
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
A series of novel N-alkyl-1-deoxynojirimycin derivatives 25 ∼ 44 were synthesised and evaluated for their in vitro α-glucosidase inhibitory activity to develop α-glucosidase inhibitors with high activity. All twenty compounds exhibited α-glucosidase inhibitory activity with IC50 values ranging from 30.0 ± 0.6 µM to 2000 µM as compared to standard acarbose (IC50 = 822.0 ± 1.5 µM). The most active compound 43 was ∼27-fold more active than acarbose. Kinetic study revealed that compounds 43, 40, and 34 were all competitive inhibitors on α-glucosidase with Ki of 10 µM, 52 µM, and 150 µM, respectively. Molecular docking demonstrated that the high active inhibitors interacted with α-glucosidase by four types of interactions, including hydrogen bonds, π–π stacking interactions, hydrophobic interactions, and electrostatic interaction. Among all the interactions, the π–π stacking interaction and hydrogen bond played a significant role in a various range of activities of the compounds.
1. Introduction
α-Glucosidase is a type of glucosidases that acts on 1,4-α-bonds, which locate on the brush edge of the small intestine and play a critical role in digestion and absorption of carbohydratesCitation1. Inhibition of α-glucosidase is one approach to delay the absorption of glucose and decrease the postprandial blood glucose levelCitation2. Therefore, α-glucosidase inhibitors are widely used for the prevention and treatment of typeII diabetes millitusCitation3. Besides, α-glucosidase participates in other physical and biological processes as well and may also be used as a therapeutic agent for other diseases, such as cancerCitation4 and HIVCitation5. Today, several types of α-glucosidase inhibitors are being clinically used for the treatment of typeII diabetes millitus, such as acarbose, voglibose, and miglitolCitation6. However, these medications also have adverse effects, including abdominal discomfort, diarrhoea, and flatulenceCitation7. So, developing novel α-glucosidase inhibitors is critical and attractive.
Iminosugars are sugars in which the endocyclic oxygen is replaced by a basic nitrogen atomCitation8. As α-glucosidase inhibitor, the best known naturally occurring iminosugar was 1-deoxynojirimycin (1-DNJ), which was first isolated from the roots of mulberry treesCitation9. 1-DNJ is currently under clinical evaluation, not only acting as an α-glucosidase inhibitorCitation10 but also a potent drug for cancerCitation11 and HIVCitation12. The accepted mechanism is that 1-DNJ inhibits α-glucosidase by competitively blocking the active site of the enzymeCitation13, and the nitrogen atom can mimic the charge of proposed transition states of oxocarbonium ion formed during hydrolysisCitation14. Pharmacokinetic studies showed that DNJ & DMJ (structurally related to DNJ) were rapidly eliminated from the body in an intact form by renal excretion, resulting in a weak effect on reducing blood sugar in vivo (Nakagawa et al.Citation15 and Faber et al.Citation16). Modification of 1-DNJ by increasing the alkalinity and introducing hydrophobic groups led to significant changes in the potency and specificity of inhibition. Ardes et al.Citation17 and Rawlings et al.Citation18 synthesised a number of deoxynojirimycin derivatives by combining different groups with a series of the N-alkyl chain length, which showed various degrees of inhibition on α-glucosidase and other enzymes. Zhang et al.Citation19 synthesised hybrids of 1-DNJ and quinazoline and obtained fifteen compounds, and some compounds exhibited significant inhibitory activities against the epidermal growth factor receptor (EGFR) tyrosine kinase and α-glucosidase. Some new N-alkyl, alkenyl, and benzyl substituted DNJ derivatives incorporating a silicon atom in the substituent were synthesised, which showed activity as potent and selective inhibitors of intestinal glucosidaseCitation20.
Here, we report for the first time the synthesis of a novel series of 1-DNJ derivatives with benzylidene acetone backbone groups (i.e. cinnamic acid, methyl 4-hydroxycinnamate, Ethyl 4′-hydroxy-3′-methoxycinnamate, and 2′-hydroxychalcone) and different length of alkyl chains, and obtained compounds 25–44. Moreover, all the compounds were evaluated for their α-glucosidase inhibitory activities. Furthermore, kinetic study and molecular docking were also performed to study the mechanism and discussed the structure–activity relationship.
2. Chemistry
The N-alkyl-deoxynojirimycin derivatives 25–44 were synthesised as shown in Scheme 1.
Scheme 1. Synthesis of intermediate products and target products of N-alkyl-deoxynojirimycin derivatives. Reagents and condition: (a) K2CO3, acetone, dibromo alkane, 65 °C, overnight or Et3N, acetone, dibromo alkane, 65 °C; (b) K2CO3, DMF, 85 °C, 6 h.
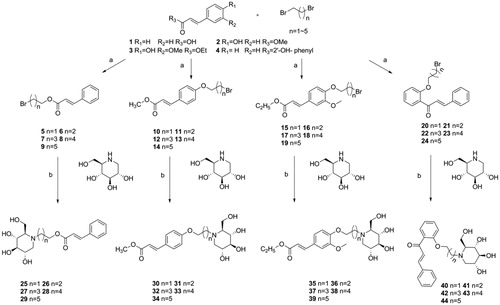
To obtain the intermediates 5–24, the cinnamic acid was reacted with dibromo alkane (i.e. 1, 2-dibromoethane, 1, 3-dibromoproane, 1, 4-dibromobutane, 1, 5-dibromopentane, and 1, 6-dibromohexane) and Et3N in acetone at 65 °C, overnight to afford compounds 5–9. Using the same method above, reactions were also carried out by changing the Et3N to K2CO3 and changing cinnamic acid to methyl 4-hydroxycinnamate, ethyl 4′-hydroxy-3′-methoxycinnamate, 2′-hydroxychalcone to afford compounds 10–14, 15–19, 20–24, respectively.
To obtain the target compounds 25–44, reactions were carried out by dissolving intermediates 5–24, 1-DNJ and K2CO3 into DMF, and stirring the mixture at 85 °C for 5–6h. The reaction was monitored by thin-layer chromatography (TLC), and the reaction products were purified with column chromatography using dichloromethane: methane = 25:2 as eluent to afford the pure compounds 25–44. The structures of all the new synthesised compounds 5–44 were characterised by HRMS, 1H and 13C NMR spectroscopy.
3. Results and discussion
3.1. In vitro α-glucosidase inhibitory activity
All the synthesised target products 25–44 were screened to check theirs in vitro α-glucosidase inhibitory activity. All the synthesised compounds show activity on α-glucosidase with IC50 ranging from 30 ± 0.60 µM to 2000 µM as compared with acarbose (IC50 = 822.0 ± 1.5 µM). The results were shown in .
Table 1. In vitro α-glucosidase inhibitory activity of compound 25–44.
Among all the tested compounds, compound 43 exhibited high α-glucosidase inhibitory activity with IC50 of 30.0 ± 0.60 µM which is ∼27-fold higher than acarbose. Similarly, compound 40 showed an excellent activity with IC50 of 160.5 ± 0.60 µM, around 5-fold better than acarbose. Others also exhibited inhibitory activities.
To better understand the structure–activity relationship, compounds were categorised into four groups “A”→“D,” cinnamic acid-1-DNJ derivatives 25–29, methyl 4-hydroxycinnamate –1-DNJ derivatives 30–34, ethyl 4′-hydroxy-3′-methoxycinnamate-1-DNJ derivatives 35–39, and 2′-hydroxychalcone-1-DNJ derivatives 40–44 belongs to “A,” “B,” “C,” and “D,” respectively. In each group, the difference among those compounds was the length of alkyl chains. The general structural formula was shown in
Figure 1. The general structural formula of N-alkyl-deoxynojirimycin derivatives.
In group “A,” compound 27 (purity: 91.5%) with a length of four carbon (n = 3) was found to be the most active compound (IC50 = 559.3 ± 0.28 µM) as compared to acarbose (IC50 = 822.0 ± 1.5 µM), neither increasing nor decreasing the length of alkyl chain would increase the inhibitory activity as observed. When the lengths of alkyl chain were two carbon (n = 1, compound 25, purity: 90.1%) and three carbon (n = 2, compound 26, purity: 93.8%), the inhibitory activities were sharply decreased; and when the lengths were five carbon (n = 4, compound 28, purity: 93.5%) and six carbon (n = 5, compound 29, purity: 93.8%), the inhibitory activities were also decreased. It indicated that the inhibitory activities were highly dependent on the length of the alkyl chain, but without a trend correlation.
In group “B,” compound 34 with the length of six carbon (n = 5) showed the most inhibitory activity (IC50 = 417.0 ± 0.14 µM), and was also the third most active compound among all the synthesised compounds. It is the same as the group “A,” the inhibitory activity was poor for the compounds with the alkyl chain containing less than four carbon. While the alkyl chain was long with more than four carbon, the inhibitory activity of the compounds increases as the length of the alkyl chain increases.
In group “C,” only compound 35 (purity: 91.2%) showed a low activity with an IC50 value of 966.2 ± 0.40 µM. Changing the alkyl chain length of the compounds led to no significant improvement in inhibitory activity. Compared with group “B,” it was suggested that the compounds without a methoxy group in the 3′-position of phenyl ring were more active than that with a methoxy group.
In the case of group “D,” all compounds were found to have excellent inhibitory activity with IC50 values between 30.0 ± 0.60 µM and 571.6 ± 0.60 µM when compared with acarbose (IC50=822.0 ± 1.5 µM). Notably, compound 43 with the length of five carbon (n = 4) displayed the highest activity with IC50 value of 30.0 ± 0.60 µM. This compound was also the most active compound among all the synthesised compounds. In addition, compound 40 with an alkyl chain of two carbon (n = 1) was the second most active compound among all the synthesised compounds (IC50 = 160.5 ± 0.60 µM). The two compounds were both active than 1-DNJ (IC50 = 222.4 ± 0.50 µM) in inhibiting α-glucosidase. Compared to group “D” with other groups, compounds in group “D” have two phenyl rings, but compounds in other groups have only one, suggesting that the number of phenyl rings plays an important role in compounds’ inhibitory activity.
3.2. Kinetic study
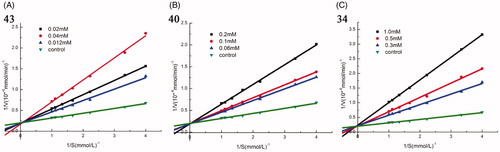
To study the inhibition mode of synthesised compounds on α-glucosidase, kinetic studies were performed with the three most active compounds 43, 40, and 34. The type of inhibition and value of Ki were determined by Lineweaver–Burk plots. As shown in , when increasing concentrations of compound 43, 40, and 34, the Vmax was not affected, while the Km increased, indicating that all these three compounds were competitive inhibitors for α-glucosidase. The Ki values of 43, 40, and 34 were 10 µM, 52 µM, and 150 µM, respectively.
Figure 2. Kinetic analysis of α-glucosidase inhibition by compounds 43, 40, and 34. (A) The Lineweaver–Burk plots in the absence and presence of different concentrations of compound 43; (B) The Lineweaver–Burk plots in the absence and presence of different concentrations of compound 40; (C) The Lineweaver–Burk plots in the absence and presence of different concentrations of compound 34.
3.3. Docking study
In order to clarify the interactions between compounds and amino acids in the substrate-binding pocket of α-glucosidase at the molecular level, a molecular docking study was carried out using Autodock VinaCitation21. Since the X-ray crystallographic structure of Saccharomyces cerevisiae α-glucosidase we used in the experiments has not been reported yet, the 3 D structure of α-glucosidase was conducted with SWISS-MODELCitation22.
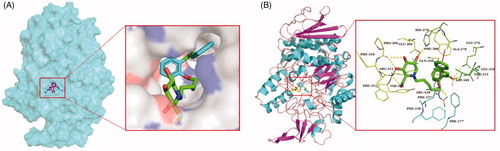
Acarbose and the most potent compounds 43, 40, and 34 were docked in the active site of the α-glucosidase. In order to explore the structure–activity relationship, compound 41 was also docked. showed the results of the molecular docking and detailed interactions, including hydrogen bonds, π–π stacking interactions, hydrophobic interactions, and electrostatic interactions. From the docking study, it was observed that acarbose () interacted with the active site of α-glucosidase via six hydrogen bonds with residues Gln350, Arg312, and Asn241. Additionally, the compound formed several electrostatic interactions with residues Phe157, Phe158, and Phe300.
Table 2. The detailed information of molecular docking results of compounds 34, 40, 41, 43, and acarbose.
The most active compound 43 was well accommodated inside the active site of α-glucosidase () and established nine hydrogen bonds with residues Glu276, Asp349, Arg439, His279, Glu304, Pro309, and Arg312 (). Additionally, the phenyl rings of the compound formed two π–π stacking interactions with Phe300 and Phe157. Furthermore, hydrophobic interactions and electrostatic interaction were observed between compound 43 and residues Leu218, Ala 278, Phe177, Phe158, and Asp349. The compound 43 has lower binding energy (−9.2 kcal/mol) than acarbose (−7.8 kcal/mol), suggesting that compound 43 was binding with enzyme more easily and strongly than acarbose.
Figure 3. (A) The predicted binding mode of compound 43 in the active site and substrate-binding pocket. (B) the interactions between 43 and modelled α-glucosidase. The yellow dashed lines represent hydrogen bonds and the red dashed lines represented π–π interactions.
The second most active compound 40, similar to compound 43, formed two π–π stacking interactions with Phe300 and Phe157 (). However, compound 40 only formed five hydrogen bonds with residues Glu276, Gln350, Asp349, and Arg439. Furthermore, several hydrophobic interactions and electrostatic interactions were also observed between compound 40 and residues Phe157, Ala278, Phe158, Phe177, Leu437, and Phe311.
Figure 4. The predicted binding mode of acarbose (A), 34 (B), 41 (C) and 40 (D) in the active site pocket. The yellow dashed lines represented hydrogen bonds and the red dashed lines represented π–π interactions.
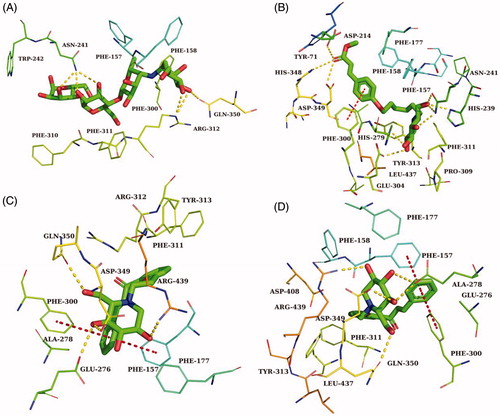
In order to explore how the length of the alkyl chain affects the activity on α-glucosidase, compound 41(n = 2), which belongs to the same group as the compounds 43(n = 4) and 40 (n = 1), was also docked. As seen in , except for the number of hydrogen bonds, other interactions were nearly the same as that for compounds 43 and 40. According to the IC50 values, structures, and docking study results, we can infer that the length of the alkyl chain of the compounds affected their α-glucosidase inhibitory activity by affecting the number of hydrogen bonds between the compounds and the enzyme.
In the case of compound 34, the third most active compound, which formed nine hydrogen bonds with the binding site residues His348, Asp349, Asp214, His279, Glu304, Asn241, His239 and also one π–π interaction with residues Phe157. In addition, several hydrophobic interactions were also observed between molecules and residues Phe158, Phe177, Tyr71, Phe300, and Leu437 (). Compared with compound 43, they both have nine hydrogen bonds, while the most significant difference between them was the number of π–π stacking interactions, the number of π–π stacking interactions for compound 34 was less than that for compound 43, suggesting that π–π stacking interaction played a significant effect on the inhibitory activity of the compounds.
Studies of the biological activity and molecular docking of these compounds showed that the inhibitory activities were highly dependent on the length of the alkyl chain, but without a trend correlation. Besides, the more phenyl rings in the molecules, the more probabilities to establish π–π stacking interactions between molecules and enzymes, which were responsible for high activity on α-glucosidase. The compounds without a methoxy group in the 3′-position of phenyl ring were more active than that with a methoxy group. The docking results were similar with previous studies. Zeng et al. synthesised a series of N-benzyl-deoxynojirimycin derivatives, and the most active compound also established a π–π stacking interactions between molecules and enzymes, which gave the most active compound a strong inhibitory activity to α-glucosidaseCitation23. Besides, the hydrogen bonds between compounds α-glucosidase and were also played important roles in high activity on α-glucosidase. Shahzad et al. synthesised a series of symmetrical salicylaldehyde-bishydrazine azo molecules. The high activity of compounds is mainly caused by hydrogen bondsCitation24, which were the same with us.
4. Conclusions
In conclusion, we synthesised a series of novel N-alkyl-1-DNJ derivatives 25–44, all the compounds were tested for their α-glucosidase inhibitory activity. Among them, the compound 43 (IC50 = 30.0 ± 0.60 µΜ) was the most active compound, which was ∼27-fold more active than acarbose (IC50 = 822.0 ± 1.5 µM) and ∼7-fold more active than 1-DNJ (IC50 = 222.4 ± 0.5 µM). The kinetic study revealed that compounds 43, 40, and 34 inhibit α-glucosidase via a competitive mechanism with Ki of 10 µΜ, 52 µΜ, and 150 µΜ, respectively. The docking study showed that hydrogen bond and π–π stacking interaction played a significant role in the anti-α-glucosidase activity of the synthesised compounds. The numbers of hydrogen bonds and π–π stacking interactions were correlated with and responsible for the compounds’ activities, and the compounds without methoxy group in the 3′-position of phenyl ring were more active than that with a methoxy group.
5. Experimental
All starting materials and reagents were purchased from commercial suppliers. α-glucosidase (EC 3.2.1.20) was purchased from Sigma-Aldrich. TLC was performed on Silica gel F-254. Melting points were measured on a microscopic melting point apparatus. The 1H NMR and 13 C NMR were measured (DMSO solution) with Bruker spectrometer (500 MHz 1H, 125 MHz 13 C). HRMS was performed on AB SCIEX Triple TOF 5600+ with electron spray ionisation (ESI) as the ion source.
5.1. General experimental procedure for the syntheses of intermediates 5–9
A solution of cinnamic acid 1 (1 mmol), Et3N(3 mmol), dibromo alkane (4 ∼ 5 mmol) in acetone was heated at 65 °C, overnight. After the reaction completed, the mixture was cooled down to room temperature. Water and ethyl acetate were added and extracted three times. The combined organic extracts were dried over Na2SO4 and then concentrated. Further purification by flash chromatography gave the title compounds.
5.1.1. 2-Bromoethyl cinnamate (5)
Yellow oil; yield: 72%; 1H NMR (500 MHz, DMSO) δ 7.77–7.72 (m, 3H), 7.45–7.43 (m, 3H), 6.68 (d, J = 16.1 Hz, 1H), 4.50 (t, J = 5.6 Hz, 2H), 3.76 (t, J = 5.6 Hz, 2H). 13C NMR (125 MHz, DMSO) δ 166.31, 145.71, 134.34, 131.11, 129.42, 128.94, 128.78, 117.93, 64.35, 31.27. HRMS (ESI) m/z [M + H]+: calcd for C11H11BrO2: 255.0015, found 255.0010.
5.1.2. 3-Bromopropyl cinnamate (6)
Yellow oil; yield: 75%; 1H NMR (500 MHz, DMSO) δ 7.74–7.69 (m, 3H), 7.45–7.39 (m, 3H), 6.64 (d, J = 16.0 Hz, 1H), 4.26 (t, J = 6.2 Hz, 2H), 3.63 (t, J = 6.6 Hz, 2H), 2.20 (p, J = 6.4 Hz, 2H). 13C NMR (125 MHz, DMSO) δ 166.57, 145.15, 134.47, 130.95, 129.37, 128.82, 118.32, 62.54, 31.82, 31.46. HRMS (ESI) m/z [M + H]+: calcd for C12H13BrO2: 269.0172, found 269.0169.
5.1.3. 4-Bromopropyl cinnamate (7)
Yellow oil; yield: 78%; 1H NMR (500 MHz, DMSO) δ 7.75–7.70 (m, 2H), 7.66 (dd, J = 16.1, 4.7 Hz, 1H), 7.44-7.42 (m, 3H), 6.63 (dd, J = 16.0, 3.2 Hz, 1H), 4.18 (dd, J = 8.4, 4.5 Hz, 2H), 3.58 (t, J = 6.6 Hz, 2H), 1.95–1.88 (m, 2H), 1.82–1.74 (m, 2H).13C NMR (125 MHz, DMSO) δ 166.64, 144.95, 134.47, 130.90, 129.36, 128.80, 118.49, 63.68, 35.07, 29.45, 27.49. HRMS (ESI) m/z [M + H]+: calcd for C13H15BrO2: 283.0328, found 283.0325.
5.1.4. 5-Bromopropyl cinnamate (8)
Yellow oil; yield: 79%; 1H NMR (500 MHz, DMSO) δ 7.74–7.69 (m, 2H), 7.66 (d, J = 16.1 Hz, 1H), 7.49–7.42 (m, 3H), 6.63 (d, J = 16.0 Hz, 1H), 4.16 (t, J = 6.5 Hz, 2H), 3.55 (t, J = 4.1 Hz, 2H), 1.83 (d, J = 7.0 Hz, 2H), 1.71–1.63 (m, 2H), 1.51–1.48 (m, 2H). 13C NMR (125 MHz, DMSO) δ 166.66, 144.88, 134.47, 130.90, 129.36, 128.79, 118.55, 64.26, 35.30, 32.32, 27.84, 24.61. HRMS (ESI) m/z [M + H]+: calcd for C14H17BrO2: 297.0485, found 297.0495.
5.1.5. 6-Bromopropyl cinnamate (9)
Yellow oil; yield: 74%; 1H NMR (500 MHz, DMSO) δ 7.76–7.69 (m, 2H), 7.66 (d, J = 16.1 Hz, 1H), 7.44–7.42 (m,3H), 6.63 (d, J = 16.0 Hz, 1H), 4.14 (t, J = 6.6 Hz, 2H), 3.52 (t, J = 6.7 Hz, 2H), 1.85–1.77 (m, 2H), 1.69–1.60 (m, 2H), 1.47–1.30 (m, 4H).13C NMR (125 MHz, DMSO) δ 166.68, 144.83, 134.49, 130.87, 129.35, 128.77, 118.59, 64.38, 35.45, 32.61, 28.54, 27.68, 25.06. HRMS (ESI) m/z [M + H]+: calcd for C15H19BrO2: 311.0641, found 311.0652.
5.2. General experimental procedure for the syntheses of intermediates 10–24
A solution of methyl 4-hydroxycinnamate 2 (1 mmol), K2CO3(2 mmol), dibromo alkane (4 ∼ 5 mmol) in acetone was heated at 65 °C, overnight. After the reaction completed, the mixture was cooled down to room temperature. Water and ethyl acetate were added and extracted three times. The combined organic extracts were dried over Na2SO4 and then concentrated. Further purification by flash chromatography gave the compounds 10–14. Replaced the Methyl 4-hydroxycinnamate with Ethyl 4′-hydroxy-3′-methoxycinnamate and 2′-Hydroxychalcone, other processes were the same, obtained compounds 15–19, 20–24, respectively.
5.2.1. Methyl (E)-3-(4-(2-bromoethoxy) phenyl) acrylate (10)
White solid; yield: 65%; m.p. 85–88 °C; 1H NMR (500 MHz, DMSO) δ 7.68 (d, J = 8.7 Hz, 2H), 7.63 (d, J = 16.0 Hz, 1H), 7.01 (d, J = 8.7 Hz, 2H), 6.51 (d, J = 16.0 Hz, 1H), 4.42–4.33 (m, 2H), 3.85–3.77 (m, 2H), 3.71 (s, 3H). 13 C NMR (125 MHz, DMSO) δ 167.34, 160.24, 144.64, 130.65, 127.57, 115.89, 115.48, 68.35, 51.78, 31.71. HRMS (ESI) m/z [M + H]+: calcd for C12H13BrO3: 285.0121, found 285.0121.
5.2.2. Methyl (E)-3-(4-(3-bromoethoxy) phenyl) acrylate (11)
White solid; yield: 68%; m.p. 77–79 °C; 1H NMR (500 MHz, DMSO) δ 7.68 (d, J = 8.6 Hz, 2H), 7.62 (d, J = 16.0 Hz, 1H), 7.00 (d, J = 8.7 Hz, 2H), 6.49 (d, J = 16.0, 1H), 4.14 (t, J = 6.0 Hz, 2H), 3.71 (s, 3H), 3.67 (t, J = 6.5 Hz, 2H), 2.29–2.22 (m, 2H).13C NMR (125 MHz, DMSO) δ 167.36, 160.71, 144.73, 130.63, 127.27, 115.68, 115.35, 65.98, 51.76, 40.53, 32.20, 31.58. HRMS (ESI) m/z [M + H]+: calcd for C13H15BrO3: 299.0277, found 299.0279.
5.2.3. Methyl (E)-3-(4-(4-bromoethoxy) phenyl) acrylate (12)
White solid; yield: 70%; m.p. 73–75 °C; 1H NMR (500 MHz, DMSO) δ 7.67 (d, J = 8.7 Hz, 2H), 7.62 (d, J = 16.0 Hz, 1H), 6.98 (d, J = 8.7 Hz, 2H), 6.49 (d, J = 16.0 Hz, 1H), 4.07 (t, J = 6.3 Hz, 2H), 3.71 (s, 3H), 3.61 (t, J = 6.7 Hz, 2H), 2.02–1.91 (m, 2H), 1.90–1.79 (m, 2H).13C NMR (125 MHz, DMSO) δ 167.38, 160.92, 144.79, 130.61, 127.06, 115.53, 115.32, 67.29, 51.75, 35.25, 29.50, 27.76. HRMS (ESI) m/z [M + H]+: calcd for C14H17BrO3: 313.0434, found 313.0432.
5.2.4. Methyl (E)-3-(4-(5-bromoethoxy) phenyl) acrylate (13)
White solid; yield: 67%; m.p. 89–91 °C; 1H NMR (500 MHz, DMSO) δ 7.66 (d, J = 8.7 Hz, 2H), 7.61 (d, J = 16.0 Hz, 1H), 6.97 (d, J = 8.7 Hz, 2H), 6.48 (d, J = 16.0 Hz, 1H), 4.03 (t, J = 6.4 Hz, 2H), 3.71 (s, 3H), 3.56 (t, J = 6.7 Hz, 2H), 1.92–1.82 (m, 2H), 1.80–1.70 (m, 2H), 1.54 (t, J = 7.6 Hz, 2H).13C NMR (125 MHz, DMSO) δ 167.39, 161.03, 144.80, 130.61, 126.98, 115.48, 115.30, 67.97, 51.75, 35.49, 32.40, 28.16, 24.71. HRMS (ESI) m/z [M + H]+: calcd for C15H19BrO3: 327.0590, found 327.0588.
5.2.5. Methyl (E)-3-(4-(6-bromoethoxy) phenyl) acrylate (14)
White solid; yield: 67%; m.p(0).76–79 °C; 1H NMR (500 MHz, DMSO) δ 7.66 (d, J = 8.4 Hz, 2H), 7.61 (d, J = 16.0 Hz, 1H), 6.97 (d, J = 8.4 Hz, 2H), 6.48 (d, J = 16.0 Hz, 1H), 4.02 (t, J = 6.3 Hz, 2H), 3.71 (s, 3H), 3.54 (t, J = 6.7 Hz, 2H), 1.88–1.78 (m, 2H), 1.77–1.65 (m, 2H), 1.50–1.39 (m, 4H).13C NMR (125 MHz, DMSO) δ 167.39, 161.06, 144.81, 130.61, 126.95, 115.46, 115.29, 68.02, 51.75, 35.55, 32.64, 28.87, 27.74, 25.09. HRMS (ESI) m/z [M + H]+: calcd for C16H21BrO3: 341.0747, found 341.0745.
5.2.6. Ethyl (E)-3-(4-(2-bromoethoxy)-3-methoxyphenyl) acrylate (15)
White solid; yield: 68%; m.p. 83–86 °C; 1H NMR (500 MHz, DMSO) δ 7.59 (d, J = 16.0 Hz, 1H), 7.39 (d, J = 1.8 Hz, 1H), 7.22 (d, J = 1.8 Hz, 1H), 7.01 (d, J = 8.3 Hz, 1H), 6.57 (d, J = 16.0 Hz, 1H), 4.40–4.31 (m, 2H), 4.18 (q, J = 7.1 Hz, 2H), 3.83 (s, 3H), 3.82–3.79 (m, 2H), 1.26 (t, J = 7.1 Hz, 3H). 13 C NMR (125 MHz, DMSO) δ 166.92, 149.92, 149.69, 144.83, 128.11, 123.17, 116.54, 113.81, 111.51, 69.04, 60.27, 56.24, 31.59, 14.71. HRMS (ESI) m/z [M + H]+: calcd for C14H17BrO4: 329.0383, found 329.0382.
5.2.7. Ethyl (E)-3-(4-(3-bromopropoxy)-3-methoxyphenyl) acrylate (16)
White solid; yield: 65%; m.p. 67–70 °C; 1H NMR (500 MHz, DMSO) δ 7.58 (d, J = 15.9 Hz, 1H), 7.37 (d, J = 1.7 Hz, 1H), 7.22 (s, 1H), 7.01 (d, J = 8.3 Hz, 1H), 6.56 (d, J = 15.9 Hz, 1H), 4.18 (q, J = 7.1 Hz, 2H), 4.12 (t, J = 6.0 Hz, 2H), 3.83 (d, J = 3.7 Hz, 3H), 3.66 (t, J = 6.5 Hz, 2H), 2.26 (t, J = 6.3 Hz, 2H), 1.26 (t, J = 7.1 Hz, 3H). 13 C NMR (125 MHz, DMSO) δ 166.94, 150.41, 149.71, 144.92, 127.73, 123.27, 116.33, 113.38, 111.24, 66.61, 60.25, 56.20, 32.32, 31.62, 14.71. HRMS (ESI) m/z [M + H]+: calcd for C15H19BrO4: 343.0540, found 343.0535.
5.2.8. Ethyl (E)-3-(4-(4-bromopropoxy)-3-methoxyphenyl) acrylate (17)
White solid; yield: 70%; m.p. 88–90 °C; 1H NMR (500 MHz, DMSO) δ 7.58 (d, J = 15.9 Hz, 1H), 7.36 (d, J = 1.5 Hz, 1H), 7.22 (d, J = 8.2 Hz, 1H), 6.99 (d, J = 8.3 Hz, 1H), 6.55 (d, J = 15.9 Hz, 1H), 4.18 (q, J = 7.1 Hz, 2H), 4.04 (t, J = 6.3 Hz, 2H), 3.82 (d, J = 3.5 Hz, 3H), 3.62 (t, J = 6.7 Hz, 2H), 2.01–1.93 (m, 2H), 1.85 (dd, J = 9.1, 5.5 Hz, 2H), 1.26 (t, J = 7.1 Hz, 3H).13C NMR (125 MHz, DMSO) δ 166.96, 150.67, 149.62, 144.99, 127.42, 123.30, 116.14, 113.12, 111.14, 67.87, 60.23, 56.18, 35.28, 29.60, 27.81, 14.71. HRMS (ESI) m/z [M + H]+: calcd for C16H21BrO4: 357.0696, found 357.0691.
5.2.9. Ethyl (E)-3-(4-(5-bromopropoxy)-3-methoxyphenyl) acrylate (18)
White solid; yield: 64%; m.p. 70–73 °C; 1H NMR (500 MHz, DMSO) δ 7.58 (d, J = 15.9 Hz, 1H), 7.35 (d, J = 1.9 Hz, 1H), 7.22 (dd, J = 8.3, 1.9 Hz, 1H), 6.98 (d, J = 8.3 Hz, 1H), 6.54 (d, J = 15.9 Hz, 1H), 4.18 (q, J = 7.1 Hz, 2H), 4.00 (t, J = 6.4 Hz, 2H), 3.81 (s, 3H), 3.56 (t, J = 6.7 Hz, 2H), 1.91–1.84 (m, 2H), 1.79–1.71 (m, 2H), 1.54 (d, J = 7.3 Hz, 2H), 1.26 (t, J = 7.1 Hz, 3H).13C NMR (125 MHz, DMSO) δ 166.97, 150.80, 149.62, 145.01, 127.32, 123.33, 116.07, 113.06, 111.13, 68.54, 60.23, 56.16, 35.50, 32.41, 28.22, 24.79, 14.71. HRMS (ESI) m/z [M + H]+: calcd for C17H23BrO4: 371.0853, found 371.0840.
5.2.10. Ethyl (E)-3-(4-(6-bromopropoxy)-3-methoxyphenyl) acrylate (19)
Colourless oil; yield: 65%; 1H NMR (500 MHz, DMSO) δ 7.58 (d, J = 15.9 Hz, 1H), 7.35 (d, J = 1.5 Hz, 1H), 7.21 (d, J = 8.2 Hz, 1H), 6.97 (d, J = 8.3 Hz, 1H), 6.54 (d, J = 15.9 Hz, 1H), 4.18 (q, J = 7.1 Hz, 2H), 3.99 (t, J = 5.8 Hz, 2H), 3.81 (s, 3H), 3.54 (t, J = 6.7 Hz, 2H), 1.87–1.78 (m, 2H), 1.77–1.67 (m, 2H), 1.44 (d, J = 2.8 Hz, 4H), 1.26 (t, J = 7.1 Hz, 3H).13C NMR (125 MHz, DMSO) δ 166.97, 150.83, 149.62, 145.02, 127.28, 123.34, 116.05, 113.01, 111.11, 68.55, 60.22, 56.15, 35.54, 32.65, 28.91, 27.73, 25.12, 14.71. HRMS (ESI) m/z [M + H]+: calcd for C18H25BrO4385.1009, found 385.0996.
5.2.11. (E)-1-(2-(2-bromoethoxy) phenyl)-3-phenylprop-2-en-1-one (20)
Yellow oil; yield: 64%; 1H NMR (500 MHz, DMSO) δ 7.77 (d, J = 3.6 Hz, 1H), 7.76 (d, J = 2.2 Hz, 1H), 7.61 (d, J = 3.7 Hz, 2H), 7.58 (dd, J = 7.5, 1.8 Hz, 1H), 7.55–7.52 (m, 1H), 7.45–7.42 (m, 3H), 7.19 (d, J = 8.4 Hz, 1H), 7.10 (t, J = 7.3 Hz, 1H), 4.50–4.45 (m, 2H), 3.83–3.79 (m, 2H).13C NMR (125 MHz, DMSO) δ 191.96, 156.95, 142.69, 135.13, 133.78, 130.86, 130.51, 129.39, 129.35, 129.08, 127.43, 121.61, 113.66, 68.95, 31.73. HRMS (ESI) m/z [M + H]+: calcd for C17H15BrO2: 331.0329, found 331.0330.
5.2.12. (E)-1-(2-(3-bromoethoxy) phenyl)-3-phenylprop-2-en-1-one (21)
Yellow oil; yield: 66%; 1H NMR (500 MHz, DMSO) δ 7.74 (d, J = 3.7 Hz, 1H), 7.73–7.72 (m, 1H), 7.56–7.51 (m, 3H), 7.46 (s, 1H), 7.45–7.42 (m, 3H), 7.20 (d, J = 8.2 Hz, 1H), 7.08 (t, J = 7.4 Hz, 1H), 4.20 (t, J = 5.8 Hz, 2H), 3.56 (t, J = 6.7 Hz, 2H), 2.23 (p, J = 6.3 Hz, 2H).13C NMR (125 MHz, DMSO) δ 192.46, 157.30, 142.71, 134.96, 133.63, 130.95, 130.17, 129.46, 129.44, 128.92, 127.43, 121.27, 113.49, 66.46, 32.31, 31.47. HRMS (ESI) m/z [M + H]+: calcd for C18H17BrO2: 345.0485, found 345.0482.
5.2.13. (E)-1-(2-(4-bromoethoxy) phenyl)-3-phenylprop-2-en-1-one (22)
Yellow oil; yield: 70%; 1H NMR (500 MHz, DMSO) δ 7.73 (d, J = 3.7 Hz, 1H), 7.72 (d, J = 2.1 Hz, 1H), 7.54 (d, J = 2.6 Hz, 1H), 7.52 (d, J = 2.3 Hz, 2H), 7.49 (s, 1H), 7.46–7.43 (m, 3H), 7.19 (d, J = 8.1 Hz, 1H), 7.07 (dd, J = 10.9, 3.9 Hz, 1H), 4.13 (t, J = 6.0 Hz, 2H), 3.42 (t, J = 6.5 Hz, 2H), 1.95–1.87 (m, 2H), 1.86–1.79 (m, 2H). 13 C NMR (125 MHz, DMSO) δ 192.42, 157.65, 142.41, 135.06, 133.66, 130.88, 130.17, 129.45, 129.35, 128.89, 127.62, 121.06, 113.52, 67.73, 34.97, 29.50, 27.93. HRMS (ESI) m/z [M + H]+: calcd for C19H19BrO2: 359.0641, found 359.0640.
5.2.14. (E)-1-(2-(5-bromoethoxy) phenyl)-3-phenylprop-2-en-1-one (23)
Pale Yellow crystals; yield: 69%; m.p. 58–60 °C; 1H NMR (500 MHz, DMSO) δ 7.72 (dd, J = 3.7, 1.9 Hz, 2H), 7.53 (s, 1H), 7.51 (d, J = 4.7 Hz, 2H), 7.49 (s, 1H), 7.44 (dd, J = 2.9, 1.9 Hz, 3H), 7.18 (d, J = 8.2 Hz, 1H), 7.06 (t, J = 7.4 Hz, 1H), 4.10 (dd, J = 5.8, 3.6 Hz, 2H), 3.33 (d, J = 5.3 Hz, 2H), 1.73–1.69 (m, 4H), 1.49–1.43 (m, 2H).13C NMR (125 MHz, DMSO) δ 192.49, 157.79, 142.23, 135.13, 133.70, 130.85, 130.17, 129.42, 129.34, 128.84, 127.76, 121.01, 113.51, 68.42, 35.00, 32.43, 28.39, 24.89.HRMS (ESI) m/z [M + H]+: calcd for C20H21BrO2: 373.0798, found 373.0796.
5.2.15. (E)-1-(2-(6-bromoethoxy) phenyl)-3-phenylprop-2-en-1-one (24)
Pale yellow crystals; yield: 65%; m.p. 7 4 ∼ 76 °C; 1H NMR (500 MHz, DMSO) δ 7.73 (d, J = 3.4 Hz, 1H), 7.71 (d, J = 2.0 Hz, 1H), 7.53 (s, 1H), 7.51 (d, J = 3.5 Hz, 2H), 7.49 (s, 1H), 7.46–7.42 (m, 3H), 7.17 (d, J = 8.1 Hz, 1H), 7.05 (t, J = 7.4 Hz, 1H), 4.09 (t, J = 6.0 Hz, 2H), 3.35 (t, J = 6.8 Hz, 2H), 1.73–1.65 (m, 2H), 1.60–1.53 (m, 2H), 1.39–1.33 (m, 2H), −1.301.26 (m, 2H).13C NMR (125 MHz, DMSO) δ 192.46, 157.84, 142.21, 135.12, 133.71, 130.87, 130.19, 129.43, 129.31, 128.83, 127.73, 120.98, 113.46, 68.50, 35.29, 32.47, 29.11, 27.77, 25.33.HRMS (ESI) m/z [M + Na]+: calcd for C21H23BrO2: 409.0774, found 409.0780.
5.3. General experimental procedure for the syntheses of target compounds 25–44
A mixture of intermedial compounds 5 (1 mmol) or other intermedial compounds (6–24, respectively), 1-DNJ (1 mmol), K2CO3 (2 mmol) in DMF was stirred at 85 °C for 6 h. After the reaction completed, the mixture was concentrated under reduced pressure at 90 °C. The product was purified by silica gel and thin layer chromatography to give the title compounds.
5.3.1. 2-(3,4,5-Trihydroxy-2-(hydroxymethyl) piperidin-1-yl) ethyl cinnamate (25)
Brown oil; yield: 33; purity: 90.1%. 1H NMR (500 MHz, DMSO) δ 7.76–7.70 (m, 2H), 7.67 (s, 1H), 7.46–7.42 (m, 3H), 7.23 (d, J = 15.5 Hz, 1H), 5.22 (s, 1H), 5.15–4.83 (m, 3H), 4.72 (s, 2H), 4.45–4.37 (m, 1H), 4.34–4.26 (m, 1H), 4.25–4.19 (m, 1H), 3.82 (d, J = 10.7 Hz, 1H), 3.67 (d, J = 4.2 Hz, 1H), 3.62 (s, 1H), 3.59–5.53 (m, 1H), 3.20 (s, 1H), 3.13–3.06 (m, 1H), 3.05–2.94 (m, 1H).13C NMR (125 MHz, DMSO) δ 166.79, 144.99, 134.57, 130.97, 129.41, 129.25, 128.83, 118.63, 79.57, 71.22, 69.82, 69.22, 65.83, 62.03, 59.86, 50.97.HRMS (ESI) m/z [M + H]+: calcd for C17H23NO6: 338.1598, found 338.1601.
5.3.2. 3-(3,4,5-Trihydroxy-2-(hydroxymethyl) piperidin-1-yl) ethyl cinnamate (26)
Brown oil; yield: 35%; purity: 93.8%. 1H NMR (500 MHz, DMSO) δ 7.76–7.79 (m, 2H), 7.66 (d, J = 16.0 Hz, 1H), 7.49–7.38 (m, 3H), 6.62 (d, J = 16.0 Hz, 1H), 4.72 (s, 3H), 4.23 (s, 1H), 4.19–4.09 (m, 2H), 3.82–3.72 (m, 1H), 3.61–3.51 (m, 1H), 3.26–3.18 (m, 1H), 3.08–3.00 (m, 1H), 2.98–2.88 (m, 1H), 2.87–2.80 (m, 1H), 2.03–1.92 (m, 2H), 1.84–1.71 (m, 2H), 1.23 (s, 2H).13C NMR (125 MHz, DMSO) δ 166.73, 144.87, 134.52, 130.92, 129.40, 128.83, 118.61, 79.59, 71.20, 69.80, 67.35, 63.25, 59.72, 57.35, 49.09, 24.67.HRMS (ESI) m/z [M + H]+: calcd for C18H25NO6: 352.1755, found 352.1766.
5.3.3. 4-(3,4,5-Trihydroxy-2-(hydroxymethyl) piperidin-1-yl) ethyl cinnamate (27)
Brown oil; yield: 38%; purity: 91.5%. 1H NMR (500 MHz, DMSO) δ 7.76–7.70 (m, 2H), 7.65 (d, J = 16.0 Hz, 1H), 7.48–7.37 (m, 3H), 6.65 (d, J = 16.0 Hz, 1H), 4.58 (s, br, 3H), 4.16 (t, J = 6.6 Hz, 2H), 3.80–3.70 (m, 1H), 3.58–3.53 (m, 2H), 3.25–3.18 (m, 1H), 3.07–3.01 (m, 1H), 2.95–2.90 (m, 1H), 2.85–2.78(m, 1H), 2.44–2.34 (m, 1H), 1.98–1.90 (m, 2H), 1.70–1.53 (m, 2H), 1.53–1.42 (m, 2H).13C NMR (125 MHz, DMSO) δ 166.75, 144.86, 134.51, 130.91, 129.39, 129.26, 128.84, 118.64, 79.71, 71.29, 69.92, 67.36, 64.56, 59.68, 57.32, 52.05, 26.77, 21.70.HRMS (ESI) m/z [M + H]+: calcd for C19H27NO6: 388.1731, found 388.1741.
5.3.4. 5-(3,4,5-Trihydroxy-2-(hydroxymethyl) piperidin-1-yl) ethyl cinnamate (28)
Brown oil; yield: 36%; purity: 93.5%. 1H NMR (500 MHz, DMSO) δ 7.76–7.70 (m, 2H), 7.66 (d, J = 16.0, 1H), 7.46–7.39 (m, 3H), 6.63 (d, J = 16.0, 1H), 4.78 (s, br, 3H), 4.29 (s, 1H), 4.15 (t, J = 6.6 Hz, 2H), 3.78–3.69 (m, 1H), 3.64–3.55 (m, 1H), 3.30–3.20 (m, 1H), 3.08 (t, J = 9.1 Hz, 1H), 2.95 (t, J = 8.8 Hz, 1H), 2.89–2.83 (m, 1H), 2.82–2.76 (m, 1H), 2.46 (s, 1H), 2.11–1.99 (m, 2H), 1.75–1.59 (m, 2H), 1.52–1.38 (m, 2H), 1.37–1.25 (m, 2H).13C NMR (125 MHz, DMSO) δ 166.75, 144.89, 134.48, 130.92, 129.40, 128.83, 118.60, 79.43, 70.93, 69.58, 67.07, 64.52, 59.18, 57.05, 52.38, 28.62, 24.41, 23.84. HRMS (ESI) m/z [M + H]+: calcd for C20H29NO6: 402.1887, found 402.1890.
5.3.5. 6-(3,4,5-Trihydroxy-2-(hydroxymethyl) piperidin-1-yl) ethyl cinnamate (29)
Brown oil; yield: 36%; purity: 93.8%. 1H NMR (500 MHz, DMSO) δ 7.75–7.69 (m, 2H), 7.65 (d, J = 16.0 Hz, 1H), 7.46–7.39 (m, 3H), 6.63 (d, J = 16.0, 1H), 4.83 (s, br, 3H), 4.39 (s, 1H), 4.14 (t, J = 6.6 Hz, 2H), 3.78–3.69 (m, 1H), 3.66–3.58 (m, 1H), 3.47–3.38 (m, 1H), 3.32–3.23 (m, 1H), 3.16–3.06 (m, 1H), 3.03–2.92 (m, 1H), 2.92–2.85 (m, 1H), 2.86–2.78 (m, 1H), 2.11 (s, 2H), 1.70–1.57 (m, 2H), 1.51–1.41 (m, 2H), 1.39–1.32 (m, 2H), 1.30–1.19 (m, 2H).13C NMR (125 MHz, DMSO) δ 166.75, 144.88, 134.48, 130.92, 129.39, 128.82, 118.60, 79.16, 70.60, 69.27, 66.89, 64.48, 58.63, 56.72, 52.43, 28.67, 26.95, 25.77, 24.42.HRMS (ESI) m/z [M + Na]+: calcd for C21H31NO6: 416.2044, found 416.2061.
5.3.6. Methyl(E)-3-(4-(2-(3,4,5-trihydroxy-2-(hydroxymethyl) piperidin-1-yl) ethoxy) phenyl) acrylate (30)
Pale brown solid; yield: 40%; m.p. 185–187 °C; purity: 90.2%. 1H NMR (500 MHz, DMSO) δ 7.66 (d, J = 6.5 Hz, 2H), 7.61 (d, J = 16, 1H), 6.97 (d, J = 8.0 Hz, 2H), 6.48 (d, J = 16.0, 1H), 4.71 (s, br, 3H), 4.28 (s, 1H), 4.13 (s, 2H), 3.71 (s, 3H), 3.59 (s, 1H), 3.27–3.13 (m, 2H), 3.03 (s, 1H), 2.95 (s, 2H), 2.86 (s, 1H), 2.18 (s, 1H), 2.09 (s, 2H).13C NMR (125 MHz, DMSO) δ 167.42, 161.09, 144.85, 130.62, 126.90, 115.42, 115.32, 79.69, 71.24, 69.89, 68.09, 67.34, 59.55, 57.24, 52.00, 51.56.HRMS (ESI) m/z [M + Na]+: calcd for C18H25NO7: 390.1523, found 390.1526.
5.3.7. Methyl(E)-3-(4-(3-(3,4,5-trihydroxy-2-(hydroxymethyl)piperidin-1-yl)ethoxy)phenyl)acrylate (31)
Pale brown solid; yield: 38%; m.p. 104–107 °C; purity: 92.2%. 1H NMR (500 MHz, DMSO) δ 7.66 (d, J = 8.6 Hz, 2H), 7.61 (d, J = 16.0 Hz, 1H), 6.97 (d, J = 8.6 Hz, 2H), 6.48 (d, J = 16.0 Hz, 1H), 4.86 (s, 3H), 4.46–4.15 (m, 1H), 4.05 (d, J = 5.6 Hz, 2H), 3.77 (d, J = 10.6 Hz, 1H), 3.71 (s, 3H), 3.64 (d, J = 22.4 Hz, 1H), 3.00 (t, J = 52.0 Hz, 4H), 2.09 (s, 2H), 1.90 (d, J = 11.2 Hz, 2H). 13 C NMR (125 MHz, DMSO) δ 167.38, 161.06, 144.81, 130.59, 126.96, 115.48, 115.34, 89.65, 79.65, 71.34, 69.88, 67.31, 66.81, 59.80, 57.52, 51.73, 49.01, 25.11.HRMS (ESI) m/z [M + H]+: calcd for C19H27NO7: 382.1860, found 382.1859.
5.3.8. Methyl(E)-3-(4-(4-(3,4,5-trihydroxy-2-(hydroxymethyl)piperidin-1-yl)ethoxy)phenyl)acrylate (32)
Pale brown solid; yield: 35%; m.p. 157–160 °C; purity: 95.2%. 1H NMR (500 MHz, DMSO) δ 7.65 (d, J = 8.7 Hz, 2H), 7.61 (d, J = 16.0 Hz, 1H), 6.97 (d, J = 8.7 Hz, 2H), 6.47 (d, J = 16.0 Hz, 1H), 4.69 (s, br, 3H), 4.25–4.09 (m, 1H), 4.06–4.00 (m, J = 6.2, 4.6 Hz, 2H), 3.75 (d, J = 11.4 Hz, 1H), 3.71 (s, 3H), 3.61–3.54 (m, 1H), 3.22 (s, 1H), 3.10–3.01 (m, 1H), 2.94 (s, 1H), 2.89–2.78 (m, 2H), 2.41 (s, 1H), 1.94 (s, 2H), 1.76–1.60 (m, 2H), 1.59–1.47 (m, 2H).13C NMR (126 MHz, DMSO) δ 167.42, 161.09, 144.85, 130.62, 126.90, 115.42, 115.32, 79.69, 71.24, 69.89, 68.09, 67.34, 59.55, 57.24, 52.00, 51.71, 26.95, 21.64.HRMS (ESI) m/z [M + Na]+: calcd for C20H29NO7: 418.1836, found 418.1840.
5.3.9. Methyl(E)-3-(4-(5-(3,4,5-trihydroxy-2-(hydroxymethyl)piperidin-1-yl)ethoxy)phenyl)acrylate (33)
Pale brown solid; yield: 37%; m.p. 85–86 °C; purity: 92.1%. 1H NMR (500 MHz, DMSO) δ 7.66 (d, J = 8.7 Hz, 2H), 7.61 (d, J = 16.0 Hz, 1H), 6.96 (d, J = 8.2 Hz, 2H), 6.48 (d, J = 16.0 Hz, 1H), 4.84–4.66 (m, 3H), 4.18 (s, 1H), 4.01 (t, J = 6.4 Hz, 2H), 3.74 (s, 1H), 3.71 (s, 3H), 3.60–3.51 (m, 1H), 3.21 (s, 1H), 3.08–3.00 (m, 1H), 2.98–2.88 (m, 1H), 2.85–2.80 (m, 1H), 2.78–2.72 (m, 1H), 2.39 (s, 1H), 1.79–1.67 (m, 2H), 1.43 (s, 2H), 1.39–1.29 (m, 2H), 1.24 (s, 2H).13C NMR (125 MHz, DMSO) δ 167.40, 161.09, 144.84, 130.62, 126.89, 115.41, 115.29, 79.69, 71.24, 69.91, 68.18, 67.23, 59.58, 57.36, 52.43, 51.80, 29.00, 24.74, 23.96.HRMS (ESI) m/z [M + H]+: calcd for C21H31NO7: 410.2173, found 410.2170.
5.3.10. Methyl(E)-3-(4-(5-(3,4,5-trihydroxy-2-(hydroxymethyl)piperidin-1-yl)ethoxy)phenyl)acrylate (34)
Pale brown solid; yield: 35%; m.p. 110–113 °C; purity: 95.5%. 1H NMR (500 MHz, DMSO) δ 7.65 (d, J = 8.6 Hz, 2H), 7.61 (d, J = 16.0 Hz, 1H), 6.97 (d, J = 8.6 Hz, 2H), 6.47 (d, J = 16.0 Hz, 1H), 4.99 (s, 4H), 4.01 (t, J = 6.2 Hz, 2H), 3.71 (s, 3H), 3.66 (s, 1H), 3.17 (s, 2H), 3.03 (s, 2H), 2.90 (s, 2H), 2.17 (d, J = 7.5 Hz, 1H), 1.74–1.69 (m, 2H), 1.46–1.38 (m, 4H), 1.32–1.27 (m, 2H).13C NMR (125 MHz, DMSO) δ 167.37, 161.10, 144.80, 132.59, 130.58, 126.92, 115.44, 115.30, 114.45, 79.71, 71.34, 69.93, 68.15, 67.17, 59.66, 57.42, 52.49, 51.72, 29.07, 27.21, 25.90, 24.98. HRMS (ESI) m/z [M + H]+: calcd for C22H33NO7: 424.2330, found424.2327.
5.3.11. Ethyl(E)-3-(3-methoxy-4-(2-(3,4,5-trihydroxy-2-(hydroxymethyl)piperidin-1-yl)ethoxy)phenyl)acrylate (35)
Pale yellow solid; yield: 32%; m.p. 146–148 °C; purity: 91.2%. 1H NMR (500 MHz, DMSO) δ 7.58 (d, J = 15.9 Hz, 1H), 7.35 (s, 1H), 7.22 (d, J = 8.2 Hz, 1H), 7.01 (d, J = 8.3 Hz, 1H), 6.55 (d, J = 15.9 Hz, 1H), 4.78 (s, 3H), 4.21–4.15 (m, 2H), 4.15–4.05 (m, 2H), 3.80 (s, 3H), 3.61 (s, 1H), 3.29–3.13 (m, 2H), 3.12–3.02 (m, 2H), 2.96 (s, 2H), 2.86 (s, 1H), 2.14 (d, J = 42.0 Hz, 2H), 1.26 (t, J = 7.1 Hz, 3H).13C NMR (125 MHz, DMSO) δ 166.99, 149.57, 145.01, 123.36, 116.15, 113.10, 111.12, 77.58, 73.23, 69.24, 67.15, 65.81, 60.25, 57.49, 56.21, 51.19, 45.16, 14.72.HRMS (ESI) m/z [M + Na]+: calcd for C20H29NO8: 434.1785, found 434.1785.
5.3.12. Ethyl(E)-3-(3-methoxy-4-(3-(3,4,5-trihydroxy-2-(hydroxymethyl)piperidin-1-yl)ethoxy)phenyl)acrylate (36)
Yellow oil; yield: 34%; purity: 90.5%.1H NMR (500 MHz, DMSO) δ 7.58 (d, J = 16.1 Hz, 1H), 7.35 (s, 1H), 7.22 (d, J = 7.9 Hz, 1H), 6.99 (d, J = 8.1 Hz, 1H), 6.55 (d, J = 15.9 Hz, 1H), 5.07 (d, J = 128.1 Hz, 3H), 4.23–4.10 (m, 2H), 4.04 (s, 2H), 3.81 (s, 3H), 3.76 (s, 1H), 3.68 (d, J = 12.7 Hz, 1H), 3.15 (d, J = 26.2 Hz, 2H), 3.05 (s, 3H), 1.95 (s, 2H), 1.23 (t, J = 7.1 Hz, 3H).13C NMR (125 MHz, DMSO) δ 166.98, 149.69, 144.98, 123.32, 116.20, 113.32, 111.25, 77.60, 73.25, 69.24, 67.13, 65.79, 60.30, 60.26, 56.23, 56.04, 51.20, 14.71, 14.54. HRMS (ESI) m/z [M + Na]+: calcd for C21H31NO8: 448.1942, found 448.1960.
5.3.13. Ethyl(E)-3-(3-methoxy-4-(4-(3,4,5-trihydroxy-2-(hydroxymethyl)piperidin-1-yl)ethoxy)phenyl)acrylate (37)
Yellow oil; yield: 35%; purity: 91.9%. 1H NMR (500 MHz, DMSO) δ 7.58 (d, J = 15.9 Hz, 1H), 7.35 (d, J = 1.4 Hz, 1H), 7.22 (d, J = 8.4, 1H), 6.98 (d, J = 8.5 Hz, 1H), 6.54 (d, J = 15.9 Hz, 1H), 4.76 (s, br, 3H), 4.22–4.14 (m, 2H), 4.03 (s, 2H), 3.81 (s, 3H), 3.76 (s, 1H), 3.75 (s, 1H), 3.62 (s, 1H), 3.26 (s, 2H), 3.10 (s, 1H), 3.03–2.83 (m, 4H), 1.71–1.66 (m, 2H), 1.56 (s, 2H), 1.26 (t, J = 7.1 Hz, 3H).13C NMR (125 MHz, DMSO) δ 167.00, 149.62, 145.06, 123.36, 116.04, 113.05, 111.09, 77.65, 73.30, 68.54, 66.75, 65.75, 60.29, 60.24, 56.15, 55.94, 51.99, 26.93, 14.72, 14.55. HRMS (ESI) m/z [M + Na]+: calcd for C22H33NO8: 440.2279, found 440.2283.
5.3.14. Ethyl(E)-3-(3-methoxy-4-(5-(3,4,5-trihydroxy-2-(hydroxymethyl)piperidin-1-yl)ethoxy)phenyl)acrylate (38)
Yellow oil; yield: 32%; purity: 90.6%. 1H NMR (500 MHz, DMSO) δ 7.61 (d, J = 15.9, 1H), 7.36 (s, 1H), 7.24 (s, 1H), 7.03 (d, J = 8.2 Hz, 1H), 6.56 (d, J = 15.9, 1H), 4.78 (s, 4H), 4.21–4.15 (m, 2H), 4.15–4.11 (m, 2H), 3.81 (s, 3H), 3.77–3.70 (m, 2H), 3.71 (s, 2H), 3.67 (s, 1H), 3.51 (s, 2H), 3.28 (s, 3H), 3.16–3.05 (m, 2H), 3.00 (s, 2H), 2.18 (s, 2H), 1.24 (t, J = 7.1 Hz, 3H).13C NMR (125 MHz, DMSO) δ 167.00, 149.57, 145.02, 123.34, 116.07, 112.99, 111.03, 77.59, 73.35, 68.52, 66.17, 65.59, 60.29, 60.26, 56.16, 55.94, 51.28.60, 23.48, 14.72, 14.55.HRMS (ESI) m/z [M + Na]+: calcd for C23H35NO8: 476.2255, found476.2256.
5.3.15. Ethyl(E)-3-(3-methoxy-4-(6-(3,4,5-trihydroxy-2-(hydroxymethyl)piperidin-1-yl)ethoxy)phenyl)acrylate (39)
Yellow oil; yield: 35%; purity: 92.1%. 1H NMR (500 MHz, DMSO) δ 7.57 (d, J = 15.9 Hz, 1H), 7.33 (d, J = 1.7 Hz, 1H), 7.20 (d, J = 8.3, 1H), 6.95 (d, J = 8.4 Hz, 1H), 6.52 (d, J = 15.9 Hz, 1H), 4.72 (s, 3H), 4.22–4.15 (m, 2H), 3.97 (t, J = 6.5 Hz, 2H), 3.81 (s, 3H), 3.74 (d, J = 10.8, 9.2 Hz, 1H), 3.58 (dd, J = 11.5, 3.0 Hz, 1H), 3.28–3.20 (m, 1H), 3.18 (s, 1H), 3.08 (t, J = 9.1 Hz, 1H), 2.94 (t, J = 8.8 Hz, 1H), 2.83 (dd, J = 11.0, 4.7 Hz, 1H), 2.80–2.72 (m, 1H), 2.46–2.37 (m, 1H), 2.04–1.93 (m, 2H), 1.77–1.65 (m, 2H), 1.47–1.34 (m, 4H), 1.29–1.20 (m, 5H).13C NMR (125 MHz, DMSO) δ 166.96, 150.89, 149.65, 144.99, 127.25, 123.29, 116.04, 113.05, 111.19, 79.57, 71.17, 69.77, 68.68, 67.07, 60.21, 59.40, 57.25, 56.17, 52.50, 49.06, 29.13, 27.17, 25.91, 24.86, 14.70.HRMS (ESI) m/z [M + H]+: calcd for C24H37NO8: 468.2592, found 468.2589.
5.3.16. (E)-3-phenyl-1-(2-(2-(3,4,5-trihydroxy-2-(hydroxymethyl)piperidin-1-yl)ethoxy)phenyl)prop-2-en-1-one (40)
Yellow oil; yield: 31%; purity: 95.1%. 1H NMR (500 MHz, DMSO) δ 7.77–7.71 (m, 2H), 7.54 (s, 1H), 7.53 (s, 2H), 7.52 (s, 1H), 7.45 (d, J = 1.9 Hz, 1H), 7.45–7.43(m, 2H), 7.23 (d, J = 8.2 Hz, 1H), 7.09–7.03 (m, 1H), 4.70 (s, br, 3H), 4.30–4.17 (m, 3H), 3.80–3.75 (m, 1H), 3.53 (d, J = 9.6 Hz, 1H), 3.22 (s, 2H), 3.04–2.98 (m, 1H), 2.93 (s, 2H), 2.13 (s, 1H), 2.09 (s, 2H).13C NMR (125 MHz, DMSO) δ 192.24, 157.83, 142.52, 135.11, 133.68, 130.84, 130.20, 129.47, 129.37, 128.95, 127.46, 121.10, 113.86, 79.53, 77.60, 73.25, 70.89, 69.24, 67.40, 65.80, 57.52, 51.50, 45.21. HRMS (ESI) m/z [M + Na]+: calcd for C23H27NO6: 436.1731, found436.1744.
5.3.17. (E)-3-Phenyl-1-(2-(3-(3,4,5-trihydroxy-2-(hydroxymethyl)piperidin-1-yl)ethoxy)phenyl)prop-2-en-1-one (41)
Yellow oil; yield: 34%; purity: 92.0%. 1H NMR (500 MHz, DMSO) δ 7.77–7.70 (m, 2H), 7.54–7.53 (m, 1H), 7.53–7.51 (m, 2H), 7.51 (s, 1H), 7.45 (d, J = 2.1 Hz, 1H), 7.44 (s, 1H), 7.32–7.24 (m, 1H), 7.20–7.16 (m, 1H), 7.08–7.03 (m, 1H), 4.66 (s, 3H), 4.14–4.10 (m, 2H), 4.09 (s, 1H), 3.68 (d, J = 11.4 Hz, 1H), 3.49 (d, J = 12.0 Hz, 1H), 3.19 (s, 1H), 3.07–2.97 (m, 1H), 2.95–2.85 (m, 2H), 2.80–2.71 (m, 1H), 2.49–2.43 (m, 1H), 2.09 (s, 2H), 1.95–1.85 (m, 2H). 13 C NMR (125 MHz, DMSO) δ 192.34, 157.84, 142.47, 135.11, 133.68, 130.85, 130.20, 129.46, 129.31, 128.87, 128.44, 127.58, 121.00, 113.65, 79.58, 71.24, 69.82, 67.57, 67.14, 59.61, 57.48, 49.20, 31.13, 25.42.HRMS (ESI) m/z [M + Na]+: calcd for C24H29NO6: 450.1887, found450.1898.
5.3.18. (E)-3-phenyl-1-(2-(4-(3,4,5-trihydroxy-2-(hydroxymethyl)piperidin-1-yl)ethoxy)phenyl)prop-2-en-1-one (42)
Yellow oil; yield: 34%; purity: 95.8%.1H NMR (500 MHz, DMSO) δ 7.74–7.68 (m, 2H), 7.56–7.49 (m, 4H), 7.48–7.42 (m, 3H), 7.19 (d, J = 8.6 Hz, 1H), 7.07–7.03 (m, 1H), 4.77–4.57 (m, 3H), 4.12 (t, J = 6.3 Hz, 3H), 3.67 (d, J = 11.4 Hz, 1H), 3.56–3.48 (m, 1H), 3.18 (s, 1H), 3.09–2.99 (m, 1H), 2.94–2.87 (m, 1H), 2.79–2.68 (m, 2H), 2.33–2.25 (m, 1H), 1.90–1.81 (m, 2H), 1.75–1.59 (m, 2H), 1.57–1.42 (m, 2H). 13 C NMR (125 MHz, DMSO) δ 192.39, 157.84, 142.43, 135.08, 133.68, 130.86, 130.19, 129.48, 129.30, 128.82, 127.58, 120.95, 113.66, 79.64, 71.22, 69.84, 68.62, 67.29, 59.47, 57.09, 51.91, 49.07, 27.15, 21.73.HRMS (ESI) m/z [M + H]+: calcd for C25H31NO6: 442.2224, found 442.2222.
5.3.19. (E)-3-phenyl-1-(2-(5-(3,4,5-trihydroxy-2-(hydroxymethyl)piperidin-1-yl)ethoxy)phenyl)prop-2-en-1-one (43)
Yellow oil; yield: 37%; purity: 95.3%. 1H NMR (500 MHz, DMSO) δ 7.75–7.69 (m, 2H), 7.55–7.49 (m, 4H), 7.47–7.42 (m, 3H), 7.18 (d, J = 8.6 Hz, 1H), 7.07–7.03 (m, 1H), 4.68 (s, 3H), 4.08 (t, J = 6.2 Hz, 2H), 3.64 (d, J = 11.0 Hz, 1H), 3.56–3.48 (m, 1H), 3.25–3.15 (m, 2H), 3.04 (t, J = 8.8 Hz, 1H), 2.91 (t, J = 8.9 Hz, 1H), 2.77–2.68 (m, 1H), 2.64–2.54 (m, 1H), 2.31–2.18 (m, 1H), 1.99–1.83 (m, 2H), 1.70 (s, 2H), 1.30 (s, 4H).13C NMR (125 MHz, DMSO) δ 192.40, 157.89, 142.28, 137.58, 135.11, 133.70, 130.86, 130.18, 129.44, 129.32, 129.00, 128.81, 127.68, 120.96, 113.59, 79.63, 71.20, 69.82, 68.71, 66.99, 59.40, 57.30, 52.31, 29.19, 24.71, 24.07.HRMS (ESI) m/z [M + H]+: calcd for C26H33NO6: 456.2381, found456.2376.
5.3.20. (E)-3-phenyl-1-(2-(6-(3,4,5-trihydroxy-2-(hydroxymethyl)piperidin-1-yl)ethoxy)phenyl)prop-2-en-1-one (44)
Yellow oil; yield: 35%; purity: 94.7%. 1H NMR (500 MHz, DMSO) δ 7.76–7.68 (m, 2H), 7.56–7.49 (m, 4H), 7.46–7.43 (m, 3H), 7.18 (d, J = 8.2 Hz, 1H), 7.08–7.02 (m, 1H), 4.72 (s, 3H), 4.09 (t, J = 6.0 Hz, 2H), 3.66 (d, J = 11.3 Hz, 1H), 3.54 (s, 1H), 3.22 (s, 1H), 3.07 (s, 1H), 2.94 (s, 1H), 2.75 (s, 1H), 2.65 (s, 1H), 2.32 (s, 1H), 1.95 (s, 2H), 1.76–1.63 (m, 2H), 1.42–1.30 (m, 2H), 1.26–1.16 (m, 2H), 1.15–1.04 (m, 2H). 13 C NMR (125 MHz, DMSO) δ 192.43, 167.48, 157.88, 142.26, 135.10, 133.85, 133.71, 131.59, 130.88, 130.18, 129.44, 129.30, 128.81, 127.72, 120.97, 113.53, 89.65, 68.68, 66.90, 56.14, 52.46, 40.97, 31.14, 29.32, 27.25, 26.18, 25.03.HRMS (ESI) m/z [M + H]+: calcd for C27H35NO6: 470.2537, found470.2524.
5.4. In vitro assay of α-glucosidase inhibitory activity
The method we used was reported before in our teamCitation23, α-glucosidase inhibitory activity was measured by using 0.1 mM phosphate buffer (pH 6.8) at 37 °C. The α-glucosidase enzyme (EC 3.2.1.20, 1 U/ml, 10 µL) in phosphate buffer was incubated with various concentrations of tested compounds (dissolved in 1% DMSO) at 37 °C for 20 min, then PNPG (10 mM, 20 µL) was added to the mixture as substrate. Finally, the absorbance was measured at 405 nm by using a spectrophotometer. The sample solution was replaced by DMSO as the control. Acarbose and 1-DNJ were used as standard drugs. The inhibition has been obtained using the formula:
5.5. Kinetic study
The kinetic analysis was carried out to ensure inhibition mode of the three most active compounds 43, 40, and 34. The 10 µL of enzyme solution (1 U/mL) was incubated with different concentrations of compound 43 (0.00 mM,0.012 mM, 0.02 mM, 0.04 mM), 40 (0.00 mM, 0.06 mM, 0.10 mM, 0.20 mM)and 34 (0.00 mM, 0.30 mM, 0.50 mM, 1.00 mM) for 20 min at 37 °C, then added different concentrations of substrate (1 mM, 0.9 mM, 0.75 mM, 0.6 mM, 0.45 mM, 0.3 mM, 0.25 mM), and change in absorbance was measured for 20 min at 405 nm by using spectrophotometer.
5.6. Molecular docking
Since the X-ray crystallographic structure of Saccharomyces cerevisiae α-glucosidase we used in the experiments has not been reported yet, the 3 D structural modelling of α-glucosidase was conducted with SWISS-MODELCitation22. The sequence in the FASTA format of α-glucosidase was download from NCBI. Isomaltase from Saccharomyces cerevisiae(PDB code 3AJ7) shows high sequence similarity (72.51%) with α-glucosidase, which structure was selected as the template for homology modelling, and the quality of the obtained homology model was verified using PROCHECKCitation25. The 3 D structures of acarbose and synthesised compounds were built by ChemBioDraw Ultra and ChemBio3D Ultra software. The AutoDock Tool 1.5.6 package was employed to generate the docking input files. Docking studies were performed using Autodock VinaCitation21. The centre of the grid boxCitation26 was placed at centre_x = 12.5825, centre_y = −7.8955, centre_z = 12.5190 with dimensions size_x = 40, size_y = 40, size_z = 40. The best-scoring poses as judged by the Vina docking score were chosen and visually analysed using PyMOL 1.8.0 software (http://www.pymol.org/).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Hirsh AJ, Yao SYM, Young JD, Cheeseman CI. Inhibition of glucose absorption in the rat jejunum: a novel action of alpha-D-glucosidase inhibitors. Gastroenterology 1997;113:205–11.
- Ross SA, Gulve EA, Wang M. Chemistry and biochemistry of type 2 diabetes. Chem Rev 2004;104:1255–82.
- Borges de Melo E, da Silveira Gomes A, Carvalho I. α- and β-Glucosidase inhibitors: chemical structure and biological activity. Tetrahedron 2006;62:10277–302.
- Mehta A, Zitzmann N, Rudd PM, et al. Alpha-glucosidase inhibitors as potential broad based anti-viral agents . FEBS Letters 1998;430:17–22.
- Clercq D. Erik Toward improved anti-HIV chemotherapy: therapeutic strategies for intervention with HIV infections. J Med Chem 1995;38:2491–517.
- Meneilly GS, Ryan EA, Radziuk J, et al. Effect of acarbose on insulin sensitivity in elderly patients with diabetes. Diabetes Care 2000;23:1162–7.
- Liu Z, Ma S. Recent advances in synthetic α-glucosidase inhibitors. ChemMedChem 2017;12:819–29.
- Compain P, Martin OR. Iminosugars: from synthesis to therapeutic applications. Chichester (UK): John Wiley and Sons; 2007.
- Yagi M, Kouno T, Aoyagi Y, Murai H. The structure of moraoline, a piperidine alkaloid from Morus species. Nippon Nogeik Kaishi 1976;50:571–2.
- Hatano A, Kanno Y, Kondo Y, et al. Use of a deoxynojirimycin-fluorophore conjugate as a cell-specific imaging probe targeting α-glucosidase on cell membranes. Bioorg Med Chem 2019;27:859–64.
- Nishimura Y. Gem-diamine 1-N-iminosugars and related iminosugars, candidate of therapeutic agents for tumor metastasis. Curr Top Med Chem 2003;3:575–91.
- Papandreou M-J. The alpha-glucosidase inhibitor 1-deoxynojirimycin blocks human immunodeficiency virus envelope glycoprotein-mediated membrane fusion at the CXCR4 binding step. Mol Pharm 2001;61:186–93.
- Wu H, Zeng W, Chen L, et al. Integrated multi-spectroscopic and molecular docking techniques to probe the interaction mechanism between maltase and 1-deoxynojirimycin, an α-glucosidase inhibitor. Int J Biol Macromol 2018;114:1194–202.
- Butters TD, van den Broek LAGM, Fleet GWJ, et al. Molecular requirements of imino sugars for the selective control of N-linked glycosylation and glycosphingolipid biosynthesis. Tetrahedron Asymmetry 2000;11:113–24.
- Nakagawa K, Kubota H, Kimura T, et al. Occurrence of orally administered mulberry 1-deoxynojirimycin in rat plasma. J Agric Food Chem 2007;55:8928–33.
- Faber ED, Oosting R, Neefjes JJ, et al. Distribution and elimination of the glycosidase inhibitors 1-deoxymannojirimycin and N-methyl-1-deoxynojirimycin in the rat in vivo. Pharm Res-Dordr 1992;9:1442–50.
- Ardes-Guisot N, Alonzi DS, Reinkensmeier G, et al. Selection of the biological activity of DNJ neoglycoconjugates through click length variation of the side chain. Org Biomol Chem 2011;9:5373–88.
- Rawlings AJ, Lomas H, Pilling AW, et al. Synthesis and biological characterisation of novel N-alkyl-deoxynojirimycin alpha-glucosidase inhibitors. Chembiochem 2009;10:1101–5.
- Zhang Y, Gao H, Liu R, et al. Quinazoline-1-deoxynojirimycin hybrids as high active dual inhibitors of EGFR and α-glucosidase. Bioorg Med Chem Lett 2017;27:4309–13.
- Lesur B, Ducep JB, Lalloz MN, et al. New deoxynojirimycin derivatives as potent inhibitors of intestinal α-glucohydrolases. Cheminform 2010;28:355–60.
- Trott O, Olson AJ. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010;31:455–61.
- Andrew W, Martino B, Stefan B, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 2018;46:W296–303.
- Zeng F, Yin Z, Chen J, et al. Design, synthesis, and activity evaluation of novel N-benzyl deoxynojirimycin derivatives for use as α-glucosidase inhibitors. Molecules 2019;24:3309.
- Shahzad D, Saeed A, Larik FA, et al. Novel C-2 symmetric molecules as α-glucosidase and α-amylase inhibitors: design, synthesis, kinetic evaluation, molecular docking and pharmacokinetics. Molecules 2019;24:1511–27.
- Kiefer F, Arnold K, Künzli M, et al. The SWISS-MODEL Repository and associated resources. Nucleic Acids Res 2009;37:D387–92.
- Wang G, Peng Z, Wang J, et al. Synthesis, in vitro evaluation and molecular docking studies of novel triazine-triazole derivatives as potential α-glucosidase inhibitors. Eur J Med Chem 2017;125:423–9.