
Abstract
The significant role of topoisomerases in the control of DNA chain topology has been confirmed in numerous research conducted worldwide. The prevalence of these enzymes, as well as the key importance of topoisomerase in the proper functioning of cells, have made them the target of many scientific studies conducted all over the world. This article is a comprehensive review of knowledge about topoisomerases and their inhibitors collected over the years. Studies on the structure–activity relationship and molecular docking are one of the key elements driving drug development. In addition to information on molecular targets, this article contains details on the structure–activity relationship of described classes of compounds. Moreover, the work also includes details about the structure of the compounds that drive the mode of action of topoisomerase inhibitors. Finally, selected topoisomerases inhibitors at the stage of clinical trials and their potential application in the chemotherapy of various cancers are described.
Introduction
In 1971 Jim Wang discovered first DNA topoisomerase I (the omega (ω) protein from Escherichia coli)Citation1,Citation2. At that time, the separation of the supercoiled and relaxed DNA was necessary to perform the activity test. To achieve this, the reaction products were run on a sucrose gradient and then the DNA contained in each fraction taken from the centrifugation tubes were collected and analysedCitation2.
In 1975, for the first time, agarose gel electrophoresis was used to differentiate between various DNA topoisomers. The use of this method greatly facilitated work for DNA topologistsCitation3. For nearly 40 years, many topoisomerases have been discovered and characterised in all three domains of life (bacteria, eukarya and archaea). The first discovery, in 1976, was bacterial DNA gyraseCitation4 and then in 1980, eukaryotic decatenase was foundCitation5.
DNA topoisomerases are a group of enzymes that control DNA topology. They are involved in many significant biological processes in all cells (e.g. DNA replication, transcription and recombination or chromosome condensation)Citation6. These enzymes bind covalently to the DNA phosphorus group, split the DNA strand or strands and finally reunite them. According to their mechanism of action, there are two main types of topoisomerases: topoisomerases I (Top I) and topoisomerases II (Top II) divided into five subfamilies (see ).
Table 1. Types and subfamilies of topoisomerases
The presence of topoisomerases and their proper functioning is one of the key elements of most processes taking place in the cell. For most of the processes requiring access to the information stored in the DNA duplex, a permanent or temporary separation of the two strands of DNA is necessary (e.g. topoisomerases enable the release of replicated chromosomes before partitioning and cell division)Citation7.
The use of cytostatic agents, which inhibit enzyme activity, leads to irreversible interruption of DNA strands (by a stable DNA-topoisomerase complex), which causes cell death. Increased topoisomerase activity observed in many cancers results in selective action of agents that are topoisomerase inhibitorsCitation8.
In the beginning, we would like to briefly describe the topoisomerases I and II and their mechanism of action. Then we will present and characterise the topoisomerases I and II inhibitors, in particular compounds in clinical trials.
Type I topoisomerases
Primarily named ω protein, topoisomerase I was discovered by James C. Wang in the 1970s. He identified the first topoisomerase I in Escherichia coliCitation2. Proteins belonging to the group of topoisomerases I were found in eukaryotic and prokaryotic organismsCitation9–12. These enzymes are responsible for relaxing negatively supercoiled DNA and allow for catenation or decatenation of broken DNACitation11,Citation13.
Topoisomerases I can be divided into three subfamilies: topoisomerases IA, topoisomerases IB and topoisomerases ICCitation14–16. For all subtypes of topoisomerases I, changing the DNA topology by breaking the phosphodiester bond between DNA strands is based on the same general mechanism. The phosphoryl group of DNA is attacked by tyrosyl group of topoisomerase I. This creates a covalent bond between the tyrosyl group and one side of the broken DNA. At the same time, the free hydroxylated strand is released and rotated. The hydroxyl end of the free strand of DNA attacks the formed phosphotyrosine bond, rebuilds the phosphodiester bond between the two strands and releases the topoisomerase to the next catalytic cycleCitation1,Citation17.
In most cases, change the topology of DNA by type I topoisomerases do not require external energy (e.g. ATP hydrolysis). Reverse gyrase is the only enzyme in topoisomerase I subfamily which needs energy from ATP hydrolysis to introduce positively supercoiled DNA. Now, one of the main challenges is to identify the way how energy stored in the DNA is converted into protein changes in the course of the reaction. Knowing this mechanism will help to fully understand not only type I topoisomerases but also other enzymes belonging to this classCitation9.
Type IA topoisomerases
Topoisomerases IA (Top IA) are a subfamily of enzymes found in all three domains of life (bacteria, archaea and eukarya). The sequence analysis has shown that all topoisomerases IA are monomeric enzymes (the exception—reverse gyrase isolated from Methanopyrus kandleri) composed of two main parts: a core (molecular weight around 67 kDa) with all the conserved domains, especially those creating the active site of the enzyme, and a carboxyl-terminal domain, very diverse in size and sequenceCitation9,Citation10.
Type IA enzymes are responsible for relaxation only negative supercoils in DNA and a single-stranded region in DNA is required for their activity. The mechanism of action of the topoisomerases IA known as “enzyme-bridged strand passage” was defined based on structural, biochemical and biophysical studies. To change DNA topology type IA enzyme causes splitting of a single strand of DNA. The enzyme attaches tyrosine to the DNA 5′-phosphoryl group with a covalent bond. Released 3′-OH end remains attached Top IA and cannot freely rotate. This result in creating a gap that allows the transport of another DNA strand. After passing the transported segment through the gap, both ends of broken DNA are religatedCitation9,Citation18,Citation19.
Examples of enzymes belonging to the Top IA subfamily are bacterial topoisomerase I, bacterial topoisomerase III, eukaryotic topoisomerase III and reverse gyraseCitation1,Citation6,Citation20.
Type IB topoisomerases
Type IB topoisomerases (Top IB) are a subfamily of enzymes presented in eukaryotes, some viruses and bacteria but not in archaeaCitation9,Citation12. As opposed to Top IA, topoisomerases IB can relax both negatively and positively supercoiled DNA with energy accumulated in DNA supercoilsCitation12. Type IB topoisomerases change DNA topology by “DNA rotation” based on the splitting of a single strand of DNA, covalent attachment of tyrosine to the 3′- phosphoryl group and release a free 5′-OH end. Temporary interruption of a single DNA strand enables the relaxation of supercoiled DNA by rotating the DNA molecule around the breakpoint. The reaction lasts until all torsional strain is removed and DNA is completely relaxed. Type IB enzymes do not require Mg2+ ions or ATP for relaxation of positive or negative supercoils in DNA ()Citation9,Citation21–23.
Figure 1. General mechanism of action of topoisomerase I (a) Top I binds to the DNA, (b) single-strand DNA (in blue) splitting, (c) controlled rotation of free DNA strand (in red), (d) religation of cleaved DNA strand.
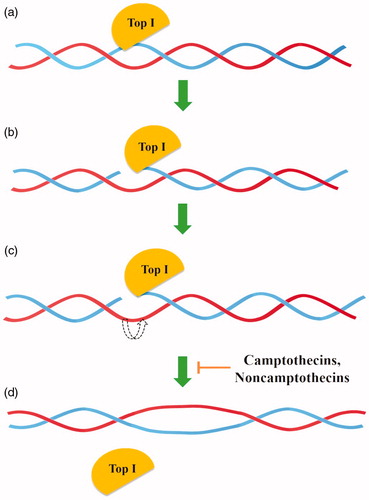
Structures of all topoisomerases IB include a highly conserved pentad of residues creating the active site (Tyr, Arg, Arg, Lys and His/Asn). The only difference in the structure of this region between eukaryotic/viral (Tyr, Arg, Arg, Lys, His) and bacterial type IB enzymes (Tyr, Arg, Arg, Lys, Asn) is a replacement of histidine with asparagine in the bacterial pentadCitation9.
The most important example of a type IB topoisomerases is the eukaryotic Top I (including human Top I), which is an important molecular target of anticancer therapies. In addition, Top IB group includes poxvirus topoisomerase and eubacterial Top IBCitation6,Citation9.
Human topoisomerase I
Human topoisomerase I (hTop I) belongs to Top IB subfamily. It is a monomeric enzyme and its molecular weight is 91 kDaCitation24. The enzyme consists of 765 amino acids which form four domains: an N-terminal domain, a linker domain, a core domain and a C-terminal domainCitation25. The detailed hTop I architecture was determined by X-ray crystallographyCitation26. There is a correlation between hTop I expression level and cell sensitivity to Top I inhibitors. While in healthy cells lower hTop I expression level has been observed, rapidly dividing cancer cells are observed to express a higher level of hTop I. Based on these results, we can assume that it is possible to target hTop I inhibitors towards cancer cells, tumours or patients with “overexpression” or increased expression of hTop ICitation24.
Type IC topoisomerases
Topoisomerase V (Top V) is a 110 kDa enzyme originally discovered in the archaeon Methanopyrus kandleri. So far, it is the only member of type IC topoisomerases subfamilyCitation27–29. Topoisomerase V is involved in DNA relaxationCitation27 and DNA repairCitation30,Citation31. Similar to type IB topoisomerases, Top V can relax negatively and positively supercoiled DNA and cleaves single strand of DNACitation13. However, structural analyses showed a novel fold in the crystal structure of topoisomerase V and probably a different location of an active site of tyrosine. This allows us to suppose that the mechanism of DNA relaxation by Top V is different than in the case of type IB topoisomerasesCitation29. Based on that, topoisomerase V was classified as a member of new topoisomerase I subfamily named type IC topoisomerasesCitation28,Citation32.
Topoisomerase I inhibitors used in the treatment of many types of cancer include compounds belong to a various class. These compounds will be presented and discussed in a separate chapter titled “Inhibitors of type I topoisomerases”.
Type II topoisomerases
Type II topoisomerases, divided into two subfamilies (topoisomerases IIA and topoisomerases IIB) are presented in various organisms. Topoisomerases IIA (Top IIA) occur in bacteria, eukaryote and also in few species of archaea, whereas topoisomerases IIB (Top IIB) are present mainly in archaea, plants and certain algae. Based on structural and phylogenetic analyses, significant differences in global architecture and biochemistry between Top IIA and Top IIB were foundCitation14,Citation33.
General mechanism of changing the topology of DNA by topoisomerases II is based on cleaving both strands of DNA duplex with Mg2+ and energy from ATP hydrolysis. Topoisomerase II covalently attaches tyrosine to the 5′ end of broken DNA, release a free 3′ end and allows to passing a second DNA duplex (the transported or T-segment) through a gap (the gate or G-segment)Citation1,Citation33–35. All enzymes belong to this family can relax both positive and negative supercoils in DNACitation1.
Due to their exceptional ability to untangle double strands of DNA, Top II is involved in many crucial nuclear processes, e.g. transcription, replication or recombinationCitation36,Citation37. The formation of double-stranded DNA breaks as a result of a loss of activity by Top II leads to cell death. Moreover, disruption of the proper functioning of the enzyme by increasing the level of DNA cleavage (both genetic and drug-induced) leads to e.g. translocations of DNACitation38.
Type IIA topoisomerases
Type IIA topoisomerases subfamily includes eukaryotic Top II, bacterial DNA gyrase, human topoisomerase II and bacterial Top IVCitation39. The sequences of all the enzymes belonging to the family of type IIA topoisomerases (Top IIA) show significant similarity, and the only differences are found in the quaternary structures of these proteins. Gyrase and topoisomerase IV (Top IV) belonging to type IIA bacterial topoisomerases are composed of two subunits: the ParE and ParC which are homologues of the GyrB and GyrA subunits of gyrase. In terms of structure, prokaryotic type IIA topoisomerases are referred to as heterotetramers in contrast to eukaryotic enzymes which belong to the group of homodimers. The N-terminal half-ends of the eukaryotic Top IIA are homologues of the GyrB/ParE subunits of gyrase and Top IV, and the central parts of the enzymes are homologues of the GyrA/ParC subunits (gyrase/Top IV). The C-terminal half-ends, unlike the N-terminal parts of the eukaryotic topoisomerases type IIA, show significant structural differences between the enzymes of different eukaryotes. Analysis of C-terminal eukaryotic and prokaryotic type IIA topoisomerases did not show similarities in the sequence of these enzymesCitation40.
Type IIA topoisomerases perform DNA double-strand cleavage based on the “two-gate” mechanism ()Citation41–43. Initially, Top IIA binds to the first DNA helix (G-segment), which will be cleaved in the further stage of the catalytic cycle. In the next step, the resulting Top IIA- G-segment complex binds to the second DNA helix (T-segment) which will be transported through the created gap. Two ATP molecules are attached to the Top IIA—G-segment–T-segment complex causing conformational changes in the enzyme. As a result of hydrolysis of one ATP molecule to ADP, in the presence of Mg2+ ions, tyrosine from both Top IIA monomers attacks the phosphodiester bond of the first DNA helix resulting in cleavage of the strand with a shift of 4-bp and becomes covalently attached to the 5′ ends of the cleaved DNA (G-segment). Thanks to the transition, it is possible to transport the T-segment through a gap resulting after breaking the G-segment. Due to the hydrolysis of the second ATP molecule, the broken strands of the G-segment are religated. After passing through the gap, the T-segment is released. Then, due to the release of ADP molecules, the DNA-topoisomerase IIA complex is transformed from a closed into an open clamp, which is connected with the release of G-segment. Top IIA is free and ready for the next enzymatic reactionCitation44–47.
Figure 2. General mechanism of action of topoisomerase II (a) topoisomerase binds to the G-segment, (b) Top IIA- G-segment complex binds to T-segment, (c) Two ATP molecules are attached to the resulting complex, (d) G-segment cleavage in presence of Mg2+ ions, (e) T-segment transport through the created gap, (f) T-segment release and religation of G-segment broken strands and (g) hydrolysis of ATP molecules and release of the G-segment.
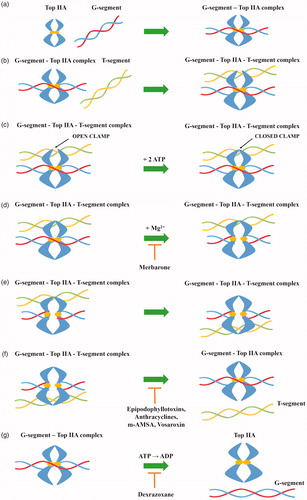
Human topoisomerase II
Human topoisomerase II (hTop II) is composed of four domains: two N-terminal domains, a core domain and C-terminal domain. There are two homodimeric isoforms of hTop II: α (molecular weight around 170 kDa) and β (molecular weight around 180 kDa)Citation48,Citation49. Their amino acid sequences are homologous in approximately 70%Citation50. Depending on the phases of the cell cycle, changes in human topoisomerase IIα (hTop IIα) and human topoisomerase IIβ (hTop IIβ) expression levels are observed. While the increased expression of hTop IIα is mainly observed in proliferating cells (the peak of expression is observed in G2-M phases), in case of hTop IIβ level of enzyme expression is constant regardless of the cell cycle stageCitation51,Citation52. hTop IIα is crucial for cell survival due to its role in chromosome condensation and segregation processes whereas hTop IIβ plays an important role in neurons developmentCitation53,Citation54.
Type IIB topoisomerases
Commonly found in Archaea (except Thermoplasmatales spp.) type IIB topoisomerases were first discovered in the hyperthermophilic archaeon Sulfolobus shibataeCitation55. The discovered enzyme was named topoisomerase VI (Top VI) and classified as a different subfamily of type II topoisomerasesCitation56. Like bacterial type IIA topoisomerases, Top VI is a heterotetramer enzyme composed of two subunits: A and B (A2B2). Structural and sequence analysis of type IIA and IIB topoisomerases did not reveal any similarities between the A subunits of both enzymes. The A subunit of Top VI shows similarity to eukaryotic Spo11 protein which is responsible for the initiation of meiotic DNA recombination as a result of the splitting of double DNA helixCitation57. The B subunit of type IIB topoisomerases, responsible for binding and hydrolysis of ATP molecules shows significant structural similarity and slight sequence similarity to the B subunit of Top IIACitation58.
Among Archaea topoisomerase VI causes relaxation of positively and negatively supercoiled DNA and is also involved in DNA replication and transcription. Initiated by the type IIB topoisomerases changes in DNA topology by the formation of breaks in double-stranded DNA occurs with a shift of 2-bp rather than 4-bp as in type IIA topoisomerasesCitation57. Recently, topoisomerase VI has been identified in the plant kingdomCitation55,Citation56,Citation59, including red and green algaeCitation60. Occurring in plants, Top VI plays an important role in the DNA endoreduplication process. Proper DNA endoreduplication is essential to maintain the correct size of plant cells and consequently the normal size of the entire plantCitation61.
Scientific papers report the identification of new type IIB topoisomerase–topoisomerase VIII (Top VIII). It is the smallest known representative of type IIB subfamily of topoisomerases. The A and B subunits of this enzyme are merged into one single protein, hence the small size of Top VIII. Top VIII occurs in genomes of few bacteria species, two bacterial plasmids and one of the Archaea plasmids. In order to determine the structure and functions Top VIII, further detailed studies of this potential new type IIB topoisomerases subfamily member are necessaryCitation62.
Topoisomerase inhibitors
Over the last few decades after discovering that doxorubicin induces DNA double helix cleavage via human topoisomerase II, scientific research based on searching of new compounds with an anticancer activity that are inhibitors of enzymes from the topoisomerases family has been intensified rapidly. To date, a significant number of topoisomerase inhibitors has been discovered and described in the scientific papers, however, in many cases, the detailed mechanism of action of these compounds is still unknown. Drugs classified as topoisomerase inhibitors can be divided into two groups: topoisomerase poisons or catalytic inhibitors. Doxorubicin, quinolones and many other anticancer or antibacterial drugs act as topoisomerases poisons. The mechanism of action of these compounds is based on the stabilisation of the formed enzyme–DNA covalent complex which prevents the religation of the cut strandCitation63. The resulting DNA damages lead to cell death through apoptosisCitation64. Catalytic inhibitors work by blocking the ability of the topoisomerase to attach to the substrate (DNA strand). Among this group there are two modes of action: the compound is binding to the DNA duplex or the topoisomerase. Regardless of the mode of action, catalytic inhibitors will lead to cell death through apoptosisCitation65.
and summarise information on important topoisomerase inhibitors and selected clinically investigated compounds, discussed in detail in this article.
Table 2. Clinically relevant topoisomerases inhibitors
Table 3. Selected topoisomerase inhibitors under clinical investigation
Inhibitors of type I topoisomerases
Mechanism of action of drugs classified as Top I inhibitors is based on the entrapment of created Top I-DNA covalent complex and prevents its cleavageCitation66–68. This leads to permanent disruption of the DNA strands and consequently, to cell cycle arrest and induction of programmed cell death-apoptosisCitation69,Citation70.
Camptothecins (CPTs)
In 1985, it was confirmed that topoisomerase I is the molecular target of camptothecin anticancer activityCitation71,Citation72. Isolated in 1966 by Wall et al. from the bark of Camptotheca acuminate tree, camptothecin is a pentacyclic ring structureCitation73. Structure–activity relationship studies revealed that modifications at C-7, C-9 or C-10 positions at quinolone moiety (A and B rings on ) increase the anticancer activity of CPTsCitation74. The E-ring, interacting with topoisomerase at three sites, is the most important part of the CPTs structure. The − OH group located at C-20 position forms a hydrogen bond with the Asp-533 side chain in the Top I polypeptide chain, and Arg-364 of Top I binds with two hydrogen bonds to the lactone part of the CPTs E-ring. Moreover, the D-ring, responsible for the stabilisation of the covalent Top I–DNA complex, is hydrogen-bonded with +1 cytosine of the uncleaved DNA chain. This H bond is located between the amino group of the +1 cytosine pyrimidine ring and the CPTs carbonyl group on the D-ring C-17 positionCitation75. The molecular mechanism of action of the drug is based on reversible induction of single-strand breakages which affects the cell replication capacity. Top I-DNA cleavable complex is stabilised by camptothecin but the resulting breaks are non-lethal to the cell due to their full reversibility. The single-stranded breaks are transformed into irreversible double-stranded breaks as a result of the cleavable complex collision with DNA replication fork. Caspase activation leads to cell death by apoptosisCitation76,Citation77. As a result of the inhibition of caspase activation, the cells are temporarily arrested in the G1 cell cycle phase, which leads to necrosisCitation78. S-phase-specific camptothecin shows its cytotoxic activity only when the DNA replication process is active. In vitro studies have shown that S-phase cells are 100–1000 times more sensitive to camptothecin than G1 or G2 cellsCitation79. Selected camptothecin derivatives used in the cancers treatment and compounds in the clinical trials () are discussed below.
Figure 3. Chemical structure of camptothecin.
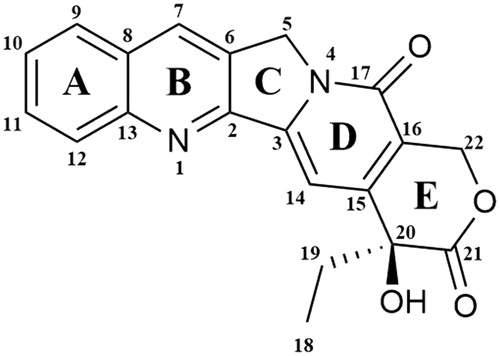
Figure 4. Chemical structures of camptothecins.
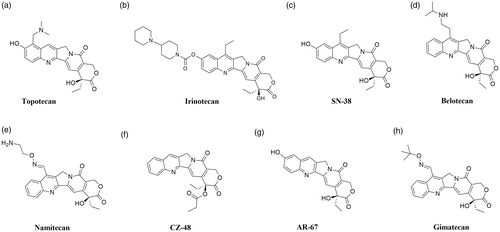
Clinically important CPTs
Topotecan (9-[(dimethylamino)methyl]-10-hydroxycamptothecin; Hycamtin®) is semisynthetic, water-soluble camptothecin (CPT) derivative and it was the first CPT derivative approved in 1996 by the US Food and Drug Administration (FDA) for clinical useCitation80. The studies conducted by Staker et al. allowed to determine the detailed mechanism of antitopoisomerase activity of topotecan (). DNA and compound intercalation occurs at the Top I-mediated DNA splitting site and is stabilised by interactions between the topotecan and −1 (upstream) and +1 (downstream) base pairs of DNA duplex. The rotation of the phosphodiester (0 P) bond results in the formation of an intercalation binding pocket. The movement of phosphodiester 0 P towards the binding pocket simultaneously stimulates energetically the release of +1 and −1 base pairs, which in turn is necessary for 0 P rotation. The transported 0 P phosphodiester is stabilised in the binding pocket as a result of the interaction between the two hydrogen bonds and the nitrogens of the main chain Gly-363 and Arg-362Citation81. The understanding of the exact method of topotecan intercalation to DNA made it possible to explain the mechanism by which this compound blocks religation of the DNA strand. As a result of binding the topotecan molecule with the DNA, the +1 (downstream) base pair is shifted by 3.6 Å and a 5′-OH of the splitted DNA strand is shifted from the phosphotyrosine by 8 Å. Due to the fact that the topotecan binding pocket is located inside the DNA substrate and, moreover, is formed after the first transesterification, it becomes understandable why camptothecin and its derivatives interact with the Top I-DNA complex and not with the enzyme itselfCitation82. Topotecan, a known topoisomerase I inhibitor, has a broad spectrum of anticancer applications. It is successfully used for the second-line treatment of metastatic ovarian cancer in over 70 countries and the treatment of relapsed platinum-sensitive small-cell lung cancer (SCLC) in more than 30 countries around the worldCitation83,Citation84. In 2006, topotecan was approved by the FDA for use to the treatment of recurrent or persistent cervical cancer not susceptible to surgery and/or radiation therapyCitation85.
Irinotecan (7-ethyl-10-[4–(1-piperidino)-1-piperidino]carbonyloxycamptothecin; CPT-11) as topotecan is a water-soluble, semisynthetic camptothecin derivative (). It is a prodrug swiftly converted in vivo by human carboxylesterase 2 (hCE2) enzyme (occurs in liver, intestinal mucosa and plasma) to metabolite named SN-38 (7-ethyl-10-hydroxycamptothecin, ) with high anticancer activity Citation86. hCE2 converts CPT-11 into its active metabolite SN-38 by hydrolysis and the metabolite shows a more than 100 times higher in vitro cytotoxicity than the parent compoundCitation87. The ionic strength, pH and protein concentration all influence the rate of hydrolysis of CPT-11 to SN-38. Both irinotecan and SN-38 occur in two forms - lactone and carboxylate. The lactone form with a closed α-hydroxy-δ-lactone ring is hydrolysed into a carboxylate form (open-ring hydroxyl acid). Based on the conducted research, it was found that the lactone form, whose antitumor activity is much more potent than the carboxylate form, is crucial for the stabilisation of the covalent Top I - DNA complexCitation88. In liver, active SN-38 is metabolised into its inactive β-glucuronide form (SN-38G) by uridine-diphosphate glucuronosyltransferase (UGT)Citation89. For the first time, irinotecan was approved in 1994 in Japan for the treatment of SCLC and non-small cell lung cancer (NSCLC), ovarian cancer and cervix cancer Citation90. Marketed by Pfizer Inc. and known as Camptosar® it is used for the treatment of colorectal cancerCitation91.
Promising active CPT derivatives
Belotecan (7-[2-(N-isopropylamlino)ethyl]-(20S)-camptothecin; CKD-602, Camtobell®) is a relatively new, semisynthetic and water-soluble CPT derivative (). It was approved in 2003 in South Korea for the treatment of patients with NSCLC and ovarian cancerCitation92. Belotecan exhibits similar efficacy profile compared to other CPT derivatives. However, its toxicity level is lower than the mentioned agentsCitation93. In 2018, Chong Kun Dang Pharmaceutical has completed the phase II clinical trial to evaluate the efficacy and safety of belotecan in comparison to topotecan in patients with relapsed SCLC (NCT01497873). Furthermore, in 2019 Lee et al. conducted in vitro and in vivo studies on the effects of CKD-602 in selected cervical cancer cell lines (CaSki, HeLa and SiHa). In CaSki-xenografted nude mice treated with CKD-602, a significant reduction of tumour volume was observedCitation94.
Namitecan (7–(2-aminoethoxy)iminomethyl-camptothecin; ST1968) is one of the CPT derivatives (). Novel topoisomerase I inhibitor was evaluated in several preclinical studies with various tumour models (including resistant to topotecan/irinotecan xenografts, paediatric tumour models and diversified squamous-cell tumour models) and a significant antitumor activity was observedCitation95. The conducted studies allowed us to determine the safety and pharmacokinetic profile of the namitecan. Furthermore, during phase I of clinical trials (NCT01748019), the anticancer activity of ST1968 in patients with endometrium and bladder cancer was observedCitation95,Citation96.
CZ-48 (camptothecin-20(S)-O-propionate hydrate) is CPT prodrug synthesised by Cao et al. in 2009. Researchers examined the anticancer activity of CZ-48 () against 21 human tumour xenografts. Obtained results showed an excellent activity of the tested compound against various cancers (i. a. bladder, breast, colon, melanoma, lung and pancreatic tumour lines). In the case of 19 from 21 tested tumour lines, notable growth inhibition (>50%) or regression was observedCitation97. Great anticancer activity and reduced toxicity compared to irinotecan and topotecan resulted in rapid clinical development of CZ-48. Cao Pharmaceuticals Inc. is currently recruiting patients with a solid tumour or malignant lymphoma of extranodal and/or solid organ site for phase I clinical trial to examine the safety of CZ-48 administered orally (NCT02575638).
AR-67 (7-tert-butyldimethylsilyl-10-hydroxycamptothecin; formerly DB-67) is a lipophilic CPT analogue synthesised by Bom et al. in the 1990s (). The scientist has designed a novel compound to improve blood stability and activity of CPT analogues. The conducted studies have shown in vitro and in vivo anticancer activity of the compound via interaction with topoisomerase I as well as improved stability of AR-67 in blood compared to clinically relevant CPTsCitation98. The information provided by Arno Therapeutics and the University of Kentucky shows that phase I of the clinical trials on the potential application of AR-67 in the treatment of patients with refractory or metastatic solid malignancies (NCT00389480 and NCT01202370) has been completed. Furthermore, in 2014 Arno Therapeutics posted information about active phase II trial of AR-67 in patients with recurrence of glioblastoma multiforme or gliosarcoma (NCT01124539) however, the current study status remains unknown.
Gimatecan (7-tert-butoxyiminomethyl-camptothecin; ST1481) is novel, semisynthetic and lipophilic CPT analogue with good oral bioavailability (). Based on the promising results of the preclinical study, gimatecan was selected for clinical developmentCitation99. Rapid absorption, compound accumulation and prolonged distribution in cancer cells are associated with Top I activity inhibition and stabilisation of the Top I DNA-Top I-gimatecan complexCitation100. High efficacy of administered orally gimatecan against a panel of subcutaneous human tumour xenograft models, metastatic and orthotopic tumour models was observedCitation101. Lee's Pharmaceutical Limited is currently recruiting patients with fallopian tube cancer, advanced ovarian epithelial cancer or primary peritoneal cancer for phase I clinical trial to investigate the safety, tolerability and pharmacokinetics of gimatecan (NCT04029909).
Noncamptothecins
Nowadays, the only group of Top I inhibitors used in the oncological treatment are camptothecins. The excellent effectiveness of these compounds is accompanied by strong side effects (e.g. diarrhoea or neutropenia) and instabilityCitation6,Citation20. As mentioned above, camptothecins are characterised by chemical instability—compounds are immediately inactivated in the blood to carboxylate. Furthermore, due to the rapid reversal of Top I-DNA covalent complexes after drug removal, long infusions are necessary to ensure the effectiveness of treatmentCitation102. Described restrictions on the use of camptothecins have prompted scientists to investigate new noncamptothecin Top I inhibitors ()Citation103,Citation104. Currently, in the clinical trials, there are compounds from indenoisoquinolinesCitation105, indolocarbazolesCitation106 and dibenzonaphthyridinonesCitation107 groups.
Figure 5. Chemical structures of noncamptothecins.
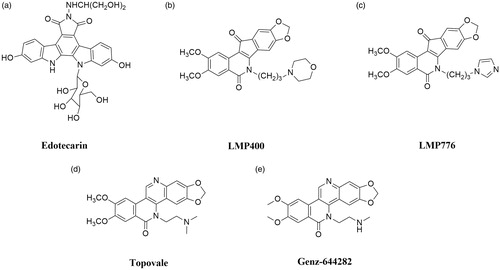
Promising active noncamptothecins
Indolocarbazoles
Edotecarin (6-N-(1-hydroxymethyl-2-hydroxy)ethylamino-12,13-dihydro-2,10-dihydroxy-13-(β-D-glucopyranosyl)-5H-indolo[2,3-a]-pyrrolo[3,4-c]-carbazole-5,7(6H)-dione; J-107088) is a synthetic analogue of natural antibiotics isolated from several Actinomycetes (). It is a derivative of a compound named NB-506. Indolocarbazole NB-506 is an anticancer compound in which mechanism of antitumor activity is based on topoisomerase I or II poisoning. Edotecarin is a strong and specific topoisomerase I inhibitor, which is more effective in inducing single-strand DNA splitting compared to CPT or NB-506Citation108. Furthermore, the stability of DNA-topoisomerase I covalent complexes formed as a result of edotecarin activity ensures longer effectiveness of the drug after the end of cell incubation with the compoundCitation106. Memorial Sloan Kettering Cancer Centre in collaboration with NCI conducted a phase I of clinical trials to evaluate the effectiveness of edotecarin combined with cisplatin in the treatment of patients with advanced or metastatic solid tumours (NCT00072332). In addition, the same collaborators completed phase II trial to study the effectiveness of treatment of women with chemoresistant locally advanced or metastatic breast cancer (NCT00070031).
Indenoisoquinolines
Indenoisoquinolines are developed in the 1990s by the NCI and represent a novel group of Top I inhibitors. These synthetic compounds are characterised by good chemical stability and better stabilisation of DNA–enzyme–drug covalent complexes than CPTsCitation103,Citation109. Among over 400 synthesised compounds, only two are currently under clinical trials: indotecan (LMP400; NSC 314622, ) and indimitecan (LMP776; NSC 725776, )Citation110. Both compounds have been studied by scientists from the U.S. NCI in the treatment of adults with relapsed solid tumours and lymphomas (NCT01051635). Based on the information provided by the National Institutes of Health Clinical Centre, it is known that phase I of clinical trials has been completedCitation111.
Dibenzonaphthyridinones
Next group of non-CPT topoisomerase I inhibitors are dibenzo[c,h][1,6]naphthyridinone derivatives. A novel group of compounds identified by LaVoie and his research team showed potent in vitro and in vivo anticancer activityCitation112,Citation113. Mechanism of topoisomerase inhibition by dibenzo[c,h][1,6]naphthyridinone derivatives is based on the stabilisation of cleavage DNA-Top I covalent complexesCitation114–116.
Topovale (8,9-dimethoxy-5–(2-N,N-dimethylaminoethyl)-2,3-methylenedioxy-5H-dibenzo[c,h][1,6]naphthyridin-6-one; ARC-111) is a synthetic compound and strong Top I inhibitor (). The efficacy of topovale was evaluated against seven cancer cell lines and the obtained results showed higher cytotoxicity compared to the SN-38 or topotecan. Furthermore, the high effectiveness of this compound in cells which expressed the efflux pumps was observedCitation14,Citation107.
Next dibenzo[c,h][1,6]naphthyridinone derivative in clinical trials is Genz-644282 (8,9-dimethoxy-5–(2-N-methylaminoethyl)-2,3-methylenedioxy-5H-dibenzo[c,h][1,6]naphthyridin-6-one). The development of this drug was based on studies of structure-activity relationship in the family of dibenzo[c,h][1,6]-naphthyridin-6-one compoundsCitation117. Studies conducted to determine the cytotoxicity of Genz-644282 () using bone marrow cells and cancer cell colony-forming assays have shown favourable cytotoxicity profile of this compound. Furthermore, based on the results obtained from studies of Genz-644282 activity in xenograft models, similar or higher activity of the tested drug was observed compared to the standard drugsCitation118. Furthermore, in 2014 the phase I of the clinical trial on safety and tolerability of the compound in the therapy of solid tumours was completed (NCT00942799).
Inhibitors of type II topoisomerases
The enzymes belonging to topoisomerases II family have the ability to achieve various conformational states as these proteins undergo significant structural changes during T-segment transport through the generated gate. In theory, trapping the enzyme in any conformational state allows manipulating of the topoisomerase II activity. To date, two types of Top II inhibitors have been identified and described: topoisomerase poisons and catalytic inhibitorsCitation119,Citation120.
Topoisomerase poisons
Topoisomerase II poisons (Top II poisons) are the first from two types of Top II inhibitors. They cause inhibition of Top II ability to religation of cleaved DNA strands. Mechanism of action of Top II poisons is based on blocking of broken DNA ends rejoining as a result of slipping Top II poison molecules between splitted nitrogenous bases. This causes DNA double-strand breakage permanent by replicating broken DNA by cells. Moreover, replication of damaged DNA leads to the activation of the DNA damage response pathway and that in turn usually leads to cancer cell death through apoptotic pathwayCitation121–123.
Epipodophyllotoxins
For almost 2000 years, podophyllotoxins have been used in traditional medicineCitation124. In the 19th century, podophyllin was identified as an effective drug used topically to treat skin cancersCitation125. Despite the moderate anticancer properties of podophyllin and its derivatives selected in clinical trials, the high toxicity of tested drugs was observedCitation126. As a result of research initiated in the 1950s by Sandoz Pharmaceuticals aimed at the synthesis of new derivatives with similar to podophyllin anticancer activity but significantly lower toxicity, in 1966 etoposide and one year later teniposide were synthesisedCitation127. Both podophyllin derivatives show a negligible affinity for DNA in the absence of Top II. Based on many studies, scientists have unequivocally concluded that drug–enzyme interactions are crucial for the proper functioning of drugs, and also indirectly participate in the formation of the Top II—DNA–compound ternary complexCitation128–130.
Clinically important epipodophyllotoxins
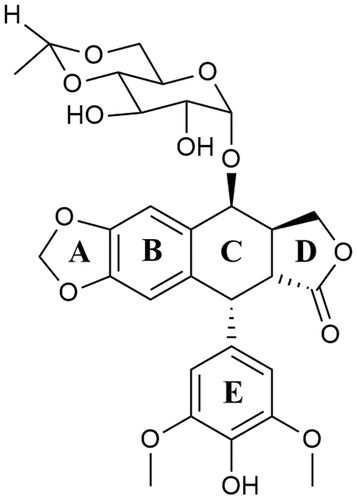
Etoposide ((8aR,9R)-5-[(7,8-dihydroxy-2-methyl-4,4a,6,7,8,8a-hexahydropyrano[3,2-d][1,3]dioxin-6-yl)oxy]-9–(4-hydroxy-3,5-dimethoxyphenyl)-5a,6,8a,9-tetrahydro-5H-[2]benzofuro[6,5-f][1,3]benzodioxol-8-one; VP-16) is a semisynthetic podophyllotoxin derivative and clinically significant anticancer compoundCitation124,Citation131. Discovered in 1966, VP-16 was approved by the FDA for cancer treatment in 1983Citation132. The anticancer activity of etoposide is based on topoisomerase II poisoning by binding to the Top II-DNA covalent complexesCitation133. This prevents religation of DNA strands, changing temporary DNA double-stranded breaks into permanentCitation134, which indirectly leads to cell death by apoptosis. The combination of the results of the studies on the binary binding of the enzyme-drug complex, the research on DNA cleavage in the Top II-DNA-compound complex and NMR analysis made it possible to determine the functions of individual structures building the etoposide—the best-known epipodophyllotoxin. Presumably, the binding of etoposide () to Top II occurs through the interaction of the enzyme with the A- and B-ring of the compound, and also through the interaction between the enzyme and the E-ring of the compound. The − OH group and the methoxyl groups on the E-ring are an important element for the proper functioning of the drug. At the same time, both mentioned above groups do not significantly affect the specificity of DNA strands splitting or creating an enzyme-drug bond. The D-ring and the glycoside at position 4 of the C-ring probably interact with DNA found in the Top II—DNA–compound ternary complex, affecting the specificity of DNA cleavage. Interestingly, both structures do not interact with the enzyme found in the binary enzyme-drug complexCitation130,Citation135. Removal of the glycoside group from the compound’s structure does not significantly affect the etoposide-induced DNA cleavage process and substituting it with, for example, a spermine fragment (as in the case of the mentioned below compound - F14512) significantly increases the potency of the parent compoundCitation136. Etoposide is used for the treatment of SCLCs, lymphomas (including non-Hodgkin’s lymphomas), acute myeloid leukaemia (AML), testicular and ovarian cancerCitation123,Citation137,Citation138.
Figure 6. Chemical structure of etoposide.
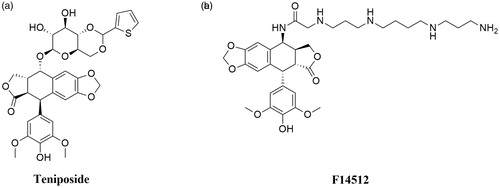
Teniposide ((5S,5aR,8aR,9R)-5-[[(2R,4aR,6R,7R,8R,8aS)-7,8-dihydroxy-2-thiophen-2-yl-4,4a,6,7,8,8a-hexahydropyrano[3,2-d][1,3]dioxin-6-yl]oxy]-9–(4-hydroxy-3,5-dimethoxyphenyl)-5a,6,8a,9-tetrahydro-5H-[2]benzofuro[6,5-f][1,3]benzodioxol-8-one; VM- 26) is a second, semisynthetic derivative of podophyllotoxin and an analogue of etoposide (). Interestingly, VM-26 was discovered and tested in clinical trials before VP-16Citation124,Citation127. The mechanism of action of teniposide is similar to etoposide. It causes DNA double-stranded breaks via stabilisation of DNA-Topoisomerase II complexesCitation139. Hypersensitivity reactions to teniposide in patients, as well as inappropriate administration of VM-26 using too low doses, caused slower development of the drug compared to etoposide. Consequently, teniposide was approved by the FDA in 1992, nearly 30 years after the first synthesis of the compoundCitation140. VM-26 is mainly used in the therapy of childhood acute lymphocytic leukaemia (ALL), glioma, central nervous system tumours and bladder cancerCitation141,Citation142.
Figure 7. Chemical structures of epipodophyllotoxins.
Promising active epipodophyllotoxin derivatives
F14512 (N-[(5S,5aS,8aR,9R)-9–(4-hydroxy-3,5-dimethoxyphenyl)-8-oxo-5a,6,8a,9-tetrahydro-5H-[2]benzofuro[5,6-f][1,3]benzodioxol-5-yl]-2-[3-[4–(3-aminopropylamino)butylamino]propylamino]acetamide) is a novel etoposide derivative developed by Barret et al. The compound was designed by the replacement of VP-16 carbohydrate group with a polycation spermine moiety (). The modification of VP-16 structure resulted in a significant increase in compound solubility, cellular uptake, and cytotoxicity of F14512Citation136. The presence of a spermine moiety in the compound molecule led to an increase in the degree of DNA binding and consequently to an enhancement of the inhibition of Top II activityCitation143. Moreover, the overexpression of polyamine transport system observed in cancer tumours was used to intensify selective drug uptake by cancer cellsCitation144,Citation145. In vitro activity of F14512 was evaluated in 29 human cancer cell lines (i.a. leukaemia, sarcoma, myeloma, prostate, pancreatic and ovarian cancer cell lines) and noteworthy cytotoxicity in 21 cell lines was observedCitation136. F14512 in vivo anticancer activity was investigated using 19 experimental models. In 13 out of 19 investigated models, F14512 exhibited a strong antineoplastic activityCitation146. Furthermore, based on the information provided by the EU Clinical Trials Register, it is known that phase I–II study to evaluate the maximum tolerated dose and the efficacy of combined therapy of F14512 and cytarabine in patients (≥60 years old) with AML has been completed (EudraCT number: 2012–005241-20).
Anthracyclines
Anthracyclines were isolated in the 1950s from Streptomyces peucetius which is one of the Actinomyces species. Obtained compounds showed antibacterial properties in vitroCitation147. For over 50 years anthracyclines combined with various cytotoxic agents or targeted agents have been the most commonly used anticancer drugs to the treatment of solid or haematological tumoursCitation148,Citation149. As topoisomerase poisons, anthracyclines stabilise DNA-topoisomerase II covalent complexes, which indirectly leads to apoptosis via double-stranded DNA breaks and inhibition of DNA transcription and replication processesCitation150. A characteristic feature of all anthracycline derivatives is that the structures of individual compounds slightly differ from one another. These slight modifications cause significant changes in compounds activity. For instance, modification of the compound’s structure by removing the 3-amino or 4-methoxy substituent significantly increases the activity of the compound. Moreover, the presence and nature of the substituent at position 3 of the sugar moiety is crucial for the selectivity of the anthracyclines DNA cleavage processCitation151.
Clinically important anthracyclines
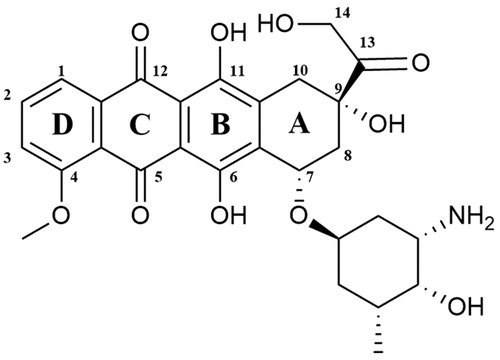
Doxorubicin ((7S,9S)-7-[(2R,4S,5S,6S)-4-amino-5-hydroxy-6-methyloxan-2-yl]oxy-6,9,11-trihydroxy-9–(2-hydroxyacetyl)-4-methoxy-8,10-dihydro-7H-tetracene-5,12-dione; Adriamycin) is a chemotherapeutic drug which belongs to the anthracyclines group. It was first extracted in the 1970s from Streptomyces peucetius var. caesiusCitation152. The doxorubicin molecule is composed of sugar and aglycone moieties. The aglycone (doxorubicinone) consists of a tetracyclic ring linked to a hydroquinone and quinone residue, a short side-chain containing a primary alcohol at C-14 atom and a carbonyl group at C-13 position (). The aminosugar (daunosamine) is linked by a glycosidic bond with the C-7 carbon located in the A-ringCitation153. The molecular docking analyses enabled the researchers to identify the mechanism of interaction between the Top II–DNA complex and DOX. The obtained results showed that the planar DOX molecule is located between the GC base pairs of cleaved DNA strands. Three H bonds are formed between topoisomerase and DOX. The −OH group of Ser-740 Top II combines with the −OH group located in position C-11 in the anthraquinone fragment and Thr-744 forms a bond with DOX carbonyl oxygen located in position C-12. The last hydrogen bond is formed between the side chain Gln-750 and the C-14 atom of DOXCitation154. A-ring and the DOX sugar moiety also interact with the DNA itself, forming H bonds between a base complement to the base at the −1 position in DNA and the −OH group in the C-9 position of the DOX A-ring. Scientists speculate that the interaction between C-9 of A-ring of DOX and DNA is crucial to stabilise the drug–molecular target complex. Proper recognition of DNA threads by compound may be associated with the formation of hydrogen bonds between DNA and daunosamine moiety. G-2 and C-1 of DNA bond with the −OH group in position 4′ and the amine group located in position 3′ of daunosamine, respectivelyCitation155. There are two main mechanisms of action of doxorubicin (DOX) which lead to the inhibition of the catalytic cycle. At the beginning of the catalytic cycle, doxorubicin can interfere with DNA binding which prevents relaxation or (de)catenation of DNA by Top II. Furthermore, DOX can prevent the religation of G-segment cleaved strandsCitation156. Despite its anticancer properties doxorubicin is responsible for serious side effect which is dose-dependent cardiotoxicityCitation157,Citation158. The exact mechanism of DOX cardiac toxicity is still unclear. However, the most probable pathway is related to the formation of iron-related free radicalsCitation159,Citation160. Strong evidence, supporting this hypothesis is the fact that dexrazoxane, a known iron chelator, is used as a doxorubicin-induced cardiotoxicity protectorCitation152,Citation161. Approved by FDA in 1974, it is used in the therapy of various types of cancer i.a. ovarian, lung, gastric and breast cancers, multiple myeloma, thyroid cancer, Hodgkin’s and non-Hodgkin’s lymphoma, paediatric cancers and sarcomaCitation152,Citation162.
Figure 8. Chemical structures of doxorubicin.
Epirubicin ((7S,9S)-7-[(2R,4S,5R,6S)-4-amino-5-hydroxy-6-methyloxan-2-yl]oxy-6,9,11-trihydroxy-9–(2-hydroxyacetyl)-4-methoxy-8,10-dihydro-7H-tetracene-5,12-dione; 4′-epidoxorubicin) is anthracycline derivative (), epimer of doxorubicinCitation163. Epirubicin (EPI) shows activity in all phases of the cell cycle, but the highest activity is observed in the S and G2 phases. The mechanism of action of EPI is similar to doxorubicin. The drug inhibits the topoisomerase II activity by stabilisation of DNA–topoisomerase II covalent complexes which prevent splitting of DNA strandsCitation164. In 1999 it was approved for clinical use in the United States. The anticancer activity of EPI was confirmed against a broad spectrum of cancers but it is mainly used against advanced breast cancerCitation165.
Figure 9. Chemical structure of anthracyclines.
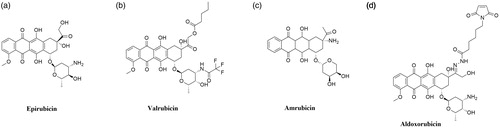
Valrubicin (N-trifluoroacetyladriamycin-14-valerate; AD-32) is anthracycline derivative and Top II inhibitor obtained by modification of DOX (). Substitutions of two side chain of DOX resulted in the formation of a novel molecule with enhanced safety profile and lack skin toxicity, which allows the topical application of the compoundCitation166,Citation167. The compound developed by Anthra Pharmaceuticals, Inc. (Valstar®) was approved by the FDA in 1998 for intravesical treatment of patients with BCG-refractory carcinoma in situCitation168. Currently ongoing clinical trials investigate the application of valrubicin i.a. in the treatment of early-stage bladder cancer (NCT00003129, phase II completed) and upper tract urothelial carcinoma (NCT01606345, phase I completed).
Promising active anthracycline derivatives
Amrubicin (9-amino-anthracycline; SM-5887) is a synthetic anthracycline derivative with potent antitumor activity based on the hTop II inhibition (). Cytotoxic activity of amrubicin results from the stabilisation of DNA–hTop II cleavable complexesCitation169,Citation170. Moreover, amrubicinol, an active amrubicin metabolite, is 5–220 times more cytotoxic than the original compoundCitation171. In Japan, amrubicin has been approved for the treatment of patients with NSCLC and SCLCCitation172. Furthermore, it is used as a therapy option in the chemotherapy of NSCLC after the 3rd-line treatmentCitation170. Phase II of clinical trials for the use of amrubicin plus pembrolizumab in the treatment of refractory SCLC (NCT03253068) is currently ongoing. In addition, research concerning the use of amrubicin for the treatment of relapsed or refractory thymic malignancies (NCT01364727, phase II completed), HER2-negative metastatic breast cancer (NCT01033032, phase I completed) and in combination with cyclophosphamide for the treatment of advanced solid organ malignancies (NCT00890955, phase I completed) is being conducted.
Aldoxorubicin ((6-maleimidocaproyl)hydrazone of doxorubicin; formerly INNO-206) is doxorubicin prodrug developed by CytRx Corporation (). The aldoxorubicin (ALDOX) molecule consists of doxorubicin conjugated with a linker (6-maleimidocaproic acid hydrazide). After intravenous injection, ALDOX is quickly and selectively attached to cysteine-34, which belongs to the group of endogenous amino acids of serum albumin. After binding to albumin, DOX is transported to the tumour. The acidic tumour environment breaks the acid-labile hydrazine bond between the linker and the drug. The release of DOX, intercalation of the drug with DNA and inhibition of topoisomerase II activity occursCitation173,Citation174. Preclinical studies performed on mouse xenograft tumour models of ovarian, breast, lung and pancreatic cancers have shown higher ALDOX anticancer activity compared to DOXCitation175,Citation176. Research conducted by Sanchez et al. demonstrated a significant in vitro and in vivo ALDOX activity against multiple myeloma cells. Moreover, an enhanced effect of combined ALDOX and bortezomib therapy of multiple myeloma compared to single-agent therapy (ALDOX or bortezomib alone) was observedCitation177. Over the last years, numerous clinical trials on the use of ALDOX in the treatment of various cancers have been initiated. Research on the use of ALDOX in the treatment of glioblastoma (NCT02014844, phase II completed), soft tissue sarcomas (NCT02049905, phase III completed) as well as in combination with gemcitabine in the treatment of metastatic solid tumours (NCT02235688, phase I completed) is ongoing. Furthermore, in May 2020 ImmunityBio, Inc. reported the launch of a clinical trial to determine the safety and efficacy of the standard chemotherapy in combination with aldoxorubicin and other drugs compared to standard chemotherapy in patients with locally advanced or metastatic pancreatic cancer (NCT04390399).
Anthracenediones
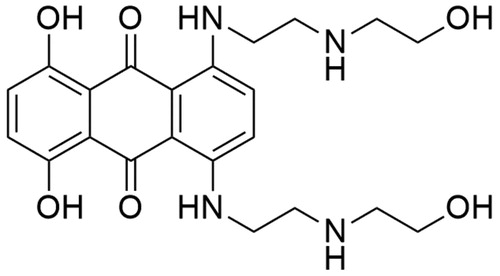
Mitoxantrone (1,4-dihydroxy-5,8-bis[2–(2-hydroxyethylamino)ethylamino]anthracene-9,10-dione; MTX) is a synthetic chemotherapeutic agent belonging to the anthracenedione derivatives ()Citation178. It was discovered in the 1970s by two independent groups of investigators: the Medical Research Division of the American CyanamidCitation179 and the Midwest Research InstituteCitation180. The search of new antineoplastic drugs began with a study of structure–activity relationships of new anthracenedione derivatives. Based on the obtained results, which revealed that the presence and composition of side-chains have a large impact on the activity of anthracenedionesCitation179,Citation181 MTX was selected for further studies as a potent anticancer compoundCitation182. Conducted in the 1990s, footprinting studies revealed that mitoxantrone-mediated DNA cleavage occurs at sites having a thymine or cytosine residue in close proximity. Additionally, the location of DNA cleavage is influenced by the presence of guanine located two nucleotides downstream—in the +2 positionCitation183. X-ray crystallographic analysis enable scientists to build the 3 D model of Top II–DNA–MTX complex. A single drug molecule is introduced exactly at both sites of splitted DNA strands. The dihydroxyanthracenedione chromophore of the MTX molecule has a dual function—through intercalation, it docks the MTX molecule in the DNA duplex and allows direct contact between the additional hydrogen bonds and amino acid residues of the protein. Two hydroxyalkylamine side-chains of the MTX molecule “surround” nucleobases located vis-a-vis the DNA breakpoint. At the same time, such method of “surrounding” DNA allows interaction with closer amino acid residues of the protein, which are responsible for further stabilisation of the complexCitation184. The anticancer activity of MTX is based on inhibition of topoisomerase II activity by stabilisation of DNA-topoisomerase II covalent complex, which leads to DNA breakages. Splitting of DNA strands results in inhibition of two significant processes: replication of DNA and transcription of RNACitation185,Citation186. MTX was approved by the FDA in 1987 and nowadays it is the only clinically approved drug from the anthracenedione derivatives groupCitation187. It is used as a chemotherapeutic drug in the treatment of acute leukaemia, lymphoma, prostate cancer and breast cancerCitation188. Furthermore, MTX is used in the treatment of multiple sclerosisCitation189.
Figure 10. Chemical structure of mitoxantrone.
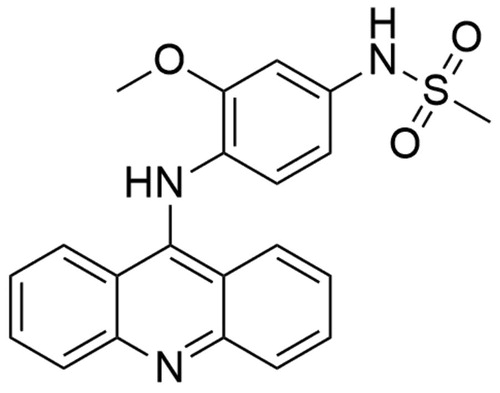
Acridines
Amsacrine (N-[4-(acridin-9-ylamino)-3methoxyphenyl]methanesulfonamide; m-AMSA) is hTop IIβ inhibitor belonging to the class of acridines. The amsacrine molecule is composed of two basic elements: the acridine group and the 4′-amino-methanesulfone-m-anisidide head group (). To determine the role of m-AMSA-mediated DNA binding in the proper functioning of the compound and to perform a comprehensive structure–activity relationship analysis, scientists analysed different amsacrine derivatives. The compounds were tested for their possibility of DNA intercalation and the ability to increase the intensity of the DNA strand cleavage process with the participation of hTop IIα and hTop IIβ. The obtained results indicate that the presence of the methoxy group in the 3′ position (m-AMSA) has a positive effect on the anti-tumour activity of the drug. In turn, changing the location of the group by placing it in the 2′ (o-AMSA) position causes inhibition of drug activity. It is probably caused by an increase in the freedom of rotation of the 4′-amino-methanesulfone-m-anisidide group, which may disrupt, e.g. the intercalation process of the 1′ substituent in the Top II–DNA–drug complexCitation190. Moro et al. conducted modelling of m-AMSA interaction with the Top II–DNA complex. Based on the obtained results, it was found that the CG and GC base pairs of DNA, which form a clamp, are 3.4 − 6.8 Å away from each other. This arrangement of base pairs allows locating the drug molecule between them. The aniline sulphonamide in position 1′ strongly binds with –OH Thr-744 group. The oxygen of Gly-747 carbonyl group and NH of sulphonamide residue form another strong bond. Moreover, hydrophobic interaction was observed between –CH3 group in position 3′ of aniline methoxyl substituent and side-chain Phe-754Citation155. The drug was approved in 1983 in CanadaCitation25 and to date is used for the treatment patients with AML and refractory ALL. The inhibitory effect of m-AMSA on hTop IIβ activity is based on the topoisomerase poisoning and increasing level of DNA- hTop IIβ covalent complexes in the cellCitation190,Citation191. Furthermore, few clinical trials are conducted to evaluate the effectiveness of combination therapy (m-AMSA with various compounds) in the treatment of several cancers (NCT03765541, NCT00003436, NCT00002719).
Figure 11. Chemical structure of amsacrine.
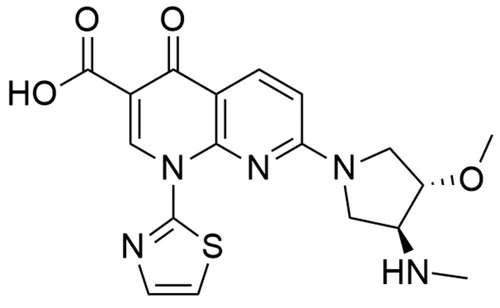
Quinolones
Vosaroxin (7-[(3S,4S)-3-methoxy-4-(methylamino)pyrrolidin-1-yl]-4-oxo-1–(1,3-thiazol-2-yl)-1,8-naphthyridine-3-carboxylic acid; formerly AG-7532, SN-595 or voreloxin) was first described in 2002. It is a synthetic, antineoplastic quinolone derivative () developed by Sunesis PharmaceuticalsCitation192,Citation193. It is the first anticancer quinolone derivative and its antineoplastic mechanism of action is based on the topoisomerase II poisoning and induction of site-specific breaks in double-stranded DNACitation194. Currently, there are ongoing clinical studies concerning the application of vosaroxin with azacitidine in treating older patients with AML (NCT03338348, University of Ulm is recruiting participants for the phase II trial). Phase I trial in the treatment of patients with myelodysplastic syndromes (NCT01913951) is ongoing. Moreover, combination therapy (vosaroxin with infusional cytarabine) is evaluated in untreated AML therapy (NCT02658487, phase II trial is ongoing).
Figure 12. Chemical structure of vosaroxin.
Catalytic inhibitors
Catalytic inhibitors are the second from two types of Top II inhibitors. Their mechanism of action is based on preventing the DNA-Top IIA bond formation, stabilisation of DNA by the formation of non-covalent complexes with Top IIA or blocking the ATPase enzyme binding siteCitation195–197.
Bisdioxopiperazines
Dexrazoxane (4-[(2S)-2–(3,5-dioxopiperazin-1-yl)propyl]piperazine-2,6-dione; ICRF-187) is an ethylenediaminetetraacetic acid (EDTA) derivative and belongs to the group of bisdioxopiperazine compoundsCitation161. To date, the ICRF-187 () is the only drug approved for the treatment of anthracycline-induced cardiotoxicity. Due to the fact that ICRF-187 is an EDTA derivative, the cardioprotective properties of this compound are attributed to its ability to chelate free iron and iron complexed by anthracyclines—this prevents the formation of cardiotoxic reactive oxygen speciesCitation198,Citation199. The bisdioxopiperazine derivative act by Top II arrestment in a specific point in the catalytic cycleCitation200. The dexrazoxane mechanism of action consists of blocking DNA dependent hydrolysis of ATP by Top II which prevents the N-terminal clamp opening and consequently blocks the release of transported DNA segmentCitation201. The ICRF-187 molecule is comprised of two piperazinedione rings bounded by a monomethyl substituted ethanediyl linker. Interactions between the enzyme and the compound occur between the Top II “tyrosine –dome” and the two ICRF-187 piperazinodione rings. The “tyrosine dome” is located approximately parallel to the piperazinodione rings of ICRF-187. Each of the Gln-365 of the Top II protomer forms a hydrophobic bond with the hydrogen of the imide dexrazoxane moiety. Furthermore, the ICRF-187 methyl side-chain forms an additional hydrophobic π-methyl bond with Tyr-28 of one of the Top II protomers. Losing this hydrophobic interaction by removing the linker group, but also replacing it with a larger side-chain significantly reduces the compound’s ability to inhibit Top II activityCitation202,Citation203.
Figure 13. Chemical structure of dexrazoxane.
Thiobarbiturates
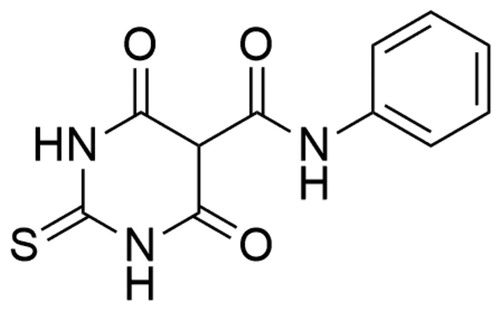
Merbarone (5-[N-phenylcarboxamido]-2-thiobarbituric acid; NSC 336628), catalytic topoisomerase II inhibitor, is a thiobarbiturate analogue (). In 1998 Fortune and Osheroff conducted biochemical studies and have classified the compound as a hTop IIα catalytic inhibitorCitation204. Moreover, in 2012 Pastor et al. carried out research on AA8 ovary fibroblast Chinese hamster cell line which showed that merbarone is not only a cytotoxic but also genotoxic compound. Besides, studies reported the induction of the endoreduplication process by compoundCitation205. The detailed mechanism of hTop IIα inhibition remains still unknown and although it is not approved as a therapeutic agent. In the hTop IIα–DNA complex, Arg-804 and Tyr-805 are important elements that allows the transesterification reaction and splitting the DNA strand at the phosphate centre. The close proximity of the phosphorus atom of the DNA sugar-phosphate backbone and hydroxyl oxygen located in the Tyr-805 side-chain determines the hTop IIα catalysed reaction. Merbarone, creating a hydrogen bond between the oxygen of the amide residue and Tyr-805 of the topoisomerase, increases the distance between the phosphorus in the DNA backbone and the hydroxyl oxygen of the Tyr-805 side-chain. Scientists presume that by increasing the distance between Tyr-805 and the hTop IIα reaction site, merbarone prevents the 5′-phosphotyrosyl bond in the hTop IIα-DNA complexCitation206. Currently, the drug is used as a tool for studies on Top IICitation191.
Figure 14. Chemical structure of merbarone.
Conclusions and perspectives
DNA replication, transcription and repair mechanisms are crucial processes occurring in each cell and topoisomerases are one of the key elements of these processes. Due to their significant biological functions, enzyme structure and mechanisms of action, these enzymes have been one of the main molecular targets in the design of new anticancer agents for nearly 30 years. Despite all the collected data about topoisomerases, there are still some elements that are unclear for us. Now, a phenomenon that poses a great challenge to scientists around the world is the developing resistance to anti-topoisomerase drugs. The main challenge is to maintain topoisomerase sensitivity to drugs, e.g. by designing new enzymes or new drug delivery systems. Furthermore, studies on structure–activity relationship or molecular docking play an important role in the drug development process. The application of computational techniques or the use of special programs to study the interactions between individual structures enables scientists to identify the key elements of the compound structure responsible for their activity. Thanks to this it is possible to modify the structures of known compounds to improve their properties or to design and synthesise new derivatives based on computer simulation results. Development of new anti-topoisomerase drugs, e.g. may allow to limit the cardiotoxic effect of anthracyclines or reduce the incidence of drug-induced secondary cancers. After all, the growing importance and rapid development of molecular genetics and molecular biology may enable the use of anti-topoisomerase agents in personalised anticancer therapy.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Forterre P, Gribaldo S, Gadelle D, Serre MC. Origin and evolution of DNA topoisomerases. Biochimie 2007;89:427–46.
- Wang JC. Interaction between DNA and an Escherichia coli protein omega. J Mol Biol 1971;55:523–33.
- Keller W. Determination of the number of superhelical turns in simian virus 40 DNA by gel electrophoresis. Proc Natl Acad Sci USA 1975;72:4876–80.
- Gellert M, Mizuuchi K, O'Dea MH, Nash HA. DNA gyrase: an enzyme that introduces superhelical turns into DNA. Proc Natl Acad Sci USA 1976;73:3872–6.
- Baldi MI, Benedetti P, Mattoccia E, Tocchini-Valentini GP. In vitro catenation and decatenation of DNA and a novel eucaryotic ATP-dependent topoisomerase. Cell 1980;20:461–7.
- Bansal S, Bajaj P, Pandey S, Tandon V. Topoisomerases: resistance versus sensitivity, how far we can go? Med Res Rev 2017;37:404–38.
- Champoux JJ. DNA topoisomerases: structure, function, and mechanism. Annu Rev Biochem 2001;70:369–413.
- E. Mutschler G, Geisslinger HK, Kroemer S, Menzel PR, 22.3. Inhibitory topoizomerazy. In: Droździk M, Kocić I, Pawlak D, editors. Farmakologia i toksykologia. 4th ed. Wrocław: MedPharm Polska; 2016. p. 880.
- Baker NM, Rajan R, Mondragón A. Structural studies of type I topoisomerases. Nucleic Acids Res 2009;37:693–701.
- Viard T, de la Tour CB. Type IA topoisomerases: a simple puzzle?. Biochimie 2007;89:456–67.
- Nagaraja V, Godbole AA, Henderson SR, Maxwell A. DNA topoisomerase I and DNA gyrase as targets for TB therapy. Drug Discov Today 2017;22:510–8.
- Capranico G, Marinello J, Chillemi G. Type I DNA topoisomerases. J Med Chem 2017;60:2169–92.
- Bush NG, Evans-Roberts K, Maxwell A. DNA topoisomerases. EcoSal Plus 2015;6.
- Pommier Y, DNA topoisomerases and cancer. New York: Springer New York; 2012.
- Kirkegaard K, Wang JC. Bacterial DNA topoisomerase I can relax positively supercoiled DNA containing a single-stranded loop. J. Mol. Biol 1985;185:625–37.
- DiGate RJ, Marians KJ. Identification of a potent decatenating enzyme from Escherichia coli. J Biol Chem 1988;263:13366–73.
- Jain C, Majumder H, Roychoudhury S. Natural compounds as anticancer agents targeting DNA topoisomerases. Curr Genomics 2017;18:75–92.
- Viard T, Cossard R, Duguet M, Bouthier De La Tour C. Thermotoga maritima-Escherichia Coli chimeric topoisomerases. Answers about involvement of the carboxyl-terminal domain in DNA topoisomerase I-mediated catalysis. J Biol Chem 2004;279:30073–80.
- Dekker NH, Rybenkov VV, Duguet M, et al. The mechanism of type IA topoisomerases. Proc Natl Acad Sci USA 2002;99:12126–31.
- Delgado JL, Hsieh C-M, Chan N-L, Hiasa H. Topoisomerases as anticancer targets. Biochem J 2018;475:373–98.
- Redinbo MR, Champoux JJ, Hol WG. Structural insights into the function of type IB topoisomerases. Curr Opin Struct Biol 1999;9:29–36.
- Koster DA, Croquette V, Dekker C, et al. Friction and torque govern the relaxation of DNA supercoils by eukaryotic topoisomerase IB. Nature 2005;434:671–4.
- Krogh BO, Shuman S. Catalytic mechanism of DNA topoisomerase IB. Mol Cell 2000;5:1035–41.
- Cinelli MA. Topoisomerase 1B poisons: Over a half-century of drug leads, clinical candidates, and serendipitous discoveries. Med Res Rev 2019;39:1294–337.
- Hevener KE, Verstak TA, Lutat KE, et al. Recent developments in topoisomerase-targeted cancer chemotherapy. Acta Pharm Sin B 2018;8:844–61.
- Redinbo MR, Stewart L, Kuhn P, et al. Crystal structures of human topoisomerase I in covalent and noncovalent complexes with DNA. Science 1998;279:1504–13.
- Slesarev AI, Stetter KO, Lake JA, et al. DNA topoisomerase V is a relative of eukaryotic topoisomerase I from a hyperthermophilic prokaryote. Nature 1993;364:735–7.
- Taneja B, Schnurr B, Slesarev A, et al. Topoisomerase V relaxes supercoiled DNA by a constrained swiveling mechanism. Proc Natl Acad Sci USA 2007;104:14670–5.
- Taneja B, Patel A, Slesarev A, Mondragón A. Structure of the N-terminal fragment of topoisomerase V reveals a new family of topoisomerases. Embo J 2006;25:398–408.
- Belova GI, Prasad R, Kozyavkin SA, et al. A type IB topoisomerase with DNA repair activities. Proc Natl Acad Sci USA 2001;98:6015–20.
- Belova GI, Prasad R, Nazimov IV, et al. The domain organization and properties of individual domains of DNA topoisomerase V, a type 1B topoisomerase with DNA repair activities. J Biol Chem 2002;277:4959–65.
- Rajan R, Taneja B, Mondragón A. Structures of minimal catalytic fragments of topoisomerase V reveals conformational changes relevant for DNA binding. Structure 2010;18:829–38.
- Schoeffler AJ, Berger JM. DNA topoisomerases: harnessing and constraining energy to govern chromosome topology. Q Rev Biophys 2008;41:41–101.
- Corbett KD, Berger JM. Structure, molecular mechanisms, and evolutionary relationships in DNA topoisomerases. Annu Rev Biophys Biomol Struct 2004;33:95–118.
- Zechiedrich EL, Christiansen K, Andersen AH, et al. Double-stranded DNA cleavage/religation reaction of eukaryotic topoisomerase II: evidence for a nicked DNA intermediate. Biochemistry 1989;28:6229–36.
- McClendon AK, Osheroff N. DNA topoisomerase II, genotoxicity, and cancer. Mutat Res 2007;623:83–97.
- Deweese JE, Osheroff N. Coordinating the two protomer active sites of human topoisomerase IIalpha: nicks as topoisomerase II poisons. Biochemistry 2009;48:1439–41.
- Deweese JE, Osheroff N. The DNA cleavage reaction of topoisomerase II: wolf in sheep's clothing. Nucleic Acids Res 2009;37:738–48.
- Chang CC, Wang YR, Chen SF, et al. New insights into DNA-binding by type IIA topoisomerases. Curr Opin Struct Biol 2013;23:125–33.
- Gadelle D, Filée J, Buhler C, Forterre P. Phylogenomics of type II DNA topoisomerases. BioEssays 2003;25:232–42.
- Roca J, Wang JC. DNA transport by a type II DNA topoisomerase: evidence in favor of a two-gate mechanism. Cell 1994;77:609–16.
- Lima CD, Mondragón A. Mechanism of type II DNA topoisomerases: a tale of two gates. Structure 1994;2:559–60.
- Berger JM, Gamblin SJ, Harrison SC, Wang JC. Structure and mechanism of DNA topoisomerase II. Nature 1996;379:225–32.
- Schmidt BH, Burgin AB, Deweese JE, et al. A novel and unified two-metal mechanism for DNA cleavage by type II and IA topoisomerases. Nature 2010;465:641–4.
- Laponogov I, Pan XS, Veselkov DA, et al. Structural basis of gate-DNA breakage and resealing by type II topoisomerases. PLoS One 2010;5: 1–8.
- Osheroff N. Eukaryotic topoisomerase II. Characterization of enzyme turnover. J Biol Chem 1986;261:9944–50.
- Pommier Y, Leo E, Zhang H, Marchand C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem Biol 2010;17:421–33.
- Kellner U, Sehested M, Jensen PB, et al. Culprit and victim - DNA topoisomerase II. Lancet Oncol 2002;3:235–43.
- Nitiss JL. DNA topoisomerase II and its growing repertoire of biological functions. Nat Rev Cancer 2009;9:327–37.
- Kaur P, Kaur V, Kaur S, DNA Topoisomerase II: promising target for anticancer drugs. In: Gandhi V, Grover R, Pathak SAB, editor. Multi-targeted approach to treatment of cancer. Springer International Publishing; 2015:323–38.
- Lepiarczyk M, Bielawska A, Sosnowska K, Bielawski K. Ludzka topoizomeraza typu II jako molekularny punkt uchwytu leków przeciwnowotworowych. Gaz Farm 2011;20:24–6.
- Linka RM, Porter ACG, Volkov A, et al. C-Terminal regions of topoisomerase IIalpha and IIbeta determine isoform-specific functioning of the enzymes in vivo. Nucleic Acids Res 2007;35:3810–22.
- Farr CJ, Antoniou-Kourounioti M, Mimmack ML, et al. The α isoform of topoisomerase II is required for hypercompaction of mitotic chromosomes in human cells. Nucleic Acids Res 2014;42:4414–26.
- Tiwari VK, Burger L, Nikoletopoulou V, et al. Target genes of topoisomerase IIβ regulate neuronal survival and are defined by their chromatin state. Proc Natl Acad Sci USA 2012;109:934–43.
- Bergerat A, Gadelle D, Forterre P. Purification of a DNA topoisomerase II from the hyperthermophilic archaeon Sulfolobus shibatae. A thermostable enzyme with both bacterial and eucaryal features. J Biol Chem 1994;269:27663–9.
- Bergerat A, De Massy B, Gadelle D, et al. An atypical topoisomerase II from archaea with implications for meiotic recombination. Nature 1997;386:414–7.
- Buhler C, Lebbink JHG, Bocs C, et al. DNA Topoisomerase VI generates ATP-dependent double-strand breaks with two-nucleotide overhangs. J Biol Chem 2001;276:37215–22.
- Corbett KD, Berger JM. Structure of the topoisomerase VI-B subunit: implications for type II topoisomerase mechanism and evolution. Embo J 2003;22:151–63.
- Hartung F, Puchta H. Molecular characterisation of two paralogous SPO11 homologues in Arabidopsis thaliana. Nucleic Acids Res 2000;28:1548–54.
- Malik S-B, Ramesh MA, Hulstrand AM, Logsdon JM. Protist homologs of the meiotic Spo11 gene and topoisomerase VI reveal an evolutionary history of gene duplication and lineage-specific loss. Mol Biol Evol 2007;24:2827–41.
- Sugimoto-Shirasu K, Stacey NJ, Corsar J, et al. DNA topoisomerase VI is essential for endoreduplication in Arabidopsis. Curr Biol 2002;12:1782–6.
- Gadelle D, Krupovic M, Raymann K, et al. DNA topoisomerase VIII: a novel subfamily of type IIB topoisomerases encoded by free or integrated plasmids in archaea and bacteria. Nucleic Acids Res 2014;42:8578–91.
- Drwal MN, Marinello J, Manzo SG, et al. Novel DNA topoisomerase IIα inhibitors from combined ligand- and structure- based virtual screening. PLoS One 2014;9:1–16.
- Pourquier P, Pommier Y, Topoisomerase I-mediated DNA damage. In: Pourquier P, Pommier Y, editors. Advances in cancer research. Vol. 80. Elsevier; 2001. p. 189–216.
- Capranico G, Binaschi M, Borgnetto ME, et al. A protein-mediated mechanism for the DNA sequence-specific action of topoisomerase II poisons. Trends Pharmacol. Sci 1997;18:323–9.
- Chen SH, Chan N-L, Hsieh T. New mechanistic and functional insights into DNA topoisomerases. Annu Rev Biochem 2013;82:139–70.
- Pommier Y. Drugging topoisomerases: lessons and challenges. ACS Chem Biol 2013;8:82–95.
- Pommier Y, Kiselev E, Marchand C. Interfacial inhibitors. Bioorg Med Chem Lett 2015;25:3961–5.
- Zhang C, Li S, Ji L, et al. Design, synthesis and antitumor activity of non-camptothecin topoisomerase i inhibitors. Bioorg Med Chem Lett 2015;25:4693–6.
- Kümler I, Brünner N, Stenvang J, et al. A systematic review on topoisomerase 1 inhibition in the treatment of metastatic breast cancer. Breast Cancer Res Treat 2013;138:347–58.
- Hsiang YH, Hertzberg R, Hecht S, Liu LF. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J Biol Chem 1985;260:14873–8.
- Hsiang YH, Liu LF. Identification of mammalian DNA topoisomerase i as an intracellular target of the anticancer drug camptothecin. Cancer Res 1988;48:1722–6.
- Wall ME, Wani MC, Cook CE, et al. Plant antitumor agents. I. The isolation and structure of camptothecin, a novel alkaloidal leukemia and tumor inhibitor from Camptotheca acuminata. J Am Chem Soc 1966;88:3888–90.
- Kacprzak KM, Chemistry and biology of camptothecin and its derivatives. In: Natural products: phytochemistry, botany and metabolism of alkaloids, phenolics and terpenes. Berlin Heidelberg: Springer; 2013. p. 643–682.
- Muqeet W, Bano Q. Camptothecin and its analogs antitumour activity by poisoning topoisomerase I, their structure activity relationship and clinical development perspective of analogs. J App Pharm 2014;6:386–95.
- Pizzolato JF, Saltz LB. The camptothecins. Lancet 2003;361:2235–42.
- Strumberg D, Pilon AA, Smith M, et al. Conversion of topoisomerase I cleavage complexes on the leading strand of ribosomal DNA into 5'-phosphorylated DNA double-strand breaks by replication runoff. Mol Cell Biol 2000;20:3977–87.
- Sané AT, Bertrand R. Caspase inhibition in camptothecin-treated U-937 cells is coupled with a shift from apoptosis to transient G1 arrest followed by necrotic cell death. Cancer Res 1999;59:3565–9.
- Li LH, Fraser TJ, Olin EJ, Bhuyan BK. Action of camptothecin on mammalian cells in culture. Cancer Res 1972;32:2643–50.
- Pommier Y. DNA topoisomerase I inhibitors: chemistry, biology, and interfacial inhibition. Chem Rev 2009;109:2894–902.
- Staker BL, Hjerrild K, Feese MD, et al. The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc Natl Acad Sci USA 2002;99:15387–92.
- Hertzberg RP, Caranfa MJ, Hecht SM. On the mechanism of topoisomerase I inhibition by camptothecin: evidence for binding to an enzyme-DNA complex. Biochemistry 1989;28:4629–38.
- Garst J. Topotecan: An evolving option in the treatment of relapsed small cell lung cancer. Ther Clin Risk Manag 2007;3:1087–95.
- Armstrong DK, Spriggs D, Levin Y, et al. Hematologic safety and tolerability of topotecan in recurrent ovarian cancer and small cell lung cancer: an integrated analysis. Oncologist 2005;10:686–94.
- Brave M, Dagher R, Farrell A, et al. Topotecan in combination with cisplatin for the treatment of stage IVB, recurrent, or persistent cervical cancer. Oncology (Williston Park, N.Y.) 2006;20:1401–10.
- Fuchs C, Mitchell EP, Hoff PM. Irinotecan in the treatment of colorectal cancer. Cancer Treat. Rev 2006;32:491–503.
- Ohtsuka K, Inoue S, Kameyama M, et al. Intracellular conversion of irinotecan to its active form, SN-38, by native carboxylesterase in human non-small cell lung cancer. Lung Cancer 2003;41:187–98.
- Mullangi R, Ahlawat P, Srinivas NR. Irinotecan and its active metabolite, SN-38: review of bioanalytical methods and recent update from clinical pharmacology perspectives. Biomed Chromatogr 2010;24:104–23.
- Haaz MC, Rivory L, Jantet S, et al. Glucuronidation of SN-38, the active metabolite of irinotecan, by human hepatic microsomes. Pharmacol Toxicol 1997;80:91–6.
- Tadokoro JI, Kakihata K, Shimazaki M, et al. Post-marketing surveillance (PMS) of all patients treated with irinotecan in Japan: clinical experience and ADR profile of 13,935 patients. Jpn J Clin Oncol 2011;41:1101–11.
- Martino E, Della Volpe S, Terribile E, et al. The long story of camptothecin: from traditional medicine to drugs. Bioorg Med Chem Lett 2017;27:701–7.
- Crul M. CKD-602 Chong Kun Dang. Curr Opin Investig Drugs 2003;4:1455–9.
- Park YH, Chung CU, Park BM, et al. Lesser toxicities of belotecan in patients with small cell lung cancer: a retrospective single-center study of camptothecin analogs. Can Respir J 2016;2016:1–8.
- Lee S, Ho JY, Liu JJ, et al. CKD-602, a topoisomerase I inhibitor, induces apoptosis and cell-cycle arrest and inhibits invasion in cervical cancer. Mol. Med 2019;25:23.
- Joerger M, Hess D, Delmonte A, et al. Phase-I dose finding and pharmacokinetic study of the novel hydrophilic camptothecin ST-1968 (namitecan) in patients with solid tumors. Invest New Drugs 2015;33:472–9.
- Joerger M, Hess D, Delmonte A, et al. Integrative population pharmacokinetic and pharmacodynamic dose finding approach of the new camptothecin compound namitecan (ST1968). Br J Clin Pharmacol 2015;80:128–38.
- Cao Z, Kozielski A, Liu X, et al. Crystalline camptothecin-20(S)-O-propionate hydrate: a novel anticancer agent with strong activity against 19 human tumor xenografts. Cancer Res 2009;69:4742–9.
- Bom D, Curran DP, Kruszewski S, et al. The novel silatecan 7-tert-butyldimethylsilyl-10-hydroxycamptothecin displays high lipophilicity, improved human blood stability, and potent anticancer activity. J Med Chem 2000;43:3970–80.
- Pratesi G, Beretta GL, Zunino F. Gimatecan, a novel camptothecin with a promising preclinical profile. Anticancer Drugs 2004;15:545–52.
- De Cesare M, Pratesi G, Perego P, et al. Potent antitumor activity and improved pharmacological profile of ST1481, a novel 7-substituted camptothecin. Cancer Res 2001;61:7189–95.
- De Cesare M. High efficacy of intravenous gimatecan on human tumor xenografts. Anticancer Res 2018;38:5783–90.
- Sheng C, Miao Z, Zhang W. New strategies in the discovery of novel non-camptothecin topoisomerase I inhibitors. Curr Med Chem 2011;18:4389–409.
- Teicher BA. Next generation topoisomerase I inhibitors: rationale and biomarker strategies. Biochem Pharmacol 2008;75:1262–71.
- Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer 2006;6:789–802.
- Kummar S, Chen A, Gutierrez M, et al. Clinical and pharmacologic evaluation of two dosing schedules of indotecan (LMP400), a novel indenoisoquinoline, in patients with advanced solid tumors. Cancer Chemother Pharmacol 2016;78:73–81.
- Saif MW, Diasio RB. Edotecarin: a novel topoisomerase I inhibitor. Clin Colorectal Cancer 2005;5:27–36.
- Li TK, Houghton PJ, Desai SD, et al. Characterization of ARC-111 as a novel topoisomerase I-targeting anticancer drug. Cancer Res 2003;63:8400–7.
- Yoshinari T, Ohkubo M, Fukasawa K, et al. Mode of action of a new indolocarbazole anticancer agent, J-107088, targeting topoisomerase I. Cancer Res 1999;59:4271–5.
- Pommier Y, Cushman M. The indenoisoquinoline noncamptothecin topoisomerase I inhibitors: update and perspectives. Mol. Cancer Ther 2009;8:1008–14.
- Cuya SM, Bjornsti M-A, van Waardenburg RCAM. DNA topoisomerase-targeting chemotherapeutics: what's new? Cancer Chemother Pharmacol 2017;80:1–14.
- A phase I study of indenoisoquinolines LMP400 and LMP776 in adults with relapsed solid tumors and lymphomas - full text view - ClinicalTrials.gov. [accessed 2020 Jun 1]. https://clinicaltrials.gov/ct2/show/NCT01051635
- Ruchelman AL, Singh SK, Ray A, et al. 5H-dibenzo[c,h]1,6-naphthyridin-6-ones: novel topoisomerase I-targeting anticancer agents with potent cytotoxic activity. Bioorg Med Chem 2003;11:2061–73.
- Ruchelman AL, Singh SK, Wu X, et al. Diaza- and triazachrysenes: potent topoisomerase-targeting agents with exceptional antitumor activity against the human tumor xenograft, MDA-MB-435. Bioorg Med Chem Lett 2002;12:3333–6.
- Zhu S, Ruchelman AL, Zhou N, et al. Esters and amides of 2,3-dimethoxy-8,9-methylenedioxy-benzo[i]phenanthridine-12-carboxylic acid: potent cytotoxic and topoisomerase I-targeting agents. Bioorg Med Chem 2005;13:6782–94.
- Satyanarayana M, Feng W, Cheng L, et al. Syntheses and biological evaluation of topoisomerase I-targeting agents related to 11-[2-(N,N-dimethylamino)ethyl]-2,3-dimethoxy-8,9-methylenedioxy-11H-isoquino[4,3-c]cinnolin-12-one (ARC-31). Bioorg Med Chem 2008;16:7824–31.
- Feng W, Satyanarayana M, Tsai YC, et al. 12-Substituted 2,3-dimethoxy-8,9-methylenedioxybenzo[i]phenanthridines as novel topoisomerase I-targeting antitumor agents. Bioorg Med Chem 2009;17:2877–85.
- Zhu S, Ruchelman AL, Zhou N, et al. 6-substituted 6H-dibenzo[c,h][2,6]naphthyridin-5-ones: reversed lactam analogues of ARC-111 with potent topoisomerase I-targeting activity and cytotoxicity. Bioorg Med Chem 2006;14:3131–43.
- Sooryakumar D, Dexheimer TS, Teicher BA, Pommier Y. Molecular and cellular pharmacology of the novel noncamptothecin topoisomerase I inhibitor Genz-644282. Mol Cancer Ther 2011;10:1490–9.
- Pogorelčnik B, Brvar M, Žegura B, et al. Discovery of mono- and disubstituted 1H-pyrazolo[3,4]pyrimidines and 9H-purines as catalytic inhibitors of human DNA topoisomerase IIα. ChemMedChem 2015;10:345–59.
- Vos SM, Tretter EM, Schmidt BH, Berger JM. All tangled up: how cells direct, manage and exploit topoisomerase function. Nat Rev Mol Cell Biol 2011;12:827–41.
- de Almeida SMV, Ribeiro AG, de Lima Silva GC, et al. Alves de Lima M do C. DNA binding and topoisomerase inhibition: how can these mechanisms be explored to design more specific anticancer agents? Biomed. Pharmacother 2017;96:1538–56.
- Nitiss JL. Targeting DNA topoisomerase II in cancer chemotherapy. Nat Rev Cancer 2009;9:338–50.
- Bailly C. Contemporary challenges in the design of topoisomerase II inhibitors for cancer chemotherapy. Chem Rev 2012;112:3611–40.
- Clark PI, Slevin ML. The clinical pharmacology of etoposide and teniposide. Clin Pharmacokinet 1987;12:223–52.
- King LS, Sullivan M. The similarity of the effect of podophyllin and colchicine and their use in the treatment of condylomata acuminata. Science 1946;104:244–5.
- Greenspan EM, Leiter J, Shear MJ. Effect of alpha-peltatin, beta-peltatin, and podophyllotoxin on lymphomas and other transplanted tumors. J Natl Cancer Inst 1950;10:1295–333.
- Hande KR. Etoposide: four decades of development of a topoisomerase II inhibitor. Eur J Cancer 1998;34:1514–21.
- Kingma PS, Burden DA, Osheroff N. Binding of etoposide to topoisomerase II in the absence of DNA: Decreased affinity as a mechanism of drug resistance. Biochemistry 1999;38:3457–61.
- Burden DA, Kingma PS, Froelich-Ammon SJ, et al. Topoisomerase II.etoposide interactions direct the formation of drug-induced enzyme-DNA cleavage complexes. J Biol Chem 1996;271:29238–44.
- Wilstermann AM, Bender RP, Godfrey M, et al. Topoisomerase II - drug interaction domains: identification of substituents on etoposide that interact with the enzyme. Biochemistry 2007;46:8217–25.
- Baldwin EL, Osheroff N. Etoposide, topoisomerase II and cancer. Curr Med Chem Anticancer Agents 2005;5:363–72.
- Stähelin HF, von Wartburg A. The chemical and biological route from podophyllotoxin glucoside to etoposide: ninth cain memorial award lecture. Cancer Res 1991;51:5–15.
- Montecucco A, Zanetta F, Biamonti G. Molecular mechanisms of etoposide. Excli J 2015;14:95–108.
- Kuruppu AI, Paranagama P, Goonasekara CL. Medicinal plants commonly used against cancer in traditional medicine formulae in Sri Lanka. Saudi Pharm J 2019;27:565–73.
- Bender RP, Osheroff N, DNA topoisomerases as targets for the chemotherapeutic treatment of cancer. In: Dai W, editor. Checkpoint responses in cancer therapy. Totowa: Humana Press; 2008. p. 57–91.
- Barret JM, Kruczynski A, Vispé S, et al. F14512, a potent antitumor agent targeting topoisomerase II vectored into cancer cells via the polyamine transport system. Cancer Res 2008;68:9845–53.
- Thakur D. Topoisomerase II Inhibitors in cancer treatment. Int J Pharm Sci Nanotechnol 2011;3:1173–81.
- Najar IA, Johri RK. Pharmaceutical and pharmacological approaches for bioavailability enhancement of etoposide. J Biosci 2014;39:139–44.
- Li J, Chen W, Zhang P, Li N. Topoisomerase II trapping agent teniposide induces apoptosis and G2/M or S phase arrest of oral squamous cell carcinoma. World J Surg Oncol 2006;4:41.
- Sun J, Wei Q, Zhou Y, et al. A systematic analysis of FDA-approved anticancer drugs. BMC Syst Biol 2017;11:87.
- Guerram M, Jiang ZZ, Zhang LY. Podophyllotoxin, a medicinal agent of plant origin: past, present and future. Chin J Nat Med 2012;10:161–9.
- Hartmann JT, Lipp HP. Camptothecin and podophyllotoxin derivatives: inhibitors of topoisomerase I and II - mechanisms of action, pharmacokinetics and toxicity profile. Drug Saf 2006;29:209–30.
- Gentry AC, Pitts SL, Jablonsky MJ, et al. Interactions between the etoposide derivative F14512 and human type II topoisomerases: implications for the C4 spermine moiety in promoting enzyme-mediated DNA cleavage. Biochemistry 2011;50:3240–9.
- Casero RA, Marton LJ. Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nat Rev Drug Discov 2007;6:373–90.
- Bombarde O, Larminat F, Gomez D, et al. The DNA-binding polyamine moiety in the vectorized DNA topoisomerase II inhibitor F14512 alters reparability of the consequent enzyme-linked DNA double-strand breaks. Mol Cancer Ther 2017;16:2166–77.
- Kruczynski A, Vandenberghe I, Pillon A, et al. Preclinical activity of F14512, designed to target tumors expressing an active polyamine transport system. Invest New Drugs 2011;29:9–21.
- Jasra S, Anampa J. Anthracycline use for early stage breast cancer in the modern era: a review. Curr Treat Options Oncol 2018;19:30.
- Nebigil CG, Désaubry L. Updates in anthracycline-mediated cardiotoxicity. Front Pharmacol 2018;9:1262.
- Aleman BMP, Moser EC, Nuver J, et al. Cardiovascular disease after cancer therapy. EJC Suppl 2014;12:18–28.
- Zunino F, Capranico G. DNA topoisomerase II as the primary target of anti-tumor anthracyclines. Anticancer Drug Des 1990;5:307–17.
- Binaschi M, Bigioni M, Cipollone A, et al. Anthracyclines: selected new developments. Curr Med Chem Anticancer Agents 2001;1:113–30.
- Thorn CF, Oshiro C, Marsh S, et al. Doxorubicin pathways: pharmacodynamics and adverse effects. Pharmacogenet Genomics 2011;21:440–6.
- Menna P, Minotti G, Salvatorelli E. In vitro modeling of the structure-activity determinants of anthracycline cardiotoxicity. Cell Biol Toxicol 2007;23:49–62.
- Pogorelcnik B, Perdih A, Solmajer T. Recent developments of DNA poisons-human DNA topoisomerase IIα inhibitors-as anticancer agents. Curr Pharm Des 2013;19:2474–88.
- Moro S, Beretta GL, Dal Ben D, et al. Interaction model for anthracycline activity against DNA topoisomerase II. Biochemistry 2004;43:7503–13.
- Lyu YL, Liu LF, 13- Doxorubicin cardiotoxicity revisited: ROS versus Top2. In: Liu X-Y, Pestka S, Shi Y-F, editors. Recent advances in cancer research and therapy. Elsevier Inc.; 2012. p. 351–369.
- Carvalho C, Santos R, Cardoso S, et al. Doxorubicin: the good, the bad and the ugly effect. Curr Med Chem 2009;16:3267–85.
- Zhang S, Liu X, Bawa-Khalfe T, et al. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med 2012;18:1639–42.
- Pugazhendhi A, Edison TNJI, Velmurugan BK, et al. Toxicity of doxorubicin (Dox) to different experimental organ systems. Life Sci 2018;200:26–30.
- Zhao L, Zhang B. Doxorubicin induces cardiotoxicity through upregulation of death receptors mediated apoptosis in cardiomyocytes. Sci Rep 2017;7:44735.
- Langer SW. Dexrazoxane for the treatment of chemotherapy-related side effects. Cancer Manag Res 2014;6:357–63.
- Drugs@FDA: FDA-Approved Drugs–Doxorubicin. [accessed 2020 Jun 19]. https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=050467
- Robert J. Epirubicin: clinical pharmacology and dose-effect relationship. Drugs 1993;45:20–30.
- Coukell AJ, Faulds D. Epirubicin. An updated review of its pharmacodynamic and pharmacokinetic properties and therapeutic efficacy in the management of breast cancer. Drugs 1997;53:453–82.
- Singh Z, Kaur H. Toxicological aspects of antineoplastic drugs doxorubicin and epirubicin. J Clin Mol Med 2019;2: 1–5.
- Israel M, Potti PG, Seshadri R. Adriamycin analogues. Rationale, synthesis, and preliminary antitumor evaluation of highly active DNA-nonbinding N-(trifluoroacetyl)adriamycin 14-O-hemiester derivatives1, 2. J Med Chem 1985;28:1223–8.
- Onrust SV, Lamb HM. Valrubicin. Drugs Aging 1999;15:69–75.
- Drug Approval Package: Valstar (Valrubicin) NDA# 20-892. [accessed 2020 Jun 9]. https://www.accessdata.fda.gov/drugsatfda_docs/nda/98/20892.cfm
- Hanada M, Mizuno S, Fukushima A, et al. A new antitumor agent amrubicin induces cell growth inhibition by stabilizing topoisomerase II-DNA complex. Jpn J Cancer Res 1998;89:1229–38.
- Sakurai R, Kaira K, Miura Y, et al. Clinical significance of topoisomerase-II expression in patients with advanced non-small cell lung cancer treated with amrubicin. Thorac Cancer 2020;11:426–35.
- Yamaoka T, Hanada M, Ichii S, et al. Cytotoxicity of amrubicin, a novel 9-aminoanthracycline, and its active metabolite amrubicinol on human tumor cells. Jpn J Cancer Res 1998;89:1067–73.
- Kurata T, Okamoto I, Tamura K, Fukuoka M. Amrubicin for non-small-cell lung cancer and small-cell lung cancer. Invest New Drugs 2007;25:499–504.
- Marrero L, Wyczechowska D, Musto AE, et al. Therapeutic efficacy of aldoxorubicin in an intracranial xenograft mouse model of human glioblastoma. Neoplasia 2014;16:874–82.
- Kratz F, Warnecke A, Scheuermann K, et al. Probing the cysteine-34 position of endogenous serum albumin with thiol-binding doxorubicin derivatives. Improved efficacy of an acid-sensitive doxorubicin derivative with specific albumin-binding properties compared to that of the parent compound. J Med Chem 2002;45:5523–33.
- Graeser R, Esser N, Unger H, et al. INNO-206, the (6-maleimidocaproyl hydrazone derivative of doxorubicin), shows superior antitumor efficacy compared to doxorubicin in different tumor xenograft models and in an orthotopic pancreas carcinoma model. Invest New Drugs 2010;28:14–9.
- Kratz F, Fichtner I, Graeser R. Combination therapy with the albumin-binding prodrug of doxorubicin (INNO-206) and doxorubicin achieves complete remissions and improves tolerability in an ovarian A2780 xenograft model. Invest New Drugs 2012;30:1743–9.
- Sanchez E, Li M, Wang C, et al. Anti-myeloma effects of the novel anthracycline derivative INNO-206. Clin Cancer Res 2012;18:3856–67.
- White RJ, Durr FE. Development of mitoxantrone. Invest New Drugs 1985;3:85–93.
- Murdock KC, Child RG, Fabio PF, et al. Antitumor agents. 1. 1,4-bis[(aminoalkyl)amino]-9,10-anthracenediones. J Med Chem 1979;22:1024–30.
- Johnson RK, Zee-Cheng RK, Lee WW, et al. Experimental antitumor activity of aminoanthraquinones. Cancer Treat Rep 1979;63:425–39.
- Zee-Cheng RKY, Cheng CC. Antineoplastic agents. Structure-activity relationship study of bis(substituted aminoalkylamino)anthraquinones. J Med Chem 1978;21:291–4.
- Halterman P, Vogelzang NJ, Farabishahadel A, Goodman OB, Mitoxantrone. In: Drug management of prostate cancer. New York: Springer; 2010. p. 125–131.
- Capranico G, De Isabella P, Tinelli S, et al. Similar sequence specificity of mitoxantrone and VM-26 stimulation of in vitro DNA cleavage by mammalian DNA topoisomerase II. Biochemistry 1993;32:3038–46.
- Wu CC, Li YC, Wang YR, et al. On the structural basis and design guidelines for type II topoisomerase-targeting anticancer drugs. Nucleic Acids Res 2013;41:10630–40.
- Damiani RM, Moura DJ, Viau CM, et al. Pathways of cardiac toxicity: comparison between chemotherapeutic drugs doxorubicin and mitoxantrone. Arch Toxicol 2016;90:2063–76.
- Rossato LG, Costa VM, De Pinho PG, et al. The metabolic profile of mitoxantrone and its relation with mitoxantrone-induced cardiotoxicity. Arch Toxicol 2013;87:1809–20.
- Drugs@FDA: FDA-Approved Drugs–Mitoxantrone. [accessed 2020 Jun 19]. https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=019297
- Enache M, Toader AM, Enache MI. Mitoxantrone-surfactant interactions: a physicochemical overview. Molecules 2016;21:1356.
- Patel KJ, Trédan O, Tannock IF. Distribution of the anticancer drugs doxorubicin, mitoxantrone and topotecan in tumors and normal tissues. Cancer Chemother. Pharmacol 2013;72:127–38.
- Ketron AC, Denny WA, Graves DE, Osheroff N. Amsacrine as a topoisomerase II poison: importance of drug-DNA interactions. Biochemistry 2012;51:1730–9.
- Murphy MB, Mercer SL, Deweese JE, Inhibitors and poisons of mammalian type II topoisomerases. In: Advances in molecular toxicology. Vol. 11. Elsevier B.V.; 2017. p. 203–240.
- Tomita K, Tsuzuki Y, Shibamori K. i, et al. Synthesis and structure-activity relationships of novel 7-substituted 1,4-dihydro-4-oxo-1-(2-thiazolyl)-1,8-naphthyridine-3-carboxylic acids as antitumor agents. Part 1. J. Med. Chem 2002;45:5564–75.
- Abbas JA, Stuart RK. Vosaroxin : a novel antineoplastic quinolone. Expert Opin Investig Drugs 2012;21:1223–33.
- Paubelle E, Zylbersztejn F, Thomas X. The preclinical discovery of vosaroxin for the treatment of acute myeloid leukemia. Expert Opin Drug Discov 2017;12:747–53.
- Li PH, Zeng P, Chen SB, et al. Synthesis and mechanism studies of 1,3-benzoazolyl substituted pyrrolo[2,3-b]pyrazine derivatives as nonintercalative topoisomerase II catalytic inhibitors. J Med Chem 2016;59:238–52.
- Deng S, Yan T, Nikolova T, et al. The catalytic topoisomerase II inhibitor dexrazoxane induces DNA breaks, ATF3 and the DNA damage response in cancer cells. Br J Pharmacol 2015;172:2246–57.
- Vann KR, Ergün Y, Zencir S, et al. Inhibition of human DNA topoisomerase IIα by two novel ellipticine derivatives. Bioorg Med Chem Lett 2016;26:1809–12.
- Jones RL. Utility of dexrazoxane for the reduction of anthracycline-induced cardiotoxicity. Expert Rev Cardiovasc Ther 2008;6:1311–7.
- Cvetković RS, Scott LJ. Dexrazoxane: a review of its use for cardioprotection during anthracycline chemotherapy. Drugs 2005;65:1005–24.
- Roca J, Ishida R, Berger JM, et al. Antitumor bisdioxopiperazines inhibit yeast DNA topoisomerase II by trapping the enzyme in the form of a closed protein clamp. Proc Natl Acad Sci USA 1994;91:1781–5.
- Jensen LH, Nitiss KC, Rose A, et al. A novel mechanism of cell killing by anti-topoisomerase II bisdioxopiperazines. J Biol Chem 2000;275:2137–46.
- Classen S, Olland S, Berger JM. Structure of the topoisomerase II ATPase region and its mechanism of inhibition by the chemotherapeutic agent ICRF-187. Proc Natl Acad Sci USA 2003;100:10629–34.
- Hasinoff BB, Patel D, Wu X. A QSAR study that compares the ability of bisdioxopiperazine analogs of the doxorubicin cardioprotective agent dexrazoxane (ICRF-187) to protect myocytes with DNA topoisomerase II inhibition. Toxicol Appl Pharmacol 2020;399:115038.
- Fortune JM, Osheroff N. Merbarone inhibits the catalytic activity of human topoisomerase IIalpha by blocking DNA cleavage. J Biol Chem 1998;273:17643–50.
- Pastor N, Domínguez I, Orta ML, et al. The DNA topoisomerase II catalytic inhibitor merbarone is genotoxic and induces endoreduplication. Mutat Res 2012;738-739: 45–51.
- Tripathi N, Guchhait SK, Bharatam PV. Pharmacoinformatics analysis of merbarone binding site in human topoisomerase IIα. J Mol Graph Model 2019;86:1–18.