
Abstract
Effective therapy of Alzheimer’s disease (AD) requires treatment with a combination of drugs that modulate various pathomechanisms contributing to the disease. In our research, we have focused on the development of multi-target-directed ligands – 5-HT6 receptor antagonists and cholinesterase inhibitors – with disease-modifying properties. We have performed extended in vitro (FRET assay) and in cellulo (Escherichia coli model of protein aggregation) studies on their β-secretase, tau, and amyloid β aggregation inhibitory activity. Within these multifunctional ligands, we have identified compound 17 with inhibitory potency against tau and amyloid β aggregation in in cellulo assay of 59% and 56% at 10 µM, respectively, hBACE IC50=4 µM, h5TH6 Ki=94 nM, hAChE IC50=26 nM, and eqBuChE IC50=5 nM. This study led to the development of multifunctional ligands with a broad range of biological activities crucial not only for the symptomatic but also for the disease-modifying treatment of AD.
1. Introduction
Alzheimer’s disease (AD) is among the top ten leading causes of death globally according to the World Health Organisation and among them, the only disease we cannot prevent, cure or even slow downCitation1. The available therapy – targeting cholinergic (donepezil, galantamine, and rivastigmine) and glutamatergic neurotransmission (memantine) – provide only a modest efficacy on cognitive impairmentCitation2. Therefore, the interest in the development of new drugs, preferentially targeting processes underlying the disease, is among urgent research goals.
AD belongs to a large group of “conformational diseases,” that are caused by misfolding, aggregation, and accumulation of certain proteins in tissues, leading to their dysfunctionCitation3. In AD, constitute proteins – amyloid β (Aβ) and tau protein – undergo changes resulting in the formation of amyloid plaques and neurofibrillary tangles (NFTs) that accumulate outside and inside neurons, respectively. The sequence of events leading to the development of the disease is still under discussion, but there is no doubt that both these proteins are necessary for the disease to developCitation4. The aim of the effective treatment directed at these proteins is to remove plaques and tangles or to stop them from developingCitation5,Citation6.
The main strategies for targeting Aβ pathology are dedicated to decrease its production by inhibition of β-secretase (BACE1), or modulation/inhibition of γ-secretase. Also, general strategies like passive and active immunisation or non-specific aggregation inhibition have been appliedCitation7. The return of monoclonal antibody aducanumab to Phase IIIb safety study for the FDA approval process has recently aroused great hopes and excitementCitation8,Citation9. It works to clear the amyloid plaques in the brain and even though the experts do not consider it as a final treatment for AD, it would be a milestone in the search for the combination therapy with anti-tau compounds. Recently, it was proven that peptide-based inhibitors of Aβ aggregation reduce aggregation and self-seeding of tau suggesting that these proteins share a common epitope thus, it is worth to control aggregation of both to stop the diseaseCitation10. It was also clarified in in vivo studies that the pathological role of interactions between Aβ and tau promotes neuronal dysfunctionCitation11. Thus, it seems reasonable to develop both Aβ and tau-targeted therapeutics.
Lack of effective therapy and the complexity of AD led to a change in the approach in the search for new anti-AD treatment. Extensive research was directed to the search for multifunctional ligands that led to the development of a large variety of compound classes targeting processes connected with causes and symptoms of AD. The identification of the most suited combination of drug targets is challenging and extremely important and should be consistent with the balanced activities against the targets of interestCitation12,Citation13. Among the anti-AD compounds in clinical trials up to 2019, the first three places belong to compounds targeting Aβ peptide, neurotransmitters system and tau proteinCitation14. Thus, a combination of these activities in one molecule may be a reasonable step towards effective slowing down of AD progression. The most explored multifunctional ligands combine anticholinesterase activity with anti-aggregation properties resulting from inhibition of β-secretase or modulation of γ-secretase and direct inhibition of processes of aggregation of Aβ and tau proteinsCitation15–18. Also, many G-protein coupled receptors are explored in combination with anticholinesterase activity, among them cannabinoid receptors CB1 and CB2Citation19–22, histamine H3 receptorsCitation23–25or serotonin 5-HT1ACitation26, 5-HT4, and 5-HT6 receptorsCitation27–31.
Here, we present the broadened in vitro and in cellulo studies on the activity of the previously published first-in-class multi-target-directed ligands () targeting serotonergic 5-HT6 receptors and cholinesterasesCitation30,Citation31. Based on the results from preclinical studies, modulation of these biological targets results in an increase of acetylcholine, glutamate, noradrenaline, and dopamine in the frontal cortex and enhanced performance in a variety of paradigms of cognitive impairment in animalsCitation32. Moreover, clinical trials showed the superiority of combination therapy with a cholinesterase inhibitor (donepezil) and 5-HT6 receptor antagonists (idalopirdine) over donepezil aloneCitation33. Guided by this evidence, we have designed multifunctional ligands − 5-HT6 antagonists and cholinesterase inhibitors – that displayed well-balanced potencies against the biological targets and proved their potential in in vivo studies.
Figure 1. Broaden screening against Aβ and tau-related targets of a library of multifunctional 5-HT6 antagonists and cholinesterase inhibitors.
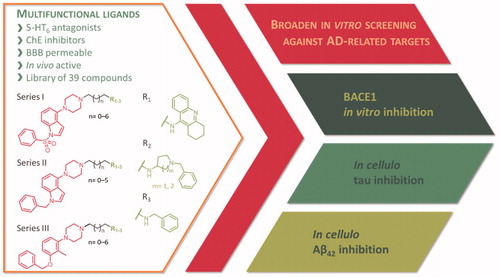
Since targeting aggregation processes may affect the processes underlying AD, we performed preliminary studies verifying the potential of these multifunctional ligands as inhibitors of Aβ and tau aggregation. Several of these compounds have already been tested for their anti-aggregating ability towards Aβ42 in vitroCitation31 using Thioflavin T (ThT) fluorescence assayCitation34. Their activity ranged from 48% to 95% at 10 µM screening concentration and, therefore, we decided to further explore their multifunctional profile and extend research on our in-house library of 39 multifunctional compounds. We have focussed on processes of aggregation by testing the compounds in cellulo in Escherichia coli cells overexpressing proteins of interest. We have verified in vitro results of Aβ42 anti-aggregating activity and tested the compounds against tau aggregation. Additionally, we have evaluated the inhibitory potential of the compounds against BACE1 – one of the key biological targets for Aβ pathology.
2. Results and discussion
2.1. Chemical profile of the compound library
The tested library of compounds () comprises three series of multifunctional ligands that contain pharmacophore fragments known for 5-HT6 receptors antagonism: 1-(phenylsulfonyl)-4-(piperazin-1-yl)-1H-indole (series I, ), 1-benzyl-4-(piperazin-1-yl)-1H-indole (series II, ), and 1–(3-(benzyloxy)-2-methylphenyl)piperazine (series III, ). These pharmacophore fragments were combined with fragments known for their effective inhibition of cholinesterases: tacrine, N-benzylpiperidine (derived from donepezil), and N-benzylpiperidine analogues: N-benzylamine and N-benzylpyrrolidine. Among the obtained multifunctional ligands, we have selected compounds excellent for further development. They displayed not only well-balanced activities against the targets of interest but also favourable preliminary ADMET and physicochemical properties. Therefore, we decided to pursue the research and to expand the panel of tests with in cellulo anti-aggregating activity and in vitro BACE1 inhibitory activity.
Table 1. Inhibition of tau and Aβ42 aggregation and BACE1 by compounds 1–16 (series I).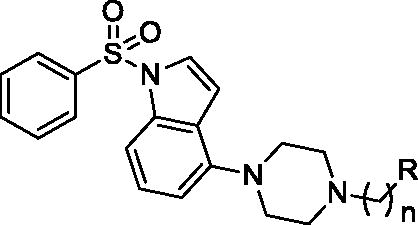
Table 2. Inhibition of tau and Aβ42 aggregation and BACE1 by compounds 17–26 (series II).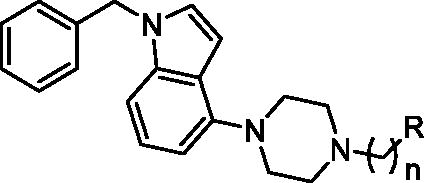
Table 3. Inhibition of tau and Aβ42 aggregation and BACE1 by compounds 27–39 (series III).
2.2. Anti-aggregating activity
We screened multifunctional ligands from our in-house library for their anti-aggregating activity against Aβ and tau in the whole bacterial cells using the Thioflavin S (ThS) fluorescence assayCitation35. Bacterial cells overexpressing different proteins provide simple models that can be easily employed to monitor the mechanisms of protein aggregation related to a variety of conformational diseasesCitation36,Citation37. In our assay, E. coli system has been implemented to study the effect of potential Aβ42 and tau aggregation inhibitors within intact cells. The advantage of the test over in vitro tests is the fact that it considers the complexity of the cellular environment that plays a crucial role in tuning protein aggregation. This assay has been validated and successfully applied to assess the anti-aggregating properties of diverse chemical structuresCitation37. Previously investigated tetrahydrobenzo[h][1,6] naphthyridine-6-chlorotacrine hybrid DP-128 was used as a reference compoundCitation38,Citation39.
At the 10 µM screening concentration all the compounds inhibited Aβ and tau aggregation with the following percentages of inhibition: 21–70% for Aβ and 12–67% for tau (, series I–III). Among them, we identified 12 inhibitors of tau and Aβ aggregation with the inhibitory potency exceeding 50%. Most compounds with such dual inhibitory activity are tacrine (5, 17, 20, 27–30) and N-benzylpiperidine (16, 37–39) derivatives. Tacrine is a widely used scaffold in the AD therapy researchCitation40. It provides compounds with reliable inhibitory activity against cholinesterases, its derivatives are repeatedly reported as Aβ aggregation inhibitorsCitation18,Citation22,Citation41,Citation42 and dual inhibitors of self-induced Aβ and tau aggregation potent in in vitro assays and living cellsCitation43. On the downside of the tacrine-based multifunctional ligands is the fact that very often they lack drug-likeness, mostly due to their high molecular weight, exceeded of limits logP values or potential toxicity. In this context, N-benzylpiperidine derived compounds, with better physicochemical properties, might be advantageous for further optimisation.
2.3. BACE1 inhibitory activity
To further explore the multifunctional profile of the ligands, we screened them for their inhibitory activity against human BACE1 using spectrofluorometric assay (fluorescence resonance energy transfer (FRET)-based). Past failures of BACE1 inhibitors in clinical trials may result from testing them at advanced stages of the disease. Therefore, BACE1 inhibitors are now being tested in earlier stages of AD and in asymptomatic subjects at risk of AD, with the hope of blocking the disease process before it becomes irreversibleCitation44.
We tested the compounds at the screening concentration of 50 µM and determined their inhibitory potency as the percentage of inhibition, which was ranging from 31% to 100% (). In the next step, we determined the IC50 values for the compounds that inhibited enzyme at the screening concentration in more than 70%. As a reference in this assay, we have used inhibitor IV (N1-[(1S,2R)-3-(cyclopropylamino)-2-hydroxy-1-(phenylmethyl)propyl]-5-[methyl(methylsulfonyl)amino]-N3-[(1R)-1-phenylethyl]-1,3-benzenedicarboxamide), which is a commercially available, cell-permeable, and potent inhibitor of BACE1Citation45,Citation46. We identified 13 BACE1 inhibitors with IC50 values ranging from 1.31 to 23.38 µM. Analysis of the determined IC50 values provides us with an interesting structure–activity relationship (SAR).
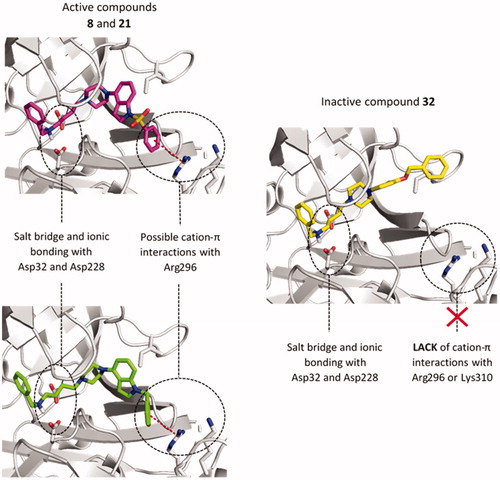
The most potent inhibitors were tacrine derivatives, with the majority of compounds’ IC50 values in the low micromolar range (1.31–8.07 µM). Again, it highlights tacrine as a privileged structure that causes multidirectional activities of its derivatives. The activity of the compounds with the tacrine moiety was maintained in all three series, although it was most stable within 1-(phenylsulfonyl)-4-(piperazin-1-yl)-1H-indole derivatives (series I, ). Compounds from series I all displayed µM potencies, while in series II compound 20, with the longest linker inhibited the enzyme in 62% and in series III 28 and 30 showed 67% and 34% of inhibition, respectively. Also, among non-tacrine derivatives, the most potent BACE1 inhibitors belong to series I: 7, 13, and 14. Importance of 1-(phenylsulfonyl)-4-(piperazin-1-yl)-1H-indole fragment for interactions with BACE1 can be observed on an example of N-benzylamine derivatives. Comparing three analogues from each series, compound 8 (series I), 21 (series II), and 32 (series III) we can see quite similar inhibitory potency for indole-based compounds IC50=19.66 µM and IC50=21.88 µM for 8 and 21, respectively, and decrease in activity for 32 − 42.8% at 50 µM concentration. These observations indicate an important role of 1-(phenylsulfonyl)-4-(piperazin-1-yl)-1H-indole, 1-benzyl-4-(piperazin-1-yl)-1H-indole and 1-(3-(benzyloxy)-2-methylphenyl)piperazine1-(3-(benzyloxy)-2-methylphenyl)piperazine fragments in the binding mode of the compounds within BACE1. Therefore, we chose compounds 8, 21, and 32 for docking studies to investigate the differences in the binding mode that could cause differences in activity.
The analysis of the predicted binding mode of compounds 8, 21, and 32 within BACE1 active site (), showed a uniform arrangement of the benzylamine fragment within subsites S3' and S2'. This position is consistent with X-ray crystallographic data on different BACE1 inhibitors containing the benzylamine fragmentCitation47,Citation48. We found the most significant changes in the arrangement of these compounds within the S3 and S4 subsites. In the case of indole derivatives (8, 21), the phenyl ring is directed towards the Arg296 and Lys310 and is engaged in a cation-π interaction. Distances between a centre of the aromatic ring and guanidine carbon of Arg296 were 5.2 Å (8) and 5.6 Å (21), respectively. In the complex used for docking, Lys 310 residue is outside the boundary for cation-π interactions (6 Å distance) (7.6 Å for comp. 8 and 7.0 Å for comp. 21), however, due to the high conformational freedom visible in many crystal structures, Lys310 may be important for binding of the active ligands. In 32, the benzyloxy fragment is mostly directed towards Ile110 where it creates weak, hydrophobic interactions. Additionally, the larger indole fragment creates more interactions with the S3 subsite than its bioisostere: methylphenol. Used GoldScore (GS) scoring function showed an approx. the 30-point difference in favour of indole derivatives than their reduced analogue (8 − 62.00, 21 − 56.55, and 32 − 30.89). These results suggest that interactions within subsides S3 and S4 cause the differences in compounds activities.
Figure 2. The predicted binding mode of compounds 8 (magenta), 21 (green), and 32 (yellow) in the binding site of BACE1. The detected cation-π interactions were marked as red lines connecting the centre of the aromatic ring and guanidine carbon of Arg296.
The influence of the linker length on the binding mode and the activity the tested compounds depends largely on the 5-HT6 and ChE-targeting fragments connected. We have observed two distinct binding modes analysing the docking results of the two most active tacrine derivatives with two (compound 1) and seven carbon atom linkers (compound 4). In both, the key factor is the formation of a salt bridge between the protonated nitrogen atom of the piperazine and the catalytic dyad (). In the case of compounds with a short linker like compound 1, we see tacrine in pockets S2' and S3' and the indole system interacting with the amino acids building the pockets S1–S3 (). This position was maintained in all analogous structures with a linker length of up to five carbon atoms. Compounds with the longer linkers (6–8 carbon atoms) show the opposite arrangement as shown on the example of compound 4 (). The indole system is now placed within pockets S2' and S3' and the linker remains rolled up in the hydrophobic space of the pocket S1 directing tacrine to the pocket S2 and S3. This change only slightly affects the formation of a salt bridge between the piperazine system and the catalytic dyad. Both arrangements are similar in terms of the number of specific bonds, such as hydrogen bonds, polar, or aromatic interactions. We have not observed such difference in the binding mode of benzylamine derivatives, which may result from the predominant role of interactions with Asp32 and Asp228.
Figure 3. Differences in the binding mode of compounds 1 (cyan), 4 (blue) and 24 (orange) within BACE1. The detected cation-π interactions were marked as red lines connecting the centre of the aromatic ring and guanidine carbon of Arg296 (4.3 Å) or Lys310 (5.6 Å).
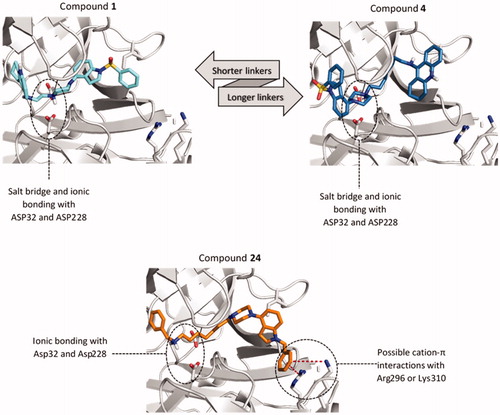
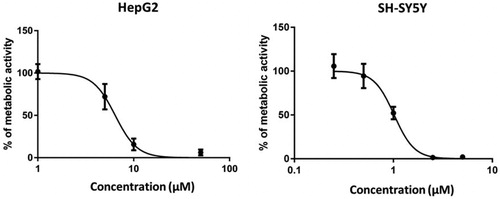
Figure 4. Compound 3 exerts concentration-dependent influence on the metabolic activity of HepG2 and SH-SY5Ycells. HepG2 and SH-SY5Ycells were incubated in the presence of increasing concentrations of compound 3 (1–100 µM) and (0.25–5 µM), respectively. After 24 h, cell viability was evaluated by MTS assay. A control group (DMSO) was considered as 100% of cell viability. Cells were treated in quadruplicate. The values are the mean ± SD of two independent experiments.
2.4. Structure–activity relationship analysis
We have described in details SAR of all the compounds in terms of their activity against 5-HT6 receptor and cholinesterases previouslyCitation30,Citation31. Briefly, SAR analysis led to the identification of a 1-(phenylsulfonyl)-4-(piperazin-1-yl)-1H-indole as a fragment that provides stable and strong affinity for 5-HT6 receptor regardless the cholinesterase-targeting fragment or the length of the linker (Ki=2–39 nM). The other 5-HT6-targeting moieties: 1-benzyl-4-(piperazin-1-yl)-1H-indole and 1-(3-(benzyloxy)-2-methylphenyl)piperazine1-(3-(benzyloxy)-2-methylphenyl)piperazine turned out to be much more sensitive to different substituents on the piperazine ring leading to decreased affinities for the receptor (Ki=72–916 nM for series II; Ki=18–845 nM for series III), with the highest affinities found for tacrine derivatives. In all three series of compounds tacrine moiety provided high, nanomolar potency towards both AChE and BuChE regardless of the linker (AChE IC50=5–57 nM and BuCHE IC50=5–45 nM). Herein, we have shown that tacrine, especially in combination with 1-(phenylsulfonyl)-4-(piperazin-1-yl)-1H-indole, contribute also to potent inhibition of BACE1 (IC50=1–8 µM). The potency against cholinestereases and BACE1 is more varied for N-benzylamine, N-benzylpiperidine, and N-benzylpyrrolidine derivatives. In terms of BACE1 inhibition some of these compounds (7, 13, or 14) show activity like those of tacrine derivatives and the others like compounds 33, 36, 38 inhibit the enzyme in only about 30% at 50 µM concentration. It also indicates the positive influence of 1-(phenylsulfonyl)-4-(piperazin-1-yl)-1H-indole fragment present in compounds 7, 13, and 14. The influence of the linker on the compounds’ activity towards BACE1 cannot be indicated, it depends on the combination of the fragments in each compound.
2.5. Cytotoxicity studies
We have determined the hepatotoxic and neurotoxic effects of compound 3 which is a representative of the most potent series and at the same time tacrine derivative, which potentially may carry a risk of hepatotoxicity. We have treated liver hepatocellular HepG2 and neuroblastoma SH-SY5Y cells with compound 3 for 24 h and determined cell viability assessed by MTS assay. We have observed a concentration-dependent decrease of cells’ viability with LC50 values of 6.4 and 1.0 µM on HepG2 and SH-SY5Y cells, respectively (). The LC50 values were significantly higher than the doses effective against 5-HT6 or cholinesterases (5-HT6 Ki =2 nM, hAChE IC50=13 nM and eqBuChE IC50=8 nM), but in the similar ranges to those active against BACE1 (IC50=7.3 µM), tau (38% at 10 µM), and amyloid β (30% at 10 µM). Therefore, in the course of the development of these compounds’ their toxicity will require further investigation.
3. Conclusions
AD is the main cause of dementia, sixth leading cause of death and from a perspective of pharmacotherapy unsolved problem and a huge challenge. Disorders in neurotransmission, especially in the cholinergic system, observed in patients with AD, led to the development of cholinesterase inhibitors as anti-AD drugs. In addition to the symptomatic intervention resulting from inhibition of cholinesterases, it was shown that they might display a disease-modifying effect due to the modulation of Aβ aggregation. Aggregation of Aβ and tau protein can be detected far ahead of the onset of disease symptoms. Therefore, inhibition or modulation of these processes is considered a major approach towards disease-modifying therapeutics.
Recently, we have developed in vitro and in vivo potent multifunctional ligands with BuChE inhibitory activity and 5-HT6 antagonism. Due to the interesting activity profile of these compounds, we decided to carry on an extended in vitro and in cellulo tests to verify their disease-modifying potential. We have tested them against crucial biological targets that contribute to the development of the disease: Aβ and tau protein aggregation and BACE1. These studies led to the emergence of a group of compounds that affect not only neurotransmission but also inhibit processes of Aβ and tau aggregation. SAR studies led to the conclusion that 1-(phenylsulfonyl)-4-(piperazin-1-yl)-1H-indole and tacrine are privileged structures that provide a broad profile of biological activities, including inhibitory potency against tau and Aβ aggregation in in cellulo assay ranging from 30% to 61% at 10 µM, hBACE1 with IC50 values from 2 to 8 µM, h5-HT6 Ki values between 2 and 36 nM, hAChE IC50 values between 1 and 46 nM and eqBuChE IC50 values in ranges from 8 to 21 nM. Also, noteworthy are non-tacrine derivatives 16 and 37–39 due to their potent anti-aggregating properties in cellulo, 57–63% against tau and 54–62% against Aβ. Compounds that inhibit both amyloid β and tau aggregation are in the minority among the multifunctional ligands against AD. Therefore, multifunctional ligands reported herein, not only potently inhibiting proteins’ aggregation but also displaying BACE1 and BuChE inhibitory potency and 5-HT6 antagonistic properties can be of great value for the development of new therapeutic strategies.
The study led to the emergence of multifunctional ligands with disease-modifying and symptomatic effects in in vitro and in cellulo models that are an excellent starting point for further search for anti-Alzheimer’s therapy.
4. Methods
4.1. Inhibition of Aβ42/tau aggregation in Escherichia coli overexpressing Aβ42/tau
4.1.1 Chemicals and bacterial culture media
The assays were conducted according to the procedure described previouslyCitation35. All general chemicals were purchased from Sigma-Aldrich (St. Louis, MO) . Reagents for bacterial media were purchased from Conda (Sevilla, Spain). M9 minimal medium for 100 mL: 10 mL of salts 10× (0.68 g of Na2HPO4, 0.30 g of KH2PO4, 0.05 g of NaCl, 0.10 g of NH4Cl), 0.2 mL of 1 M MgSO4, 0.2 mL of 50 mM CaCl2, 2.5 mL of 20% glucose, and 87.1 mL of H2O.
4.1.2. Aβ42/tau aggregation inhibition assay in Escherichia coli
E. coli competent cells BL21 (DE3) were transformed with the pET28a vector (Novagen, Inc., Madison, WI) carrying the DNA sequence of Aβ42 or with pTARA containing the RNA polymerase gene of T7 phage (T7RP) under the control of the promoter pBAD, then with pRKT42 vector encoding four repeats of tau protein in two inserts. For overnight culture preparation, 10 ml of M9 minimal medium containing 50 μg/mL of kanamycin (Aβ42) or 50 μg/mL of ampicillin and 12.5 μg/mL of chloramphenicol (tau) were inoculated with a colony of BL21 (DE3) bearing the plasmid Aβ42 or tau plasmids to be expressed at 37 °C and 180 rpm using an incubator shaker (Ovan, Barcelona, Spain). After overnight growth, the OD600 was usually 2 − 2.5. For expression of Aβ42 peptide/tau protein, 200 μL of overnight culture were transferred into Eppendorf tubes of 1.5 mL containing 790 μL of fresh M9 minimal medium with 50 μg/mL of kanamycin1 mM of isopropyl 1-thio-β-D-galactopyranoside (IPTG) (Aβ42) or 50 μg/mL of ampicillin, 12.5 μg/mL of chloramphenicol, 0.25% of arabinose (tau), and 25 μM of ThS and 10 μL of reference, tested compounds (1 mM in DMSO, final concentration: 10 μM) or DMSO. The samples were grown for 24 h at 37 °C and 180 rpm using an incubator shaker (Ovan). The samples with DMSO were prepared as a negative control (maximum level of Aβ42/tau, 0% of inhibition). In the positive controls, the M9 medium without IPTG (minimal level of Aβ42) or arabinose (minimal level of tau) was used. For absorbance and fluorescence determination, 200 μL of each Eppendorf tube was transferred in triplicate into 96-well plate. Finally, the fluorescence emission at 520 nm, when exciting at 440 nm and absorbance at 600 nm was recorded using the multi-mode microplate reader (Beckman Counter, Brea, CA). The absorbance at 600 nm (OD600) of these samples was assessed to check the bacterial growth and potential intrinsic toxicity of the tested compounds. Final data are the average of three independent experiments. DP-128 was used as a reference compoundCitation38.
4.2. In vitro inhibition of human BACE1
BACE1 FRET Assay Kit (Panvera, Madison, WI) was performed according to the manufacturer’s protocol with small modifications. The Kit includes purified baculovirus-expressed BACE1 and specific peptide substrate (Rh-EVNLDAEFK-quencher), based on the “Swedish” mutant of APP. The analysis was carried, using 384-well black microplates and a microplate reader (EnSpire Multimode; PerkinElmer, Waltham, MA). The wavelength was optimised for the 553 nm excitation and 576 nm emission. Stock solutions of all test compounds were prepared in DMSO and further diluted with assay buffer (50 mM sodium acetate; pH 4.5). In the first step, 10 μL of BACE1 substrate was mixed with 10 μL of test compound (or assay buffer; i.e. blank sample), then 10 μL of the enzyme (1 U/mL) was added to start the reaction. After 60 min of incubation at 25 °C, 10 μL of stop solution (2.5 M sodium acetate) was applied to stop the reaction. The fluorescence signal was read at 576 nm. Percent of inhibition was calculated from [1 − (S60 − S0)/(C60 − C0)] × 100, where S0 and S60 are fluorescence intensities of the test sample (enzyme, substrate, test compound) at the beginning of the reaction and after 60 min, respectively, while C0 and C60 are analogical fluorescence intensities of the blank sample (enzyme, substrate, buffer). All the compounds were tested at a screening concentration of 50 μM. Each compound was analysed in triplicate. Commercially available BACE1 Inhibitor IV (Calbiochem, Merck; Nottingham, UK) was used as the reference compound. The IC50 values were calculated using nonlinear regression (GraphPad Prism 5 [GraphPad Software, San Diego, CA, 5.00]) by plotting the residual enzyme activities against the applied inhibitor concentration.
4.3. Cytotoxicity studies for compound 3
4.3.1. Cell culture and treatments
The human liver carcinoma HepG2 cell line was purchased from American Type Culture Collection (LGC Standards, UK). The human neuroblastoma SH-SY5Y cell line was purchased from American Type Culture Collection (CRL-2266, Manassas, VA). The cells were grown in Advanced Dulbecco’s modified Eagle’s medium (Sigma, St. Louis, MO) supplemented with 10% foetal bovine serum (FBS) (HyClone, Logan, UT), 2 mM L-glutamine, 50 U/mL penicillin and 50 μg/mL streptomycin (Sigma, St. Louis, MO) in a humidified atmosphere of 95% air and 5% CO2 at 37 °C, and grown to 80% confluence. Compound 3 was prepared as a stock solution of 20 mM in DMSO and was used at concentrations of 1–100 µM in case of HepG2 treatment, and at concentrations of 0.25–5 µM in case of SH-SY5Y treatment.
4.3.2. Cell viability assay
HepG2 and SH-SY5Y cells were seeded in 96-well plates (2 × 104/well) and assessed by MTS ([3–(4,5-dimethylthiazol-2-yl)-5–(3-carboxymethoxyphenyl)-2–(4-sulfophenyl)-2H-tetrazolium, inner salt) assay for their response to compound 3. Cells were treated as described above, and cell viability was assessed after 24 h using the CellTiter 96® Aqueous One Solution Cell Proliferation Assay (Promega, Madison, WI), following the manufacturer’s instructions. Absorbance was measured with an automatic microplate reader (Tecan Safire, Switzerland) at a wavelength of 492 nm. Results are presented as a percentage of the control (DMSO).
4.4. Molecular modelling studies
All tested compounds were prepared in LigPrep (Schrödinger) and exported to mol2 files. Docking studies were performed with GOLD 5.3 (CCDC). Before docking, BACE1 protein (PDB code 4D8C) was prepared using Hermes 1.7 (CCDC). All histidine residues were protonated at Nε, the hydrogen atoms were added, a ligand and water molecules were removed. The binding sites were defined as all amino acid residues within a radius of 15 Å from ligand (BXD) in the BACE1 complex. We applied the automatic settings of the genetic algorithm for very flexible ligands (number of operations equal 125,000). We received 10 conformations by the ligand sorted by the GoldScore scoring function. Results of docking were visualised with PyMOL 0.99rc6 (DeLano Scientific LLC, San Francisco, CA).
Disclosure statement
No potential conflict of interest was by reported the author(s).
Additional information
Funding
References
- Prince M, Comas-Herrera A, Knapp M, et al. World Alzheimer report: improving healthcare for people living with dementia: coverage, quality and costs now and in the future. 2016. Available from: http://eprints.lse.ac.uk/67858/. [accessed 20 Dec 2019].
- Knight R, Khondoker M, Magill N, et al. A systematic review and meta-analysis of the effectiveness of acetylcholinesterase inhibitors and memantine in treating the cognitive symptoms of dementia. Dement Geriatr Cogn Disord 2018;45:131–51.
- Carrell RW, Lomas DA. Conformational disease. Lancet 1997;350:134–8.
- van der Kant R, Goldstein LSB, Ossenkoppele R. Amyloid-β-independent regulators of tau pathology in Alzheimer disease. Nat Rev Neurosci 2020;21:21–35.
- Cummings J, Lee G, Ritter A, et al. Alzheimer’s disease drug development pipeline: 2019. Alzheimers Dement 2019;5:272–93.
- Yiannopoulou KG, Papageorgiou SG. Current and future treatments for Alzheimer’s disease. Ther Adv Neuro Diso 2013;6:19–33.
- Godyń J, Jończyk J, Panek D, Malawska B. Therapeutic strategies for Alzheimer’s disease in clinical trials. Pharmacol Rep 2016;68:127–38.
- Huang LK, Chao SP, Hu CJ. Clinical trials of new drugs for Alzheimer disease. J Biomed Sci 2020;27:18.
- Schneider L. A resurrection of aducanumab for Alzheimer’s disease. Lancet Neurol 2020;19:111–2.
- Griner SL, Seidler P, Bowler J, et al. Structure-based inhibitors of amyloid beta core suggest a common interface with tau. Elife 2019;8:e46924.
- Busche MA, Wegmann S, Dujardin S, et al. Tau impairs neural circuits, dominating amyloid-β effects, in Alzheimer models in vivo. Nat Neurosci 2019;22:57–64.
- Bolognesi ML. Harnessing polypharmacology with medicinal chemistry. ACS Med Chem Lett 2019;10:273–5.
- Albertini C, Salerno A, de Sena Murteira Pinheiro P, Bolognesi ML. From combinations to multitarget‐directed ligands: a continuum in Alzheimer’s disease polypharmacology. Med Res Rev 2020;1–28. DOI:10.1002/med.21699
- Liu PP, Xie Y, Meng XY, Kang JS. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct Target Ther 2019;4:29.
- Wichur T, Więckowska A, Więckowski K, et al. 1-Benzylpyrrolidine-3-amine-based BuChE inhibitors with anti-aggregating, antioxidant and metal-chelating properties as multifunctional agents against Alzheimer’s disease. Eur J Med Chem 2020;187:111916.
- Sang Z, Wang K, Shi J, et al. Apigenin-rivastigmine hybrids as multi-target-directed liagnds for the treatment of Alzheimer’s disease. Eur J Med Chem 2020;187:111958.
- Costa GP, Baldinotti RSM, Fronza MG, et al. Synthesis, molecular docking, and preliminary evaluation of 2-(1,2,3-triazoyl)benzaldehydes as multifunctional agents for the treatment of Alzheimer’s disease. ChemMedChem 2020;15:610–22.
- Chalupova K, Korabecny J, Bartolini M, et al. Novel tacrine-tryptophan hybrids: multi-target directed ligands as potential treatment for Alzheimer’s disease. Eur J Med Chem 2019;168:491–514.
- Montanari S, Mahmoud AM, Pruccoli L, et al. Discovery of novel benzofuran-based compounds with neuroprotective and immunomodulatory properties for Alzheimer’s disease treatment. Eur J Med Chem 2019;178:243–58.
- Dolles D, Nimczick M, Scheiner M, et al. Aminobenzimidazoles and structural isomers as templates for dual-acting butyrylcholinesterase inhibitors and hCB2 R ligands to combat neurodegenerative disorders. ChemMedChem 2016;11:1270–83.
- González-Naranjo P, Pérez-Macias N, Campillo NE, et al. Cannabinoid agonists showing BuChE inhibition as potential therapeutic agents for Alzheimer’s disease. Eur J Med Chem 2014;73:56–72.
- Scheiner M, Dolles D, Gunesch S, et al. Dual-acting cholinesterase-human cannabinoid receptor 2 ligands show pronounced neuroprotection in vitro and overadditive and disease-modifying neuroprotective effects in vivo. J Med Chem 2019;62:9078–102.
- Malek R, Arribas RL, Palomino-Antolin A, et al. New dual small molecules for Alzheimer’s disease therapy combining histamine H3 receptor (H3R) antagonism and calcium channels blockade with additional cholinesterase inhibition. J Med Chem 2019;62:11416–22.
- Bajda M, Łażewska D, Godyń J, et al. Search for new multi-target compounds against Alzheimer’s disease among histamine H3 receptor ligands. Eur J Med Chem 2020;185:111785.
- Ghamari N, Dastmalchi S, Zarei O, et al. In silico and in vitro studies of two non-imidazole multiple targeting agents at histamine H3 receptors and cholinesterase enzymes. Chem Biol Drug Des 2020;95:279–90.
- Li X, Wang H, Xu Y, et al. Novel vilazodone-tacrine hybrids as potential multitarget-directed ligands for the treatment of Alzheimer’s disease accompanied with depression: design, synthesis, and biological evaluation. ACS Chem Neurosci 2017;8:2708–21.
- Hatat B, Yahiaoui S, Lecoutey C, et al. A novel in vivo anti-amnesic agent, specially designed to express both acetylcholinesterase (AChE) inhibitory, serotonergic subtype 4 receptor (5-HT4R) agonist and serotonergic subtype 6 receptor (5-HT6R) inverse agonist activities, with a potential interest against Alzheimer’s disease. Front Aging Neurosci 2019;11:148.
- Rochais C, Lecoutey C, Hamidouche K, et al. Donecopride, a Swiss army knife with potential against Alzheimer's disease. Br J Pharmacol 2020;177:1988–2005.
- Lecoutey C, Hedou D, Freret T, et al. Design of donecopride, a dual serotonin subtype 4 receptor agonist/acetylcholinesterase inhibitor with potential interest for Alzheimer’s disease treatment. Proc Natl Acad Sci USA 2014;111:3825–30.
- Więckowska A, Kołaczkowski M, Bucki A, et al. Novel multi-target-directed ligands for Alzheimer’s disease: combining cholinesterase inhibitors and 5-HT6 receptor antagonists. Design, synthesis and biological evaluation. Eur J Med Chem 2016;124:63–81.
- Więckowska A, Wichur T, Godyń J, et al. Novel multitarget-directed ligands aiming at symptoms and causes of Alzheimer’s disease. ACS Chem Neurosci 2018;9:1195–214.
- Khoury R, Grysman N, Gold J, et al. The role of 5 HT6-receptor antagonists in Alzheimer’s disease: an update. Expert Opin Investig Drugs 2018;27:523–33.
- Wilkinson D, Windfeld K, Colding-Jørgensen E. Safety and efficacy of idalopirdine, a 5-HT 6 receptor antagonist, in patients with moderate Alzheimer’s disease (LADDER): a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol 2014;13:1092–9.
- Levine H. Thioflavine T interaction with synthetic Alzheimer’s disease beta-amyloid peptides: detection of amyloid aggregation in solution. Protein Sci 1993;2:404–10.
- Pouplana S, Espargaro A, Galdeano C, et al. Thioflavin-S staining of bacterial inclusion bodies for the fast, simple, and inexpensive screening of amyloid aggregation inhibitors. Curr Med Chem 2014;21:1152–9.
- Ami D, Natalello A, Lotti M, Doglia SM. Why and how protein aggregation has to be studied in vivo. Microb Cell Fact 2013;12:17.
- Caballero AB, Espargaró A, Pont C, et al. Bacterial inclusion bodies for anti-amyloid drug discovery: current and future screening methods. Curr Protein Pept Sci 2019;20:563–76.
- di Pietro O, Pérez-Areales FJ, Juárez-Jiménez J, et al. Tetrahydrobenzo[h][1,6]naphthyridine-6-chlorotacrine hybrids as a new family of anti-Alzheimer agents targeting β-amyloid, tau, and cholinesterase pathologies. Eur J Med Chem 2014;84:107–17.
- Espargaró A, Medina A, di Pietro O, et al. Ultra rapid in vivo screening for anti-Alzheimer anti-amyloid drugs. Sci Rep 2016;6:23349.
- Sameem B, Saeedi M, Mahdavi M, Shafiee A. A review on tacrine-based scaffolds as multi-target drugs (MTDLs) for Alzheimer’s disease. Eur J Med Chem 2017;128:332–45.
- Zha X, Lamba D, Zhang L, et al. Novel tacrine-benzofuran hybrids as potent multitarget-directed ligands for the treatment of Alzheimer’s disease: design, synthesis, biological evaluation, and x-ray crystallography. J Med Chem 2016;59:114–31.
- Fancellu G, Chand K, Tomás D, et al. Novel tacrine-benzofuran hybrids as potential multi-target drug candidates for the treatment of Alzheimer’s disease. J Enzyme Inhib Med Chem 2020;35:211–26.
- Lv P, Xia CL, Wang N, et al. Synthesis and evaluation of 1,2,3,4-tetrahydro-1-acridone analogues as potential dual inhibitors for amyloid-beta and tau aggregation. Bioorg Med Chem 2018;26:4693–705.
- Panza F, Lozupone M, Solfrizzi V, et al. BACE inhibitors in clinical development for the treatment of Alzheimer's disease. Expert Rev of Neurother 2018;18:847–57.
- ben Halima S, Mishra S, Raja KMP, et al. Specific inhibition of β-secretase processing of the Alzheimer disease amyloid precursor protein. Cell Rep 2016;14:2127–41.
- Stachel SJ, Coburn CA, Steele TG, et al. Structure-based design of potent and selective cell-permeable inhibitors of human β-secretase (BACE-1). J Med Chem 2004;47:6447–50.
- Rueeger H, Lueoend R, Rogel O, et al. Discovery of cyclic sulfone hydroxyethylamines as potent and selective β-site APP-cleaving enzyme 1 (BACE1) inhibitors: structure-based design and in vivo reduction of amyloid β-peptides. J Med Chem 2012;55:3364–86.
- Rueeger H, Rondeau JM, McCarthy C, et al. Structure based design, synthesis and SAR of cyclic hydroxyethylamine (HEA) BACE-1 inhibitors. Bioorganic Med Chem Lett 2011;21:1942–7.