
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Carbonic anhydrases (CAs) are metalloenzymes responsible for the reversible hydration of carbon dioxide to bicarbonate, a fundamental reaction involved in various physiological and pathological processes. In the last decades, CAs have been considered as important drug targets for different pathologies such as glaucoma, epilepsy and cancer. The design of potent and selective inhibitors has been an outstanding goal leading to the discovery of new drugs. Among the different strategies developed to date, the design of carbohydrate-based CA inhibitors (CAIs) has emerged as a versatile tool in order to selectively target CAs. The insertion of a glycosyl moiety as a hydrophilic tail in sulfonamide, sulfenamide, sulfamate or coumarin scaffolds allowed the discovery of many different series of sugar-based CAIs, with relevant inhibitory results. This review will focus on carbohydrate-based CAIs developed so far, classifying them in glycosidic and glycoconjugated inhibitors based on the conjugation chemistry adopted.
1. Introduction
Carbonic anhydrase (CA) is a family of metalloenzymes responsible for the catalysis of the reversible hydration/dehydration of carbon dioxide to bicarbonate. So far, eight genetically distinct CA families are known being present in living organisms, denominated as α-, β-, δ-, γ-, ζ-, η-, θ- and ι-CAs. The last class was only recently discoveredCitation1,Citation2. This classification is based on their structural fold and generally aligns with the host organism: α-CAs are mainly expressed in vertebrates, β-CAs in bacteria and fungi, δ and ζ in marine diatoms and η in protozoaCitation3.
α-CAs are widely expressed in mammals and have been classified in 16 isoenzymes, which differ not only in their subcellular localisation, catalytic activity, and susceptibility to different classes of inhibitors but also in enzymatic efficiency.
The classification of α-CAs includes: cytosolic isozymes (CA I, CA II, CA III, CA VII, CA XIII), membrane-bound isoenzymes (CA IV, CA IX, CA XII, CA IV, CA XV), mitochondrial (CA VA and CA VB) and secreted isoforms (CA VI, primarily in saliva). Additionally, there are three isoforms CA VIII, CA X, CA XI, that are not catalytically active called CA-related proteins (CARPs)Citation4. The α-CAs use as metal cofactor necessary for enzyme catalysis the Zn2+ ion.
CAs are very efficient catalyst for the reversible hydration of carbon dioxide to bicarbonate and a proton (CO2 + H2O ↔ HCO3− + H+), one of the simplest chemical reactions connected with vital processes. Principally, two steps constitute the reaction mechanism:
the nucleophile attack of carbon dioxide by a zinc-bound hydroxyl, obtaining the key intermediate zinc-bound bicarbonate, subsequently replaced by a water molecule
the regeneration of the zinc-bound hydroxyl, through proton transfer (EquationEquation (1)
(1)
This equilibrium contributes to a range of physiological functions that involve the production, transport or consumption of CO2, H+ and HCO3−. In fact, CA role is significative in controlling critical physiological processes such as respiration and acid–base regulation, electrolyte secretion, bone resorption, calcification and biosynthetic reactions, which require bicarbonate as a substrate (lipogenesis, gluconeogenesis, and ureagenesis).
In the last years, targeting tumour-associated carbonic anhydrases has been considered an innovative approach to the development of alternative cancer therapies. A lot of effort has been directed to study the involvement of CAs in tumour progression. Actually, CA IX and XII are the ones predominantly expressed in poorly vascularised hypoxic tumours and have minimal expression in healthy cellsCitation6,Citation7.
Generally, pH in cancer cells is regulated by a highly sophisticated molecular mechanism able to maintain a slightly alkaline intracellular pH and an acidic extracellular pH. This balance is fundamental for cell proliferation in hypoxia conditions and for developing metastasisCitation8.
The elevate neoplastic cell metabolism causes an increased extracellular acid production compared to healthy cells. A fundamental CA role is to catalyse the hydration of tumour generated CO2, producing HCO3− and H+. The effective consequence is an improvement of [H+] in the extracellular space, lowering extracellular pH and promoting tumour growth and invasion in hypoxic conditions.
The importance of modulating the hypoxic tumour microenvironment has prompted researchers to investigate CA IX and XII as target enzymes for therapeutic intervention in cancer.
2. CA inhibitors: general features
So far, the inhibition of CA activity using small molecules has been deeply studied. The design and synthesis of carbonic anhydrase inhibitors (CAIs) led to developing different classes of compounds. Nowadays, the most active classes of CAIs identified are sulfonamides and their bioisosters, sulfamides and sulfamatesCitation9. These compounds usually present a strong activity for CAs but not a proper selectivity for a specific disease-associated enzyme isoform. In fact, the recurring challenge tackled in the field of CA inhibition is the development of isoform-specific inhibitors. Analysing the crystal structure of all 16 α-CA isoforms is possible to observe a significant structural conservation in the catalytic binding site, which makes very difficult to design selective-isoform inhibitors. Nevertheless, an α-CA selective pocket has been identified through comparative studies at the edge of the binding site close to the zinc ionCitation10.
Three crucial structural elements are necessary to design an effective CAI: a zinc-binding group (ZBG), a hydrophobic moiety and a “tail” (). The ZBG is a group able to interact both with the catalytic zinc ion and with the residues Thr199 and Glu106, conserved in the active site of all α-CAs. The hydrophobic moiety is an organic scaffold such as an aromatic or heteroaromatic ring, presenting the tail directly attached. Both polar and lipophilic groups are acceptable motives as tail units, which can interact with hydrophobic or hydrophilic halves in the binding site.
Figure 1. Schematic representation of zinc-binding CAI general structure in the CA binding site.
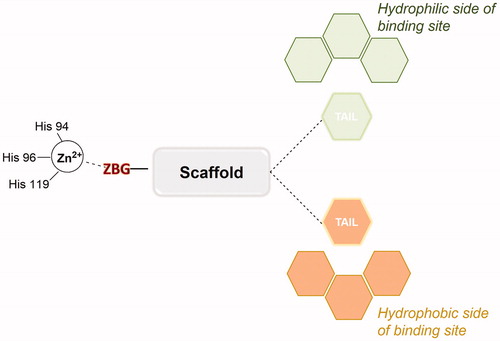
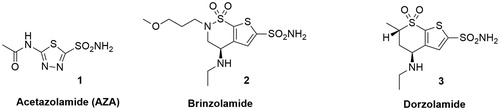
As regards the three elements of the pharmacophore, two common but also complementary approaches have been applied to CAI design in the last decade: the “ring” approach and the “tail” approachCitation11. The ring approach consists of investigating several ring systems containing a sulfonamide ZBG or other chelating groups. This method was used for the successful discovery of the antiglaucoma drugs acetazolamide (AZA) 1, brinzolamide 2 and dorzolamide 3 (). This strategy allowed to identify inhibitors with promising interactions with the CA binding site but with low water solubility. 5-Acetamido-1,3,4-thiadiazole-2-sulfonamide or Acetazolamide (AZA, ) is considered the reference CA inhibitor and it is now approved for the treatment of a range of conditions including glaucoma, epilepsy and altitude sicknessCitation12.
Figure 2. Chemical structures of Acetazolamide (AZA) 1, Brinzolamide 2 and Dorzolamide 3.
Therefore, the introduction of the “tail approach” was proposed. In this method, the inhibitors presented a general scaffold containing a ZBG, commonly an aromatic or heteroaromatic sulfonamide, with different tails appended. The tails originally were water solubilising groups, which could be functionalised, such as amino, imino or hydroxyl groups. Later, the structure of the tails was modified, incorporating diversified moieties (metal-coordinating groups, positively charged functions etc) able to bind a region in the edge of the active site cavity. This area presents the highest variability among the different isoforms, allowing the compounds to discriminate the various CAs as isoenzyme selective CAIsCitation13. Furthermore, modifications of the tail can modulate the physicochemical properties such as solubility and enzyme-binding ability. Additionally, the ring and tail approaches have been combined to develop selective isoform inhibitors. This technique proposed a combinatorial approach presenting a ring system that strongly binds the active site while the tail region of the compound interacts with some isoform-specific residuesCitation11. Furthermore, recently even three tails have been introduced in the molecules of CAIs with the aim to improve the fitting of the ligand with the subpockets which differentiate the various CA isoformsCitation14.
In this context, carbohydrates, having a wide range of chemotypes, constitute a successful example of “hydrophilic tails”. In fact, glycosylated moieties were incorporated into different CAI scaffolds as the hydrophilic functions, leading to highly successful CAIs able to target the tumour associated CA IX/XIICitation15. The insertion of a carbohydrate residue in a CAI scaffold was called “the sugar approach” and allowed the development of numerous potent inhibitors of potential clinical use.
3. CAIs with carbohydrate scaffold: the sugar approach
In the next paragraphs the sugar approach, applied to the identification of CAIs in the last 15 years, will be discussed. In general, the insertion of a carbohydrate moiety has been developed to have a limited membrane permeability. In fact, these compounds were designed to selectively target cancer relevant CA isoenzymes which present an extracellular active site, like CA IX and CA XII, over other intracellular isoenzymes.
We classified all the inhibitors in two major groups, glycosides and glycoconjugates, on the basis of the linkage between the sugar and the ZBG. Glycosidic inhibitors are molecules presenting the ZBG as substituent in anomeric or C-6 position of the sugar while glycoconjugated inhibitors present a linker between the sugar and the aromatic scaffold containing the ZBG.
3.1. Glycosidic CA inhibitors
3.1.1. Topiramate analogues
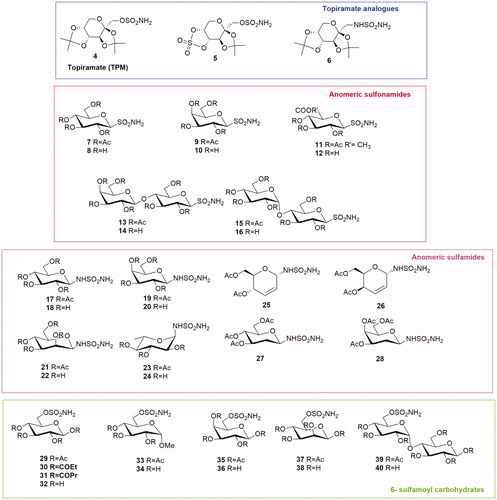
Topiramate (TPM) 4 () is a well-known broad-spectrum drug used in epilepsy as monotherapy or adjunctive therapyCitation16. Its structure, designated chemically as 2,3:4,5-di-O-isopropylidene-β-d-fructopyranose sulfamate, resulted particularly interesting. In fact, the monosaccaridic portion bearing the sulfamate moiety is responsible for anticonvulsant properties. This molecule, presenting the fundamental components of a CAI structure, was tested as CA inhibitor but data appeared rather controversial. The first results were reported by Maryannoff et al.Citation17 and TPM presented a micromolar activity against CA II. Curiously, in the following papers by the same group the inhibition constant decreased up to submicromolar rangeCitation18–19.
Figure 3. General structures of Topiramate analogues and anomeric sulfonamides, anomeric sulfamides and 6-sulfamoyl carbohydrates.
Afterwards, Supuran et al.Citation20 reported TPM as a low nanomolar inhibitor of several CAs using a different assay, the most reliable carbon dioxide hydration technique via stopped-flow apparatus. These data were also confirmed by crystallographic studiesCitation21.
Starting from TPM structure two analogues were reported (5 and 6, ) and the good interaction of TPM structure with the active site was confirmed also by X-ray crystallographic analysis. In fact, the deprotonated sulfamate moiety chelated with a tetrahedral geometry the catalytic zinc ion.
3.1.2. Anomeric sulfonamides
Anomeric sulfonamides are a class of glycosides presenting a primary sulfonamide group in the anomeric position of a carbohydrate. Poulsen’s group proposed a series of anomeric sulfonamides as CAIs inhibitors (7–16, )Citation22. The tricky synthesisCitation23 of S-glycosyl sulfonamides is based on the oxidation step of 2,4-dimethoxybenzyl protected sulfenamides. In these sulfonamides the aromatic moiety of the classical zinc binding sulfonamide is absent, replaced by a hydrophilic mono- or disaccharide. The saccharide portion is directly attached to the sulfonamide group affording a series of S-glycosyl primary sulfonamides (7–16). The anomeric sulfonamide CAIs were tested by the CO2 hydration assay on human CA I, II, IX and XII showing a Ki in the micromolar range and no isoenzyme selectivity. The X-ray crystallography study of this series of compounds demonstrated that the glycosyl moiety weakly interacts with the enzyme active site. Moreover, the carbohydrate was not involved in a significant configuration change of active site residues.
The same group studied also the physicochemical profile of these molecules, analysing the topological polar surface. All compounds fell within the range indicative of molecules with poor membrane permeability. Therefore, even if the reported compounds showed no selectivity for specific CAs, their physicochemical properties could reveal a preferential inhibition of transmembrane CAs.
3.1.3. Anomeric sulfamides
Anomeric sulfamides are glycosylamine carrying a sulfonyl group in anomeric position. Colinas et al.Citation24 reported the synthesis and biological evaluation of a series of N-glycosyl sulfamides presenting a sulfamide moiety directly attached to monosaccharides. The per-o-acetylated glycoconjugates derived from d-glucose, d-galactose, d-mannose and d-rhamnose provided a new class of β-sulfamide glycosides (17–24, ). Enzyme inhibition data revealed that the protected derivatives were micromolar inhibitor of human CA I, II and nanomolar inhibitors of CA IX with a good selectivity over CA I and CA II. On the other hand, deacetylated sulfamide glycosides were weak inhibitors of all tested CAs. The physicochemical profile (topological polar surface area and lipophilicity) of derivatives 17–24 showed a poor level of membrane permeability as expected.
The same group reported also a series of N-glycosyl sulfamides based on O-glycal structure (25–28, )Citation25. The stereoselectivity, obtained using different catalysts, afforded α-derivatives 25 and 26, and β-N-2-deoxy-glycosyl sulfamides 27 and 28. These compounds were potent inhibitors of human CA II, IX and XII (Ki in nanomolar range) selective over CA I (micromolar range). Particularly interesting is the different activity of epimers: the erythro derivative 25 showed a nanomolar inhibition for CA IX (9 nM) and a six-fold selectivity over CA II probably due to a clash interaction of the 4-acetyl moiety with CA II aminoacid residues. On the other hand, its threo analogue 26 did not show any selectivity.
3.1.4. 6-Sulfamoyl carbohydrates
In 2011 Poulsen’s group developed a new series of carbohydrate CAIs constituted by mono-and disaccharides presenting a sulfamoyl function on the C-6 hydroxyl group (29–40, )Citation26. The sulfamates 29–40 were constituted by the monosaccharides d-glucose, d-galactose, d-mannose, methyl-α-glucopyranoside or the disaccharide maltose. The enzyme inhibition data, obtained by the CA catalysed hydration of CO2, revealed that the new compounds were weak inhibitors of the off-target CA I (micromolar range). Moreover, the sulfamate series behaved as effective CA II inhibitors in the low nanomolar range, especially the acetylated mannose derivative 37 (Ki=11.3 nM). The best results were obtained testing the new molecules on CA IX with a Ki from 7.8 nM to 86 nM. The glucose analogues 29–32 were also selective for CA IX in the range of 8 to 14 fold compared to CA II.
The authors designed the acylated derivatives as orally available drugs and supposed that they could be potential prodrugs. In a following paper the same research group demonstrated that the previously described acylated carbohydrate-based sulfamates 29, 30, 33 and 35 were simultaneously inhibitors and substrates of human CA II, as confirmed by X-ray crystallographic studiesCitation27. As expected, the X-ray structures displayed the canonical CA protein-sulfamate interactions. Surprisingly, hydrolysis of the acylated protecting group in anomeric position was observed for all the compounds analysed. C-1 and C-2 are the first positions to lose the protecting groups and only for d-galactose derivative 35 also the hydrolysis of C-3 occurred. Furthermore, the acylated hydrolysis of anomeric position included an inversion of configuration from the equatorial configuration of compounds 29, 30, 33 and 35 to the observed axial configuration.
3.2. Glycoconjugated CA inhibitors
3.2.1. Topiramate derivatives
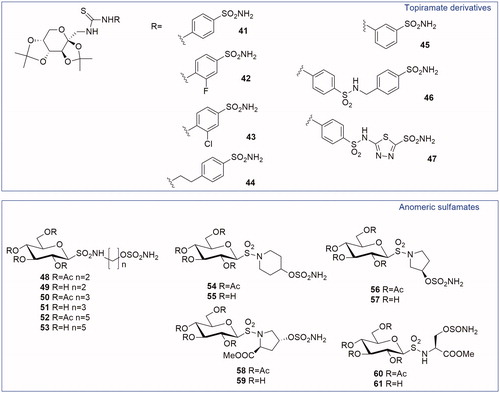
On the basis of the promising results previously described for Topiramate analogues (3.1.1 paragraph), in 2007 a series of CAIs incorporating the protected fructopyranose scaffold present in TPM was reportedCitation28. In this case, the fructopyranose was inserted as a tail on aromatic/heteroaromatic sulfonamide by means of a thioureido functionality (41–47, ). These new compounds were active against CA II and VII (Ki ranging from 6 to 750 nM). Compounds 42 and 46 were tested by MES test in mice, through intraperitoneal injection of 50 mg/kg, showing a better anticonvulsant activity than TPM. Moreover, all the TPM derivatives were efficient inhibitors of CA IX (Ki = 10–4500 nM), with compounds 44 (Ki = 10 nM) and 46 (Ki = 11 nM) showing the best inhibitory activity.
Figure 4. General structures of Topiramate thioureido-derivatives and anomeric sulfamates.
3.2.2. Anomeric sulfamates
Anomeric sulfamates are a class of compounds reported by Moeker et al.Citation29 in 2014. They have a glucose moiety substituted in anomeric position, via a sulfonamide functionality, with a variable linker region presenting a primary sulfamate as ZBG (48–61, ). The linker is constituted by linear alkyl chains or by cyclic systems (piperidine or pyrrolidine) to prove that a different binding to CA active site was possible by changing the linker length, steric bulk and stereochemistry close to the ZBGCitation30.
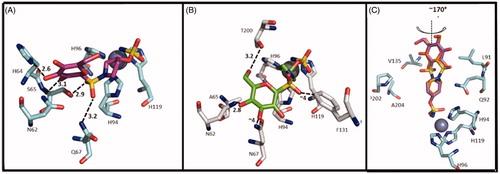
The purpose of the study was to move away from the classical CAI structure towards novel chemical entities with alternative pharmacophores. All compounds were tested on human CA I, II, IX and XII showing a weak inhibition of CA I and a good activity on CA IX and CA XII up to subnanomolar range. Most of the sulfamates showed an excellent CA IX inhibition with Ki values of 1.9–2.4 nM including the acetylated derivatives 52, 54, 56 and 58 and the deacetylated 51, 53, 55 and 57. In particular, the SAR analysis for CA IX isoform revealed that the steric bulk of the ring system, able to separate the saccharide moiety from the ZBG, was preferred over a short linear alkyl chain. The X-ray crystallographic analysis reported two similar binding modes for compound 57 with CA II and CA IX (). This compound preferably interacts with the hydrophilic pocket of CA binding site. In CA II and CA IX the hydrophilic pocket is generally conserved, explaining the nanomolar binding affinities of 57 for both isoforms. In contrast, compound 55 presented a selectivity for CA IX over CA II. In fact, the piperidine derivative 55 interacts with the hydrophobic pocket of CA IX, which has been identified as one of the selective pockets in CA IX active site ().
Figure 5. Interactions at the binding site in the structure of CA IX/57 complex (A), CA II/57 complex (B) and CA IX/55 complex (C) Citation29. A) CA IX mimic (cyan) and 57 (magenta) (PDB ID: 4R5A). B) CA II (grey) and 57 (green) (PDB ID: 4R59). C) Overlay of the two conformations of 55 (purple and orange) with CA IX mimic. (PDB ID: 4R5B).
3.2.3. S-glycosyl sulfonamides and sulfenamides
In 2010 Lopez et al.Citation31 reported the synthesis and biological evaluation of a class S-glycosyl sulfenamides and S-glycosyl sulfonamides (62–91, ). The synthesis of these compounds proposed the direct connection between the anomeric position of a carbohydrate and the SO2NH2 group. The anomeric S-glycosyl sulfenamides have been obtained by reaction of sugar thioacetates with either primary or secondary amines. The following oxidation process of the sulfenamides gave the desired sulfonamides. Per-O-acetylated sugar precursors derived from monosaccharides d-glucose, d-galactose, d-glucuronic acid methyl ester, and disaccharides lactose and maltose. The enzymatic inhibitory activity of the glycoconjugates was determined by the CA catalysed hydration of CO2. The majority of compounds reported a micromolar activity for CA I. On the contrary, all derivatives were good CA IX inhibitors (Ki = 79–106 nM) and potent CA XII inhibitors (Ki sulfonamides 10 nM; Ki sulfenamides 8.5–16.7 nM). Fundamental is the role of the carbohydrate in determining the selectivity for the extracellular active site of CA IX and CA XII. In fact, the lipophilic properties of cell membranes behave as a physical barrier minimising the passive membrane permeability of polar inhibitors. This behaviour promoted the preferential inhibition of extracellular CAs.
Figure 6. General structures of S-glycosyl sulfonamides and sulfenamides and thioureido-glycoconjugates.
3.2.4. Thioureido glycoconjugates
A library of 32 novel glycoconjugated thiourea-bridged benzene sulfonamides has been reported by Moeker et al. in 2012Citation32. The thiourea linker has been obtained by reaction of glycosyl isothiocyanates with a panel of simple benzene sulfonamides comprising either a free amine or a hydrazide (compounds 92–123, ). The thioureido compounds were screened for their enzymatic activity on five human CA isozymes: CA I, II and membrane-associated isozymes IX, XII and XIV. A number of inhibitors were identified as potent inhibitors of CAs (with Ki in the nanomolar range) targeting transmembrane anchored CAs with very good selectivity over off-target cytosolic isozymes I and II. In particular, four potent CA IX selective inhibitors were identified (Ki < 10 nM): the acetylated compound 96 and the deacetylated compounds 101, 105 and 113. The most impressive was the maltose-based compound 113 with a Ki of 2.1 nM against CA IX (with a 2029-fold selectivity over CA I and 129-fold selectivity over CA II). Moreover, the majority of the S-glycosyl compounds were efficient CA XIV inhibitors, Ki of <10 nM, with the only outliers 94 and 102, with a Ki of 126 and 807 nM. The best CA XIV inhibitors identified were compounds 96, 101, 105, 113, 117 and 119. Interestingly, analysing the results for CA XIV, the galactose-based compounds 94, 95, 102, 103 and 111 presented a better inhibition profile with respect to the other CAs. This may indicate that this isozyme is more responsive to subtle variations in stereochemistry compared to other CAs.
The molecules in this study also provided candidates with the possibility of dual-action for inhibition of extracellular CAs (IX, XII and XIV)—either directly as free sugars (membrane impermeable) or indirectly as acetylated sugar prodrugs, becoming free sugars upon non-specific esterase hydrolysis. An additional physicochemical feature of the free sugar thioureido-glycoconjugates 92–123 was a high water solubility (>20 mg/mL), calculated in silico as cLogP values.
3.2.5. Amido phenyl sulfonamides
In 2020 a new series of carbohydrate-based sulfonamides was reported (compounds 124–131, )Citation33. The conjugation was achieved with a classical amide bond between the proper aminosugar and the p-sulfamoyl benzoic acid. The compounds were tested in vitro against a panel of CAs showing a good activity (Kis nM-µM range) for the target antiglaucoma CA II. Moreover all these compounds showed great water solubility, reporting a pH of their solution in water of 7. This property is relevant for eye application of drugs in the treatment of glaucoma, compared to the slightly acid solution of the reference compound Dorzolamide (pH = 5.5).
Figure 7. General structures of amido phenyl sulfonamide glycoconjugates.
Compounds 126, 127, 129 had nanomolar activity for CA II and, formulated as 1% water solution and administrated topically to hypertensive rabbits, they were able to lower intraocular pressure in glaucomatous animals better than dorzolamide.
3.2.6. Glycosyl triazole CA inhibitors: the “click tailing” strategy
Click chemistry has been successfully applied to generate libraries of glycosyl triazoles as CA inhibitors using the so-called “click tailing” strategy. The 1,3-dipolar cycloaddition reaction (1,3-DCR) of an organic azide with a terminal alkyne, the Huisgen’s reaction, affords a mixture of 1,4- and 1,5-disubstituted-1,2,3-triazoles. The regioselectivity can be achieved by using catalysts as copper or rutheniumCitation34.
The Cu(I) catalysis allows to obtain a regiospecifically oriented 1,4-disubstituted-1,2,3-triazole. Sharpless discovered a dramatic rate enhancement (107 fold) using the Cu catalysis, with an easy purification step. In fact, this reaction has rapidly become the premier click chemistry reaction (CuAAC)Citation35. On the contrary, the regioselective synthesis of the other possible products of 1,3-DCR, the 1,5-disubstituted-1,2,3-triazoles, could be achieved by ruthenium catalysis (RuAAC)Citation36.
In the last decade, the click reaction was widely reported as medicinal chemistry synthetic strategyCitation37, due to its high yields and easy purification step. Moreover, a lot of literature reported the click chemistry reaction as a powerful tool to synthesise glycoconjugated hit compoundsCitation38–40. In fact, the 1,2,3-triazole is a non-classical peptide bond bioisostere and may interact with the biological targetsCitation41.
In the next paragraphs, the application of the “click tailing” approach in order to obtain potent and selective CA inhibitors will be analysed and detailed. In the last decade, more than 100 different compounds have been synthesised using this synthetic approach and screened on CAs.
3.2.6.1. 1,4-disubstituted triazole glycoconjugates
3.2.6.1.1. Anomeric 1,4-disubstituted triazole sulfonamide glycoconjugates
Wilkinson et al.Citation42–44 reported the first glycoconjugated libraries of CA inhibitors (132–155 Series 1 and 156–171 Series 2, ) using the copper-catalysed click chemistry reaction. In this first series of compounds the carbohydrate was connected on its anomeric position, starting from the corresponding azide precursors (mono- and di-saccharides). The 1,2,3-triazole linked the anomeric azides with either an ester or amide functionality, or directly with the aromatic sulfonamide fragment. Two libraries of glycosyl triazoles were screened using the CO2 hydration assay for CA I, CA II and transmembrane CA IX, CA XII and CA XIV. The consistent potency and selectivity of the majority of these compounds confirmed the effectiveness of the carbohydrate tails. In fact, click tailed glycoconjugates were able to impart selectivity for the isozymes differently from the non-selective parent benzenesulfonamide scaffolds. There are numerous glycosyl triazoles identified in these libraries presenting a low Ki. In particular, the methyl-β-D-Glucoronate triazole 146 of Series 1 was found a nanomolar inhibitor of tumour-associated CA IX (Ki = 23 nM), showing a strong selectivity over CA II. Moreover, the methyl-β-D-Glucoronate triazoles 162 and 163 of Series 2 were proven to induce potent inhibition of CA IX (with a Ki 9.9 and 8.4 nM respectively). The best results in CA inhibition were achieved by the mannose derivatives 168 and 169 presenting a Ki around 5 nM. It is possible to affirm that the glucoronate and mannose tail fragments are important candidates for SAR investigation targeting CA IX isoenzyme.
Figure 8. General structures of anomeric 1,4-disubstituted triazole sulfonamide glycoconjugates.
In 2013 Poulsen’s groupCitation45 described new glycoconjugated CA inhibitors (172–195, ), exploiting the anomeric position of differently acylated glucose and galactose scaffolds conjugated with 3- or 4-ethynyl benzenesulfonamide through CuAAC. Both the glucose (171–183) and the galactose (184–195) series were synthesised with variable acyl groups on the hydroxyl function: acetyl, propionyl, butanoyl, pentanoyl and 3-methyl pentanoyl groups. The CA enzymes inhibition profile for all compounds was determined against CA I, CA II, CA IX, CA XII and CA XIV using a stopped-flow instrument. The SAR analysis between the glucose and galactose series revealed that the structure of the carbohydrate moiety could affect CA inhibition profile. The stereochemical difference between these epimers must lead to a different interaction in the binding site, confirmed by the fact that the glucose series presented a better inhibition profile for the target CA IX. There was no consistent SAR trend for the different acyl groups, meaning that each glycoconjugate could act as a prodrug for the corresponding deprotected analogue.
In fact, the fully deprotected compounds 177 and 183 were the most active on the target CA IX and selective over the off-target CA II compared to the acylated analogues. The unmasked glycoconjugates were selective towards overexpressed CA IX and CA XII thanks to a combination of limited membrane permeability and improved Ki. Therefore, the acylated derivative prodrugs should provide good oral absorption and low CA inhibition. ADME analysis confirmed the degradation of the acyl group of compounds 172–174 in human plasma and human liver microsomes (HLMs), as a result of compounds susceptibility to hydrolytic enzymes such as microsomal esterases. This process produced the fully deacylated compound 177. Moreover, the glucose-derivative 177 presented a very good value of cLogP and a negligible membrane permeability confirmed by Caco-2 cell model.
3.2.6.1.2. The O-glycosyl and the S-glycosyl 1,4-disubstituted triazole sulfonamides
As mentioned before the glycoconjugated CA inhibitors were readily prepared in a parallel fashion using click tailing strategy. Poulsen’s group reported a series of 1,2,3-triazole-tethered glycoconjugates possessing an O-glycoside functionality or S-glycoside functionality (thio, sulfinyl and sulfonyl) connected with benzenesulfonamide fragments. The first to be described was the O-glycoside seriesCitation46 (196–205, ), where the para-benzenesulfonamide was connected to 1,2,3-triazole-tethered O-glycoside tails by CuAAC reaction. The sugar panel encompassed monosaccharide derivatives such as pyranose glucose (196–197), galactose (198–199), arabinose (200–201), a furanose ribose (202–203) as well as a disaccharide derivative, maltose (204–205). All these compounds were either micromolar or low-nanomolar inhibitors of CA I, while against CA II and IX their inhibition constants were in the range of 6.8–53 and 9.7–107 nM respectively. Regarding the tumour associated CA IX, the most potent inhibitor of this series of glycoconjugates was the galactose derivative 198 with a Ki of 9.7 nM.
Figure 9. General structures of O-Glycosyl and the S-glycosyl 1,4-disubstituted triazoles, 1,4-disubstituted triazole saccharin-glycoconjugates and 1,5-disubstituted glycosyl triazoles.
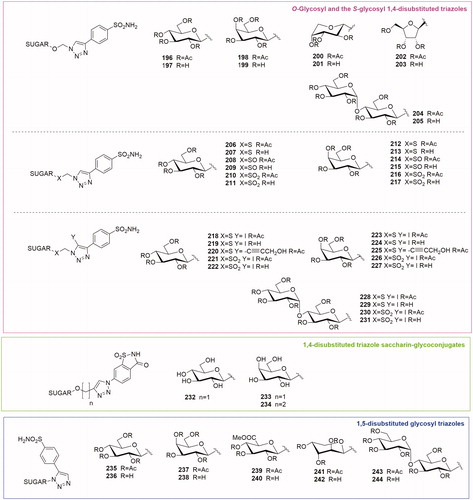
Subsequently, the same research group published the S-glycoside analogues (206–217, )Citation47. The replacement of the natural occurring O-glycosidic linkage by S-glycosides is an approach to enhance the stability of the glycosidic linkage towards enzymatic hydrolysis whilst retaining vital molecular recognition interactions with the biological target. A panel of S-, sulfinyl and sulfonyl propynyl glycosides (d-glucose, d-galactose) were conjugated to a para azido benzene sulfonamide by click chemistry reaction. The S-propynyl carbohydrates afforded all the thio-derivatives after conjugation (206, 207, 212 and 213). A partial oxidation of the S-propynyl sugars gave the SO-sulfinyl precursors and, after coupling reaction, their corresponding sulfinyl glycoconjugates (208, 209, 214 and 215). Otherwise, a total oxidation of S-propynyl sugar generated the SO2-sulfonyl sugar precursors and their glycoconjugated derivatives (210, 211, 216 and 217).
The second series of compounds was investigated by enzymatic assays both in the per-O-acetylated form and in the free hydroxyl form. Glycosides were potent inhibitors of CA II (Ki 2.9–9.9 nM), IX (Ki 6.1–9.9 nM) and XII (Ki 8.4–11.9 nM) with Ki values narrowly distributed—and just one outlier to this trend—compound 207 with a Ki of 257 nM against CA IX. The oxidation state of the sulfur had no impact on the CA inhibition constants. CA I inhibition by these S-glycoside inhibitors was typically weaker (10–20-fold) than for all other isozymes with Kis in the mid-high nM range (60–117 nM). Galactose compounds (212–217, Kis 60–62 nM) were marginally stronger CA I inhibitors than glucose compounds (206–211, Kis 101–114 nM).
Comparison of CA inhibition of the O-glycoside series (196–205) with the thio series (206–217) demonstrated that the replacement of the oxygen atom by a sulfur atom (compounds 206, 207, 212 and 213), SO (compounds 208, 208, 214 and 215) and SO2 (compounds 210, 211, 216 and 217) typically gave weaker inhibitors for CA I, while CA II inhibition was similar, and CA IX inhibition was generally stronger. This trend indicated that the sulfur analogues are able to selectively target tumour associated CA IX with respect to the oxygen analogues.
The third series of compounds, published in 2011 by Morris et al.Citation48, presented an S- and SO2 glycoside linked to an unusual 1,4-disubstituted triazole scaffold. In fact, in this series the H atom at the 5-position of the triazole moiety is replaced by an iodine atom (218–231, ) in order to build a SAR and a structure property relationship (SPRs) and to have a strategic intermediate for further developing the library. The synthesis of this class of compounds was based on a variation of the classical CuAAC reaction using the CuI/NMB catalytic system. This modification allows the in situ generation of I+ and facilitates the formation of the 5-iodo-1,4-disubstituted 1,2,3-triazoles. Moreover, the synthesis of the 1,4,5-trisubstituted triazoles 220 and 225 was achieved by a Sonogashira reaction catalysed by PdCl2(PPh3)2 introducing a hydroxyl alkyne into the 5-iodo-1,4-disubstituted triazole scaffold. Similarly to acetazolamide (AZA), used as reference compound, the S-glycosides 210–223 were potent inhibitors of CA II, IX and XII, most of them within a nanomolar range (5.3–112 nM), with only few exceptions. The two alkynyl derivatives 218 and 231 demonstrated a good isoenzyme selectivity for the cancer associated CAs. In fact, compound 225 presented a 24-fold selectivity for CA XII (Ki = 9.0 nM) over CA II. The most active compounds were selected for further cell-based studies on two fibroblast cell lines (one lacking endogenous expression of CA IX and one overexpressing CA IX). This assay allowed to discriminate between the protective role of CA IX in an acidic tumour microenvironment and a general cytotoxicity. Compounds 218 and 219 were able to selectively block CA IX-induced survival. Moreover, extended studies with the per-acetylated analogue 218 revealed its ability to decrease membrane-associated CA activity to a similar level as that obtained in cells lacking CA IX. The acetylated glucose inhibitor 218 tested on human colon carcinoma cell model LS174Tr cells gave better results than the reference compound AZA. In fact, AZA was not as effective as compound 218 in reducing CA IX activity or reducing the proliferation of colon carcinoma cells.
3.2.6.1.3. The 1,4-disubstituted triazole saccharin glycoconjugates
Artificial sweeteners were recently reported to bind selectively CA IX with nanomolar activityCitation49,Citation50. Among them, saccharin has been used as a ZBG in drug development using the tail approachCitation51. The saccharin-based compounds displayed over 1000-fold selectivity for CA IX over off-target isoforms CA I and CA II. The affinity and selectivity of saccharin as CA IX inhibitor were modulated by linking it to a hydrophobic and hydrophilic portion by Moeker et al.Citation52 Among these derivatives one hydrophilic glycoconjugate 232 (), endowed with a glucose moiety linked to saccharin by a 1,4-disubstituted triazole, showed a potent activity for CA IX (Ki 49.5 nM) and an extremely poor inhibition of the other CAs (Ki> 50000 nM) compared to non-glycoconjugated derivatives and saccharin itself. On the basis of compound 232 structure, Murray et al.Citation53 reported the design, synthesis and biological evaluation of two new glycoconjugated saccharin derivatives (233–234, ) replacing the β-glucose with a β-galactose (233) and increasing the length of the triazole spacer by introducing an ethyl linker (234). Enzymatic inhibition assays revealed that the galactose derivative 233 had a nanomolar activity on CA IX (IC50 = 206 nM) comparable with that of the glucose derivative 232 (IC50 = 197 nM). The linker elongation in compound 234 enhanced the inhibitory activity (IC50 = 25 nM) and doubled the selectivity ratio between CA IX and CA II.
3.2.6.2. 1,5-disubstituted triazole sulfonamide glycoconjugates
Salmon et al. in 2011Citation54 reported the only carbohydrate-based triazole series obtained by applying the reverse regioselectivity ruthenium-catalysed azide-alkyne cycloaddition (RuAAC). This reaction allowed to generate a library of 1-(β-D-glycosyl)-5-benzenesulfonamide-1,2,3-triazole compounds 235–244, . The glycosyl azide included derivatives of d-glucose, d-galactose, d-glucuronic acid methyl ester, d-arabinose and d-maltose. The regioselective synthesis of 1,5-disubstituted-1,2,3-triazoles was achieved by RuAAC using [Cp/RuCl(cod)] as catalyst. All the compounds were tested by a stopped flow enzymatic assay and a number of 1,5-disubstituted triazole inhibitors (compounds 236–239, 243 and 244) were able to block CA IX with inhibition constants lower than 10 nM. The per-acetylated glucose derivative 235 (CA IX Ki = 32.6 nM) possessed a very good selectivity for CA IX over off-target CA I (42-fold selectivity) and CA II (5-fold selectivity).
A comparison between the CA IX inhibition by the new 1,5-disubstituted triazoles (235–244 Ki 7.0–68.3 nM) and by their 1,4-disubstituted counterparts (156–171, ), showed that the first ones were better CA IX inhibitors.
3.2.7. Glycosyl conjugates of coumarins
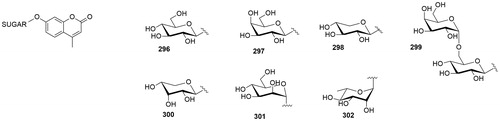
A series of 7-glycosylated 4-methyl coumarins (296–302, ) incorporating various glycosyl moieties was reported by Touisni et al.Citation55 This new series of compounds was inspired by the 7-hydroxy coumarin (umbelliferone) structure, which was reported to be a selective, though not very potent (Ki in the submicromolar range) inhibitor of tumour associated CA IX and XIICitation56. The sugar approach was applied to the umbelliferone structure to obtain glycosyl conjugates of coumarins through glycosylation reaction using mono- and disaccharides functionalised as trichloroacetimidates. These compounds were poor inhibitors of off-target CA I and CA II (Ki in the micro-millimolar range) but the majority of them were nanomolar inhibitor of CA IX and CA XII. Moreover, the 7-mannosyl-4-methylumbelliferone 301 was tested on 4T1 syngeneic mouse mammary tumour cells and it significantly inhibited the growth of primary tumours at a 30 mg/kg dose. Compound 301 could be an interesting candidate to develop conceptually novel anticancer drugs.
Figure 10. General structure of coumarin glycosylated CA inhibitors.
3.2.8. C-glycosyl CA inhibitors
The replacement of the naturally occurring O-glycoside by a C-glycoside bond was often reported in synthesis of glycosyl compounds in order to investigate different biological applications. In fact, this isosteric substitution is able to improve the compound stability towards enzymatic degradation of glycosidic bond, maintaining the molecular recognition for biological targetsCitation57,Citation58,. In the last years different series of C-glycosides were evaluated as CA inhibitors, as reviewed by Martinez et al.Citation59
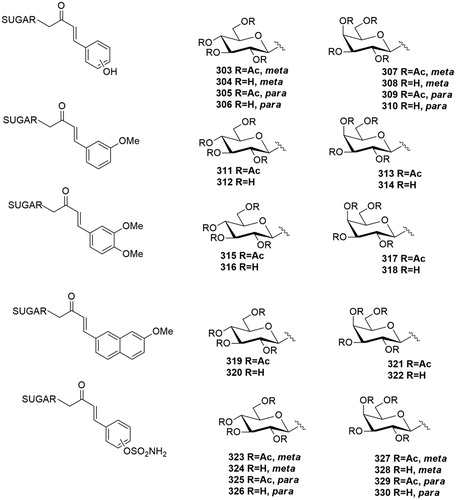
In 2013 Riafrecha al.Citation60 reported the first series of C-cinnamoyl glycosides as CA inhibitors (303–310, ), a small series of phenolic derivatives where the carbohydrate was tethered to a phenol CA pharmacophore through a carbon chain. The synthesis was performed by aldol condensation of β-C-glucosyl and β-C-galactosyl ketones with the proper aldehyde, followed by a basic hydrolysis. This is the first study in which α-CAs have been investigated for their interaction with a C-glycoside. The C-cinnamoyl glycosides were generally effective CA inhibitors, with inhibition constants in the low micromolar range against CA I, II, IV, VA, VB, VI, VII, IX, XII, XIII, XIV and ineffective inhibitors of CA III. Neither the stereochemistry of the sugar or the hydroxyl position in the aromatic ring affected the inhibition profile.
Figure 11. General structure of C-glycosyl CA inhibitors.
Later onCitation61, this C-cinnamoyl glycoside series has been tested as Mycobacterium tuberculosis β-CA inhibitors. The inhibitory activity of C-glycosides 303–310 against human CA II and the purified M. tubercolosis β-CA revealed that all compounds were able to inhibit both of them with Kis in the µM range. The anti-tubercolar activity of these compounds was evaluated by using fresh cultures of M. turbercolosis H37Rv and compound 303 was identified as the first mtCA inhibitor with antimycobacterial activity.
Moreover, this series of compounds was tested for the inhibition of β-CA from Brucella suis, a Gram negative pathogen responsible for the most widespread bacterial zoonosis, known as brucellosisCitation62. The majority of these compounds showed micromolar or submicromolar activity and an excellent selectivity for pathogen over human enzymes. The better inhibitory profile against β-CAs over α-CAs was reported by protected 3-hydroxyphenyl derivatives 303, 304, 307 and 308.
In 2014 the same group reportedCitation63 a series of C-glycosides (311–322, ) containing the methoxyaryl scaffold linked to a C-glucosyl and C-galactosyl portion as human CAI, II, IX and XII inhibitors. This study identified as highly selective hCA IX and hCA XII inhibitors the protected derivatives 313 and 321, and the deprotected 312, 320, 314 and 322. Comparing the methoxyaryl series (311–322) with the phenol one (303–310), a significant improvement of the selectivity profile for the tumour associated hCA (IX and XII) was reported. In fact, in the first series the inhibition profile was flat and there were only small variations in the Ki values across all the hCAs studied.
The methoxyphenyl series was tested against a panel of CAs encompassing the human α-CAs I and II, the fungal C. neoformans β-CAS and the pathogenic Brucella suis enzymesCitation64. The deprotected glycosides incorporating the 6-methoxy-2-naphthyl moiety (320 and 322) were able to preferentially inhibit pathogens CAs over human CAs, showing the best activity and selectivity profile.
In addition, the antioxidant activity of both these series was evaluated through DPPH and ABTS radical scavenging assaysCitation65. All the deprotected derivatives exhibited better activity than the per-O-acetylated analogues and the phenolic series reported better antioxidant activity compared to the methoxyaryl one. The 3-hydroxyphenyl glycoside 303, exhibited the best profile with both the antioxidant assays.
Recently, the same research group reportedCitation66 another C-glycosyl series bearing a sulfamoyl group on the aromatic scaffold (323–330, ). This small series was tested on human CA I, II, IV and IX. Peracetylated derivatives (323, 325, 327 and 329) were nanomolar inhibitor of CA II isoform. The deprotected analogues (324, 328 and 330) were micromolar inhibitors of CA II, except C-glucosyl derivative 326, which was nanomolar inhibitor of this isoform. Furthermore, all the peracetylated C-glycosides (323, 325, 327 and 329) revealed a micromolar CA IX activity and a good selectivity profile over to the other CAs. The compound presenting the best selectivity profile for CA IX over CA II was the p-substituted sulfamoyl C-glycoside 326, with a selectivity ratio II/IX of 5.66.
3.2.9. Glycoconjugated CA inhibitors: the “dual-tail” approach
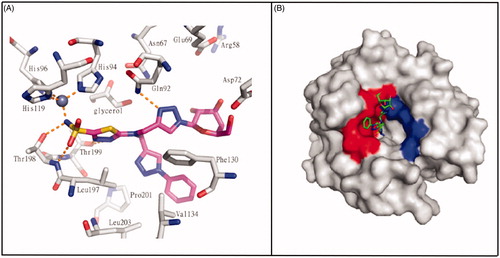
The “dual-tail” approach is a strategy employed to obtain potent CAIs by matching simultaneously the hydrophobic and hydrophilic halves of the CA active site, and maintaining the ZBG. This method was firstly reported by Tanpure et al.Citation67 who proposed a series of compounds presenting a phenyl moiety as hydrophobic tail connected with a glucose fragment as hydrophilic tail. The design of these compounds was based on AZA structure, by adding dual tail groups to AZA primary scaffold to obtain derivatives 331–333 (). The tail combination included two hydrophilic units in compound 331 or one hydrophobic and one hydrophilic moieties in compound 332. In addition, a corresponding single tail analogue, compound 333, was prepared for comparison. These dual-tail derivatives showed a decreased activity with respect to AZA for CA II and CA I, as confirmed by the X-ray crystallographic analysis of compound 332 in complex with CA II. In fact, compound 332 makes multiple interactions with the protein as it extends out from the catalytic pocket (). In the surface representation of CA II and compound 332, the hydrophobic half of CA II (red, ) dominates the binding orientation with compound 332, while the carbohydrate moiety does not interact with the hydrophilic half of the binding site (blue, ).
Figure 12. General structure of dual-tail CA inhibitor glycoconjugates.
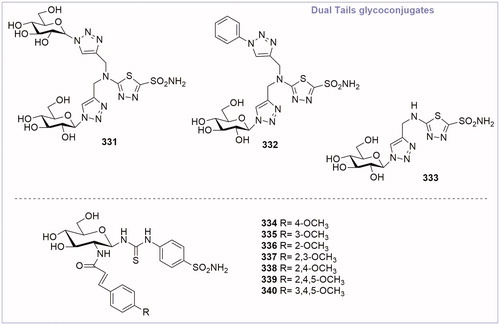
Figure 13. (A) Interactions at the binding site in the structure of CA II/332(magenta) complex (PDB ID: 4CQ0). (B) Surface representation of CA II/332 complex. The hydrophobic half of CA II is red and the hydrophilic half is blue to highlight the different interactionsCitation67.
Afterwards, Hou et al. in 2017Citation68 revisited the dual-tail approach choosing different tails and varying the type of connection. Cinnamic acid was chosen as hydrophobic tail because it was found to be a novel structural motif for CA inhibition by Maresca et al.Citation69 Amino glucosamine moiety was inserted as hydrophilic tail and the connection was achieved by thioureido and amido linkage. The ZBG was represented by a sulfanilamide function. Seven compounds (334–340, ) were synthesised and tested. Biological data revealed that the majority of compounds effectively inhibited CA II and IX in the range of 7–152 nM. Compound 338 (CA IX IC50 = 7 nM) was the best glycoconjugated CA IX inhibitor, revealing better activity than the reference compound (AZA IC50 = 30 nM). Moreover, compound 338 was tested on cancer cell models (MDA-MB-231 and HT-29 cell lines) by in vitro assay, to evaluate its effect on hypoxia-induced extracellular acidosis. The glycoconjugated CA IX inhibitor 338 was able to significantly reduce the extracellular acidification of cancer cells in 0.1 and 1 mM concentrations showing an increased antiproliferative activity with respect to AZA. These results demonstrated the efficacy of the dual approach to simultaneously matching the hydrophobic and hydrophilic halves of the active site by linking hydrophobic and hydrophilic fragments.
4. Summary and outlook
CAs are a family of metalloenzymes able to catalyse the reversible hydration of carbon dioxide to bicarbonate and a proton. Thus, members of this enzyme superfamily (comprising 16 isoforms in humans) are primary regulators of pH within and outside the cell. Differing in catalytic activity and subcellular localisation, CA isoforms represent important biological targets with diverse therapeutic potential in pathologies like cancer, epilepsy and glaucoma.
So far, the inhibition of CA activity using small molecules has been largely investigated. The design and synthesis of carbonic anhydrase inhibitors (CAIs) got much success in developing different chemical classes of compounds. However, the high structural homology among the different CA active sites has made very difficult to obtain isoform-selective inhibitors.
The CAI pharmacophore is constituted by three elements: a zinc-binding group, an aromatic/heterocyclic ring system and a “tail”, important to discriminate among the different isoforms. In fact, two common but also complementary approaches have been applied to CAIs design: the ring approach and the tail approach. In the last 15 years, the insertion of carbohydrates, mono and di-saccharides as a hydrophilic tail in CAI structure has been termed the “sugar approach”. In this review, we described all the carbohydrate-based CA inhibitors present in literature and their biological activity. We classified the compounds according to the kind of linkage between the sugar and the zinc-binding group in: glycosidic and glycoconjugated CAIs. Most of them are potent and selective inhibitors of tumour-associated CA IX and CA XII, presenting Ki values in the nanomolar and sub nanomolar range. In and are summarised the CA IX inhibitory activity (Ki, nM) and selectivity over CA I of selected compounds i.e. the most representative derivative for each class of CA glycosidic and glycoconjugated inhibitors. The presence of an extended hydrophilic moiety limits the activity of these CAIs towards those CAs endowed with an extracellular active site such as the membrane-bound isoforms IX and XII. These enzymes are overexpressed in many hypoxic tumour types but their presence is limited to few normal tissues, thus their selective inhibition would lead to reduced side effects. Some of these tumour-directed CAIs have been tested on cell-based models of tumour proliferation with encouraging results. For this reason, the research needs to progress with further in vitro and in vivo studies on these classes of compounds which represent a potentially effective and safer alternative to current cancer treatments.
Table 1. Chemical structure, CA IX inhibitory activity, CA I/ CA IX selectivity profile of the best compound for each class of CA glycosidic inhibitors
Table 2. Chemical structure, CA IX inhibitory activity, CA I/ CA IX selectivity profile of the best compound for each class of CA glycoconjugated inhibitors
Disclosure statement
The authors declare no conflict of interest.
Additional information
Funding
References
- Jensen EL, Clement R, Kosta A, et al. A new widespread subclass of carbonic anhydrase in marine phytoplankton. Isme J 2019;13:2094–106.
- Del Prete S, Nocentini A, Supuran CT, et al. Bacterial ι-carbonic anhydrase: a new active class of carbonic anhydrase identified in the genome of the Gram-negative bacterium Burkholderia territorii. J Enzyme Inhib Med Chem 2020; 35:1060–8.
- (a) Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81. (b) Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31:345–60. (c) Supuran CT. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin Drug Discov 2017;12:61–88.
- (a) Alterio V, Di Fiore A, D’Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68. (b) Supuran CT. Exploring the multiple binding modes of inhibitors to carbonic anhydrases for novel drug discovery. Expert Opin Drug Discov 2020;15:671–86.
- Nocentini A, Supuran CT. Advances in the structural annotation of human carbonic anhydrases and impact on future drug discovery. Expert Opin Drug Discov 2019;14:1175–97.
- Chiche J, Ilc K, Laferrière J, et al. Hypoxia-inducible carbonic anhydrase IX and XII promote tumor cell growth by counteracting acidosis through the regulation of the intracellular pH. Cancer Res 2009;69:358–68.
- Annan DA, Maishi N, Soga T, et al. Carbonic anhydrase 2 (CAII) supports tumor blood endothelial cell survival under lactic acidosis in the tumor microenvironment. Cell Commun Signal 2019;17:169.
- Monti SM, Supuran CT, De Simone G. Anticancer carbonic anhydrase inhibitors: a patent review (2008–2013). Expert Opin Ther Pat 2013;23:737–49.
- Supuran CT. Carbonic anhydrase inhibitors and their potential in a range of therapeutic areas. Expert Opin Ther Pat 2018;28:709–12.
- Pinard MA, Mahon B, McKenna R. Probing the surface of human carbonic anhydrase for clues towards the design of isoform specific inhibitors. Biomed Res Int 2015;2015:453543.
- Bozdag M, Ferraroni M, Nuti E, et al. Combining the tail and the ring approaches for obtaining potent and isoform-selective carbonic anhydrase inhibitors: solution and X-ray crystallographic studies. Bioorg Med Chem 2014;22:334–40.
- Kayser B, Dumont L, Lysakowski C, et al. Reappraisal of acetazolamide for the prevention of acute mountain sickness: a systematic review and meta-analysis. High Alt Med Biol 2012;13:82–92.
- (a) Supuran CT, Scozzafava A, Briganti F, et al. Carbonic anhydrase inhibitors - part 78(#). Synthesis of water-soluble sulfonamides incorporating beta-alanyl moieties, possessing long lasting-intraocular pressure lowering properties via the topical route. Eur J Med Chem 2000;35:309–21. (b) Wang Z, Qin Y, Wang P, et al. Sulfonamides containing coumarin moieties selectively and potently inhibit carbonic anhydrases II and IX: design, synthesis, inhibitory activity and 3D-QSAR analysis. Eur J Med Chem 2013;66:1–11.
- Bonardi A, Nocentini A, Bua S, et al. Sulfonamide inhibitors of human carbonic anhydrases designed through a three-tails approach: improving ligand/isoform matching and selectivity of action. J Med Chem 2020;63:7422–44.
- (a) Winum JY, Poulsen SA, Supuran CT. Therapeutic applications of glycosidic carbonic anhydrase inhibitors. Med Res Rev 2009;29:419–35. (b) Colinas PA. Novel glycomimetics: anomeric and N-Glycosyl Sulfonamides. Curr Org Chem 2012;16:1670–9. (c) Winum JY, Colinas PA, Supuran CT. Glycosidic carbonic anhydrase IX inhibitors: a sweet approach against cancer. Bioorg Med Chem 2013;21:1419–26.
- Latini G, Verrotti A, Manco R, et al. Topiramate: its pharmacological properties and therapeutic efficacy in epilepsy. Mini Rev Med Chem 2008;8:10–23.
- Maryanoff BE, Nortey SO, Gardocki JF, et al. Anticonvulsant O-alkyl sulfamates. 2,3:4,5-Bis-O-(1-methylethylidene)-beta-D-fructopyranose sulfamate and related compounds. J Med Chem 1987;30:880–7.
- Maryanoff BE, McComsey DF, Costanzo MJ, et al. Comparison of sulfamate and sulfamide groups for the inhibition of carbonic anhydrase-II by using topiramate as a structural platform. J Med Chem 2005;48:1941–7.
- Klinger AL, McComsey DF, Smith-Swintosky V, et al. Inhibition of carbonic anhydrase-II by sulfamate and sulfamide groups: an investigation involving direct thermodynamic binding measurements. J Med Chem 2006;49:3496–500.
- Winum JY, Temperini C, El Cheikh K, et al. Carbonic anhydrase inhibitors: clash with Ala65 as a means for designing inhibitors with low affinity for the ubiquitous isozyme II, exemplified by the crystal structure of the topiramate sulfamide analogue. J Med Chem 2006;49:7024–31.
- Casini A, Antel J, Abbate F, et al. Carbonic anhydrase inhibitors: SAR and X-ray crystallographic study for the interaction of sugar sulfamates/sulfamides with isozymes I, II and IV. Bioorg Med Chem Lett 2003;13:841–5.
- Lopez M, Paul B, Hofmann A, et al. S-glycosyl primary sulfonamides–a new structural class for selective inhibition of cancer-associated carbonic anhydrases. J Med Chem 2009;52:6421–32.
- Lopez M, Drillaud N, Bornaghi F, et al. Synthesis of S-glycosyl primary sulfonamides. J Org Chem 2009;74:2811–6.
- (a) Colinas PA, Tempera CA, Rodriguez OM, et al. Stereoselective synthesis of Novel N-β-glycosyl sulfonamides by sulfonamidoglycosylation of per-O-acetylated Sugars. Synthesis 2009;24:4143–8. (b) Rodriguez OM, Maresca A, Tempera CA, et al. N-β-glycosyl sulfamides are selective inhibitors of the cancer associated carbonic anhydrase isoforms IX and XII. Bioorg Med Chem Lett 2011;21:4447–50.
- Colinas PA, Bravo RD, Vullo D, et al. Carbonic anhydrase inhibitors. Inhibition of cytosolic isoforms I and II, and extracellular isoforms IV, IX, and XII with sulfamides incorporating sugar moieties. Bioorg Med Chem Lett 2007;17:5086–90.
- Lopez M, Trajkovic J, Bornaghi LF, et al. Design, synthesis, and biological evaluation of novel carbohydrate-based sulfamates as carbonic anhydrase inhibitors. J Med Chem 2011;54:1481–9.
- Lopez M, Vu H, Wang CK, et al. Promiscuity of carbonic anhydrase II. Unexpected ester hydrolysis of carbohydrate-based sulfamate inhibitors. J Am Chem Soc 2011;133:18452–62.
- Winum JY, Thiry A, Cheikh KE, et al. Carbonic anhydrase inhibitors. Inhibition of isoforms I, II, IV, VA, VII, IX, and XIV with sulfonamides incorporating fructopyranose-thioureido tails. Bioorg Med Chem Lett 2007;17:2685–91.
- Moeker J, Mahon BP, Bornaghi LF, et al. Structural insights into carbonic anhydrase IX isoform specificity of carbohydrate-based sulfamates. J Med Chem 2014; 57:8635–45.
- Mahon BP, Lomelino CL, Ladwig J, et al. Mapping selective inhibition of the cancer-related carbonic anhydrase IX using structure-activity relationships of glucosyl-based sulfamates. J Med Chem 2015; 58:6630–8.
- Lopez M, Bornaghi LF, Innocenti A, et al. Sulfonamide linked neoglycoconjugates-a new class of inhibitors for cancer-associated carbonic anhydrases. J Med Chem 2010; 53:2913–26.
- Moeker J, Teruya K, Rossit S, et al. Design and synthesis of thiourea compounds that inhibit transmembrane anchored carbonic anhydrases. Bioorg Med Chem 2012;20:2392–404.
- Hou Z, Li C, Liu Y, et al. Design, synthesis and biological evaluation of carbohydrate-based sulphonamide derivatives as topical antiglaucoma agents through selective inhibition of carbonic anhydrase II. J Enzyme Inhib Med Chem 2020;35:383–90.
- Rostovtsev VV, Green LG, Fokin VV, et al. A stepwise huisgen cycloaddition process: copper(I)‐catalyzed regioselective “ligation” of azides and terminal alkynes. Angew Chem 2002;41:2596–9.
- (a) Himo B, Lovell T, Hilgraf R, et al. Copper(I)-catalyzed synthesis of azoles. DFT study predicts unprecedented reactivity and intermediates. J Am Chem Soc 2005;127:210–6. (b) Worrell BT, Malik JA, Fokin VV. Direct evidence of a dinuclear copper intermediate in Cu(I)-catalyzed azide-alkyne cycloadditions. Science 2013;340:457–60.
- Boren BC, Narayan S, Rasmussen LK, et al. Ruthenium-catalyzed azide-alkyne cycloaddition: scope and mechanism. J Am Chem Soc 2008;130:8923–30.
- Jiang X, Hao X, Jing L, et al. Recent applications of click chemistry in drug discovery. Expert Opin Drug Discov 2019;14:779–89.
- Nuti E, Cuffaro D, D'Andrea F, et al. Sugar-based arylsulfonamide carboxylates as selective and water-soluble matrix metalloproteinase-12 inhibitors. ChemMedChem 2016;11:1626–37.
- D'Andrea F, Nuti E, Becherini S, et al. Design and synthesis of ionic liquid-based Matrix Metalloproteinase Inhibitors (MMPIs): a simple approach to increase hydrophilicity and to develop MMPI-coated gold nanoparticles. ChemMedChem 2019;14:686–98.
- Tripathi RP, Dwivedi P, Sharma A, et al. Triazolyl glycoconjugates in medicinal chemistry. In: Witczak ZJ, Bielski R, eds. Click chemistry in glycoscience. Hoboken (NJ): John Wiley and Sons; 2013.
- Massarotti A, Aprile S, Mercalli V, et al. Are 1,4- and 1,5-disubstituted 1,2,3-triazoles good pharmacophoric groups? ChemMedChem 2014;9:2497–508.
- Wilkinson BL, Bornaghi LF, Houston TA, et al. A novel class of carbonic anhydrase inhibitors: glycoconjugate benzene sulfonamides prepared by "click-tailing". J Med Chem 2006;49:6539–48.
- Wilkinson BL, Bornaghi LF, Houston TA, et al. Inhibition of membrane-associated carbonic anhydrase isozymes IX, XII and XIV with a library of glycoconjugate benzene sulfonamides. Bioorg Med Chem Lett 2007;17:987–92.
- Wilkinson BL, Innocenti A, Vullo D, et al. Inhibition of carbonic anhydrases with glycosyltriazole benzene sulfonamides. J Med Chem 2008;51:1945–53.
- Carroux CJ, Rankin GM, Moeker J, et al. A prodrug approach toward cancer-related carbonic anhydrase inhibition. J Med Chem 2013;56:9623–34.
- Wilkinson BL, Bornaghi LF, Houston TA, et al. Carbonic anhydrase inhibitors: inhibition of isozymes I, II, and IX with triazole-linked O-glycosides of benzene sulfonamides. J Med Chem 2007;50:1651–7.
- Singer M, Lopez M, Bornaghi LF, et al. Inhibition of carbonic anhydrase isozymes with benzene sulfonamides incorporating thio, sulfinyl and sulfonyl glycoside moieties. Bioorg Med Chem Lett 2009;19:2273–6.
- Morris JC, Chiche J, Grellier C, et al. Targeting hypoxic tumor cell viability with carbohydrate-based carbonic anhydrase IX and XII inhibitors. J Med Chem 2011;54:6905–18.
- Murray AB, Lomelino CL, Supuran CT, et al. “Seriously sweet”: acesulfame K exhibits selective inhibition using alternative binding modes in carbonic anhydrase Isoforms. J Med Chem 2018;61:1176–−81.
- Lomelino CL, Murray AB, Supuran CT, et al. Sweet binders: carbonic anhydrase IX in complex with sucralose. ACS Med Chem Lett 2018;9:657–61.
- Mahon BP, Hendon AM, Driscoll JM, et al. Saccharin: a lead compound for structure-based drug design of carbonic anhydrase IX inhibitors. Bioorg Med Chem 2015;23:849–54.
- Moeker J, Peat TS, Bornaghi LF, et al. Cyclic secondary sulfonamides: unusually good inhibitors of cancer-related carbonic anhydrase enzymes. J Med Chem 2014;57:3522–31.
- Murray AB, Quadri M, Li H, et al. Synthesis of saccharin-glycoconjugates targeting carbonic anhydrase using a one-pot cyclization/deprotection strategy. Carbohydr Res 2019;476:65–70.
- Salmon AJ, Williams ML, Maresca A, et al. Synthesis of glycoconjugate carbonic anhydrase inhibitors by ruthenium-catalysed azide-alkyne 1,3-dipolar cycloaddition. Bioorg Med Chem Lett 2011;21:6058–61.
- Touisni N, Maresca A, McDonald PC, et al. Glycosyl coumarin carbonic anhydrase IX and XII inhibitors strongly attenuate the growth of primary breast tumors. J Med Chem 2011;54:8271–7.
- Maresca A, Scozzafava A, Supuran CT. 7,8-disubstituted- but not 6,7-disubstituted coumarins selectively inhibit the transmembrane, tumor-associated carbonic anhydrase isoforms IX and XII over the cytosolic ones I and II in the low nanomolar/subnanomolar range. Bioorg Med Chem Lett 2010;20:7255–8.
- Compain P. Glycomimetics: design, synthesis, and therapeutic applications. Molecules 2018;23:1658.
- Asensio JL, Cañada FJ, Cheng X, et al. Conformational differences between O- and C-glycosides: the alpha-O-man-(1–>1)-beta-Gal/alpha-C-Man-(1–>1)-beta-Gal case–a decisive demonstration of the importance of the exo-anomeric effect on the conformation of glycosides. Chem Eur J 2000;6:1035–41.
- Martínez HL, Riafrecha LE, Colinas PA. C-cinnamoyl glycosides: an emerging "Tail" for the development of selective carbonic anhydrase inhibitors. Curr Med Chem 2019;26:2601–8.
- Riafrecha LE, Rodríguez OM, Vullo D, et al. Synthesis of C-cinnamoyl glycosides and their inhibitory activity against mammalian carbonic anhydrases. Bioorg Med Chem 2013;21:1489–94.
- Buchieri MV, Riafrecha LE, Rodríguez OM, et al. Inhibition of the β-carbonic anhydrases from Mycobacterium tuberculosis with C-cinnamoyl glycosides: identification of the first inhibitor with anti-mycobacterial activity. Bioorg Med Chem Lett 2013;23:740–3.
- Riafrecha LE, Vullo D, Ouahrani-Bettache S, et al. Inhibition of β-carbonic anhydrases from Brucella suis with C-cinnamoyl glycosides incorporating the phenol moiety. J Enzyme Inhib Med Chem 2015;30:1017–20.
- Riafrecha LE, Rodríguez OM, Vullo D, et al. Attachment of carbohydrates to methoxyaryl moieties leads to highly selective inhibitors of the cancer associated carbonic anhydrase isoforms IX and XII. Bioorg Med Chem 2014;22:5308–14.
- Riafrecha LE, Vullo D, Supuran CT, et al. C-glycosides incorporating the 6-methoxy-2-naphthyl moiety are selective inhibitors of fungal and bacterial carbonic anhydrases. J Enzyme Inhib Med Chem 2015;30:857–61.
- Zaro MJ, Bortolotti A, Riafrecha LE, et al. Anti-tubercular and antioxidant activities of C-glycosyl carbonic anhydrase inhibitors: towards the development of novel chemotherapeutic agents against Mycobacterium tuberculosis. J Enzyme Inhib Med Chem 2016;31:1726–30.
- Riafrecha LE, Bua S, Supuran CT, et al. Improving the carbonic anhydrase inhibition profile of the sulfamoylphenyl pharmacophore by attachment of carbohydrate moieties. Bioorg Chem 2018;76:61–6.
- Tanpure RP, Ren B, Peat TS, et al. Carbonic anhydrase inhibitors with dual-tail moieties to match the hydrophobic and hydrophilic halves of the carbonic anhydrase active site. J Med Chem 2015;58:1494–501.
- Hou Z, Lin B, Bao Y, et al. Dual-tail approach to discovery of novel carbonic anhydrase IX inhibitors by simultaneously matching the hydrophobic and hydrophilic halves of the active site. Eur J Med Chem 2017;132:1–10.
- Maresca A, Akyuz G, Osman SM, et al. Inhibition of mammalian carbonic anhydrase isoforms I-XIV with a series of phenolic acid esters. Bioorg Med Chem 2015;23:7181–8.