly expressed in the substantia nigra of PD patients. Thus, inhibition of MAO-B is effective in alleviating the symptoms of PD patients. Indeed, several substances that can effectively ameliorate emerging motor symptoms of PD are being used nowadays by physicians, amongst them levodopa, dopamine agonists, and MAO-B inhibitors. However, mainly due to their favourable safety profile in addition to their putative neuroprotective capabilities, MAO-B inhibitors may constitute a preferable therapeutic option for early PDCitation10,Citation11.
MAO-B is inhibited by different potent agents such as selegiline (I, ), rasagiline (II), safinamide (III), and sembragiline (IV), the most commonly used therapies for PD and Alzheimer’s disease (AD) via blocking dopamine degradation in the nigrostriatal pathway. However, numerous undesirable side effects of these inhibitors have been confirmed in long-term treatment of PD such as hallucinations, headaches, production of neurotoxic metabolites, sodium channel blockade, calcium channel modulation, and inhibition of stimulated release of glutamateCitation12–24. Not only for that reason, but also since there is no disease-modifying treatment has been discovered yet for PD, there is still a vital need to develop novel selective MAO-B inhibitors as promising therapeutically active candidates for PD patients. Hence, this encouraged our institute to launch a discovery project aiming at identification of novel promising MAO-B inhibitors. As a result, various active small molecules have been recently reported including unsaturated ketonesCitation25, oxazolopyridines and thiazolopyridinesCitation26, biaryl derivativesCitation27, in addition to indole-substituted benzothiazoles and benzoxazolesCitation28. Moreover, other research groups reported diverse MAO-B inhibitors belonging to different chemical scaffolds such as chalcones, pyrazoles, chromones, coumarins, xanthines, isatin derivatives, thiazolidindiones, (thiazol-2-yl)hydrazones, 1,5-diphenylpenta-2,4-dien-1-ones, in addition to indazole- and indole-5-carboxamidesCitation29–34.
Figure 1. Chemical structures of well-known MAO-B inhibitors.
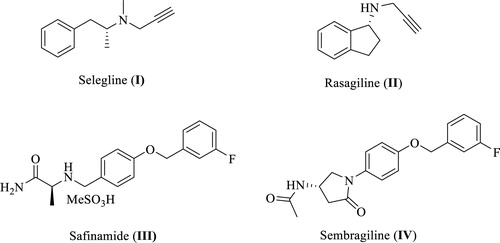
In the past few years, the indole moiety attracted the attention of medicinal chemists as a biologically accepted pharmacophore possessing wide spectrum of biological activitiesCitation35,Citation36. However, only few indole-based MAO-B inhibitors with high efficacy and promising safety profile were found in literature such as indole-based chalcones, indole-5-carboxamides, in addition to indole-substituted benzothiazoles and benzoxazolesCitation28,Citation33,Citation37,Citation38. One of these studies was carried out by Tzvetkov et al who preliminary prepared some indazole-based compounds (V, VI, and VII, ) to be evaluated over human MAO-B. While compound V was totally inactive, the biological results of VI and VII (MAO-B IC50=2590 and 3300 nM, respectively), coupled with their moderate selectivity values, were promising enough to pursue further structural modifications based on incorporation of a reverse amide linker to dramatically increase the activity and afford new highly potent indazole- and indole-5-carboxamidesCitation33. However, in their report, they mainly focussed on development of free NH indazole-based analogues with only two potent derivatives belonging to the indole scaffold. Accordingly, the current study took the optimization task for compounds VI and VII as a starting point towards novel N-substituted indole-based derivatives with potential inhibition against MAO-B.
Figure 2. Rational design of the newly synthesized analogues (series A and B).
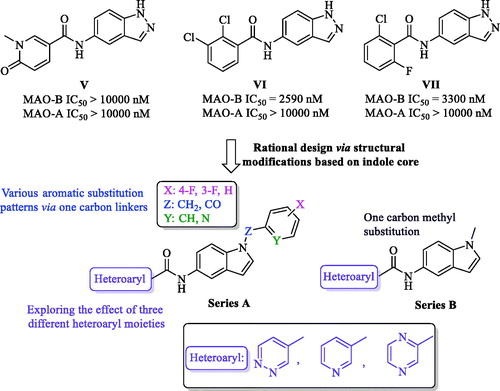
The docking model of the most potent indazole derivative discovered by Tzvetkov et al proposed the binding site of MAO-B having some space remains unoccupied and, in addition, the indazole free NH was predicted not to be incorporated in any type of H-bond interaction with the binding site. By searching literature, the scaffold modification of compounds VI and VII into indole core retaining the same original amide linker at position 5 was found to be a completely new approach and not to be yet studied. Inspired by these observations, diverse chemical structural alterations on compounds VI and VII were planned as depicted in ; two novel series were designed via replacing the indazole core with indole scaffold while keeping the original amide spacer of VI and VII into 5-position of the indole, which have, to the best of our knowledge, not been reported so far. In addition, aiming to design our derivatives armed with a potential capability to bind into the predicted unoccupied space (in the binding site of MAO-B reported by Tzvetkov et al), the free NH of the indole moiety was substituted by one carbon spacer (Z = CH2 or CO) connecting the indole scaffold to various aromatic substitution patterns in series A, while in series B, the indole nitrogen was linked to one carbon methyl group. Moreover, we incorporated three different six-membered aromatic heterocycles (pyridine, pyridazine, or pyrazine), not only anticipated to interact with the binding site of MAO-B in a similar fashion to the substituted aryl moieties in V and VI, but also, targeting possible extra-interaction(s) owing to the heteroatom(s) in these six-membered systems. Accordingly, in this study, we present the complete synthetic routes of the newly synthesized compounds through microwave heating technology, the enzymatic inhibitory activities and selectivity values over MAO enzymes, structure–activity relationship (SAR), a detailed kinetic study, in addition to computational docking models of the most active ligands with MAO enzymes.
2. Materials and methods
2.1. Chemistry
General: All solvents and reagents were obtained from commercial suppliers and used without further purification. Microwave-assisted synthetic procedure was applied in a Biotage Initiator + apparatus operating in single mode; the microwave cavity producing controlled irradiation at 2.45 GHz (Biotage AB, Uppsala, Sweden). The reactions were carried out in sealed vessels by employing magnetic stirring with maintaining the desired temperature for the programmed time period. TLC was performed using glass sheets pre-coated with silica gel 60 F254 purchased by Merck (Kenilworth, NJ). The NMR spectra were obtained on Bruker Avance 400 (Billerica, MA). Column chromatography was carried out on Merck Silica Gel 60 (230–400 mesh) (Kenilworth, NJ). High-resolution spectra were performed on Waters ACQUITY UPLC BEH C18 1.7 μ–Q-TOF SYNAPT G2-Si High Definition Mass Spectrometry.
2.1.1. General procedure for synthesis of compounds 2a–c
For compounds 2a–2c; NaH (60% in oil, 48.0 mg, 1.2 mmol) was added to a solution of 5-nitroindole (1, 162.2 mg, 1.0 mmol) dissolved in DMF (11 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 10 min. The appropriate reagent (1.0 mmol; 4-fluorobenzyl bromide for 2a, 3-fluorobenzoyl chloride for 2b, and 2-(chloromethyl)pyridine hydrochloride for 2c) was added to the mixture. The mixture was then stirred at 100 °C for 24 h, cooled to room temperature, and partitioned between EA and water using aqueous NaHCO3. The organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. Finally, the residue was purified by column chromatography (SiO2, EA/n-Hex) to get the target intermediate.
2.1.1.1. 1-(4-Fluorobenzyl)-5-nitro-1H-indole (2a)
Yellow solid, yield: 91%, mp: 126.4–127.0 °C, 1H NMR (400 MHz, CDCl3) δ 5.34 (s, 2H), 6.74 (s, 1H), 7.03 (d, J = 8.1 Hz, 2H), 7.08 (s, 2H), 7.28 (d, J = 10.6 Hz, 2H), 8.08 (d, J = 8.6 Hz, 1H), 8.60 (s, 1H). ReportedCitation39.
2.1.1.2. (3-Fluorophenyl)(5-nitro-1H-indol-1-yl)methanone (2b)
Yellowish white solid, yield: 67%, mp: 151.0–152.0 °C, 1H NMR (400 MHz, CDCl3) δ 6.79 (d, J = 3.7 Hz, 1H), 7.35–7.40 (m, 1H), 7.47–7.50 (m, 2H), 7.54–7.60 (m, 2H), 8.25 (dd, J = 2.2 Hz, 9.1 Hz, 1H), 8.47–8.51 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 109.24, 116.52, 116.53 (JC–F = 24.1 Hz), 117.20, 119.86 (JC–F = 21.1 Hz), 120.27, 125.06 (JC–F = 3.0 Hz), 130.06, 130.69 (JC–F = 5.0 Hz), 130.79, 135.29 (JC–F = 8.1 Hz), 138.97, 144.55, 162.51 (JC–F = 249.5 Hz), 167.08.
2.1.1.3. 5-Nitro-1-(pyridin-2-ylmethyl)-1H-indole (2c)
Yellow solid, yield: 24%, 1H NMR (400 MHz, CDCl3) δ 5.49 (s, 2H), 6.76–6.80 (m, 2H), 7.20–7.23 (m, 1H), 7.33 (d, J = 9.0 Hz, 1H), 7.37 (d, J = 3.2 Hz, 1H), 7.59 (td, J = 1.6 Hz, 7.7 Hz, 1H), 8.08 (dd, J = 2.1 Hz, 9.1 Hz, 1H), 8.61 (d, J = 1.9 Hz, 2H). ReportedCitation40,Citation41.
2.1.2. Synthesis of 1-methyl-5-nitro-1H-indole (2d)
K2CO3 (55.3 mg, 0.24 mmol) was added to a solution of 5-nitroindole (1, 162.2 mg, 1.0 mmol) in DMF (5 mL). Dimethyl carbonate (DMC) (0.17 mL, 2.0 mmol) was added and the reaction mixture was refluxed for 3 h, then cooled to 4–5 °C, followed by adding ice-cold water to the mixture. Precipitated solid was filtered and washed with water. Yellow solid, yield: 83%, mp: 164.7–165.9 °C, 1H NMR (400 MHz, CDCl3) δ 3.85 (s, 3H), 6.66 (d, J = 2.8 Hz, 1H), 7.21 (d, J = 2.9 Hz, 1H), 7.33 (d, J = 9.0 Hz, 1H), 8.11 (d, J = 9.0 Hz, 1H), 8.57 (s, 1H). ReportedCitation42.
2.1.3. General procedure of compound 3a–d
Each of compounds 2a−d (1.0 mmol) was dissolved in EtOH/H2O (5/2), followed by adding NH4Cl (267.5 mg, 5.0 mmol) and iron powder (558.5 mg, 10.0 mmol). The reaction mixture was refluxed for 1 h and filtered on celite then washed with EtOH. The residue was extracted using EA, water and aqueous NaHCO3. The organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified by column chromatography (SiO2, EA/n-Hex) to get the target intermediate.
2.1.3.1. 1-(4-Fluorobenzyl)-1H-indol-5-amine (3a)
Brown solid, yield: 80%, mp: 76.2–77.4 °C, 1H NMR (400 MHz, DMSO-d6) δ 4.49 (s, 2H), 5.27 (s, 2H), 6.18 (d, J = 2.8 Hz, 1H), 6.49 (dd, J = 1.8 Hz, 8.6 Hz, 1H), 6.69 (d, J = 1.6 Hz, 1H), 7.12 (t, J = 9.0 Hz, 3H), 7.20 (t, J = 8.5 Hz, 2H), 7.28 (d, J = 3.0 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 48.80, 99.98, 104.01, 110.64, 112.32, 115.65 (JC–F = 21.1 Hz), 128.94, 129.41 (JC–F = 8.0 Hz), 129.80, 130.07, 135.35 (JC–F = 3.0 Hz), 142.00, 161.79 (JC–F = 243.5 Hz). ReportedCitation43,Citation44.
2.1.3.2. (5-Amino-1H-indol-1-yl)(3-fluorophenyl)methanone (3b)
Orange solid, yield: 91%, mp: 91.7–92.9 °C, 1H NMR (400 MHz, DMSO-d6) δ 5.06 (s, 2H), 6.54 (d, J = 3.7 Hz, 1H), 6.68 (dd, J = 2.1 Hz, 8.7 Hz, 1H), 6.77 (d, J = 2.0 Hz, 1H), 7.19 (d, J = 3.7 Hz, 1H), 7.49–7.66 (m, 4H), 8.01 (d, J = 8.7 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 104.60, 109.35, 113.38, 116.07 (JC–F = 23.1 Hz), 116.81, 118.88 (JC–F = 21.1 Hz), 125.23 (JC–F = 3.0 Hz), 128.06, 128.23, 131.36 (JC–F = 8.1 Hz), 132.34, 137.14 (JC–F = 7.0 Hz), 146.25, 162.17 (JC–F = 245.5 Hz).
2.1.3.3. 1-(Pyridin-2-ylmethyl)-1H-indol-5-amine (3c)
Light orange solid, yield: 72%, mp: 94.1–95.3 °C, 1H NMR (400 MHz, DMSO-d6) δ 4.51 (s, 2H), 5.38 (s, 2H), 6.22 (s, 1H), 6.49 (d, J = 8.2 Hz, 1H), 6.71 (s, 1H), 6.86 (d, J = 7.7 Hz, 1H), 7.07 (d, J = 8.5 Hz, 1H), 7.26–7.30 (m, 2H), 7.69 (t, J = 6.8 Hz, 1H), 8.54 (d, J = 3.5 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 51.69, 100.09, 104.01, 110.57, 112.34, 121.39, 122.91, 129.30, 129.76, 130.26, 137.45, 142.05, 149.55, 158.41. ReportedCitation45.
2.1.3.4. 1-Methyl-1H-indol-5-amine (3d)
Brown solid, yield: 78%, mp: 103.0–103.9 °C, 1H NMR (400 MHz, DMSO-d6) δ 3.68 (s, 3H), 4.48 (s, 2H), 6.12 (d, J = 2.7 Hz, 1H), 6.56 (dd, J = 2.0 Hz, 8.6 Hz, 1H), 6.70 (d, J = 1.8 Hz, 1H), 7.09–7.12 (m, 2H). ReportedCitation46,Citation47.
2.1.4. General procedure of compounds 4a–l
To an MW vial, were successively added compound 3 (0.1 mmol), HATU (41.8 mg, 0.11 mmol), DIPEA (0.05 mL, 0.27 mmol), the appropriate heteroaryl carboxylic acid derivative (0.11 mmol) and DMF (12 mL) at room temperature. The MW vial was sealed and heated under MW conditions for 45 min at 116 °C. The reaction mixture was extracted using EA, water and brine. The organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified by column chromatography (SiO2, EA/n-Hex) to get the desired target compound.
2.1.4.1. N-(1-(4-fluorobenzyl)-1H-indol-5-yl)pyridazine-4-carboxamide (4a)
Yellowish green solid, yield: 46%, mp: 212.6–213.1 °C, 1H NMR (400 MHz, DMSO-d6) δ 5.42 (s, 2H), 6.52 (d, J = 2.6 Hz, 1H), 7.15 (t, J = 8.8 Hz, 2H), 7.26 (t, J = 8.0 Hz, 2H), 7.41 (d, J = 8.0 Hz, 1H), 7.48 (d, J = 8.6 Hz, 1H), 7.54 (d, J = 2.8 Hz, 1H), 8.04 (s, 1H), 8.13 (d, J = 2.9 Hz, 1H), 9.48 (d, J = 4.7 Hz, 1H), 9.67 (s, 1H), 10.59 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 48.89, 101.82, 110.57, 113.13, 115.80 (JC–F = 21.1 Hz), 116.40, 124.92, 128.58, 129.53 (JC–F = 8.1 Hz), 130.39, 130.99, 132.98, 133.59, 134.93 (JC–F = 3.0 Hz), 149.47, 152.54, 161.89 (JC–F = 243.5 Hz), 162.29.
2.1.4.2. N-(1-(4-fluorobenzyl)-1H-indol-5-yl)pyrazine-2-carboxamide (4b)
Light yellow solid, yield: 73%, mp: 161.5–162.0 °C, HPLC purity: 6.02 min, 100%, 1H NMR (400 MHz, DMSO-d6) δ 5.42 (s, 2H), 6.51 (d, J = 3.0 Hz, 1H), 7.15 (t, J = 8.9 Hz, 2H), 7.26–7.30 (m, 2H), 7.46 (d, J = 8.8 Hz, 1H), 7.53–7.56 (m, 2H), 8.16 (d, J = 1.5 Hz, 1H), 8.82 (t, J = 1.5 Hz, 1H), 8.93 (d, J = 2.4 Hz, 1H), 9.32 (d, J = 1.2 Hz, 1H), 10.58 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 48.88, 101.76, 110.48, 113.00, 115.78 (JC–F = 21.1 Hz), 116.53, 128.55, 129.55 (JC–F = 9.1 Hz), 130.27, 130.87, 133.48, 134.95 (JC–F = 4.0 Hz), 143.65, 144.35, 145.86, 147.92, 161.60, 161.88 (JC–F = 243.5 Hz). HRMS (ESI) m/z calcd. for C20H15FN4O [M + H]+: 347.1308. Found: 347.1306.
2.1.4.3. N-(1-(4-fluorobenzyl)-1H-indol-5-yl)nicotinamide (4c)
White solid, yield: 53%, mp: 200.0–200.4 °C, 1H NMR (400 MHz, DMSO-d6) δ 5.42 (s, 2H), 6.50 (d, J = 3.0 Hz, 1H), 7.15 (t, J = 8.9 Hz, 2H), 7.25–7.28 (m, 2H), 7.44 (dd, J = 8.8 Hz, 17.8 Hz, 2H), 7.53 (d, J = 3.0 Hz, 1H), 7.56–7.59 (m, 1H), 8.04 (s, 1H), 8.31 (d, J = 8.0 Hz, 1H), 8.76 (d, J = 3.5 Hz, 1H), 9.13 (d, J = 1.4 Hz, 1H), 10.34 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 48.87, 101.72, 110.42, 113.08, 115.77 (JC–F = 22.1 Hz), 116.61, 123.93, 128.58, 129.51 (JC–F = 9.1 Hz), 130.20, 131.37, 131.49, 133.39, 134.98 (JC–F = 3.0 Hz), 135.78, 161.88 (JC–F = 242.5 Hz), 164.06.
2.1.4.4. N-(1-(3-fluorobenzoyl)-1H-indol-5-yl)pyridazine-4-carboxamide (4d)
Yellowish white solid, yield: 75%, mp: 196.7–196.9 °C, 1H NMR (400 MHz, DMSO-d6) δ 6.84 (d, J = 3.6 Hz, 1H), 7.44 (d, J = 3.6 Hz, 1H), 7.54–7.71 (m, 5H), 8.16–8.18 (m, 1H), 8.23 (s, 1H), 8.33 (d, J = 8.9 Hz, 1H), 9.52 (d, J = 4.9 Hz, 1H), 9.70 (s, 1H), 10.85 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 109.54, 113.17, 116.26, 116.51, 118.57, 119.37 (JC–F = 20.1 Hz), 125.01, 125.55 (JC–F = 3.0 Hz), 129.45, 131.40, 131.45, 131.53, 132.75 (JC–F = 5.0 Hz), 135.08, 136.57 (JC–F = 7.04 Hz), 149.46, 152.58, 162.20 (JC–F = 245.5 Hz), 162.75, 167.18.
2.1.4.5. N-(1-(3-fluorobenzoyl)-1H-indol-5-yl)pyrazine-2-carboxamide (4e)
White solid, yield: 94%, mp: 221.3–222.2 °C, HPLC purity: 6.00 min, 100%, 1H NMR (400 MHz, DMSO-d6) δ 6.81 (d, J = 2.8 Hz, 1H), 7.43 (d, J = 3.0 Hz, 1H), 7.56 (t, J = 8.4 Hz, 1H), 7.61–7.67 (m, 3H), 7.86 (d, J = 8.9 Hz, 1H), 8.28 (d, J = 8.8 Hz, 1H), 8.32 (s, 1H), 8.85 (s, 1H), 8.96 (s, 1H), 9.34 (s, 1H), 10.85 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 109.54, 113.16, 116.32, 116.37 (JC–F = 23.1 Hz), 118.77, 119.36 (JC–F = 20.1 Hz), 125.55, 129.33, 131.33, 131.49 (JC–F = 8.1 Hz), 132.61, 134.97, 136.59 (JC–F = 8.1 Hz), 143.72, 144.49, 145.65, 148.13, 162.11, 162.20 (JC–F = 245.5 Hz), 167.13. HRMS (ESI) m/z calcd. for C20H13FN4O2 [M + H]+: 361.1101. Found: 361.1095.
2.1.4.6. N-(1-(3-fluorobenzoyl)-1H-indol-5-yl)nicotinamide (4f)
White solid, yield: 57%, mp: 212.9–214.2 °C, 1H NMR (400 MHz, DMSO-d6) δ 6.82 (d, J = 3.6 Hz, 1H), 7.43 (d, J = 3.6 Hz, 1H), 7.56–7.73 (m, 6H), 8.23 (d, J = 1.5 Hz, 1H), 8.30 (d, J = 8.9 Hz, 1H), 8.35 (d, J = 7.9 Hz, 1H), 8.78–8.79 (m, 1H), 9.16 (d, J = 1.5 Hz, 1H), 10.58 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 109.57, 113.07, 116.36 (JC–F = 24.1 Hz), 116.38, 118.63, 119.34 (JC–F = 21.1 Hz), 123.99, 125.51, 129.29, 131.12, 131.37, 131.49 (JC–F = 8.1 Hz), 132.46, 135.64, 135.92, 136.63 (JC–F = 7.0 Hz), 149.15, 152.55, 162.20 (JC–F = 245.5 Hz), 164.47, 167.15.
2.1.4.7. N-(1-(pyridin-2-ylmethyl)-1H-indol-5-yl)pyridazine-4-carboxamide (4g)
Yellow solid, yield: 85%, mp: 141.8–143.0 °C, 1H NMR (400 MHz, DMSO-d6) δ 5.52 (s, 2H), 6.54 (d, J = 2.7 Hz, 1H), 6.96 (d, J = 7.8 Hz, 1H), 7.28 (t, J = 5.3 Hz, 1H), 7.39–7.45 (m, 2H), 7.53 (d, J = 2.9 Hz, 1H), 7.71 (t, J = 6.9 Hz, 1H), 8.06 (s, 1H), 8.13–8.14 (m, 1H), 8.55 (d, J = 4.4 Hz, 1H), 9.49 (d, J = 5.1 Hz, 1H), 9.67 (s, 1H), 10.61 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 51.70, 101.85, 110.57, 113.13, 116.44, 121.53, 123.10, 124.93, 128.55, 130.77, 131.01, 132.98, 133.84, 137.59, 149.48, 149.72, 152.55, 157.89, 162.29.
2.1.4.8. N-(1-(pyridin-2-ylmethyl)-1H-indol-5-yl)pyrazine-2-carboxamide (4h)
Light yellow solid, yield: 39%, mp: 152.0–152.9 °C, 1H NMR (400 MHz, DMSO-d6) δ 5.51 (s, 2H), 6.52 (d, J = 3.0 Hz, 1H), 6.96 (d, J = 7.8 Hz, 1H), 7.26–7.29 (m, 1H), 7.41 (d, J = 8.8 Hz, 1H), 7.51–7.53 (m, 2H), 7.71 (td, J = 1.6 Hz, 7.6 Hz, 1H), 8.15 (d, J = 1.5 Hz, 1H), 8.54 (d, J = 4.6 Hz, 1H), 8.81 (d, J = 1.5 Hz, 1H), 8.92 (d, J = 2.4 Hz, 1H), 9.31 (s, 1H), 10.57 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 51.70, 101.80, 110.47, 113.00, 116.56, 121.55, 123.09, 128.52, 130.67, 130.89, 133.74, 137.59, 143.66, 144.36, 145.87, 147.93, 149.71, 157.93, 161.62.
2.1.4.9. N-(1-(pyridin-2-ylmethyl)-1H-indol-5-yl)nicotinamide (4i)
Light brown solid, yield: 38%, mp: 142.9–144.3 °C, 1H NMR (400 MHz, DMSO-d6) δ 5.52 (s, 2H), 6.52 (d, J = 2.9 Hz, 1H), 6.95 (d, J = 7.8 Hz, 1H), 7.29 (t, J = 4.9 Hz, 1H), 7.41 (s, 2H), 7.52 (d, J = 2.9 Hz, 1H), 7.56–7.59 (m, 1H), 7.70–7.74 (m, 1H), 8.05 (s, 1H), 8.31 (d, J = 7.8 Hz, 1H), 8.55 (d, J = 4.2 Hz, 1H), 8.76 (d, J = 3.5 Hz, 1H), 9.13 (s, 1H), 10.32 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 51.71, 101.76, 110.41, 113.10, 116.65, 121.51, 123.08, 123.93, 128.55, 130.59, 131.37, 131.52, 133.64, 135.79, 137.58, 149.07, 149.70, 152.31, 157.96, 164.08.
2.1.4.10. N-(1-methyl-1H-indol-5-yl)pyridazine-4-carboxamide (4j)
Yellowish green solid, yield: 28%, mp: 181.1–182.0 °C, 1H NMR (400 MHz, DMSO-d6) δ 3.81 (s, 3H), 6.45 (d, J = 3.0 Hz, 1H), 7.36 (d, J = 3.0 Hz, 1H), 7.47 (s, 2H), 8.04 (s, 1H), 8.15 (dd, J = 2.3 Hz, 5.3 Hz, 1H), 9.50 (dd, J = 1.0 Hz, 5.3 Hz, 1H), 9.69 (t, J = 1.0 Hz, 1H), 10.61 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 33.03, 100.94, 110.08, 112.94, 116.15, 124.91, 128.20, 130.77, 130.94, 133.03, 134.35, 149.49, 152.53, 162.25.
2.1.4.11. N-(1-methyl-1H-indol-5-yl)pyrazine-2-carboxamide (4k)
Brown solid, yield: 63%, mp: 172.5–174.0 °C, 1H NMR (400 MHz, DMSO-d6) δ 3.80 (s, 3H), 6.44 (s, 1H), 7.34 (s, 1H), 7.44 (d, J = 7.8 Hz, 1H), 7.61 (d, J = 7.1 Hz, 1H), 8.15 (s, 1H), 8.83 (s, 1H), 8.94 (s, 1H), 9.32 (s, 1H), 10.57 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 33.01, 100.90, 110.00, 112.72, 116.22, 128.19, 130.67, 130.83, 134.26, 143.66, 144.34, 145.94, 147.90, 161.59.
2.1.4.12. N-(1-methyl-1H-indol-5-yl)nicotinamide (4l)
White solid, yield: 22%, mp: 164.2–164.9 °C, 1H NMR (400 MHz, DMSO-d6) δ 3.80 (s, 3H), 6.43 (d, J = 2.9 Hz, 1H), 7.34 (d, J = 3.0 Hz, 1H), 7.42–7.49 (m, 2H), 7.56–7.60 (m, 1H), 8.03 (d, J = 1.2 Hz, 1H), 8.32–8.34 (m, 1H), 8.77 (dd, J = 1.4 Hz, 4.8 Hz, 1H), 9.15 (d, J = 1.7 Hz, 1H), 10.33 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 33.01, 100.84, 109.91, 112.89, 116.37, 123.92, 128.21, 130.74, 131.27, 131.41, 134.16, 135.77, 149.07, 152.27, 164.03.
2.2. Monoamine oxidase enzyme assay
The inhibitory activity of MAO-A, B enzyme of the compounds was evaluated based on previously described methodCitation25. The MAO activity was measured in relative fluorescence units (RFUs) evoked by peroxidase-catalyzed oxidation of Amplex Red® to resorufin, in which H2O2 generated by MAO reaction was used as the electron donor. Human recombinant MAO-A (hMAO-A) and MAO-B (hMAO-B) enzyme expressed in insect cells were obtained from Sigma-Aldrich (St. Louis, MO). The 2 μL of test compound in DMSO (final concentration: 1 nM to 10 μM) was treated with 98 μL of hMAO enzyme solution in 50 mM sodium phosphate buffer (pH 7.4, final protein amounts: ∼1.25 μg protein/well for MAO-A and ∼2.5 μg protein/well for MAO-B) on 96-well black plate and incubated for 15 min at 37 °C. Then, 100 μL of reaction working solution which is mixed solution of 400 μM Amplex Red® (Cayman, Ann Arbor, MI, final concentration: 200 μM), 2 U/mL horseradish peroxidase (Sigma-Aldrich, St. Louis, MO, final concentration: 1 U/mL) and 2 mM substrate (p-tyramine for MAO-A, benzylamine for MAO-B, Sigma-Aldrich, St. Louis, MO, final concentration: 1 mM) in 50 mM sodium phosphate buffer (pH 7.4) were added and incubated for 20 min at 37 °C in the dark. The fluorescent intensity was quantified using a microplate reader (SpectraMax®i3, Molecular Devices, Sunnyvale, CA) with an excitation at 545 nm and an emission at 590 nm. The 50% inhibitory concentrations (IC50) of compounds were determined as the mean ± SEM in triplicate from the dose–response inhibition curves using SigmaPlot® 13.0 (Palo Alto, CA).
2.3. Kinetic study of MAO-B inhibition mode
The type of MAO-B inhibition of compound 4e was determined by the Michaelis–Menten kinetic study. The catalytic rates of hMAO-B enzyme in the absence or in the presence of three different concentrations (10, 30, and 100 nM) of compound 4e were measured at seven different concentrations of the benzylamine (0.065, 0.125, 0.25, 0.5, 1, 2, and 4 mM). The corresponding progression curves and the Lineweaver–Burk plots were generated using GraphPad Prism 7 (La Jolla, CA). The maximal velocity (Vmax), the Michaelis constant (Km), and the inhibition constant (Ki) were calculated using SigmaPlot® 13.0 (Palo Alto, CA).
2.4. Molecular modelling study
Crystal structure of MAO-A (PDB ID: 2Z5X)Citation48 and MAO-B (PDB code: 2V5Z)Citation31 protein downloaded from protein data bank (www.pdb.org). MAO-B structure (PDB code: 2V5Z) is complexed with safinamide and MAO-A structure (PDB ID: 2Z5X) complexed with harmine (7-methoxy-1-methyl-9h-beta-carboline). Protein structures prepared using the protein preparation wizardCitation49 of Schrodinger 2018 suite of the package (Schrödinger LLC, New York, NY) at the default setting and 7.4 pH value. All the inhibitors were sketched using ChemDraw Professional 16.0 and saved as structure data file format and imported to Ligprep module. Ligprep module of Schrodinger was used to prepare all ligands, and further geometry optimized. Re-docking X-ray ligands confirmed the reproducibility of the docking program (data not shown). All minimised conformations of ligands were docked into the safinamide binding site using Glide's standard precision moduleCitation50 and generated 10 poses for each ligand. The docking figures were rendered using the Discovery Studio Client 2019 package (Dassault Systèmes; BIOVIA. Discovery Studio Modeling Environment; Release 2019; Dassault Systèmes: San Diego, CA, 2019). We selected the docked poses with more negative Glide docking scores and significant interactions with MAO-B protein.
3. Results and discussion
3.1. Chemical synthesis
The newly synthesized target compounds 4a−l were prepared as outlined in Scheme 1. Starting from the commercially available 5-nitroindole (1), substitution on position N1 of the indole core was carried out using 4-fluorobenzyl bromide for 2a, 3-fluorobenzoyl chloride for 2b, and 2-(chloromethyl)pyridine hydrochloride for 2c, in presence of NaH (60% in oil) and dimethylformamide (DMF) solvent to yield analogues 2a−cCitation39–41. Dimethyl carbonate reagent was used as a methyl group donor to the amine moiety in compound 1 by heating both in presence of potassium carbonate as a basic catalyst to give compound 2dCitation42. Preparation of intermediates 3a−d, required for synthesis of the final analogues, was performed by reduction of 5-nitro group in 2a−d using iron powder as a dissolving metal in a slightly acidic medium (ammonium chloride)Citation51. With intermediates in hand, the last step in the synthetic route was achieved via amide coupling reactions between the free amine groups in 3a−d and the appropriate heteroaryl carboxylic acid in presence of DIPEA and HATU to afford the target small molecules 4a−l (full chemical structures, ).
Scheme 1. Reagents and conditions: (a) appropriate halide derivative, NaH (60% in oil), DMF, 0–100 °C, 24 h; (b) dimethyl carbonate, K2CO3, DMF, reflux, 3 h; (c) NH4Cl, Fe, EtOH/H2O, reflux, 1 h; (d) appropriate heteroaryl carboxylic acid, DIPEA, HATU, DMF, MW, 116 °C, 45 min.
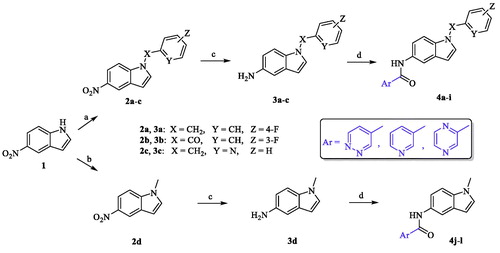
Table 1. Inhibitory effects of the synthesized compounds against MAO-B.
3.2. MAO-B assay
3.2.1. Inhibition of MAO-B by the prepared compounds 4a–4l at single dose of 10 µM
The target compounds 4a−l were biologically evaluated for their potential inhibitory activities over human MAO-B recombinant enzyme. The enzyme inhibition assay of the tested compounds was conducted using Amplex® Red reagent technology which offers quantitative enzyme activity sensitive detection with extended dynamic range compared to classic colorimetric oxidase assays in addition to its advantage of the fluorescence emission outside the range of compound autofluorescence (if any). Since Amplex® Red reagent is a colourless substrate able to react with H2O2 with a 1:1 stoichiometry to yield highly fluorescent resorufin, the elicited inhibitory activities of the tested compounds over MAO-B were evaluated via spectrophotometrical measurement and the fluorescence rate of the resorufin dye formation was detected. As mentioned previously, all the target compounds were designed to incorporate the same amide moiety in VI and VII at position 5 of the indole scaffold connecting one of three different heteroaromatic cycles with the indole core which is substituted with either aromatic or aliphatic moiety. First, a set of nine compounds (4a−i, series A) was prepared possessing varied aromatic functional groups on N1 of the indole. By analyzing the obtained preliminary biological results of series A (), it was confirmed that our rational design of retaining the original amide spacer of VI and VII with the N-substituted indole core is still able to yield even more active MAO-B inhibitors (4b and 4e). In addition, we noticed that the benzyl and benzoyl moieties, bearing the electron-withdrawing 3- or 4-fluoro groups in 4a−f, exerted better inhibition than the pyridylmethyl substituent (4g−i). Also, while modest inhibition values were elicited by analogues 4a, 4c, 4d, and 4f possessing either benzyl or benzoyl moiety on the indole ring (13.15 ± 3.32%, 11.24 ± 0.24%, 19.08 ± 0.47%, and 30.29 ± 0.24%, respectively), it was noted that only two heteroaryl cycles (pyridine and pyridazine) attached to the amide spacer were represented in the chemical structures of these four compounds. Interestingly, analogues 4b and 4e having the 2-pyrazinyl moiety and 4-fluorobenzyl moiety or 3-fluorobenzoyl moiety, respectively, were the most active compounds in this series with inhibition values of 88.49 ± 0.13% for 4b and 66.39 ± 0.79% for 4e. However, as mentioned, when the substitution on N1 of the indole scaffold was changed to the pyridylmethyl group, the inhibitory activity over MAO-B was dramatically decreased to disclose low active derivatives 4g, 4h, and 4i (1.83 ± 0.75%, 2.38 ± 0.66%, and 1.81 ± 0.85%, respectively). It is also noteworthy to mention that the three different heteroaryl cycles incorporated in our design (pyridazine, pyrazine, and pyridine) were represented in each of the last three compounds. Thus, taking into consideration the significant difference of the biological activity between compounds 4b and 4h possessing the same heteroaryl cycle (2-pyrazinyl), we have concluded that not only the type of the heteroaryl cycle attached to the amide linker is an essential factor to design a highly active MAO-B inhibitor, but also the nature of N1 substituent of the indole core; both are necessary elements to acquire a higher MAO-B inhibitory effect.
Next, we totally changed the aromatic substitution on N1 of the indole moiety to methyl group to acquire series B (4j−l). It was noted that the inhibitory activities elicited by compounds 4j−l significantly increased compared to those of 4g−i having pyridylmethyl group (9.52 ± 0.61%, 49.68 ± 0.68%, and 11.57%±0.9 for compounds 4j, 4k, and 4l, respectively). It was important to keep the three heteroarylcycles fully represented in this series (4j−l) in order to outline a fine-tuned interpretation about their inhibitory effect over MAO-B. Interestingly, compound 4k possessing 2-pyrazinyl ring attached to the amide linker showed higher inhibitory effect than compounds 4j and 4l, which reconfirms the value of the 2-pyrazinyl heterocycle as essential factor to obtain higher inhibitory effect over MAO-B.
3.2.2. Dose dependent assay of the most active analogues 4b and 4e over MAO-B
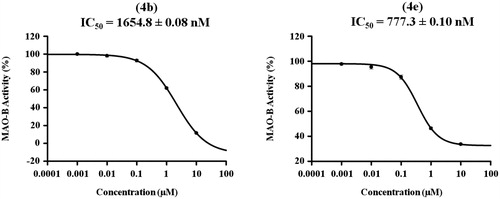
The inhibitory potencies (IC50 values) of compounds producing inhibition of MAO-B activity by more than 50% were determined using five doses assay with 10, 1, 0.1, 0.01, and 0.001 μM concentrations of the tested compounds. Consequently, analogues 4b and 4e were further evaluated to determine their IC50 over MAO-B in triplicate from the dose response inhibition curves using Sigma-Plot software version 13.0 as indicated in . While compound 4b exerted low micromolar IC50 value of 1654.8 ± 0.08 nM, a higher potency in sub-micromolar range elicited by analogue 4e (777.3 ± 0.10 nM).
Figure 3. Dose dependent assay of compounds 4b and 4e over MAO-B.
3.2.3. Selectivity assay of compounds 4b and 4e
It is known that non-selective (MAO-A/B combined) inhibitors lead to several problems when taken concomitantly with tyramine-containing foods (such as cheese) since the drug's inhibition of MAO-A causes a dangerous elevation of serum tyramine levels, which can cause hypertensive symptomsCitation52. On the other side, selective MAO-B inhibitors can bypass this problem by preferentially inhibiting MAO-B. Thus, to check the selectivity of the most active derivatives for the MAO-B isoform, a selectivity assay was performed by testing analogues 4b and 4e over MAO-A at a high dose concentration of 100 µM followed by detection of their IC50 values. As presented in , both derivatives exhibited very weak inhibitory effect over MAO-A even with a highly concentrated dose of 100 µM (12.95 ± 1.09% and 4.76 ± 0.59% for 4b and 4e, respectively). To calculate the selectivity index (SI: the selectivity for the MAO-B isoform given as the ratio of IC50 (MAO-A)/IC50 (MAO-B)), IC50 values of both compounds have been identified and found to be more than 100 µM. Consequently, the selectivity indices were calculated as illustrated in . While compound 4b showed selectivity index > 60 for MAO-B isoform, the most active derivative 4e has a higher SI value (>120). Thus, compared to the modest selectivity index of rasagiline (II, calculated SI > 50Citation33) both 4b and 4e were more selective for MAO-B. Accordingly, compound 4e was selected for further evaluation to define its kinetic mode of interaction with the human MAO-B enzyme.
Table 2. Inhibitory effects of the compounds 4b and 4e against MAO-A.
3.2.4. Kinetic study to define the interaction mode of compound 4e with MAO-B
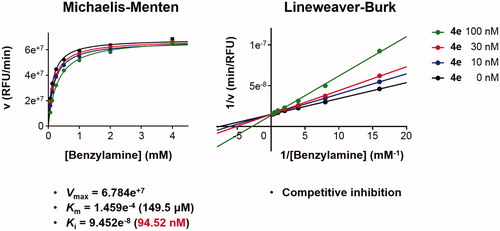
Detailed substrate-dependent kinetic experiments were carried out to examine the mode of MAO-B inhibition of compound 4e. The initial rates of MAO-B inhibition in the absence and presence of 4e were measured at various concentrations of benzylamine, a selective substrate for MAO-B, and both Michaelis–Menten and the Lineweaver–Burk plots are depicted in . The Lineweaver–Burk plots for three different concentration of 4e were linear and intersected at the y-axis. The maximal velocity (Vmax), the Michaelis constant (Km), and the inhibition constant (Ki) were calculated using SigmaPlot® (Palo Alto, CA) (Vmax = 6.784e+7, Km = 1.459e−4, and Ki = 9.452e−8). Hence, it can be stated that compound 4e is competitive, and accordingly, reversible MAO-B inhibitor.
Figure 4. Type of inhibition of MAO-B by compound 4e. The catalytic rates were measured at different concentrations of benzylamine (0.065, 0.125, 0.25, 0.5, 1, 2, and 4 mM) in the absence and in the presence of different concentrations (10, 30, and 100 nM) of compound 4e. The Vmax, Km, and Ki values were calculated using SigmaPlot®.
3.3. Molecular docking
In order to provide deeper insights into the SAR of the new series, the most active compounds 4b and 4e were selected to be docked into the active binding region of MAO-B. Therefore, the docking studies were conducted in order to determine the in silico binding modes of these compounds, evaluate the contribution of the substitution on N1 of the indole moiety, understand the essential incorporation effect of 2-pyrazinyl heterocycle to MAO-B enzyme activity, gain insights for the selectivity of this class of compounds for MAO-B isoform, and finally to prove the non-covalent reversible binding of this class of inhibitors to MAO-B. The X-ray crystal structures of MAO-B (PDB ID: 2V5Z)Citation31 were obtained from Protein Data Bank (www.pdb.org). The docked model of the most active compounds 4b and 4e is demonstrated in ; 4b (A) and 4e (B), while shows their two-dimensional (2D) interaction model; 4b (A) and 4e (B).
Figure 5. The docked model of the most active compounds 4b (A) and 4e (B) into MAO-B binding pocket.
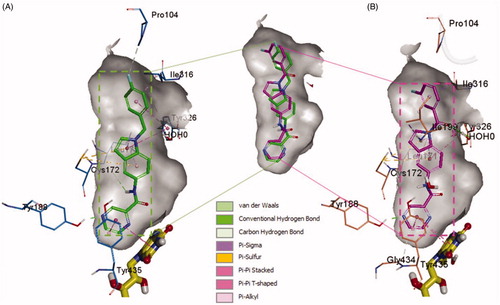
Figure 6. Two-dimensional (2D) interaction model for 4b (A) and 4e (B) with MAO-B.
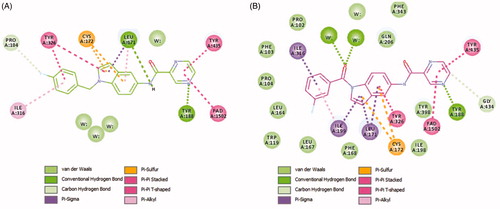
For compound 4b (Glide score −10.62) (), the central indole ring interacted via π–π-stacking, π–sulphur, and π–sigma interactions with Tyr326, Cys172, and Leu171, respectively. The pyrazine ring interacts via an H-bond, π–π stacking, and T-shaped π–π-stacking interactions with Tyr188, and Tyr435, and fused ring of FAD, respectively. The amide “NH” forms an H-bond with the main chain carbonyl of Leu171 while 4-fluorobenzyl moiety forms π–sigma and π–π-stacking interactions with the Ile316 and Tyr326, respectively. It was noticed that Fluorine atom at the para position contributed to week carbon–hydrogen bond with Pro104. Interestingly, a similar binding pose of 4e (Glide score −10.79) was observed in the binding pocket of MAO-B with a shifted orientation (). The indole ring packed at the centre of the binding cavity having π-sigma interactions with Ile199, Leu171, and T-shaped π stacking interaction with Tyr326, respectively. Cys172 holds the indole ring by two π–sulphur bonds. 3-fluorobenzoyl group is shifted towards a more hydrophobic pocket in line with Pro104, Ile199, Ile316, and Tyr326. The oxygen of this carbonyl spacer involved in two H-bond interactions with a water molecule (distance ∼2.6 Å). Whereas, the CH2 group in 4b does not involve in any H-bond interactions. This could highlight the higher potency of 4e over 4b. The carbonyl linker at N1 position has improved the binding efficacy as well as potency. Pyrazine ring placed near to FAD ring and showed two π–π stacking bonds with FAD and Tyr435. The nitrogen atom in pyrazine ring (at 4-position) established a strong H-bond bond with Tyr188 (distance 1.9 Å).
From the docked poses of 4b and 4e, it was concluded that to get higher potency, compounds should possess 1,4-pyrazine and m-fluorobenzoyl moieties. From the SAR study, we can see that compounds 4a and 4c possess pyridazine and pyridine rings show 13.15% and 11.24% of MAO-B inhibition compared with 4b (88.49%) and 4e (66.39%). In case of compounds 4d and 4f, both have m-fluorobenzoyl substitution at indole “N,” but 4f shows more significant inhibition of MAO-B. It also suggests that the pyridine ring is more tolerable than pyridazine. On the other side, when the fluorobenzyl is replaced by 2-methylpyridine (4g, 4h, and 4i), a significant decrease in the % MAO-B inhibition was noticed. The reason for the reduced MAO-B inhibitory activity could be the relative higher polarity of 2-methylpyridine compared to the fluorobenzyl moiety. The surrounding region at this substitution is highly hydrophobic lined by the residues Pro104, Trp119, Leu164, Leu167, Ile199, and Ile316. The incorporation of more hydrophobic ligand substitution may enhance the % MAO-B inhibition, and polar substitution might lead to a further reduction in % MAO-B inhibition. Further, removal of p-fluorophenyl from the compounds 4a, 4b, and 4c obtained compounds 4j, 4k, and 4l displayed a reduction in % MAO-B inhibition. This suggests the importance of p-fluorophenyl moiety in the MAO-B inhibition, because this part of ligand is docked at the upper pocket of MAO-B, where it holds π–π-stacking, π–sulphur, π–sigma interactions with Tyr326, Cys172, and Leu171. Removal of such interactions (4j–4l) results in deterioration of the % inhibition of MAO-B.
4b and 4e ligands were docked inside the MAO-A protein (PDB ID: 2Z5X)Citation48. For 4b ligand (glide score −6.65), the pyrazine ring formed strong π–π-stacked interactions with FAD and Tyr444 ( and ). The amide linker showed no H-bond interactions with the surrounding residue; it established only one week carbon hydrogen bond with Tyr407. The central indole ring has week π–alkyl interactions with Ile180, Ile335. Fluorobenzyl group has strong π–π-stacked and week π–alkyl contact with Phe208 and Cys323, Ile325, respectively. Fluorine substitution on para position has two halogen interactions with Gly110, and Ala111. Similarly, ligand 4e (glide score −4.20) has a week π–alkyl interaction between indole core and Ile180, and Ile335 ( and ). The amide linker had no strong interactions with the surrounding residues. The pyrazine ring showed π–π-stacked interactions with Tyr444 and π–π-T-shaped contact with FAD. Upper hydrophobic pocket fluorobenzyl group showed π–alkyl interaction with Leu97, Cys32, and π–π-stacked interactions with Phe208. Fluorine atom has only one-halogen bond interactions with Ala111. Carbonyl linker did not show any contribution to protein-ligand interactions. Compared to MAO-B protein, ligands 4e and 4b have weaker interaction patterns inside MAO-A binding pocket.
Figure 7. Binding orientation of ligand 4b and 4e inside the MAO-A protein structure. Ligand 4b (A) and 4e (B) shown in orange and blue colour stick format. FAD molecule shown in yellow stick format. Hydrophobic surface area shown around the ligand.
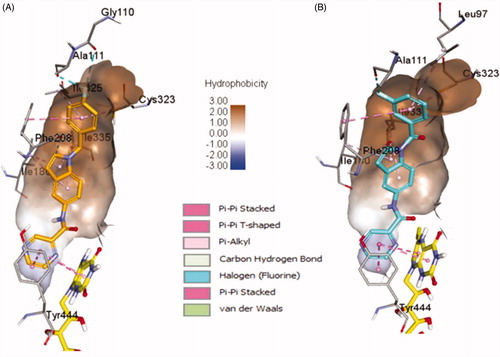
Figure 8. 2D interactions of ligands 4b and 4e inside MAO-A binding pocket. 4b (A) and 4e (B) ligands shown in orange and blue colour, respectively.
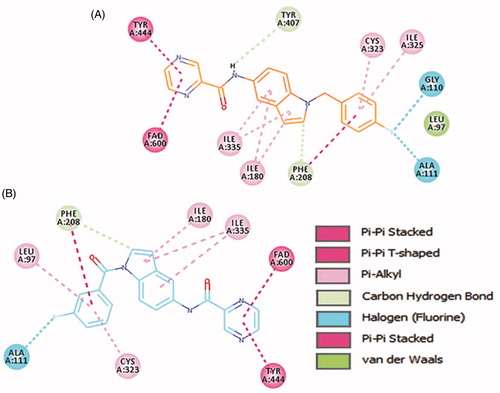
Overall, MAO-A and MAO-B proteins have a higher sequence identity and similarity. The two crystal structures of MAO-A (PDB ID: 2Z5X) and MAO-B (PDB ID: 2V5Z) have 73.3% sequence identity. It was noted that the binding site residues are also very similar except for some residues. Similar and variable binding site residues of MAO-B/MAO-A are listed as follows: Leu88/Leu97, Pro102/Ala111, Gly101/Gly110, Pro104/Pro113, Leu171/Ile180, Cys172/Asn181, Tyr188/Tyr197, Ile199/Phe208, Thr314/Cys323, Ile316/Ile235, Tyr326/Ile335, Tyr398/Tyr407, and Tyr435/Tyr444. Important Leu171 (MAO-B) residue involves in H-bond interactions with amide linker of the ligand and reliable π–sigma contacts with the central indole ring, which is replaced with Ile180 (MAO-A) that only constitutes few week pi-alkyl interactions with the ligand. Another side similar aromatic residues Tyr188/Tyr197; Tyr188 (MAO-B) has strong H-bond with pyrazine nitrogen while Tyr197 in MAO-A does not show any promising interaction with the ligand moieties. Cys172 in MAO-B has also significant π-sulphur interaction with the central indole ring, while in MAO-A protein, it was replaced by Asn181 which does not interact at all with the ligand molecules. Overall, in the docking study, 4b and 4e ligand showed higher docking score inside MAO-B protein. Additionally, the abundant aromatic interactions (π–π, π–sigma, and π–sulphur) and strong H-bond interactions mediated by pyrazine nitrogen, carbonyl oxygen, and carboxamide nitrogen inside MAO-B protein make ligands 4b and 4e to be more selective towards MAO-B protein.
4. Conclusions
In summary, we present simple and convenient chemical synthetic route using microwave conditions towards synthesis of new promising MAO-B inhibitors. Our results investigated in vitro MAO-B inhibitory activity of two indole-based series originally designed from two indazole-containing lead compounds (VI and VII); by retaining their original amide spacer while applying scaffold modification into an indole core. Moreover, various substitution patterns were incorporated on the free NH group of the indole aiming to obtain a new lead with potential MAO-B inhibitory activity. Interestingly, amongst all members, compound 4e displayed a promising profile of high potency, selectivity, in addition to a competitive MAO-B mode of action. As a result, our group is currently performing further optimization studies of compound 4e focussing on computer-aided lead optimization to advance more potent indole-based derivative as a step towards a new potential clinical candidate for MAO-B inhibition.
Disclosure statement
The authors declare no conflict of interest.
Additional information
Funding
References
- Schapira AH, Bezard E, Brotchie J, et al. Novel pharmacological targets for the treatment of Parkinson's disease. Nat Rev Drug Discov 2006;5:845–54.
- Barker RA, Stacy M, Brundin P. A new approach to disease-modifying drug trials in Parkinson's disease. J Clin Investig 2013;123:2364–5.
- Sveinbjornsdottir S. The clinical symptoms of Parkinson's disease. J Neurochem 2016;139:318–24.
- Greenamyre JT, Hastings TG. Biomedicine. Parkinson's-divergent causes, convergent mechanisms. Science (New York, N.Y.) 2004;304:1120–2.
- Alexander GE. Biology of Parkinson's disease: pathogenesis and pathophysiology of a multisystem neurodegenerative disorder. Dial Clin Neurosci 2004;6:259–80.
- Moore DJ, West AB, Dawson VL, Dawson TM. Molecular pathophysiology of Parkinson's disease. Annu Rev Neurosci 2005;28:57–87.
- Kalgutkar AS, Dalvie DK, Castagnoli N, Taylor TJ. Interactions of nitrogen-containing xenobiotics with monoamine oxidase (MAO) isozymes A and B: SAR studies on MAO substrates and inhibitors. Chem Res Toxicol 2001;14:1139–62.
- Silverman RB. Radical ideas about monoamine oxidase. Acc Chem Res 1995;28:335–42.
- Shih JC, Chen K, Ridd MJ. Monoamine oxidase: from genes to behavior. Annu Rev Neurosci 1999;22:197–217.
- Robottom BJ. Efficacy, safety, and patient preference of monoamine oxidase B inhibitors in the treatment of Parkinson's disease. Patient Prefer Adherence 2011;5:57–64.
- Lohle M, Reichmann H. Controversies in neurology: why monoamine oxidase B inhibitors could be a good choice for the initial treatment of Parkinson's disease. BMC Neurol 2011;11:112.
- Rascol O, Perez-Lloret S, Ferreira JJ. New treatments for levodopa-induced motor complications. Mov Disord 2015;30:1451–60.
- Robakis D, Fahn S. Defining the role of the monoamine oxidase-B inhibitors for Parkinson's disease. CNS Drugs 2015;29:433–41.
- Fernandez HH, Chen JJ. Monoamine oxidase-B inhibition in the treatment of Parkinson's disease. Pharmacotherapy 2007;27:174S–85S.
- Finberg JP. Update on the pharmacology of selective inhibitors of MAO-A and MAO-B: focus on modulation of CNS monoamine neurotransmitter release. Pharmacol Ther 2014;143:133–52.
- Duty S, Jenner P. Animal models of Parkinson's disease: a source of novel treatments and clues to the cause of the disease. Br J Pharmacol 2011;164:1357–91.
- Fabbri M, Rosa MM, Abreu D, Ferreira JJ. Clinical pharmacology review of safinamide for the treatment of Parkinson's disease. Neurodegener Dis Manage 2015;5:481–96.
- Caccia C, Maj R, Calabresi M, et al. Safinamide: from molecular targets to a new anti-Parkinson drug. Neurology 2006;67:S18–S23.
- Caccia C, Salvati P, Rossetti S, Anand R. 2.207 Safinamide: beyond MAO-B inhibition. Parkinson Relat Disord 2007;13:S99.
- Chazot PL. Safinamide for the treatment of Parkinson's disease, epilepsy and restless legs syndrome. Curr Opin Investig Drugs 2000;8:570–9.
- Pevarello P, Bonsignori A, Dostert P, et al. Synthesis and anticonvulsant activity of a new class of 2-[(arylalky)amino]alkanamide derivatives. J Med Chem 1998;41:579–90.
- Borroni E, Bohrmann B, Grueninger F, et al. Sembragiline: a novel, selective monoamine oxidase type B inhibitor for the treatment of Alzheimer's disease. J Pharmacol Exp Ther 2017;362:413–23.
- Nave S, Doody RS, Boada M, et al. Sembragiline in moderate Alzheimer's disease: results of a randomized, double-blind, placebo-controlled phase II trial (MAyflOwer RoAD). J Alzheimers Dis 2017;58:1217–28.
- Jo S, Yarishkin O, Hwang YJ, et al. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat Med 2014;20:886–96.
- Choi JW, Jang BK, Cho NC, et al. Synthesis of a series of unsaturated ketone derivatives as selective and reversible monoamine oxidase inhibitors. Bioorg Med Chem 2015;23:6486–96.
- Park HR, Kim J, Kim T, et al. Oxazolopyridines and thiazolopyridines as monoamine oxidase B inhibitors for the treatment of Parkinson's disease. Bioorg Med Chem 2013;21:5480–7.
- Yeon SK, Choi JW, Park JH, et al. Synthesis and evaluation of biaryl derivatives for structural characterization of selective monoamine oxidase B inhibitors toward Parkinson's disease therapy. Bioorg Med Chem 2018;26:232–44.
- Nam MH, Park M, Park H, et al. Indole-substituted benzothiazoles and benzoxazoles as selective and reversible MAO-B inhibitors for treatment of Parkinson's disease. ACS Chem Neurosci 2017;8:1519–29.
- Carradori S, Silvestri R. New frontiers in selective human MAO-B inhibitors. J Med Chem 2015;58:6717–32.
- Reis J, Cagide F, Chavarria D, et al. Discovery of new chemical entities for old targets: insights on the lead optimization of chromone-based monoamine oxidase B (MAO-B) inhibitors. J Med Chem 2016;59:5879–93.
- Binda C, Wang J, Pisani L, et al. Structures of human monoamine oxidase B complexes with selective noncovalent inhibitors: safinamide and coumarin analogs. J Med Chem 2007;50:5848–52.
- Desideri N, Fioravanti R, Proietti Monaco L, et al. Alcaro, 1,5-diphenylpenta-2,4-dien-1-ones as potent and selective monoamine oxidase-B inhibitors. Eur J Med Chem 2013;59:91–100.
- Tzvetkov NT, Hinz S, Küppers P, et al. Indazole- and indole-5-carboxamides: selective and reversible monoamine oxidase B inhibitors with subnanomolar potency. J Med Chem 2014;57:6679–703.
- Carradori S, Secci D, Bolasco A, et al. Patent-related survey on new monoamine oxidase inhibitors and their therapeutic potential. Expert Opin Ther Pat 2012;22:759–801.
- Sharma V, Kumar P, Pathak D. Biological importance of the indole nucleus in recent years: a comprehensive review. J Heterocycl Chem 2010;47:491–502.
- Zdrazil B, Guha R. The rise and fall of a scaffold: a trend analysis of scaffolds in the medicinal chemistry literature. J Med Chem 2018;61:4688–703.
- Balsa D, Fernandez-Alverez E, Tipton KF, Unzeta M. Monoamine oxidase inhibitory potencies and selectivities of 2-[N-(2-propynyl)-aminomethyl]-1-methyl indole derivatives. Biochem Soc Trans 1991;19:215–8.
- Sasidharan R, Manju SL, Ucar G, et al. Identification of indole-based chalcones: discovery of a potent, selective, and reversible class of MAO-B inhibitors. Archiv Der Pharm 2016;349:627–37.
- Gurkan AS, Karabay A, Buyukbingol Z, et al. Syntheses of novel indole lipoic acid derivatives and their antioxidant effects on lipid peroxidation. Archiv Der Pharm 2005;338:67–73.
- Smaill JB, Gonzales AJ, Spicer JA, et al. Tyrosine kinase inhibitors. 20. Optimization of substituted quinazoline and pyrido[3,4-d]pyrimidine derivatives as orally active, irreversible inhibitors of the epidermal growth factor receptor family. J Med Chem 2016;59:8103–24.
- Li B, Ma J, Xie W, et al. Regioselective C2 oxidative olefination of indoles and pyrroles through cationic rhodium(III)-catalyzed C–H bond activation. Chemistry 2013;19:11863–8.
- Jiang X, Tiwari A, Thompson M, et al. A practical method for N-methylation of indoles using dimethyl carbonate. Org Process Res Dev 2001;5:604–8.
- Carter MC, Cockerill GS, Guntrip SB, et al. Preparation of 6-(thiomorpholinomethylfuranyl)-4-quinazolinamines as protein tyrosine kinase inhibitors. UK: Glaxo Group Ltd.; 2000.
- Cho I-H, Lim J-W, Noh J-Y, et al. Preparation of 1H-indoles as a highly selective cyclooxygenase-2 inhibitors. S. Korea: Cheil Jedang Corporation; 2003.
- T, Ishikawa K, Miwa M, Seto, et al. Fused pyrimidines as growth factor receptor tyrosine kinase inhibitors, their preparation, pharmaceutical compositions, and use in therapy. Japan: Takeda Pharmaceutical Company Limited; 2007.
- Ple P, Jung FH. Preparation of quinazoline derivatives for use in treatment of cell proliferative disorders or disease assocd. with angiogenesis and/or vascular permeability. Sweden/UK: AstraZeneca AB/AstraZeneca UK Limited; 2006.
- Thomson SP, Davies RT, Allanson NM, et al. Preparation of indolizinyloxoacetamides and related compounds as medical and agrochemical fungicides. Manchester (UK): F2G Ltd.; 2006.
- Son SY, Ma J, Kondou Y, et al. Structure of human monoamine oxidase A at 2.2-A resolution: the control of opening the entry for substrates/inhibitors. Proc Natl Acad Sci USA 2008;105:5739–44.
- Sastry GM, Adzhigirey M, Day T, et al. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des 2013;27:221–34.
- Friesner RA, Banks JL, Murphy RB, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 2004;47:1739–49.
- Ramadas K, Srinivasan N. Iron-ammonium chloride – a convenient and inexpensive reductant. Synth Commun 1992;22:3189–95.
- Happe K. Monoamine oxidase inhibitors. In: Enna SJ, Bylund DB, eds. xPharm: the comprehensive pharmacology reference. New York: Elsevier; 2007:1–3.