
Abstract
In this study, some benzimidazole-oxadiazole derivatives were synthesised and tested for their in vitro anticancer activities on five cancer cell lines, including HeLa, MCF7, A549, HepG2 and C6. Their structures were elucidated by IR, 1H-NMR, 13C-NMR, 2 D-NMR and HRMS spectroscopic methods. Among all screened compounds; 5a, 5b, 5d, 5e, 5k, 5l, 5n and 5o exhibited potent selective cytotoxic activities against various tested cancer cell lines. Especially, compounds 5l and 5n exhibited the most antiproliferative activity than Hoechst 33342 and doxorubicin against HeLa cell line, with IC50 of 0.224 ± 0.011 µM and 0.205 ± 0.010 µM, respectively. Furthermore, these potent lead cytotoxic agents were evaluated in terms of their inhibition potency against Topoisomerase I and it was determined that selected compounds inhibited the Topoisomerase I. Docking studies were performed and probable interactions in the DNA-Topo I enzyme complex was determined.
1. Introduction
DNA topoisomerases (Topo) are ubiquitous enzymes that are required for proper chromosome structure and segregation. These enzymes play important roles in triggering, controlling, and modifying topological DNA problems during cell proliferation, differentiation, and survival. Thus, topo enzymes possesses immense importance in almost all stages of the cell cycle. As a result, the role of mammalian DNA topoisomerases as molecular targets for anticancer drugs has been explored, and it was found that topoisomerase inhibition could curb cancerous cell growth across a variety of cell linesCitation1–8.
Human Topo are divided into two classes: Topoisomerase I (topo I), breaks only one strand of DNA and Topoisomerase II (topo II), breaks both strands of DNACitation9–11. Several Topo poisons have been approved for the chemotherapeutic treatment of different cancer types in recent decades, including the Topo I inhibitors camptothecin, topotecan and the Topo-II-targeting drugs etoposide and amsacrinCitation12,Citation13.
In the field of anticancer drugs, DNA topoisomerase I has been reported to be an important target for anticancer agents but topoisomerase inhibitors have several barriers and limitations. These include poor solubility, accumulation in target tissues or organs and prolonged half-life in plasma as well as drug efflux from the target cells and cancer cell resistanceCitation14–17.
Benzimidazoles represent a structurally unique class of Topo I poisons that act as DNA minor groove binders such as Hoescht 33258 and Hoechst 33342 ()Citation18–20. These drugs are known to hinder the breakage/reunion reaction of topoisomerase I, in which the enzyme is reversibly trapped in a state where the DNA is cleaveCitation6. Furthermore, benzimidazole is known to be a versatile scaffold that possess potential anticancer, antitumor and antiproliferative activitiesCitation21–27. Similarly, oxadiazole has engrossed significant attention of medicinal chemists owing to their wide range of useful pharmacological actions particularly cytotoxic activities against DNA topoisomerase ICitation28–31.
Figure 1. Hoechst 33258 and Hoechst 33342 compounds.
In the light of this information, it is aimed to develop new anticancer effective compounds in this research. To this aim, Hoechst compounds have been identified as precursor compounds. In addition to the hydroxy (–OH) and ethoxy (–OC2H5) substituents in the para position of the phenyl ring in the Hoechst compounds, the methoxy (–OCH3) substituent was also used for synthesised compounds. Instead of bisbenzimidazole, the 1,3,4-oxadiazole ring attached to the benzimidazole ring was synthesised. Methyl, ethyl, pyridine, pyrimidine and phenyl substituents were used for the fourth position of piperazine. The cytotoxicity of these synthesised compounds to different cell lines, and Topo I inhibitory activities were evaluated. Besides different mechanisms such as Flow cytometric analysis, DNA synthesis inhibition assay was also studied in our research. In order to determine the possible interactions of compound, showed high activity, docking studies have been performed.
2. Experimental
2.1. Chemistry
Whole chemicals employed in the synthetic procedure were purchased from Sigma-Aldrich Chemicals (Sigma-Aldrich Corp., St. Louis, MO, USA) or Merck Chemicals (Merck KGaA, Darmstadt, Germany). Melting points of the obtained compounds were determined by MP90 digital melting point apparatus (Mettler Toledo, OH, USA) and were uncorrected. The IR spectra were obtained on a Shimadzu, IR Prestige-21 (Shimadzu, Tokyo, Japan). 1H NMR, HSQC, HMBC, COSY and 13C NMR spectra of the synthesised compounds were registered by a Bruker 300 MHz and 75 MHz digital FT-NMR spectrometer (Bruker Bioscience, Billerica, MA, USA) in DMSO-d6, respectively. Splitting patterns were designated as follows: s: singlet; d: doublet; t: triplet; m: multiplet in the NMR spectra. Coupling constants (J) were reported as Hertz. M + 1 peaks were determined by Shimadzu LC/MS ITTOF system (Shimadzu, Tokyo, Japan). All reactions were monitored by thin-layer chromatography (TLC) using Silica Gel 60 F254 TLC plates (Merck KGaA, Darmstadt, Germany).
2.1.1. General procedure for the synthesis of the compounds
2.1.1.1. Synthesis of 2-(4-substitutedphenyl)-1H-benzo[d]imidazole-6-carboxylic acid derivatives (1a-1c)
A mixture of 4-substituted benzaldehyde (0.03 mol) and sodium disulphide (0.03 mol, 5.7 g) in DMF were treated under microwave irradiation (Anton-Paar Monowave 300) at 240 °C and 10 bar for 5 min. After cooling the mixture, 3,4-diamino benzoic acid (0.03 mol, 4.56 g) was added and kept under the same reaction conditions. The progress of the reaction was monitored by TLC. After completion of the reaction, the mixture was poured into crushed ice. The solid obtained was filtered, washed with water, dried and recrystallized from EtOH.
2.1.1.2. Synthesis of 2-chloro-1-(4-sübstitüepiperaz-1-yl) ethan-1-one derivatives (1d-1h)
Chloroacetyl chloride (0.014 mol, 1.056 ml) in THF (15 ml) was taken into ice bath. 4-Substituted piperazine derivatives (0.012 mol) and TEA (0.0132 mol, 1.90 ml) in THF (50 ml) were added dropwise to this solution. After completion of dropping, the reaction mixture was stirred at room temperature for 1 h. The precipitated product was filtered washed with water and dried.
2.1.1.3. Synthesis of Methyl 2-(4-substitutedphenyl)-1H-benzo[d]imidazole-6-carboxylate derivatives (2a-2c)
2–(4-Substitutedphenyl)-1H-benzo[d]imidazole-6-carboxylic acid (1a-1c) derivative compound (0.025 mol) was dissolved in methanol and several drops of sulphuric acid were added into the mixture. The mixture was refluxed for 72 h, and the precipitate was filtered off.
2.1.1.4. Synthesis of 2-(4-Substitutedphenyl)-1H-benzo[d]imidazole-6-carbohydrazide derivatives (3a-3c)
Methyl 2–(4-substitutedphenyl)-1H-benzo[d]imidazole-6-carboxylate (2a-2c) (0.018 mol) and excess of hydrazine hydrate (5 ml) in EtOH (15 ml) were mixed in a vial (30 ml) of microwave synthesis reactor (Anton-Paar Monowave 300). The reaction mixture was heated at 240 °C and 10 bar for 10 min. After cooling, the mixture was poured into iced water, the product was filtered, washed with water, dried, and recrystallized from EtOH.
2.1.1.5. Synthesis of 2-((4-Substituted phenyl)-(6-(5-mercapto-1,3,4-oxadiazol-2-yl)-1H-benzo [d]imidazole derivatives (4a-4c)
2–(4-Substitutedphenyl)-1H-benzo[d]imidazole-6-carbohydrazide (3a-3c) derivative (0.01 mol) was dissolved in a solution of NaOH (0.01 mol, 0.4 g) in ethanol and carbon disulphide (0.01 mol, 0.60 ml) was added in the mixture. The mixture was refluxed for 8 h. When the reaction was finished, the solution was cooled and acidified to pH 4–5 with concentrated hydrochloric acid solution to obtained target compound.
2.1.1.6. General synthesis method of target compounds (5a-5o)
2-((4-Substitutedphenyl)-(6–(5-mercapto-1,3,4-oxadiazol-2-yl)-1H-benzo[d]imidazole (4a-4c) (0.001 mol), potassium carbonate (0.001 mol, 0.138 g) and 2-chloro-1-(4-substitutedepiperazin-1-yl) -ethane-1-on (0.0015 mol) derivative were dissolved in acetone and refluxed for 6 h. After TLC control, the solvent was evaporated, the residue was washed with water, dried and then recrystallized from ethanol to afford final compounds (5a-5o).
Methyl 2-(4-hydroxyphenyl)-1H-benzo[d]imidazole-6-carboxylate (2a)
Yield: 75%, M.P. = 306.1–307.5 °C. FTIR (ATR, cm−1): 3334 (N-H), 1687 (C=O), 844 (1,4-disubstituted benzene). 1H-NMR (300 MHz, DMSO-d6): δ = 3.88 (3H, s, –CH3), 6.95 (2H, d, J = 8.70 Hz, 1,4-disubstituted benzene), 7.62 (1H, d, J = 8.34 Hz, benzimidazole-C4), 7.82 (1H, dd, J1=8.43 Hz, J2=1.26 Hz, benzimidazole-C5), 8.04 (2H, d, J = 8.64 Hz, 1,4-disubstituted benzene), 8.14 (1H, s, benzimidazole-C7), 10.05 (1H, s, N–H). 13C-NMR (75 MHz, DMSO-d6): δ = 52.41, 107.48, 111.94, 116.26 (2C), 120.89, 123.40, 126.28, 129.02, 130.54 (2C), 134.46, 154.79, 160.14, 167.28. HRMS (m/z): [M + H]+ calcd for C15H13N2O3: 269.0910; found: 269.0921.
Methyl 2-(4-methoxyphenyl)-1H-benzo[d]imidazole-6-carboxylate (2 b)
Yield: 68%, M.P.=230.4–231.7 °C. FTIR (ATR, cm−1): 3250 (N–H), 1695 (C=O), 839 (1,4-disubstituted benzene). 1H-NMR (300 MHz, DMSO-d6): δ = 3.85 (3H, s, –OCH3), 3.87 (3H, s, –COOCH3), 7.13 (2H, d, J = 8.85 Hz, 1,4-disubstituted benzene), 7.65 (1H, br.s., benzimidazole-C–H), 7.83 (1H, d, J = 7.95 Hz, benzimidazole C–H), 8.14 (2H, d, J = 8.85 Hz, 1,4-disubstituted benzene), 8.22 (1H, br.s., benzimidazole, C-H). 13C-NMR (75 MHz, DMSO-d6): δ = 52.43, 55.87, 111.49, 113.08, 115.31 (2C), 118.69, 120.49, 122.45, 123.29, 123.56, 128.88 (2C), 130.43, 161.55, 167.26. HRMS (m/z): [M + H]+ calcd for C16H15N2O3: 283.1083; found: 283.1077.
Methyl 2-(4-ethoxyphenyl)-1H-benzo[d]imidazole-6-carboxylate (2c)
Yield: 60%, M.P.= 252.8–254.4 °C. FTIR (ATR, cm−1): 3477 (N-H), 1645 (C=O), 844 (1,4-disubstituted benzene). 1H-NMR (300 MHz, DMSO-d6): δ = 1.36 (3H, t, J = 6.96 Hz, –CH3), 3.87 (3H, s, –CH3), 4.11 (2H, q, J = 6.96 Hz, -CH2), 7.11 (2H, d, J = 8.88 Hz, 1,4-disubstituted benzene), 7.63 (1H, d, J = 8.43 Hz, benzimidazole-C4), 7.83 (1H, dd, J1=8.40 Hz, J2=1.59 Hz, benzimidazole-C5), 8.12 (2H, d, J = 8.88 Hz, 1,4-disubstituted benzene), 8.1 (1H, s, benzmidazole-C7), 13.07 (1H, s, N-H). 13C-NMR (75 MHz, DMSO-d6): δ = 15.04, 52.38, 63.81, 114.39, 115.33, 116.22 (2C), 118.31, 119.34, 122.31, 123.53, 128.49 (2C), 128.88, 154.40, 160.84, 167.26. HRMS (m/z): [M + H]+ calcd for C17H17N2O3: 297.1232; found: 297.1234.
2-((4-Hydroxyphenyl)-(6-(5-mercapto-1,3,4-oxadiazol-2-yl)-1H-benzo[d]imidazole (4a)
Yield: 81%, M.P.= 226.8–228.3 °C. FTIR (ATR, cm−1): 3483 (N-H), 1645 (C=O), 844 (1,4-disubstituted benzene). 1H-NMR (300 MHz, DMSO-d6):δ = 7.06 (2H, d, J = 8.76 Hz, 1,4-disubstituted benzene), 7.89 (2H, m, benzimidazole-C–H), 8.10 (1H, s, benzimidazole-C-H), 8.24 (2H, d, J = 8.70 Hz, 1,4-disubstituted benzene), 10.75 (1H, s, O-H). 13C-NMR (75 MHz, DMSO-d6): δ = 112.11, 115.27, 115.48, 116.86 (2C), 118.85, 122.86, 130.64 (2C), 134.41, 136.72, 152.40, 160.92, 162.58, 177.83. HRMS (m/z): [M + H]+ calcd for C15H11N4O2S: 311.0595; found: 311.0597.
2-((4-Methoxyphenyl)-(6-(5-mercapto-1,3,4-oxadiazol-2-yl)-1H-benzo[d]imidazole (4b)
Yield: 78%, M.P.= 274.4–276.5 °C. FTIR (ATR, cm−1): 3381 (N-H), 1633 (C=O), 837 (1,4-disubstituted benzene). 1H-NMR (300 MHz, DMSO-d6): δ = 3.90 (3H, s, –OCH3), 7.24–7.28 (2H, m, 1,4-disubstituted benzene), 7.89 (1H, s, benzimidazole C-H), 8.12 (1H, s, benzimidazole C–H), 8.32 (2H, d, J = 8.70 Hz, 1,4-disubstituted benzene), 8.40 (1H, s, benzimidazole C-H). 13C-NMR (75 MHz, DMSO-d6): δ = 56.25, 111.07, 114.23, 115.49, 115.76 (2C), 119.97, 122.60, 130.24 (2C), 130.49, 133.74, 152.25, 160.86, 162.73, 177.22. HRMS (m/z): [M + H]+ calcd for C16H13N4O2S: 325.0748; found: 325.0754.
2-((4-Ethoxyphenyl)-(6–(5-mercapto-1,3,4-oxadiazol-2-yl)-1H-benzo[d]imidazole (4c)
Yield: 84%, M.P.= 288.2–289.9 °C. FTIR (ATR, cm−1): 3402 (N-H), 1624 (C=O), 846 (1,4-disubstituted benzene). 1H-NMR (300 MHz, DMSO-d6): δ = 1.38 (3H, t, J = 6.90 Hz, -CH3), 4.17 (2H, q, J = 6.96 Hz, -CH2), 7.23 (2H, d, J = 8.28 Hz, 1,4-disubstituted benzene), 7.91–7.93 (2H, m, benzimidazole-C-H), 8.12 (1H, s, benzimidazole-C-H), 8.24 (2H, d, J = 7.47 Hz, 1,4-disubstituted benzene). 13C-NMR (75 MHz, DMSO-d6): δ = 14.92, 64.35, 112.17, 115.55, 115.96, 116.14 (2C), 119.30, 123.28, 130.57 (2C), 133.78, 135.98, 151.83, 155.14, 162.99, 177.88. HRMS (m/z): [M + H]+ calcd for C17H15N4O2S: 339.0910; found: 339.0910.
2-((5–(2-(4-Hydroxyphenyl)-1H-benzo[d]imidazol-6-yl)-1,3,4-oxadiazol-2-yl)thio)-1–(4-methyl- piperazin-1-yl) -ethane-1-on (5a)
Yield: 65%, M.P.= 161.3–163.4 °C. FTIR (ATR, cm−1): 3618 (N-H), 1645 (C=O), 840 (1,4-disubstituted benzene). 1H-NMR (300 MHz, DMSO-d6): δ = 2.76 (3H, s, -CH3), 3.35 (8H, br.s., piperazine), 4.64 (2H, s, -CH2), 6.96 (2H, d, J = 8.64 Hz, 1,4-disubstituted benzene), 7.71 (1H, d, J = 8.28 Hz, benzimidazole-C4), 7.80 (1H, dd, J1=8.40 Hz, J2=1.15 Hz, benzimidazole-C5), 8.09 (2H, d, J = 8.61 Hz, 1,4-disubstituted benzene), 8.13 (1H, s, benzimidazole-C7), 10.20 (1H, s, O-H). 13C-NMR (75 MHz, DMSO-d6): δ = 36.76, 42.47, 45.75, 52.23, 52.44, 60.63, 112.96, 115.85, 116.47 (2C), 117.52, 118.66, 121.59, 129.67 (2C), 153.79, 161.14, 163.26, 165.68, 166.19, 167.30, 169.40. HRMS (m/z): [M + H]+/2 calcd for C22H23N6O3S: 226.0804; found: 226.0810.
2-((5–(2-(4-Hydroxyphenyl)-1H-benzo[d]imidazol-6-yl)-1,3,4-oxadiazol-2-yl)thio)-1–(4-ethyl- piperazin-1-yl) -ethane-1-on (5 b)
Yield: 78%, M.P.= 199.9–201.2 °C. FTIR (ATR, cm−1): 3311 (N-H), 1645 (C=O), 839 (1,4-disubstituted benzene). 1H-NMR (300 MHz, DMSO-d6): δ = 1.01 (3H, t, J = 7.16, CH3), 2.34–2.51 (6H, m, CH2, piperazine), 3.48–3.50 (4H, m, piperazine), 4.56 (2H, s, CH2), 6.90 (2H, d, J = 8.70 Hz, 1,4-disubstituted benzene), 7.67 (1H, d, J = 8.46 Hz, benzimidazole-C4), 7.74 (1H, dd, J1=8.39 Hz, J2:1.65 Hz, benzimidazole-C5), 8.06 (2H, d, J = 8.70 Hz, 1,4-disubstituted benzene), 8.09 (1H, s, benzimidazole-C7). 13C-NMR (75 MHz, DMSO-d6): δ = 12.32, 37.04, 42.23, 45.85, 51.91, 52.31, 52.79, 113.41, 116.30, 116.45, 116.70 (2C), 120.38, 129.02 (2C), 155.20, 155.45, 161.21, 163.08, 165.06, 165.89, 166.58, 168.44. HRMS (m/z): [M + H]+ calcd for C23H25N6O3S: 465.1692; found: 465.1703.
2-((5–(2-(4-Hydroxyphenyl)-1H-benzo[d]imidazol-6-yl)-1,3,4-oxadiazol-2-yl) thio)-1–(4-(phenyl) -piperazin-1-yl) -ethane-1-on (5c)
Yield: 72%, M.P.= 271.1–273.4 °C. FTIR (ATR, cm−1): 3365 (N-H), 1645 (C=O), 840 (1,4-disubstituted benzene). 1H-NMR (300 MHz, DMSO-d6): δ = 3.13–3.26 (4H, m, piperazine), 3.63–3.70 (4H, m, piperazine), 4.64 (2H, s, -CH2), 6.82 (1H, t, J = 7.23 Hz, phenyl C-H), 6.99–6.93 (4H, m, 1,4-disubstituted benzene, phenyl C-H), 7.24 (2H, t, J = 7.29 Hz, phenyl C-H), 7.66 (1H, br.s., benzimidazole-C4), 7.81 (1H, d, J = 8.34 Hz, benzimidazole-C5), 8.01–8.06 (3H, m, 1,4-disubstituted benzene, benzimidazole-C7), 10.10 (1H, s, O-H), 13.09 (1H, s, N-H).13C-NMR (75 MHz, DMSO-d6): δ = 37.03, 42.06, 45.66, 48.63, 48.97, 116.21 (2C), 116.28, 116.37, 116.77 (2C), 119.18, 119.85, 120.83, 121.11, 128.86, 129.03 (2C), 129.48 (2C), 151.15, 159.93, 160.18, 162.08, 163.22, 165.26, 166.46. HRMS (m/z): [M + H]+ calcd for C27H25N6O3S: 513.1699; found: 513.1703.
2-((5-(2-(4-Hydroxyphenyl)-1H-benzo[d]imidazol-6-yl)-1,3,4-oxadiazol-2-yl)thio)-1-(4-(pyridin-2-yl)-piperazin-1-yl)-ethane-1-on (5d)
Yield: 69%, M.P.= 247.6–248.9 °C. FTIR (ATR, cm−1): 3628 (N-H), 1645 (C=O), 840 (1,4-disubstituted benzene). 1H-NMR (300 MHz, DMSO-d6): δ = 3.56–3.66 (8H, m, piperazine), 4.65 (2H, s, CH2), 6.68–6.69 (2H, m, pyridine C-H), 6.99 (2H, d, J = 8.73 Hz, 1,4-disubstituted benzene), 7.57–7.58 (2H, m, pyridine C-H), 7.72 (1H, d, J = 8.37 Hz, benzimidazole-C4), 7.82 (1H, dd, J1=5.40 Hz, J2=1.53 Hz, benzimidazol-C5), 8.11–8.14 (2H, m, 1,4-disubstituted benzene, benzimidazole-C7), 10.26 (1H, s, O-H). 13C-NMR (75 MHz, DMSO-d6): δ = 37.17, 41.83, 44.74, 44.99, 45.41, 107.88, 113.21, 113.79, 114.59, 116.27 (2C), 116.73, 117.11, 120.67, 120.74, 129.13 (2C), 138.24, 147.83, 154.66, 158.94, 160.34, 163.22, 165.40, 165.49, 166.47. HRMS (m/z): [M + H]+ calcd for C26H24N7O3S: 514.1636; found: 514.1656.
2-((5–(2-(4-Hydroxyphenyl)-1H-benzo[d]imidazol-6-yl)-1,3,4-oxadiazol-2-yl)thio)-1-(4-(pyrimidine-2-yl)-piperazin-1-yl)-ethane-1-on (5e)
Yield: 78%, M.P.= 181.4–183.2 °C. FTIR (ATR, cm−1): 3587 (N-H), 1645 (C=O), 840 (1,4-disubstituted benzene). 1H-NMR (300 MHz, DMSO-d6): δ = 3.60 (2H, br.s, piperazine), 3.67 (2H, br.s., piperazine), 3.79 (2H, br.s., piperazine), 3.90 (2H, brs., piperazine), 4.64 (2H, s, CH2), 6.74 (1H, J = 4.80, pyrimidine), 7.09 (2H, J = 8.76 Hz, 1,4-disubstituted benzene), 7.92 (1H, d, J = 8.55 Hz, benzimidazole-C4), 8.03, (1H, dd, J1=8.60 Hz, J2=1.17 Hz, benzimidazole-C5), 8.21 (1H, s, benzimidazole-C7), 8.35 (2H, d, J = 8.73 Hz, 1,4-disubstituted benzene), 8.44 (2H, d, J = 4.83 Hz, pyrimidine). 13C-NMR (75 MHz, DMSO-d6): δ = 37.43, 41.82, 43.67, 43.95, 45.37, 110.86, 111.87, 113.24, 115.16, 117.01 (2C), 120.25, 124.09, 131.19 (2C), 132.41, 134.34, 151.27, 158.26, 159.99, 163.40, 164.30, 165.12, 165.37. HRMS (m/z): [M + H]+ calcd for C25H23N8O3S: 515.1610; found: 515.1608.
2-((5-(2-(4-Methoxyphenyl)-1H-benzo[d]imidazol-6-yl)-1,3,4-oxadiazol-2-yl) thio)-1–(4-methyl- piperazin-1-yl)-ethane-1-on (5f)
Yield: 74%, M.P.= 156.6–158.7 °C. FTIR (ATR, cm−1): 3446 (N-H), 1645 (C=O), 835 (1,4-disubstituted benzene). 1H-NMR (300 MHz, DMSO-d6): δ = 2.78 (3H, s, -CH3), 3.43 (6H, br.s., piperazine), 3.65 (2H, br.s., piperazine), 3.87 (3H, s, –OCH3), 4.64 (2H, s, -CH2), 7.17 (2H, d, J = 8.91 Hz, 1,4-disubstituted benzene), 7.78 (1H, d, J = 8.46 Hz, benzimidazole-C4), 7.87 (1H, dd, J1= 8.43 Hz, J2= 1.50 Hz benzimidazole-C5), 8.16 (1H, br.s. benzimidazole-C7), 8.25 (2H, d, J = 8.82 Hz, 1,4-disubstituted benzene), 11.38 (1H, s, N-H).13C-NMR (75 MHz, DMSO-d6): δ = 36.82, 42.40, 42.79, 45.66, 52.19, 52.42, 56.00, 113.15, 113.44, 115.09 (2C), 115.98, 117.70, 120.46, 121.70, 124.59, 129.56 (2C), 153.34, 162.18, 163.31, 165.67, 166.15. HRMS (m/z): [M + H]+ calcd for C23H25N6O3S: 465.1703; found: 465.170.
2-((5-(2-(4-Methoxyphenyl)-1H-benzo[d]imidazol-6-yl)-1,3,4-oxadiazol-2-yl)thio)-1–(4-ethyl-piperazin-1-yl)-ethane-1-on (5 g)
Yield: 66%, M.P.= 180.4–181.9 °C. FTIR (ATR, cm−1): 3334 (N-H), 1645 (C=O), 840 (1,4-disubstituted benzene). 1H-NMR (300 MHz, DMSO-d6): δ = 1.01 (3H, t, J = 7.08 Hz, CH3), 2.34 (2H, m, CH2), 3.85 (3H, s, OCH3), 3.47–3.53 (8H, m, piperazine), 4.56 (2H, s, CH2), 7.13 (2H, d, J = 8.79 Hz, 1,4-disubstituted benzene), 7.71 (1H, d, J = 8.40 Hz, benzimidazole-C4), 7.80 (1H, dd, J1=8.42 Hz, J2=1.44 Hz benzimidazole-C5), 8.13 (1H, br.s., benzimidazole-C7), 8.17 (2H, d, J = 8.88 Hz, 1,4-disubstituted benzene). 13C-NMR (75 MHz, DMSO-d6): δ = 12.36, 37.10, 42.23, 45.85, 51.92, 52.32, 52.80, 55.85, 114.88 (2C), 119.77, 120.02, 120.63, 122.75, 128.71, 128.93 (2C), 153.30, 154.66, 161.25, 161.42, 163.19, 165.05, 166.48. HRMS (m/z): [M + H]+ calcd for C24H27N6O3S: 479.1872; found: 479.1860.
2-((5-(2-(4-Methoxyphenyl)-1H-benzo[d]imidazol-6-yl)-1,3,4-oxadiazol-2-yl)thio)-1–(4-(phenyl)-piperazin-1-yl)-ethane-1-on (5 h)
Yield: 77%, M.P.= 166.7–169.2 °C. FTIR (ATR, cm−1): 3367 (N-H), 1645 (C=O), 840 (1,4-disubstituted benzene). 1H-NMR (300 MHz, DMSO-d6): δ = 3.19 (4H, br.s., piperazine), 3.67 (4H, br.s., piperazine), 3.85 (3H, s, -OCH3), 4.63 (2H, s, -CH2), 6.81 (1H, t, J = 7.14 Hz, phenyl C-H), 6.97 (2H, d, J = 7.92 Hz, phenyl C-H), 7.14 (2H, d, J = 8.79 Hz, 1,4-disubstituted benzene), 7.23 (2H, d, J = 7.47 Hz, phenyl C-H), 7.76 (1H, d, J = 8.46 Hz, benzimidazole C4), 7.80 (1H, dd, J1= 8.37 Hz, J2= 1.20 Hz, benzimidazole-C5), 8.16 (1H, br.s. benzimidazole-C7), 8.29 (2H, d, J = 8.70 Hz, 1,4-disubstituted benzene), 10.72 (1H, s, N-H). 13C-NMR (75 MHz, DMSO-d6): δ = 42.23, 45.85, 51.92, 52.32, 52.80, 55.85, 114.88 (2C), 116.12, 116.63 (2C), 117.06, 119.77, 120.02, 120.63, 122.75, 122.86 128.71 (2C), 128.93 (2C), 153.30, 154.66, 161.25, 161.42, 163.19 165.05, 166.48. HRMS (m/z): [M + H]+ calcd for C28H27N6O3S: 527.1849; found: 527.1860.
2-((5-(2-(4-Methoxyphenyl)-1H-benzo[d]imidazol-6-yl)-1,3,4-oxadiazol-2-yl)thio)-1–(4-(pyridin-2-yl)-piperazin-1-yl)-ethane-1-on (5ı)
Yield: 72%, M.P.= 168.3–170.1 °C. FTIR (ATR, cm−1): 3350 (N-H), 1651 (C=O), 829 (1,4-disubstituted benzene). 1H-NMR (300 MHz, DMSO-d6): δ = 3.52–3.54 (2H, m, piperazine), 3.61–3.65 (6H, m, piperazine), 3.83 (3H, s, -OCH3), 4.62 (2H, s, CH2), 6.65–6.69 (1H, m, pyridine C-H), 6.85–6.87 (1H, m, pyridine C-H), 7.07 (2H, d, J = 8.91 Hz, 1,4-disubstituted benzene), 7.53–7.58 (1H, m, pyridine C-H), 7.64 (1H, s, benzimidazole-C4), 7.70 (1H, dd, J1=8.37 Hz, J2=1.50 Hz, benzimidazole-C5), 8.09 (1H, s, benzimidazole-C7), 8.12–8.14 (1H, m, pyridine C-H), 8.20 (2H, d, J = 8.85 Hz, 1,4-disubstituted benzene). 13C-NMR (75 MHz, DMSO-d6): δ = 37.12, 41.83 44.72, 44.97, 45.42, 55.75, 107.77, 113.80, 114.03, 114.61, 114.82, 115.23 (2C), 116.07, 119.53, 124.75, 128.70 (2C), 128.80, 138.12, 142.46, 148.04, 159.09, 160.87, 162.71, 165.43, 167.01. HRMS (m/z): [M + H]+ calcd for C27H26N7O3S: 528.1825; found: 528.1812.
2-((5-(2-(4-Methoxyphenyl)-1H-benzo[d]imidazol-6-yl)-1,3,4-oxadiazol-2-yl)thio)-1–(4-(pyrimidin-2-yl)-piperazin-1-yl)-ethane-1-on (5j)
Yield: 80%, M.P.= 118.7–120.3 °C. FTIR (ATR, cm−1): 3406 (N-H), 1627 (C=O), 839 (1,4-disubstituted benzene). 1H-NMR (300 MHz, DMSO-d6): δ = 3.64 (2H, br.s, piperazine), 3.72–3.76 (6H, m, piperazine), 3.82 (3H, s, -OCH3), 4.62 (2H, s, CH2), 6.68 (1H, t, J = 4.71, pyrimidine), 7.12 (2H, J = 8.91 Hz, 1,4-disubstituted benzene), 7.70 (1H, d, J = 8.31 Hz, benzimidazole C4), 7.79 (1H, dd, J1= 8.43 Hz, J2= 1.47 Hz, benzimidazole-C5), 8.13 (1H, br.s. benzimidazole-C7), 8.20 (2H, J = 8.85 Hz, 1,4-disubstituted benzene), 8.40 (2H, d, J = 4.74 Hz, pyrimidine). 13C-NMR (75 MHz, DMSO-d6): δ = 37.22, 41.94, 43.77, 45.53, 52.71, 55.71, 110.54, 110.91, 110.96, 114.09, 114.52 (2C), 116.10, 119.28, 125.13, 128.82 (2C), 158.43, 158.46, 160.74, 161.53, 161.62, 162.61, 165.49, 167.55, 168.04. HRMS (m/z): [M + H]+ calcd for C26H25N8O3S: 529.1755; found: 529.1765.
2-((5-(2-(4-Ethoxyphenyl)-1H-benzo[d]imidazol-6-yl)-1,3,4-oxadiazol-2-yl)thio)-1–(4-methyl-piperazin-1-yl)-ethane-1-on (5k)
Yield: 79%, M.P.= 181.2–183.5 °C. FTIR (ATR, cm−1): 3360 (N-H), 1645 (C=O), 833 (1,4-disubstituted benzene). 1H-NMR (300 MHz, DMSO-d6): δ = 1.37 (3H, t, J = 6.81 Hz, -CH3), 2.75 (3H, s, -CH3), 3.39 (4H, br.s., piperazine), 3.60 (4H, br.s., piperazine), 4.47–4.50 (2H, m, -CH2), 4.66 (2H, s, -CH2), 7.21 (2H, d, J = 8.67 Hz, 1,4-disubstituted benzene), 7.88 (1H, d, J = 8.67 Hz, benzimidazole-C4), 7.99 (1H, d, J = 8.55 Hz, benzimidazole-C5), 8.22 (1H, s, benzimidazole-C7), 8.38 (2H, d, J = 8.16 Hz, 1,4-disubstituted benzene), 11.55 (1H, s, N-H). 13C-NMR (75 MHz, DMSO-d6): δ = 14.96, 36.87, 39.11, 42.38, 42.74, 45.66, 52.18, 64.25, 112.48, 115.79 (2C), 117.36, 119.21, 121.51, 123.22, 126.03 (2C), 128.45, 130.51, 151.88, 162.62, 163.81, 165.63, 165.80. HRMS (m/z): [M + H]+ calcd for C24H27N6O3S: 479.1873; found: 479.1860.
2-((5-(2-(4-Ethoxyphenyl)-1H-benzo[d]imidazol-6-yl)-1,3,4-oxadiazol-2-yl)thio)-1–(4-ethyl-piperazin-1-yl)-ethane-1-on (5 l)
Yield: 77%, M.P.= 109.4–110.8 °C. FTIR (ATR, cm−1): 3429 (N-H), 1653 (C=O), 827 (1,4-disubstituted benzene). 1H-NMR (300 MHz, DMSO-d6): δ = 1.19–1.21 (3H, m, -CH3), 1.36 (3H, t, J = 6.90 Hz, -CH3), 2.93–2.98 (2H, m, -CH2), 3.80–3.90 (8H, m, piperazine), 4.10–4.13 (2H, m, -CH2), 4.62 (2H, s, -CH2), 7.10 (2H, d, J = 8.91 Hz, 1,4-disubstituted benzene), 7.73 (1H, d, J = 8.40 Hz, benzimidazole-C4), 7.81 (1H, dd, J1=8.37 Hz, J2=1.50 Hz, benzimidazole-C5), 8.13 (1H, s, benzimidazole-C7), 8.22 (2H, d, J = 8.82 Hz, 1,4-disubstituted benzene). 13C-NMR (75 MHz, DMSO-d6): δ = 15.06, 36.81, 37.88, 43.45, 50.57, 50.98, 51.22, 60.65, 63.81, 113.08, 115.28, 116.74 (2C), 120.76, 122.30, 125.99, 127.43, 129.02 (2C), 132.31, 154.34, 160.80, 163.07, 165.51, 165.61. HRMS (m/z): [M + H]+ calcd for C25H29N6O3S: 493.2019; found: 493.2016.
2-((5-(2-(4-Ethoxyphenyl)-1H-benzo [d] imidazol-6-yl)-1,3,4-oxadiazol-2-yl) thio)-1–(4-(phenyl)-piperazin-1-yl)-ethane-1-on (5 m)
Yield: 70%, M.P.= 262.5–263.9 °C. FTIR (ATR, cm−1): 3367 (N-H), 1645 (C=O), 840 (1,4-disubstituted benzene). 1H-NMR (300 MHz, DMSO-d6): δ = 1.35 (3H, t, J = 6.93 Hz, -CH3), 2.32 (2H, br.s., piperazine), 2.41 (2H, br.s., piperazine), 3.52 (4H, br.s., piperazine), 4.11 (2H, q, J = 6.96 Hz, –CH2), 4.53 (2H, s, –CH2), 7.15 (2H, d, J = 8.94 Hz, 1,4-disubstituted benzene), 7.44–7.47 (3H, m, phenyl C-H), 7.66–7.69 (2H, m, phenyl C-H), 7.76 (1H, d, J = 8.46 Hz, benzimidazole-C4), 7.81–7.85 (1H, m, benzimidazole-C5), 8.15 (1H, br.s. benzimidazole-C7), 8.28 (2H, d, J = 8.85 Hz, 1,4-disubstituted benzene), 10.70 (1H, s, N-H). 13C-NMR (75 MHz, DMSO-d6): δ = 36.75, 42.72, 45.68, 50.38, 50.84, 55.91, 59.07, 114.95, 115.83 (2C), 117.01, 121.02, 121.93, 128.76 (2C), 129.03, 129.09, 129.18, 129.83, 130.20, 131.01, 131.89, 154.05, 161.69, 162.99, 163.10, 165.59, 166.41, 169.44. HRMS (m/z): [M + H]+ calcd for C29H29N6O3S: 541.2002; found: 541.2016.
2-((5-(2-(4-Ethoxyphenyl)-1H-benzo[d]imidazol-6-yl)-1,3,4-oxadiazol-2-yl) thio-1–(4-(pyridin-2-yl)-piperazin-1-yl)-ethane-1-on (5n)
Yield: 71%, M.P. = 168.3–170.4 °C. FTIR (ATR, cm−1): 3361 (N-H), 1635 (C=O), 840 (1,4-disubstituted benzene). 1H-NMR (300 MHz, DMSO-d6): δ = 1.37 (3H, t, J = 6.93 Hz, –CH3), 3.52–3.54 (2H, m, piperazine), 3.59–3.65 (6H, m, piperazine), 4.13 (2H, q, J = 6.99 Hz, -CH2), 4.65 (2H, s, –CH2), 6.66–6.70 (1H, m, pyridine C–H), 6.88 (1H, d, J = 8.61 Hz, pyridine-C–H), 7.11 (2H, d, J = 8.85 Hz, 1,4-disubstituted benzene), 7.54–7.60 (1H, m, benzimidazole-C4), 7.80 (2H, m, pyridine C–H, benzimidazole C–H), 8.06–8.15 (2H, m, pyridine C–H, benzimidazole C–H), 8.18 (2H, d, J = 8.49 Hz, 1,4-disubstituted benzene), 13.50 (1H, s, N–H). 13C-NMR (75 MHz, DMSO-d6): δ = 15.07, 37.18, 41.84, 44.72, 44.98, 45.42, 63.81, 107.80, 113.82, 114.63 (2C), 115.31, 116.84, 119.76, 122.26, 123.67, 125.16, 128.96, 130.02 (2C), 138.15, 141.90, 148.04, 159.08, 160.83, 164.04, 165.40, 166.45. HRMS (m/z): [M + H]+ calcd for C28H28N7O3S: 542.1982; found: 542.1969.
2-((5-(2-(4-Ethoxyphenyl)-1H-benzo[d]imidazol-6-yl)-1,3,4-oxadiazol-2-yl)thio)-1–(4-(pyrimidine-2-yl)-piperazin-1-yl)-ethane-1-on (5o)
Yield: 67%, M.P. = 118.7–120.5 °C. FTIR (ATR, cm−1): 3334 (N-H), 1645 (C=O), 871 (1,4-disubstituted benzene). 1H-NMR (300 MHz, DMSO-d6): δ = 1.37 (3H, t, J = 6.90 Hz, -CH3), 3.59 (2H, br.s., piperazine), 3.65 (2H, br.s., piperazine), 3.75–3.76 (4H, m, piperazine), 4.11 (2H, q, J = 6.99 Hz, –CH2), 4.64 (2H, s, –CH2), 6.67 (1H, t, J = 4.77 Hz, pyrimidine), 8.09 (2H, d, J = 8.73 Hz, 1,4-disubstituted benzene), 7.70 (1H, d, J = 7.70 Hz, benzimidazole-C4), 7.79 (1H, dd, J1 = 8.40 Hz, J2 = 1.59 Hz, benzimidazole-C5), 7.89 (1H, s, benzimidazole-C7), 8.15 (2H, d, J = 8.73 Hz, 1,4-disubstituted benzene), 8.40 (2H, d, J = 4.71 Hz, pyrimidine). 13C-NMR (75 MHz, DMSO-d6): δ = 15.08, 37.25, 41.95, 43.41, 43.68, 45.53, 63.76, 110.99, 115.23 (2C), 116.27, 119.73, 119.96, 120.37, 122.71, 123.06, 128.72 (2C), 128.91, 153.41, 158.48, 160.54, 162.13, 163.06, 165.46, 166.66, 179.63. HRMS (m/z): [M + H]+ calcd for C27H27N8O3S: 543.1907; found: 543.1921.
2.2. 2 D NMR
2D NMR studies including HSQC (Heteronuclear single-quantum correlation spectroscopy), HMBC (Heteronuclear multiple-bond correlation spectroscopy) and COSY (Correlation Spectroscopy) were performed for compound 5e ( and Supporting information).
Table 1. Hydrogen and carbon values of compound 5e detected by 2 D NMR.
2.3. Biological activity
2.3.1. Cytotoxicity test
The anticancer activity of compounds 5a–5o were screened according to the MTT assays. The MTT assays were performed as previously describedCitation32,Citation33. Anticancer activity of final compounds was assessed against five different cancer cell lines A549 (lung carcinoma cell line), HeLa (cervical cell line), MCF-7 (human breast adenocarcinoma cell line), HepG2 (human liver carcinoma cell line) and C6 (rat glioma cell line) cell lines as well as NIH3T3 (mouse embryo fibroblast cell line). Doxorubicin and Hoechst 33342 were used as the reference drugs in the MTT assays.
2.2.2. DNA synthesis inhibition assay
The BrdU cell proliferation method was performed to analyse the effects of the active compounds on proliferation of cancer cells. Cancer cells were seeded into the 96-well plates at a density of 1 × 104 cells. Compounds were added into the each well at three different concentrations (IC50/2, IC50 and 2xIC50) and the plates were incubated for 24 h. At the end of the incubation period, BrdU solution was added and cells were reincubated for 2 h at 37 °C. Anti-BrdU-POD (100 ml) was added and the mixture was incubated for 90 min. Microplates were washed with PBS for three times. After adding substrate solution, the mixture was incubated for 15 min. OD of the wells were determined at 492 nm. Proliferation of control cells was assessed as 100% and growth inhibition % of cells, treated with test compounds and cisplatin were calculatedCitation34.
2.2.3. Flow cytometric analysis
Death pathway of the carcinogenic cell lines was detected by Annexin V-FITC Apoptosis Detection Kit (BD, Pharmingen) as reported previously describedCitation35. Doxorubicin and compounds, which possess the highest cytotoxic activity, were used at their IC50 concentration. FCS Express software was used to display the percent of normal and apoptotic cells at different stages. In the diagrams, Q1, Q2, Q3, and Q4 demonstrates the necrotic cells (positive for PI and negative for annexin/FITC), late apoptotic or necrotic cells (positive for annexin and PI), live cells (negative for annexin and PI), and apoptotic cells (negative for PI and positive for annexin), respectively. The experiments were carried out in triplicates.
2.2.4. DNA topoisomerase I assay
In this study, topoisomerase I assay kit (TG1018‐2; TopoGen) was used to determine if synthesised compounds showed topoisomerase I inhibition. The topoisomerase I inhibition activities of final compounds were measured as relaxation of supercoiled plasmid DNA using agarose gel electrophoresis and camptothecin was used as a positive control. The assay was carried out in a final volume of 20 µl reaction volume containing 2 µl of 10xTGS Buffer, 6 µl water, 2 µl supercoiled plasmid DNA, 2 µl test compound, 2 µl of Top1, 2 ul 10% SDS, 2 µl proteinase K, 2 µl DNA loading dye. After incubation of reaction mixtures at 37 °C for 30 min, electrophoresis was done on a 1% agarose gel at a potential 50 V for 75 min using 1xTAE buffer.
2.3. Molecular docking
A structure-based in silico docking method was applied to determine the binding and interaction modes of compound 5n that show significant anticancer activity in the series, in the active region of the DNA-Topoisomerase I enzyme complex. A protein–ligand interaction analysis was performed on the DNA-Topoisomerase I enzyme complex by using X-ray crystal structure retrieved from Protein Data Bank server (www.pdb.org) (PDB Code: 1T8I)Citation35.
The chemical structures of ligands were drawn using the Schrödinger MaestroCitation36 interface and then were submitted to the Protein Preparation Wizard protocol of the Schrödinger Suite 2016 Update 2Citation37. Then the ligands were prepared by the LigPrep 3.8Citation38 to set to protonation states at pH 7.4 ± 1.0 and the atom types, correctly. Bond orders were assigned, and hydrogen atoms were added to the structures. The grid generation was formed using Glide 7.1Citation39. Flexible docking runs were performed with single precision docking mode (SP).
3. Result and discussion
3.1. Chemistry
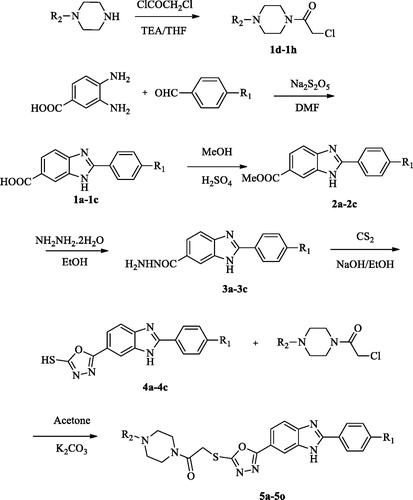
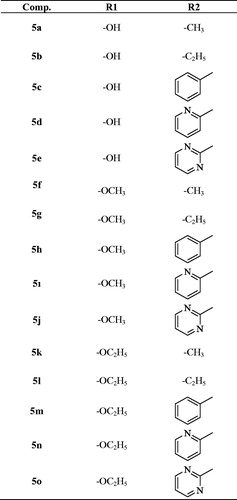
The protocol adopted for synthesis of compounds (5a-5o) was shown in Scheme 1. In the first step, 2–(4-substitutedphenyl)-1H-benzo[d]imidazole-6-carboxylic acid (1a-1c) derivatives were obtained by the reaction of 4-substituted benzaldehyde with 3,4-diamino benzoic acid using sodium bisulphite in DMF. The compound (1a-1c) was converted to a methyl ester (2a-2c) by simple esterification reaction followed by treatment with hydrazine hydrate to obtain 2–(4-substitutedphenyl-1H-benzo[d]imidazole-6-carbohydrazide (3a-3c). The reaction of the hydrazide derivative (3a-3c) with carbon disulfide in boiling ethanol and KOH gave 2-((4-substitutedphenyl)-(6–(5-mercapto-1,3,4-oxadiazol-2-yl)-1H-benzo[d]imidazole derivative (4a-4c). At the last reaction step, the compound (4a-4c) was reacted with acetylated piperazine derivatives in acetone to produce 2-((5–(2-(4-substitutedphenyl)-1H-benzo[d]imidazol-6-yl)-1,3,4-oxadiazol-2-yl) thio)-1– (4-sübstitüepiperazin- 1-yl)-ethane-1-on derivatives (5a-5o).
After the isolation and purification, the structures of newly synthesised compounds (5a-5o) were characterised by using various modern analytical techniques like IR, 1H NMR, 13C NMR, 2 D-NMR and HRMS.
In general, for all the synthesised compounds, the stretching bands for C=O and N-H were observed between 1627–1653 cm−1 and 3311–3628 cm−1, respectively. The stretching absorption belonging to 1,4-disubstituted benzene was determined at 827–871 cm−1.
In the 1H-NMR spectra, protons of piperazine was seen between 2.32–3.90 ppm. The –CH3 group protons of compounds 5a, 5f and 5k on para position of piperazine ring were observed at 2.75–2.78 ppm as asinglet. Again, in position para of the piperazine ring, methyl protons from the -C2H5 protons of compounds 5b, 5g and 5l were observed in the range of 1.01–1.19 ppm, and -CH2 protons in the range of 2.34–2.78 ppm. The aromatic protons of the phenyl (5d, 5l and 5n), pyridine (5d, 5l ve 5n) and pyrimidine (5e, 5j and 5o) substituent attached to the piperazine ring were observed in the range of 6.81–7.69, 6.65–8.14 and 6.67–8.44 ppm, respectively.
The –CH3 group protons of compounds 5f, 5g, 5h, 5ı and 5j on phenyl ring were observed at 3.82–3.87 ppm ppm as a singlet. The -OC2H5 group of compounds 5k, 5l, 5m, 5n ve 5o on phenyl ring, –OCH2 protons were observed at 4.11–4.13 ppm as a quartette while for other compounds (5k, 5l,) –OCH2 protons were appeared as multiplet. The protons of CH3 group of ethyl substituent were observed 1.35–1.37 ppm as a singlet. Methylene protons between C=O and -S group were recorded as a singlet peak between 4.53–4.65 ppm.
A broad singlet due to NH proton of the benzimidazole ring was recorded at 10.70–13.50 ppm. The aromatic protons of 4-substitutedphenyl assigned at 6.90–8.09 and 8.06–8.38 as two doublets. The benzimidazole protons were visualised in the form of doublet, doublet’s doublet and one singlet in 1H NMR spectra at around 7.64–7.92, 7.74–8.03 and 7.89–8.22 due to H-4, H-5, and H-7 protons.
The 13C NMR spectra showed the peaks between 165.61–169.44 ppm due to carbonyl group (C=O). The carbons of the piperazine ring were observed at 39.11–55.91 ppm. The 13CNMR spectra of all the derivatives showed carbon values in the predictable regions, while the HRMS analysis confirmed the mass with the calculated values of the target compounds. In addition for structure elucidations using routine spectroscopic methods, 2 D NMR studies including HMBC, HSQC and COSY were performed for compound 5e as seen in and supporting information.
3.2. Biological activity
3.2.1. Cytotoxicity assay
The benzimidazole-1,3,4-oxadiazole derivatives (5a-5o) were tested their cytotoxic effects in vitro using five cancer cell line A549 (lung carcinoma cell line), HeLa (cervical cell line), MCF-7 (human breast adenocarcinoma cell line), HepG2 (human liver carcinoma cell line) and C6 (rat glioma cell line) cell lines as well as NIH3T3 (mouse embryo fibroblast cell line). Doxorubicin and Hoechst 33342 were used for this study as a reference drugs. The IC50 values, defined as the half maximal inhibitory concentration, were summarised in .
Table 2. IC50 values (µM) of the compounds 5a-5o, doxorubicin and Hoechst 33342 against A549, MCF-7, C6, HepG2, HeLa and NIH3T3.
Investigations of the cytotoxic activity against A549, MCF-7, C6, HepG-2 and HeLa indicated that the HeLa was the most sensitive cell line to the influence of the new derivatives. It was important to note that compounds 5l and 5n exhibited better antiproliferative activity than Hoechst 33342 and doxorubicin against HeLa cell line, with IC50 of 0.224 ± 0.011 µM and 0.205 ± 0.010 µM, respectively. The above MTT assay results confirmed our expectation that compounds 5l and 5n may exhibit effective activity and lower toxicity and thus possibly better therapeutic potency than Hoechst 33342. Furthermore, compound 5b exhibited prominent activity against HeLa cell line with IC50 0.698 ± 0.032 µM and shows poor activity against the other cell lines with IC50 ≥100 µM
Compound 5a emerged as potential compound against both cell lines with IC50 5.704 ± 0.254 µM against MCF-7 cell line and IC50 5.695 ± 0.283 µM against HepG2 cell line. IC50 values of compounds 5d, 5e and 5o for A549 cell line were determined as 9.148 ± 0.391, 7.318 ± 0.352 and 6.442 ± 0.287 µM, respectively. Only compound 5n displayed most promising cytotoxic activity against A549 cell line with an IC50 value of 7.388 ± 0.325 µM. Besides, among compounds 5a-5o, compound 5k exhibited the most promising anticancer activity against C6 cell line with IC50 values of 3.204 ± 0.152 µM.
It is crucial that an anticancer agent affects the cancer cell line but having minimal or no side-effect on healthy cells. For this purpose, the cytotoxic effects of the active compounds on the NIH3T3 cell line were investigated ().
According to all results, it can be concluded that 4-hydroxyphenyl and 4-ethoxyphenyl enhanced anticancer activity more than 4-methoxyphenyl. The presence of methyl and ethyl group at the 4th position of the piperazine scaffold also increased anticancer activity, whereas phenyl moiety at the 4th position of the piperazine ring led to a significant drop in anticancer activity. However, compounds carrying pyridine and pyrimidine substituents have also been found to be active.
3.2.2. DNA synthesis inhibition assay
According to the MTT assay, compound 5n for A549 cell line; compounds 5a, 5d, 5e, 5o for MCF-7 cell line; compound 5k for C6 cell line; compound 5a for HepG2 cell line; compounds 5b, 5l and 5n for HeLa cell line were selected for the DNA synthesis inhibition assay. A549, C6, MCF-7, HepG2 and HeLa cells were incubated with three different concentrations (IC50/2, IC50, and IC50/2) of the compounds for 24 and 48 h time periods. The tested compounds showed time-and dose-dependent inhibitory activity on DNA synthesis ofthe tumour cells. Doxorubicin was used as positive control.
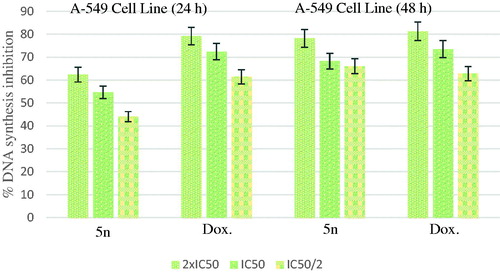
shows the DNA % synthesis inhibitory activity of the compound 5n and standard drug on A549 cells. Compound 5n was found to have 78.17, 68.25 and 66.00% DNA synthesis inhibition at 14.77, 7.39 and 5.39 µg/mL doses after 48 h of incubation, whereas doxorubicin was found to have 81.22, 73.43 and 62.78% inhibition at 24.84, 12.42 and 6.21 µg/mL doses, respectively.
Figure 2. % DNA synthesis inhibition activities of compound 5n and doxorubicin against A549 cell line.
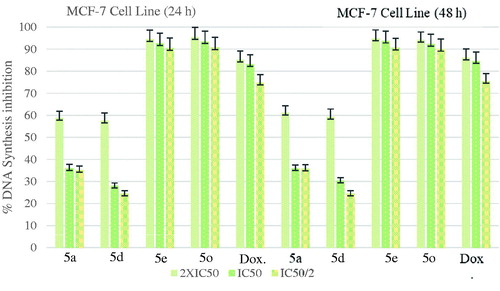
The DNA % synthesis inhibitory activity of the compounds 5a, 5d, 5e, 5o, and standard drug on MCF7 cells is seen in . Among of the compounds, compound 5e was determined as the most active compounds. Compound 5e was observed to have an approximate inhibitory activity with a value of 93.81% at 7.318 µg/mL concentration whereas doxorubicin had 84.60% inhibition at 10.525 µg/mL concentration after 48-h incubation.
Figure 3. % DNA synthesis inhibition activities of compounds 5a, 5d, 5e, 5o and doxorubicin against MCF-7 cell line.
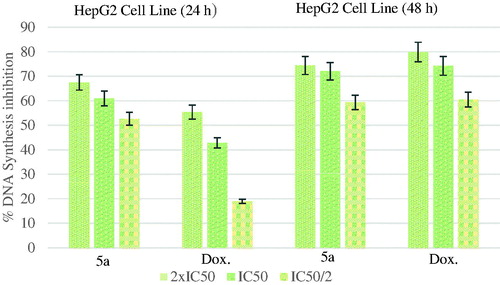
shows the DNA % synthesis inhibitory activity of the compound 5a and standard drug on HepG2 cells. Compound 5a had 60.89 and 72.05% inhibitory activity after 24- and 48-h incubation, at 5.695 µg/mL whereas doxorubicin had 42.80 and 74.22% inhibitions at 16.482 µg/mL.
Figure 4. % DNA synthesis inhibition activities of compound 5a and doxorubicin against HepG2 cell line.
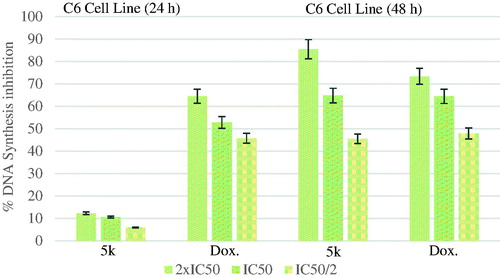
The DNA % synthesis inhibitory activity of the compound 5k and standard drug on C6 cells is seen . DNA % inhibition was increased with the increasing incubation period (24 and 48 h) for the compound 5k and doxorubicin. This increase can be clearly appreciated in particular for the compound 5k. Measurement of the inhibition for the compound 5k at lower dose (IC50/2) after 48 h incubation was eight times higher than at 24 h of measurement.
Figure 5. % DNA synthesis inhibition activities of compound 5k and doxorubicin against C6 cell line.
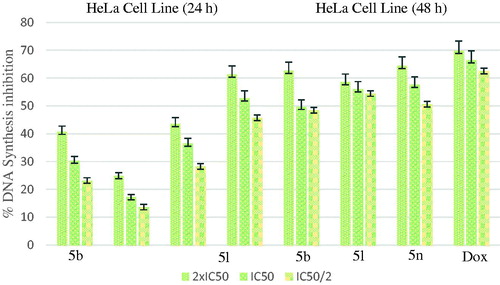
shows the DNA % synthesis inhibitory activity of the compounds 5b, 5l, 5n and standard drug on HeLa cells. DNA % inhibition was increased with the increasing incubation period (24 and 48 h) for all of the compounds. Compounds 5b, 5l, 5n were found to have 49.70, 56.01 and 57.65% DNA synthesis inhibition at 0.698, 0.224 and 0.205 µg/mL doses after 48 h of incubation whereas doxorubicin was found to have 66.48% inhibition at 14.280 µg/mL doses, respectively.
Figure 6. % DNA synthesis inhibition activities of compounds 5 b, 5 l, 5n and doxorubicin against HeLa cell line.
3.2.3. Flow cytometric analysis
Annexin V/PI staining on tested cancer cells treated with the active compounds was performed to investigate induction of apoptosis. Usually, four quadrant images were generated by flow cytometry: Q1 shows damaged cells due to any technical factor rather than compound, Q2 area shows late apoptotic cells, Q3 denotes normal cells while Q4 illustrates early apoptotic cells. According to the MTT assay, compound 5n for A549 cell line; compounds 5a, 5d, 5e, 5o for MCF-7 cell line; compound 5k for C6 cell line; compound 5a for HepG2 cell line; compounds 5b, 5l and 5n for HeLa cell line were selected for the flow cytometric analysis. Furthermore, flow cytometry studies were performed for reference drug doxorubicin. Flow cytometric analysis diagrams of tested compounds and doxorubicin at IC50/2, IC50, and 2xIC50 concentrations on cancer cells were presented in .
Figure 7. The flow cytometric analysis diagram of compound 5n and doxorubicin for A549 cell line.
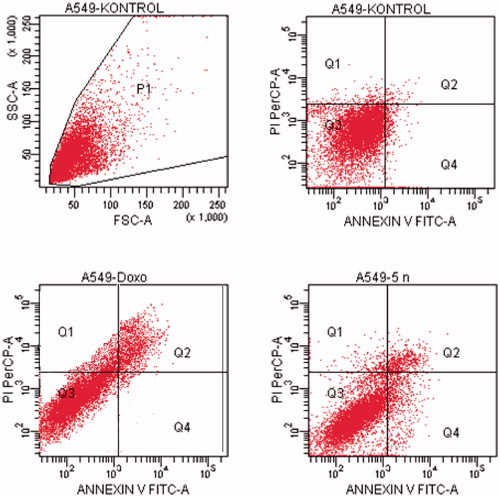
Figure 8. The flow cytometric analysis diagram of compounds 5d, 5e, 5o and doxorubicin for MCF-7 cell line.
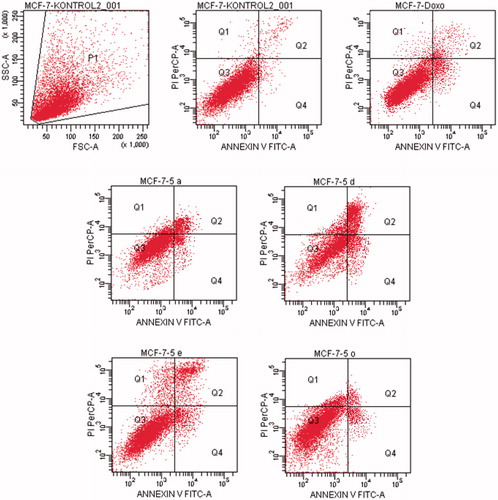
Figure 9. The flow cytometric analysis diagram of compound 5k and doxorubicin for C6 cell line.
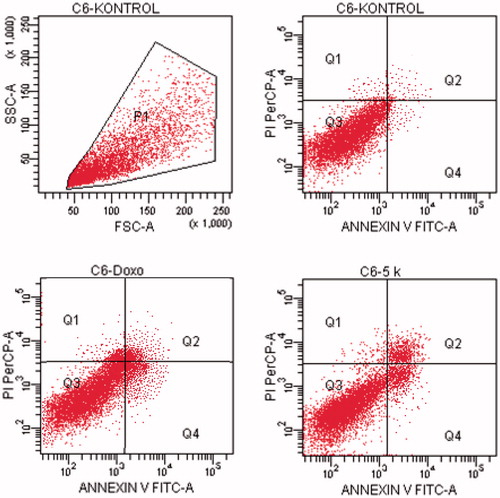
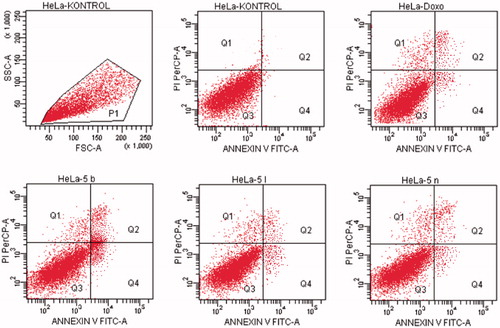
Figure 10. The flow cytometric analysis diagram of compound 5a and doxorubicin for HepG2 cell line.
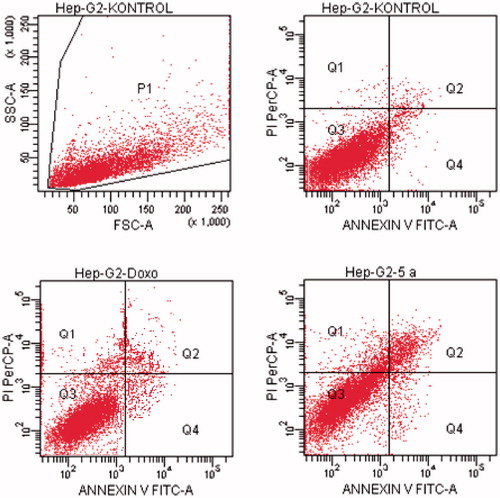
Figure 11. The flow cytometric analysis diagram of compounds 5 b, 5 l, 5n and doxorubicin for HeLa cell line.
Flow cytometric analysis diagram of doxorubicin with compound 5n was presented in for A549 cell line. Accordingly, the percentage of apoptotic cells at IC50 concentrations was calculated as 19.2% for doxorubicin and 13% for compound 5n.
Flow cytometric analysis diagram of doxorubicin with compounds 5a, 5d, 5e and 5o in MCF-7 cell line was presented in . Compound 5d caused the highest percentage of apoptosis (early + late apoptotic cells) with 22.3% whereas doxorubicin caused 12.7% on MCF-7 cells. The other compounds 5a, 5e and 5o possessed 15.4, 13.1 and 8.7% apoptotic cell percentages.
Flow cytometric analysis diagram of doxorubicin with compound 5k was presented in for C6 cell line. Accordingly, the percentage of apoptotic cells at IC50 concentrations was calculated as 13.7% for doxorubicin and 12.3% for compound 5k.
For HepG2 cell line, the percentage of apoptotic cells at IC50 concentrations was calculated as 9.4% for doxorubicin and 13.2% for compound 5a (). As a result, it is observed that compound 5a induces apoptotic cell death more than doxorubicin in HepG2 cell line in IC50 concentrations and 24 h incubation time.
Flow cytometric analysis diagram of doxorubicin and compounds 5b, 5l, 5n on HeLa cell line was presented in . For HeLa cells, compound 5b showed the highest percentage of 17.4% whereas doxorubicin caused 17.4% apoptotic cells. The other tested compounds 5l and 5n followed it with 8.6 and 15.2% percentages.
3.2.4. DNA topoisomerase I assay
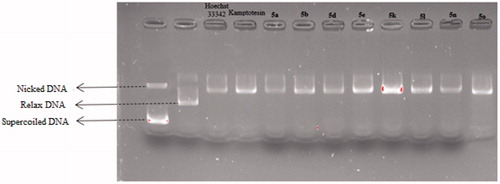
To determine whether the DNA intercalation properties of compounds 5a, 5b, 5d, 5e, 5k, 5l, 5n and 5o might result in Topo I inhibition, agarose gel electrophores is assay was performed to evaluate the Topo I inhibitory activity of them via agarose-gel electrophoresis and CPT was used as the positive control. The TopoGEN Topoisomerase I Drug Screening Kit were optimised and implemented to accomplish this goal.
The topo I DNA relaxation data demonstrated that the supercoiled kDNA migrates faster on the agarose gel than the relaxed topoisomers (nicked) caused by the full activity of topo I. Therefore, the appearance of relaxed topoisomers along with residual supercoiled form indicates the partial activity of topoisomerase I and thus its inhibition by the test compounds. In a previous studies, it was said that the compounds poisoning topoisomerase caused the formation of nick bands in such gel appearances, while the formation of Supercoiled bands was said to be caused by catalytic inhibitors that bound to either DNA or enzymeCitation40,Citation41.
The results in indicated that compounds 5a, 5b, 5d, 5e, 5k, 5l, 5n and 5o exhibited potent topo I inhibition activity. By the comparison of the effective inhibited concentrations of camptothecin with these compounds, it could be concluded that these compounds exhibited similar topo I inhibition activity with camptothecin.
Figure 12. Topo I activity of compounds 5a, 5 b, 5d, 5e, 5k, 5 l, 5n, 5o, Hoechst 33342 and camptothecin.
3.3. Molecular docking
In order to determine the possible interactions of compound, showed high activity, docking studies have been performed by using high-resolution crystal structure of the DNA-Topoisomerase I enzyme complex (PDB Code: 1T8I)Citation35,Citation42–45. Hoechst 33342 molecule was included in the docking study with the relevant crystal structure before performing the analysis of binding modes of selected compound which has displayed significant topoisomerase I enzyme activity (5n). Hence, the docking protocol was verified by examining and comparing binding modes and interactions of Hoechst 33342.
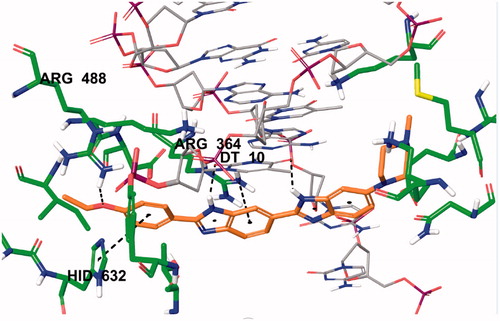
Two-dimensional interaction pose of Hoechst 33342 with DNA-Topoisomerase I enzyme complex active region is given in . The oxygen atom in the ethoxy group in the structure forms a hydrogen bond with amino Arg488. The phenyl ring near to ethoxy group establishes π-π interaction with imidazole ring of Hid632. The benzimidazole ring adjacent to phenyl ring has been found to be very important for both polar and apolar interactions. Benzene and imidazole structures of benzimidazole ring forms cation-π interactions separately with amino of Arg364. Each of the nitrogen atoms located in the same position of successive benzimidazole rings in the structure of Hoechst 33342 creates a hydrogen bond with carbonyl of thymine (DT10) in the D chain of DNA-Topoisomerase enzyme complex. In addition, these interactions analyse of Hoechst 33342 were found to be similar and compatible with the literature dataCitation3.
Figure 13. Three-dimensional interaction of Hoechst 33342 with the DNA-Topoisomerase I enzyme complex active site.
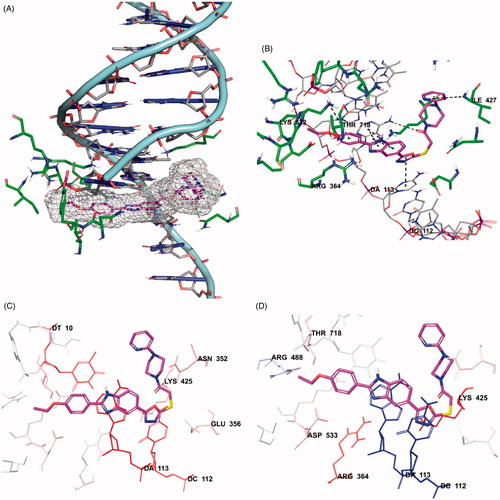
The docking poses of compound 5n was given in . When the docking poses of this compound was examined, it was seen that this compound interact with both amino acids in the enzyme active region and certain regions of the DNA contained in the complex crystal.
Figure 14. Docking poses of compound 5n. (A) Placement of the compound 5n on the DNA-Topoisomerase I enzyme complex active site. (B) The interacting mode of compound 5n in the active region of DNA-Topoisomerase I enzyme. (C) Van der Waals interaction of DNA-Topoisomerase I enzyme complex active site of compound 5n. (D) Electrostatic interaction of DNA-Topoisomerase I enzyme complex active site of compound 5n.
Scheme 1. Synthesis way of the compounds 5a-5o.
Scheme 1. Continued.
Thus, it was observed that it cause damage in DNA chain and prevent the binding of topoisomerase enzyme to DNA (). Also, when looking at the chemical structure of the compound, it has been seen that benzimidazole, oxadiazole and pyridine structures show similar interactions. Benzimidazole ring system has attracted attention in terms of polar and apolar interactions. π–π interactions were observed separately between purine ring of adenine (DA113) in the DNA chain and benzene and imidazole rings of benzimidazole in the compound 5n. Also, in this compound, phenyl ring of benzimidazole forms π–π interaction with Arg364. The nitrogen atom in the benzimidazole ring is found to be important for polar interactions.
The oxadiazole ring in the structure of compound 5n has been shown to contribute to binding to DNA-Topoisomerase I enzyme complex active site (). The oxadiazole ring forms π–π interaction with pyrimidine of cytosine (DC112) in DNA. It has been determined that carbonyl group in the chemical structures of selected compound is especially important in binding to the DNA chain (). In compound 5n, carbonyl group forms a hydrogen bond with amino group in the purine ring of adenine (DA113) of DNA. The oxygen atom of ethoxy group in the para position for compound 5n forms a hydrogen bond with hydroxyl of Thr718 in both compounds. An additional cation–π interaction is observed between amino group of Lys532 and para-ethoxypheny. It is observed that the chemical structure of selected compound whose molecular docking studies are carried out include pyridine ring adjacent to the piperazine ring. It has been observed that nitrogen atoms in this ring systems are very effective and important in binding to related region. As it is known, nitrogen atom in these ring systems have the ability to make hydrogen bonds by acting as an electron donor by keeping the unpaired electron pairs on them outside the ring without including them in the ring electron system. They strengthen the binding profiles of the compound by forming hydrogen bonds with the active region thanks to their properties. Hydrogen bond formation with amino group of Ile427 is observed in the compound 5n. Van der Waals and electrostatic interactions of selected compound with DNA-Topoisomerase I enzyme complex active region is given in . According to the docking analysis procedure followed in Van der Waals interactions, red and pink coloured amino acids and nucleic acids indicate strong van der Waals interactions. Hereunder, it is seen that Asn352, Glu356, Lys425, Lys532, Asp533 and Ile535 amino acids and DT10, DC112 and DA113 nucleic acids in DNA play an important role in terms of van der Waals interactions. Strong electrostatic interactions are similarly represented by red- and blue-coloured amino acids and nucleic acids according to the docking analysis procedure. In terms of electrostatic interactions, it is seen that Glu356, Arg364, Lys425, Arg488, Lys532, Asp533 and Thr718 amino acids and DT10, DC112 and DA113 nucleic acids in DNA play an important role.
presents the binding affinity and interacting residues of compound 5n and Hoechst. The binding affinity was calculated according to the parameters of docking score, Glide gscore and Glide emodel, and is as follow: for compound 5n −7.409, −7.889, −100.082 kcal/mol and for Hoechst −4.373, −5.989, −66.563 kcal/mol. As can be seen from this table, compound 5n showed more potent binding affinity than Hoechst.
Table 3. Binding affinity and interacting residues of compound 5n and Hoechst.
4. Conclusion
In conclusion, 15 novel benzimidazole-1,3,4-oxadiazole derivatives were synthesised and evaluated for their anticancer effects on A549, MCF-7, C6, HepG2 and HeLa cell lines. The results of this preliminary cytotoxicity screening using the MTT assay suggested that many of them (5a, 5b, 5d, 5e, 5k, 5l, 5n and 5o) displayed potent antitumor activity in different cancer cell lines. Especially, compounds 5l (0.224 µM) and 5n (0.205 µM) displayed potent and selective anticancer activity against HeLa cell line compared to doxorubicin (14.280 µM) and Hoechst 33342 (0.306 µM). Further detailed biological studies including flow cytometric analysis, DNA synthesis inhibition assay and Topo I inhibition studies delivered promising results. Docking studies of compound 5n and Hoechst 33342 were performed and probable interactions in the DNA-Topo I enzyme complex was determined. According to all in vitro and in silico studies, novel benzimidazole-1,3,4-oxadiazole derivatives could be served as a novel promising scaffold for the development of new chemotherapeutic agent.
Supplemental Material
Download PDF (5.7 MB)Disclosure statement
The authors declare no conflicts of interest.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Singh R, Pandey S, Sur S, et al. PPEF: A bisbenzimdazole potent antimicrobial agent interacts at acidic triad of catalytic domain of E. coli topoisomerase IA. Biochim Biophys Acta Gen Subj 2019;1863:1524–35.
- Liang X, Wu Q, Luan S, et al. A comprehensive review of topoisomerase inhibitors as anticancer agents in the past decade. Eur J Med Chem 2019;171:129–68.
- Bansal S, Sur S, Tandon V. Benzimidazoles: Selective inhibitors of topoisomerase I with differential modes of action. Biochemistry 2019;58:809–17.
- Halawa AH, Elgammal WE, Hassan SM, et al. Synthesis, anticancer evaluation and molecular docking studies of new heterocycles linked to sulfonamide moiety as novel human topoisomerase types I and II poisons. Bioorg. Chem 2020;98:103725.
- Mohamady S, Gibriel AA, Ahmed MS, et al. Design and novel synthetic approach supported with molecular docking and biological evidence for naphthoquinone-hydrazinotriazolothiadiazine analogs as potential anticancer inhibiting topoisomerase-IIB. Bioorg. Chem 2020;96:103641.
- Issar U, Arora R, Kumari T, et al. Combined pharmacophore-guided 3D-QSAR, molecular docking, and virtual screening on bis-benzimidazoles and ter-benzimidazoles as DNA–topoisomerase I poisons. Struct. Chem 2019;30:1185–201.
- Shu B, Yu Q, Hu DX, et al. Synthesis and biological evaluation of novel indole-pyrazoline hybrid derivatives as potential topoisomerase 1 inhibitors. Bioorg Med Chem Lett 2020;30:126925.
- Sharma PC, Bansal KK, Sharma A, et al. Thiazole-containing compounds as therapeutic targets for cancer therapy. Eur J Med Chem 2020;188:112016.
- Lee JF, Chang TY, Liu ZF, et al. Design, synthesis, and biological evaluation of heterotetracyclic quinolinone derivatives as anticancer agents targeting topoisomerases. Eur. J. Med. Chem 2020;190:112074.
- Joshi G, Kalra S, Yadav UP, et al. E-pharmacophore guided discovery of pyrazolo [1,5-c] quinazolines as dual inhibitors of topoisomerase-I and histone deacetylase. Bioorg. Chem 2020;94:103409.
- K Kathiravan M, N Kale A, Nilewar S. Discovery and development of topoisomerase inhibitors as anticancer agents. Mini Rev Med Chem 2016;16:1219–29.
- Aichinger G, Lichtenberger FB, Steinhauer TN, et al. The Aza-Analogous Benzo [c] phenanthridine P8-D6 Acts as a Dual Topoisomerase I and II Poison, thus Exhibiting Potent Genotoxic Properties. Molecules 2020;25:1524.
- Lian C, Cao S, Zeng W, et al. RJT-101, a novel camptothecin derivative, is highly effective in the treatment of melanoma through DNA damage by targeting topoisomerase 1. Biochem Pharmacol 2020;171:113716.
- Dehshahri A, Ashrafizadeh M, Afshar EG, et al. Topoisomerase inhibitors: Pharmacology and emerging nanoscale delivery systems. Pharmacol. Res 2020;151:104551.
- Nunhart P, Konkoľová E, Janovec L, et al. Fluorinated 3,6,9-trisubstituted acridine derivatives as DNA interacting agents and topoisomerase inhibitors with A549 antiproliferative activity. Bioorg Chem 2020;94:103393.
- Liu XW, Tang YC, Liu NY, et al. Topo I inhibition, DNA photocleavage, Molecular docking and cytotoxicities of two new phenanthroline‐based ruthenium complexes. Appl Organomet Chem 2020;34:e5312.
- Alonso C, Fuertes M, Martín-Encinas E, et al. Novel topoisomerase I inhibitors. Syntheses and biological evaluation of phosphorus substituted quinoline derivates with antiproliferative activity. Eur J Med Chem 2018;149:225–37.
- Sović I, Jambon S, Pavelić SK, et al. Synthesis, antitumor activity and DNA binding features of benzothiazolyl and benzimidazolyl substituted isoindolines. Bioorg Med Chem 2018;26:1950–60.
- Sheng C, Miao Z, Zhang W. New strategies in the discovery of novel non-camptothecin topoisomerase I inhibitors. Curr Med Chem 2011;18:4389–409.
- Galal SA, Hegab KH, Hashem AM, et al. Synthesis and antitumor activity of novel benzimidazole-5-carboxylic acid derivatives and their transition metal complexes as topoisomerease II inhibitors. Eur J Med Chem 2010;45:5685–91.
- Refaat HM. Synthesis and anticancer activity of some novel 2-substituted benzimidazole derivatives. Eur J Med Chem 2010; 45:2949–56.
- Romero-Castro A, León-Rivera I, Ávila-Rojas LC, et al. Synthesis and preliminary evaluation of selected 2-aryl-5(6)-nitro- 1H-benzimidazole derivatives as potential anticancer agents. Arch Pharm Res 2011;34:181–9.
- Abonia R, Cortés E, Insuasty B, et al. Synthesis of novel 1,2,5-trisubstituted benzimidazoles as potential antitumor agents. Eur J Med Chem 2011;46:4062–70.
- Rashid M, Husain A, Mishra R, et al. Design and synthesis of benzimidazoles containing substituted oxadiazole, thiadiazole and triazolo-thiadiazines as a source of new anticancer agents. Arab J Chem 2019;12:3202–24.
- Sunke R, Babu PV, Yellanki S, et al. Ligand-free MCR for linking quinoxaline framework with a benzimidazole nucleus: a new strategy for the identification of novel hybrid molecules as potential inducers of apoptosis. Org Biomol Chem 2014;12:6800–5.
- Rashid M, Husain A, Mishra R. Synthesis of benzimidazoles bearing oxadiazole nucleus as anticancer agents. Eur J Med Chem 2012;54:855–66.
- Rao VM, Rao AS, Rani SS, et al. Ultrasound assisted cu-catalyzed synthesis of 1,2-disubstituted benzimidazoles as potential antibacterial agents. Mini Rev Med Chem 2018;18:1233–9.
- Formagio ASN, Tonin LTD, Foglio MA, et al. Synthesis and antitumoral activity of novel 3-(2-substituted-1,3,4-oxadiazol-5-yl) and 3-(5-substituted-1,2,4-triazol-3-yl) β-carboline derivatives. Bioorg Med Chem 2008;16:9660–7.
- Glomb T, Szymankiewicz K, Świątek P. Anti-cancer activity of derivatives of 1,3,4-oxadiazole. Molecules 2018;23:3361.
- Neeraja P, Srinivas S, Mukkanti K, et al. 1H-1,2,3-Triazolyl-substituted 1,3,4-oxadiazole derivatives containing structural features of ibuprofen/naproxen: Their synthesis and antibacterial evaluation. Bioorg Med Chem Lett 2016;26:5212–7.
- Mochona B, Mazzio E, Gangapurum M, et al. Synthesis of some benzimidazole derivatives bearing 1,3,4-oxadiazole moiety as anticancer agents. Chem Sci Trans 2015;4:534–40.
- Çevik UA, Osmaniye D, Çavuşoğlu BK, et al. Synthesis of novel benzimidazole–oxadiazole derivatives as potent anticancer activity. Med Chem Res 2019;28:2252–61.
- Evren AE, Yurttaş L, Eksellı B, et al. Novel Tri-substituted Thiazoles Bearing Piperazine Ring: Synthesis and Evaluation of their Anticancer Activity. Lett Drug Des. Discov 2019;16:547–55.
- Acar Çevik U, Sağlık BN, Korkut B, et al. Antiproliferative, cytotoxic, and apoptotic effects of new benzimidazole derivatives bearing hydrazone moiety. J Heterocyc Chem 2018;55:138–48.
- Staker BL, Feese MD, Cushman M, et al. Structures of three classes of anticancer agents bound to the human topoisomerase I-DNA covalent complex. J Med Chem 2005;48:2336–45.
- Maestro, version 10.6, Schrödinger, LLC, New York, NY, 2016.
- Schrödinger LLC, New York, NY, 2016.
- LigPrep, version 3.8, Schrödinger, LLC, New York, NY, 2016.
- Glide, version 7.1, Schrödinger, LLC, New York, NY, 2016.
- Oksuzoglu E, Tekıner-Gulbas B, Alper S, et al. Some benzoxazoles and benzimidazoles as DNA topoisomerase I and II inhibitors. J Enzyme Inhib Med Chem 2008;23:37–42.
- Foto E, Özen Ç, Zilifdar F, et al. Benzoxazines as new human topoisomerase I inhibitors and potential poisons. DARU J Pharma Sci 2019;28:65–73.
- Xu Y, Wu L, Rashid HU, et al. Novel indolo-sophoridinic scaffold as Topo I inhibitors: Design, synthesis and biological evaluation as anticancer agents. Eur J Med Chem 2018;156:479–92.
- Xu Y, Jing D, Chen R, et al. Design, synthesis and evaluation of novel sophoridinic imine derivatives containing conjugated planar structure as potent anticancer agents. Bioorg. Med. Chem 2018;26:4136–44.
- Arepalli SK, Lee C, Sim S, et al. Development of 13H-benzo [f] chromeno [4,3-b][1,7] naphthyridines and their salts as potent cytotoxic agents and topoisomerase I/IIα inhibitors. Bioorg. Med. Chem 2018;26:5181–93.
- Arthur DE, Uzairu A. Molecular docking study and structure‐based design of novel camptothecin analogues used as topoisomerase I inhibitor. J Chin Chem Soc 2018;65:1160–78.