
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
The synthesis and carbonic anhydrase (CA; EC 4.2.1.1) activating effects of a series of oxime ether-based amino alcohols towards four human (h) CA isoforms expressed in human brain, hCA I, II, IV and VII, are described. Most investigated amino alcohol derivatives induced a consistent activation of the tested CAs, with KAs spanning from a low micromolar to a medium nanomolar range. Specifically, hCA II and VII, putative main CA targets when central nervous system (CNS) diseases are concerned, were most efficiently activated by these oxime ether derivatives. Furthermore, a multitude of selective hCA VII activators were identified. As hCA VII is one of the key isoforms involved in brain metabolism and other brain functions, the identified potent and selective hCA VII activators may be considered of interest for investigations of various therapeutic applications or as lead compounds in search of even more potent and selective CA activators.
1. Introduction
Activators of the metalloenzymes carbonic anhydrases (CAs; EC 4.2.1.1, CAAs) have been lately going through a second youth in drug discovery processesCitation1. Early evidence of the CA activation efficacy of amines, such as histamine, dating back to the 1940s was thereafter long debated up, and even considered as an experimental artefactCitation2,Citation3. In the early 1990s, the combination of highly purified enzymes and precise techniques, such as the stopped flow assay, put an end to the long controversy, testifying the undeniable existence of CAAsCitation4–7. CAAs intervene in the second and rate determining step of the catalytic mechanism of carbon dioxide reversible hydration (EquationEquations 1(1)
(1) and Equation2
(2)
(2) ), that is the regeneration of the catalytically active, zinc hydroxide species, by a proton transfer reaction from the Zn2+ – bound water molecule to the external medium (EquationEquation 2
(2)
(2) )Citation6–8. This process is assisted by active site residues acting as proton shuttle, such as His residues placed in the middle or at the entrance of the active site cavity of the α-class human CAsCitation4.
(1)
(1)
(2)
(2)
(3)
(3)
The activators non-competitively activate the CAs through the formation of a ternary complex consisting of the enzyme, the substrate, and the activator. CAAs bind apart from the metal-coordination system, namely at the middle edge of CAs active site cavity, where they assist the proton shuttlingCitation9–11. As a proof of this phenomenon, it was demonstrated that efficient activators possess pKa values in the range of 6–8, values similar to the pKa of the His imidazole moietyCitation7.
It is seemingly odd that CAs, among the most efficient enzymes in Nature, may be activated for biomedical purposesCitation8. However, genetic deficiencies of several CA isoforms (e.g. human CA I, II, IV, VA, XII and XIV) have been reported in the last decades, associated to diseases such as osteopetrosis, cerebral calcifications, retinal problems, hyperammonaemia, hyperchlorhydrosisCitation12–15, and the loss of function of these enzymes would in principle be treatable with CA selective activatorsCitation8. In addition, there is evidence that CAs activation improves memory deficits, cognitive performance and learningCitation16–18, being nine of the fifteen known human CA isoforms present in brainCitation19,Citation20. Contrariwise, other evidence supported that CA inhibitors (CAIs) impair memory in human, according to studies on the CAIs topiramate and acetazolamide during acute high-altitude exposureCitation21,Citation22. Thus, CAs represent a crucial family of new targets for improving cognition, but also in therapeutic areas, such as phobias, obsessive-compulsive disorder, generalised anxiety, and post-traumatic stress disorders, for which few effective therapies are available. In fact, in a recent paper, one of our groups showed that the CAA D-phenylalanine and the CAI acetazolamide are respectively able to reinforce and impair extinction memory, that is, a new memory trace that inhibits the expression of the memory of a traumatic eventCitation23. In this golden period for CAAs, the discovery of new brain isoform selective CAAs (as well as CAIs) is invaluable to elucidate the role of CA isoforms in brain processes. In addition, CAAs are considered relevant in artificial tissues and in CO2 capture and sequestration processesCitation1,Citation24.
Here, we extend the knowledge of CAA chemotypes by describing the synthesis and CA activating effects of a series of oxime ether based amino alcohols towards four hCA isoforms expressed in human brain.
2. Material and methods
2.1. Chemistry
1H and 13 C NMR spectra were recorded on a Bruker Avance III HD 400 MHz spectrometer. Chemical shifts (δ) are reported in parts per million and coupling constants (J) are reported in hertz (Hz). 13 C NMR spectra were fully decoupled. The following abbreviations were used to explain multiplicities: singlet (s), doublet (d), triplet (t), double doublet (dd), broad (br), and multiplet (m). Chromatographic separations were performed on silica gel columns by flash column chromatography (Kieselgel 40, 0.040−0.063 mm, Merck). Reactions were followed by thin-layer chromatography (TLC) on Merck aluminium silica gel (60 F254) sheets that were visualised under a UV lamp. Evaporation was performed in vacuo (rotating evaporator). Sodium sulphate was always used as the drying agent. Commercially available chemicals were purchased from Sigma-Aldrich.
2.1.1. General procedure for the synthesis of amino alcohols 1–13
A solution of the proper oxime (17–27) (10 mmol) in anhydrous DMF (10 ml) was added portionwise to a solution of MeONa in MeOH (30 ml), prepared from anhydrous MeOH (30 ml) and Na (11 mmol). The reaction mixture was stirred at 60 °C for 1 h and then cooled at rt. Epichlorohydrin (0.86 ml, 11 mmol) dissolved in anhydrous DMF (10 ml) was added dropwise, and the resulting mixture was stirred for 1 h at rt, poured into water (100 ml) and extracted with CHCl3 (2 × 100mL). The organic phases were combined, washed with water (2 × 200 ml), dried (Na2SO4) filtered and evaporated under reduced pressure. The crude was distilled in vacuo affording an oil corresponding to the proper epoxide (28–38, yield 75–85%). NMR data of 28–35 were in accordance with those reported in literatureCitation25–33.
(E)-4-Chlorobenzaldehyde O-oxiran-2-ylmethyl oxime (36): yield 76%; 1H NMR (400 MHz, CDCl3): δ 8.40 (s, 1H), 7.35 (m, 2H), 7.30 (m, 2H), 4.38 (dd, J = 12.3 Hz, J = 3.5 Hz, 1H), 4.13 (dd, J = 12.3 Hz, J = 3.5 Hz, 1H), 3.28–3.32 (m, 1H), 2.88 (dd, J = 5.2 Hz, J = 4.2 Hz, 1H), 2.70 (dd, J = 5.2 Hz, J = 4.2 Hz, 1H).
(E)-2-Chlorobenzaldehyde O-oxiran-2-ylmethyl oxime (37): yield 82%; 1H NMR (400 MHz, CDCl3): δ 8.34 (s, 1H), 7.37 (m, 2H), 7.28–7.24 (m, 2H), 4.35 (dd, J = 12.3 Hz, J = 3.5 Hz, 1H), 4.06 (dd, J = 12.3 Hz, J = 3.5 Hz, 1H), 3.30–3.34 (m, 1H), 2.90 (dd, J = 5.2 Hz, J = 4.2 Hz, 1H), 2.76 (dd, J = 5.0 Hz, J = 4.2 Hz, 1H).
(E)-3-Chlorobenzaldehyde O-oxiran-2-ylmethyl oxime (38): yield 79%; 1H NMR (400 MHz, CDCl3): δ 8.51 (s, 1H), 7.34–7.28 (m, 1H), 7.26–7.23 (m, 3H), 4.32 (dd, J = 12.1 Hz, J = 3.7 Hz, 1H), 4.18 (dd, J = 12.1 Hz, J = 3.7 Hz, 1H), 3.31–3.35 (m, 1H), 2.88 (dd, J = 5.0 Hz, J = 4.1 Hz, 1H), 2.67 (dd, J = 5.0 Hz, J = 4.1 Hz, 1H).
A stirred solution of epoxide (28–38, 10 mmol) in dry benzene (6 ml) was treated with an excess of isopropylamine or tert-butylamine (50 mmol). The reaction mixture was stirred for 12 h at 90 °C, and then evaporated. The crude was dissolved in a mixture of MeOH/EtOH 3:7 (20 ml) and treated with 1.2 equivalent of the proper organic acid (oxalic, malic or fumaric acids) to give the corresponding amino alcohol (1–13) as a white solid salt.
(E)-2,3-Dihydro-1H-inden-1-one O-(3-(tert-butylamino)-2-hydroxypropyl) oxime maleate 1: Maleate salt. 1H NMR (400 MHz, CDCl3): δ 9.51 (s, 1H), 7.94 (s, 1H), 7.61 (d, J = 7.6 Hz, 1H), 7.34–7.31(m, 2H), 7.26–7.23 (m, 1H), 6.18 (s, 2H), 4.43–4.38 (m, 1H), 4.29 (dd, J = 4.08 Hz, J = 12 Hz, 1H), 4.19 (dd, J = 6.0 Hz, J = 11.6 Hz, 1H), 3.21–3.28 (m, 1H); 3.05–3.02 (m, 3H), 2.90–2.87 (m, 2H), 1.43 (s, 9H). 13 C NMR (100 MHz, DMSO-d6): δ 167.7, 163.5, 148.9, 136.6, 135.8, 131.1, 127.5, 126.4, 121.4, 75.7, 66.1, 56.8, 44.4, 28.5, 26.8, 25.45.
(E)-Benzaldehyde O-(3-(tert-butylamino)-2-hydroxypropyl) oxime maleate 2: Maleate salt. 1H NMR (400 MHz, DMSO-d6): δ 8.32 (s, 1H), 7.65–7.62 (m, 2H), 7.45–7.44 (m, 3H), 6.02 (s, 2H), 4.14–4.13 (m, 2H), 3.70–3.67 (m, 1H), 3.11–3.08 (m, 1H), 2.88–2.83 (m, 1H), 1.28 (s, 9H). 13 C NMR (100 MHz, DMSO-d6): δ 167.2, 149.5, 136.1, 131.7, 130.1, 128.8, 126.9, 75.4, 65.5, 56.4, 43.9, 25.0.
Propan-2-one O-(3-(tert-butylamino)-2-hydroxypropyl) oxime oxalate 3: oxalate salt. 1H NMR (400 MHz, DMSO-d6): δ 4.00–3.87 (m, 3H), 2.99 (dd, J = 2.4 Hz, J = 12.4 Hz, 1H), 2.76 (dd, J = 9.1 Hz, J = 12.4 Hz, 1H), 1.81 (s, 3H), 1.26 (s, 9H). 13 C NMR (100 MHz, DMSO-d6): δ 164.7, 155.0, 74.4, 65.5, 56.0, 44.1, 25.0, 21.2.
(E)-4-Methoxybenzaldehyde O-(3-(tert-butylamino)-2-hydroxypropyl) oxime maleate 4: Maleate salt. 1H NMR (400 MHz, DMSO-d6): δ 8.24 (s, 1H), 7.58 (m, 2H), 7.02 (m, 2H), 6.01 (s, 2H), 4.27–4.07 (m, 6H), 3.33 (s, 3H), 3.15–3.05 (m, 1H), 2.94–2.89 (m, 1H), 1.27 (s, 9H). 13 C NMR (100 MHz, DMSO-d6): δ 165.4, 161.2, 149.4, 128.9, 124.7, 114.8, 75.8, 65.9, 56.5, 55.7, 44.6, 25.4.
(E)-3-Methoxybenzaldehyde O-(3-(tert-butylamino)-2-hydroxypropyl) oxime oxalate 5: Oxalate salt. 1H NMR (400 MHz, DMSO-d6): δ 8.27 (s, 1H), 7.35 (t, J = 7.6 Hz, 2H), 7.21–7.17 (m, 2H), 7.01 (dd, J = 1.6 Hz, J = 8 Hz, 1H), 4.12–4.05 (m, 3H), 3.77 (s, 3H), 3.09–3.06 (m, 1H), 2.86–2.81 (m, 1H), 1.28 (s, 9H). 13 C NMR (100 MHz, DMSO-d6): δ 165.4, 159.9, 149.8, 133.6, 130.4, 119.9, 116.5, 112.1, 76.1, 65.9, 56.5, 55.6, 44.6, 25.4.
(E)-2-Methoxybenzaldehyde O-(3-(tert-butylamino)-2-hydroxypropyl) oxime oxalate 6: Oxalate salt. 1H NMR (400 MHz, DMSO-d6): δ 8.42 (s, 1H), 7.65 (dd, J = 1.6 Hz, J = 8 Hz, 2H), 7.43 (m, 1H), 7.10 (d, J = 8 Hz, 1H), 6.99 (t, J = 7.2 Hz, 1H), 4.11–4.05 (m, 3H), 3.83 (s, 3H), 3.07 (bd, J = 12 Hz, 1H), 2.86–2.83 (m, 1H), 1.28 (s, 9H). 13 C NMR (100 MHz, DMSO-d6): δ 165.2, 157.7, 145.2, 132.2, 126.3, 121.1, 120.0, 112.4, 76.0, 65.6, 56.2, 50.2, 47.3, 19.2.
(E)-3-Chlorobenzaldehyde O-(3-(tert-butylamino)-2-hydroxypropyl) oxime oxalate 7: Oxalate salt. 1H NMR (400 MHz, DMSO-d6): δ 8.32 (s, 1H), 7.68 (m, 1H), 7.60–7.59 (m, 1H), 7.50–7.47 (m, 2H), 4.15–4.05 (m, 3H), 3.08–3.05 (bd, J = 12 Hz, 1H), 2.83 (dd, J = 8.8 Hz, J = 12.4, 1H), 1.27 (s, 9H). 13 C NMR (100 MHz, DMSO-d6): δ 165.4, 148.6, 134.4, 131.3, 130.3, 126.9, 125.9, 76.3, 65.9, 56.5, 44.6, 25.5.
(E)-4-Chlorobenzaldehyde O-(3-(tert-butylamino)-2-hydroxypropyl) oxime oxalate 8: Oxalate salt. 1H NMR (400 MHz, DMSO-d6): δ 8.32 (s, 1H), 7.68 (m, 1H), 7.65 (m, 2H), 7.51 (m, 2H), 4.14–4.09 (m, 3H), 3.09–3.05 (bd, J = 12.4 Hz, 1H), 2.83 (m, 1H), 1.27 (s, 9H). 13 C NMR (100 MHz, DMSO-d6): δ 165.4, 148.6, 134.4, 131.2, 130.2, 126.9, 125.9, 76.3, 65.9, 56.5, 44.6, 25.4.
Cyclohexanone O-(2-hydroxy-3-(isopropylamino)propyl) oxime oxalate 9: Oxalate salt. 1H NMR (400 MHz, DMSO-d6): δ 3.90–3.83 (m, 3H), 3.01–2.98 (m, 1H), 2.80–2.77 (m, 1H), 2.67–2.63 (m, 1H), 2.40–2.38 (m, 2H), 2.12 (t, J = 6.8 Hz, 2H), 1.61–1.48 (m, 6H), 1.08 (d, J = 6.4 Hz, 6H). 13 C NMR (100 MHz, DMSO-d6): δ 165.35, 160.0, 75.5, 67.0, 49.4, 31.8, 27.0, 25.7, 25.2.
(E)-1,7,7-Trimethylbicyclo[2.2.1]heptan-2-one O-(2-hydroxy-3-(isopropylamino)propyl) oxime fumarate 10: Fumarate salt. 1H NMR (400 MHz, DMSO-d6): δ 6.47 (s, 1H), 4.00–3.91 (m, 2H), 3.87–3.82 (m, 1H), 3.24–3.21 (m, 1H), 2.96–2.93 (m, 1H), 2.78–2.76 (m, 1H), 2.50–2.40 (m, 2H), 1.93 (dd, J = 2.4 Hz, J = 17.6 Hz, 1H), 1.86 (t, J = 3.2 Hz, 1H), 1.78–1.74 (m, 1H), 1.71–1.64 (m, 1H), 1.32–1.28 (m, 1H), 1.18 (t, J = 6.0 Hz, 6H), 0.91 (s, 3H), 0.87 (s, 3H), 0.71 (s, 3H). 13 C NMR (100 MHz, DMSO-d6): δ 169.2, 168.3, 135.5, 75.3, 65.9, 51.8, 49.7, 48.2, 43.5, 33.9, 27.2, 19.6, 19.5, 18.9, 18.7, 11.6.
Propan-2-one O-(2-hydroxy-3-(isopropylamino)propyl) oxime oxalate 11: Oxalate salt. 1H NMR (400 MHz, DMSO-d6): δ 4.04–3.99 (m, 1H), 3.94 (dd, J = 5.36 Hz, J = 11.2 Hz, 1H), 3.86 (dd, J = 5.32 Hz, J = 11.2 Hz, 1H), 3.34–3.29 (m, 2H), 3.00 (dd, J = 2.64 Hz, J = 12.56 Hz, 1H), 2.85–2.80 (m, 1H), 1.80 (s, 6H), 1.21 (t, J = 6.04 Hz, 6H). 13 C NMR (100 MHz, DMSO-d6): δ 165.4, 155.4, 75.0, 66.0, 50.1, 47.5, 21.7, 19.2, 18.5.
(E)-4-Chlorobenzaldehyde O-(2-hydroxy-3-(isopropylamino)propyl) oxime oxalate 12: Oxalate salt. 1H NMR (400 MHz, DMSO-d6): δ 8.31 (s, 1H), 7.65 (m, 2H), 7.51 (m, 2H), 4.13–4.10 (m, 3H), 3.33–3.28 (m, 1H), 3.08–3.04 (m, 1H), 2.91–2.86 (m, 1H), 1.21 (t, J = 6.4 Hz, 6H). 13 C NMR (100 MHz, DMSO-d6): δ 165.5, 148.6, 134.4, 134.0, 131.2, 130.2, 127.7, 125.9, 76.4, 65.6, 50.1, 47.3, 19.24, 18.7.
(E)-2-Chlorobenzaldehyde O-(2-hydroxy-3-(isopropylamino)propyl) oxime oxalate 13: Oxalate salt. 1H NMR (400 MHz, DMSO-d6): δ 8.49 (s, 1H), 7.81 (dd, J = 1.6 Hz, J = 7.6 Hz, 1H), 7.55 (dd, J = 1.2 Hz, J = 8 Hz, 1H), 7.48 (dt, J = 1.6 Hz, J = 7.2 Hz, 1H), 7.41 (m, 1H), 4.16–4.11 (m, 3H), 3.34–3.31 (m, 1H), 3.10–3.07 (m, 1H), 2.93–2.88 (m, 1H), 1.22 (t, J = 6.4 Hz, 6H). 13 C NMR (100 MHz, DMSO-d6): δ 165.25, 146.2, 133.2, 132.2, 130.4, 129.6, 128.1, 127.7, 76.5, 65.6, 50.2, 47.2, 19.2, 18.6.
2.1.2. General procedure for the synthesis of aminoalcohols 14–16
Epichlorohydrin (0.88 ml,11.2 mmol) was added dropwise to a stirred solution of N-hydroxy-5-norbornene-2,3-dicarboximide (2 g, 11.2 mmol) and Et3N (3.12 ml, 22.4 mmol) in anhydrous DMF (8 ml). After stirring for 18 h at rt the reaction mixture was poured into water (50 ml) and extracted with CHCl3 (2×50mL). The organic phases were combined, washed with water (2 × 100 ml), dried (Na2SO4) filtered and evaporated under reduced pressure. The crude epoxide 39 was purified by crystallisation with n-hexane. NMR data were in accordance with those reported in literatureCitation25.
A stirred solution of epoxide 39 (611 mg, 2.6 mmol) in dry EtOH (8 ml) was treated with an excess of iPrNH2 (0.43 ml, 5 mmol) or t-BuNH2 (0.52 ml, 5 mmol). The reaction mixture was stirred at 50 °C for 4 h, and then the solvent was evaporated. The crude was constituted principally by the amino alcohol 40 (iPr) or 41(tBu) and it was used in the next reaction step without further purification.
A solution of the proper amino alcohol 40 or 41 (1.56 mmol) in 7.8 ml of NH3-MeOH 7 N was stirred rt for 2 h. The resulting mixture was filtered and evaporated. The crude was purified by flash chromatography with EtOAc/MeOH/Et3N (8: 1.5: 0.5) affording derivative 42 or 43 (60–63% yield). NMR data were in accordance with those reported in literatureCitation25.
A solution of p-, m-, or o- hydroxybenzaldehyde (6.75 mmol) in EtOH (11.4 ml) and the proper oxyamine 42 or 43 (6.75 mmol) was refluxed for 12 h. After cooling, the mixture was evaporated. The crude was dissolved in a mixture of MeOH/EtOH 3:7 (20 ml) and treated with 1.2 equivalent of the proper organic acid (oxalic, malic or fumaric acids) affording the amino alcohols (14–16, 70–80% yields) as white solids after crystallisation from MeOH/Et2O.
(E)-2-Hydroxybenzaldehyde O-(2-hydroxy-3-(isopropylamino)propyl) oxime maleate 14: Maleate salt. 1H NMR (400 MHz, DMSO-d6): δ 9.99(bs, 1H), 8.43 (s, 1H), 8.29 (bs, 1H), 7.54 (d, J = 7.6 Hz, 2H), 7.27 (m, 1H), 6.90 (d, J = 7.8 Hz, 1H), 6.85 (t, J = 7.6 Hz, 1H), 6.01 (s, 2H), 5.76 (m, 1H), 4.12–4.07 (m, 3H), 3.33–3.30 (m, 1H), 3.09–3.07 (m, 1H), 2.90–2.89 (m, 1H), 1.22 (m, 6H). 13 C NMR (100 MHz, DMSO-d6): δ 167.6, 156.5, 147.4, 132.0, 127.4, 119.9, 117.8, 116.7, 75.9, 65.7, 50.3, 47.1, 19.3, 18.5.
(E)-3-Hydroxybenzaldehyde O-(3-(tert-butylamino)-2-hydroxypropyl) oxime fumarate 15: Fumarate salt. 1H NMR (400 MHz, DMSO-d6): δ 8.17 (s, 1H), 7.21 (t, J = 7.6 Hz, 1H), 7.04–7.00 (m, 2H), 6.82–6.80 (m, 2H), 6.40 (s, 1H), 4.07–4.06 (m, 2H), 4.02–3.94 (m, 1H), 2.89–2.86 (m, 1H), 2.71–2.66 (m, 1H), 1.18 (s, 9H). 13 C NMR (100 MHz, DMSO-d6): δ 169.8, 158.2, 149.7, 133.5, 130.3, 118.6, 117.7, 113.3, 76.4, 66.6, 54.5, 45.0, 26.5.
(E)-4-Hydroxybenzaldehyde O-(3-(tert-butylamino)-2-hydroxypropyl) oxime fumarate 16: 1H NMR (400 MHz, DMSO-d6): δ 8.12 (s, 1H), 7.42 (m, 2H), 6.80 (m, 2H), 6.40 (s, 1H), 4.02–3.97 (m, 3H), 3.04–3.00 (m, 1H), 2.87–2.83 (m, 1H), 2.70–2.65 (m, 1H), 1.10 (d, J = 6.4 Hz, 6H).13C NMR (100 MHz, DMSO-d6): δ 169.0, 159.8, 149.3, 129.0, 123.1, 116.1, 76.4, 66.9, 49.4, 48.9, 21.1, 20.7.
2.2. Carbonic anhydrase activation
A stopped-flow methodCitation34 has been used for assaying the CA catalysed CO2 hydration activity with Phenol red as indicator, working at the absorbance maximum of 557 nm, following the initial rates of the CA-catalysed CO2 hydration reaction for 10–100 s. For each activator, at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of activator (0.1 mM) were prepared in distilled-deionised water and dilutions up to 0.1 nM were done thereafter with the assay buffer. The activation constant (KA), defined similarly with the inhibition constant (KI), was obtained by considering the classical Michaelis–Menten equation (EquationEquation 4(4)
(4) ), which has been fitted by nonlinear least squares by using PRISM 3:
(4)
(4)
where [A]f is the free concentration of activator.
Working at substrate concentrations considerably lower than KM ([S]≪KM), and considering that [A]f can be represented in the form of the total concentration of the enzyme ([E]t) and activator ([A]t), the obtained competitive steady-state equation for determining the activation constant is given by EquationEquation (5)(5)
(5) :
(5)
(5)
where v0 represents the initial velocity of the enzyme-catalysed reaction in the absence of an activatorCitation35–37. Enzyme concentrations in the assay system were in the range of 9–12 nM.
3. Results and discussion
3.1. Chemistry
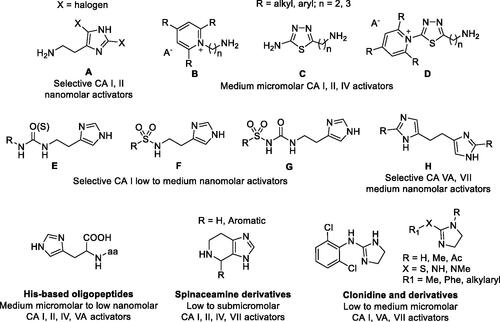
As mimic of the main proton shuttling residue (i.e. histidine), histamine () is the main lead for designing CAAsCitation8. The binding mode of histamine with hCA II was elucidated by X-ray crystallography ()Citation6. In the CAA/enzyme adduct, a complex network of H-bonds involve the Zn-bound water molecule, His64 and the imidazole ring of the activator, located far away from the metal ion (). Successive X-ray crystallographic studies showed that many other amines and amino acids () bind in this area and share flexible tails decorated with protonable moieties ()Citation38–40. No isoform-selective CAAs were detected so far among these amines and amino acid derivatives, except for few exceptions (e.g. histamine shows a 10 nM KAs against hCAs VA and XIV but is a micromolar activator of the remaining isoforms)Citation1. The molecule of histamine has been extensively modified (), including substituents on the imidazole C atoms (A)Citation41,Citation42, replacing the imidazole ring with other heterocycles, such as 2,4,6-trisubstituted pyridinium (B), 1,3,4-thiadiazole (C) or a combination of these two ring systems (D)Citation43,Citation44 and functionalising the NH2 group, as in carboxamides/ureas/thioureas (E)Citation45, sulphonamides (F)Citation46, arylsulfonylureas (G)Citation47, bis-histamine (H)Citation48,Citation49, oligopeptidesCitation49,Citation50, or imidazole/imidazoline derivatives of the alkaloyd spinaceamineCitation51,Citation52 and drug clonidineCitation53 () were reported to possess improved CA activatory profile when potency and isoform selectivity are concerned.
Figure 1. Natural amino acids and amines investigated for the activation of catalytically active hCA isoforms.
Figure 2. Superimposition of CAA – hCA II complexes as determined by X-ray crystallography. (A) Surface view: the hydrophobic half of the active site is coloured red; HisCitation64 is coloured green and the hydrophilic half in blue. (B) Ribbon active site view. The activators are histamine, in green (PDB 1AVN); L-His, in magenta (PDB 2ABE); L-Phe, in gold (PDB 2FMG); adrenaline, in cyan (PDB 2HKK).
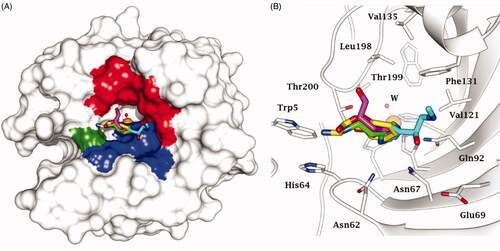
Figure 3. Histamine-based carbonic anhydrase activators.
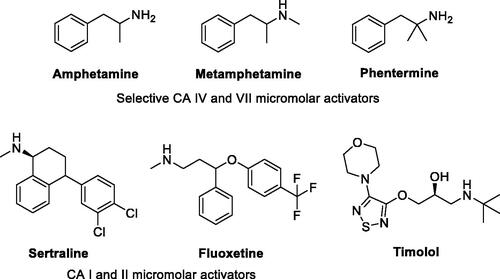
Interestingly, pharmacological agents born for other medical applications () showed also to possess more or less relevant CA-activating effects, among which psychoactive compounds of the amphetamine and methamphetamine familyCitation54, the selective serotonin reuptake inhibitors fluoxetine, sertraline and citalopramCitation55, the phosphodiesterase IV inhibitor sildenafilCitation56, and the β-blocker amino alcohol derivative timololCitation57. Timolol was taken as lead compound in the present study to design a new series of uncommon CAAs, which do not bear imidazole like scaffolds, intesively explored in the field. The enzyme kinetic method showed that timolol noncompetitively activates hCA I and II by forming of a ternary complex consisting of the enzyme, the substrate, and timolol. Docking studies were used to point out that timolol also binds at the entrance of the active site cavity nearby the proton shuttle residue His64Citation57. Thus, a series of aminoalcohol oxime ether derivatives previously shown to possess β-blocking or analgesic/antiarrhythmic activity (compounds 1–16)Citation25–32, was investigated for the activation of a panel of brain hCAs.
Figure 4. Other pharmacological agents showing hCAs activating properties.
Compounds 1–6 and 9–16 were prepared as previously described (Schemes 1 and 2)Citation25–32. Here, an updated synthetic procedure is reported. Moreover, new 1H NMR and 13 C NMR spectra of final compounds are provided in the experimental section. A fully detailed description is provided for the synthesis of new compounds 7 and 8, following the same synthetic procedure.
Scheme 1. Preparation of Amino alcohols 1–13. Reagents and conditions: (i) epichlorohydrin, MeONa/MeOH, dry DMF, 60 °C, 1 h; (ii) iPrNH2 or tBuNH2, Benzene, 90 °C, 12 h.
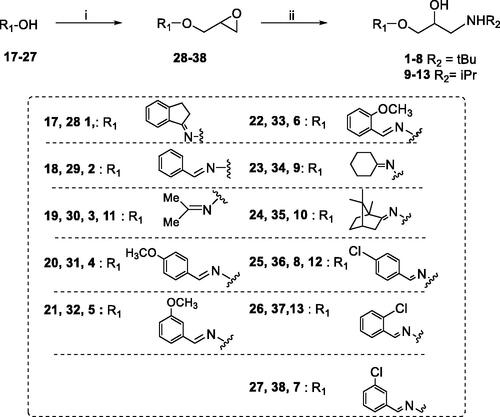
Amino alcohols 1–13 were synthesised as reported in Scheme 1Citation26–32. Oximes 17–27 were treated with epichlorohydrin in strong basic conditions affording epoxides 28–38. The amino alcohols 1–13 were obtained by reaction of oxime ethers 28–38 with an excess of the proper amine, iPrNH2 or tBuNH2. The final compounds were purified by crystallisation as organic salts.
The phenolic derivatives 14–16 were prepared as described in Scheme 2. N-hydroxy-5-norbornene-2,3-dicarboximide was reacted with epichlorohydrin to give the epoxide 39. The treatment of 39 with an excess of iPrNH2 or tBuNH2 afforded amino alcohol 40 or 41. Oxyamines 42 and 43 were obtained by aminolysis with NH3-MeOH 7 N of 40 or 41.
Scheme 2. Preparation of Amino alcohols 14–16. Reagents and conditions: (i) epichlorohydrin, Et3N, dry DMF, rt, 18 h; (ii) iPrNH2 or tBuNH2, Benzene, 50 °C, 4 h; (iii) NH3 MeOH 7 N, rt, 2 h; (iv) o-, m-, p-hydroxybenzaldehyde, EtOH, 90 °C, 12 h.
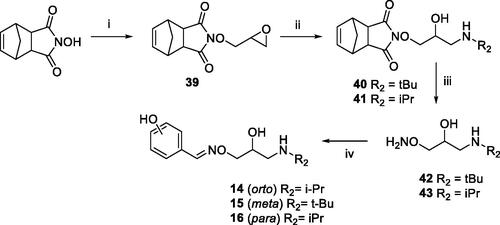
The condensation between derivative 42 or 43 and o-, m- or p-hydroxybenzaldehyde afforded the desired amino alcohols 14–16, purified by crystallisation as organic salts.
All the final compounds have been achieved as organic salt oximes presenting E configuration. The configuration of the C=N bound for compounds 1, 2, 4–8, 12 and 13 was assigned by analogy considering the E configuration of starting oximes 17, 18, 20–22, 25–27. As previously reported, the configuration of E-oximes 17, 18, 20–22, 25–27 was stable under the following reaction conditions.
The E configuration of amino alcohols 14–16 was conferred by comparison between the 1H signals of the iminic protons of compounds 14–16 with the same signals of final compounds 1, 2, 4–8, 12 and 13. The chemical shift of iminic proton signal resulted around δ 8.05–8.50 ppm for all derivatives as usually reported for oximes with E configuration.
3.2. Carbonic anhydrase activation
Amino alcohols 1–16 were here assayed for their activating properties of 4 catalytically active and physiologically relevant hCA isoforms expressed in human brain, that are: the cytosolic hCA I, II, and VII, and the membrane associated hCA IVCitation58. In the CNS context, hCA I is expressed in the motor neurons in human spinal cordCitation59. The physiologically dominant isoform hCA II is located both in the choroid plexus, and in oligodendrocytes, myelinated tracts, astrocytes and myelin sheaths in the vertebrates brainCitation60. hCA IV is located on the luminal surface of cerebral capillaries, associated with the blood–brain barrier, and expressed in layers III and VI in the cortex, thalamus and hippocampusCitation60,Citation61. CA VII is expressed in the cortex, hippocampus and thalamusCitation62,Citation63. CA VII might be considered a brain-associated CA as it is predominantly expressed in the brain, and absent in most other tissues. CA VII is also considered a key molecule in age-dependent neuronal pH regulation.
The CA activation data of these 4 isoforms with amino alcohols 1–16 and histamine as standard CAA are shown in . The following structure–activity relationship (SAR) can be worked out:
Table 1. Activation data of human CA isoforms I, II, IV and VII with amino alcohols 1–16 and histamine as reference CAA by a stopped flow CO2 hydrase assayCitation34. 
A special mention should be done for phenol derivatives 14–16 which, uniquely among amino alcohols 1–16, did not produce any activation of the tested hCAs. In fact, it is worth stressing that CAI properties, rather than as CAAs, are commonly ascribed to the phenolic chemotype. Phenols have been thoroughly reported to anchor to the zinc-bound nucleophile (water molecule or hydroxide ion), that is, by one of the four CA inhibition mechanisms known to date. Surprisingly, neither a significant inhibition was detected by treating CA I, II, IV and VII with phenols 14–16. One could speculate that the CAI efficacy of 14–16 is counterpoised by their CAA action, hindering its detection by the Stopped Flow assay.
The cytosolic isoform hCA I was moderately activated by all remaining amino alcohols, that are 1–13, with KA values in the range of 0.92–12.1 µM. The tert-butylamino derivatives 1–8 induce a slightly greater activation of CA I than isopropylamines 9–13. In the 1–8 subset, it can be noted that the o- or p-substitution of the aromatic ring bearing the oxime ether group lower the KA values 4, 6, 8 up to fivefold with respect to 2 (KAs of 1.37, 1.26 and 0.92 vs 4.25 µM). In contrast, m-substitutions are deleterious for the binding to the target as in 5 and 7 (KAs of 7.10 and 6.02 µM). The cyclisation of the oxime ether group to the aromatic ring as in 1 led to the second-best CA I activation after 8 (KAs of 0.94 vs 0.92 µM).
The physiologically most relevant isoform hCA II was activated by most tested amino alcohols more than hCA I (KAs in the range 0.079–7.19 µM), except for compounds 4 and 13 (KAs of 6.35 vs 5.90 µM). Surprisingly, the m-anisole derivative 5 reported a 100-fold enhancement of activation efficacy of hCA II when compared to hCA I (KAs of 0.079 vs 7.10 µM). Most remaining tert-butylamino derivatives showed submicromolar KA values, whereas all compounds of the subset 9–13 activate hCA II in a low micromolar range (KAs in the range 1.94–5.90 µM). All CAAs reported here were more active towards hCA II than histamine, which is a quite weak activator of this isoform with a KA of 125 µM.
No submicromolar KA values were measured for amino alcohols 1–13 as hCA IV activators. Indeed, all KAs are in a rather flat low micromolar range (KAs in the range 1.01–12.9 µM), making such a membrane associated isozyme the less activated by the assayed derivatives. Notably, the reference CAA histamine even less activates hCA IV with a KA of 25.3 µM. No significant differences exist between tert-butyl and isopropyl derivatives as for the activation of hCA IV.
The other cytosolic isoform investigated here, hCA VII, was rather potently activated, that is chiefly in a submicromolar range, by most of the compounds reported in this work (KAs in the range 0.082–8.86 µM). All derivatives showed much better activation profile than the reference histamine (KA of 37.6 µM) towards hCA VII. Contrariwise to the other hCAs tested, the most efficient CAAs are the isopropylamino derivatives 9 and 10, ethers of secondary oxime with an aliphatic pendant showing medium nanomolar KAs values of 82 and 91 nM, respectively. Only the oxime ethers of p-anisaldehyde and acetone in the isopropylamino and tert-butylamino series, respectively, that are compounds 4 and 11, show KAs steadily in a low micromolar range. As this isoform is one of the most widely spread in the brain, and probably involved in crucial metabolic/pH regulation processes, these results are of interest in the search of more effective CA VII activators than the currently available such derivatives.
4. Conclusions
In the present study, we described the synthesis and CA activating effects of a series of oxime ether-based amino alcohols towards four hCA isoforms expressed in human brain, that are CA I, II, IV and VII. Except for the phenolic compounds 14–16, all amino alcohol derivatives from this study induce a consistent activation of the tested CAs, with KAs spanning from a low micromolar to a medium nanomolar range. Specifically, hCA II and VII, probable main CA targets when CNS diseases are concerned,Citation58 are most potently activated by these oxime ether derivatives. With KAs of 79 and 420 nM towards hCA II and VII, respectively and KAs for CA I and IV settling in a range 10- to 100-fold higher, derivative 5 from the tert-butylamines series turned out as the most potent and selective hCA II activator of the study. On the other hand, a multitude of selective hCA VII activators were identified. Specifically, a mention should be done for derivatives 9 and 10, from the isopropylamines series, showing a 10- to 100-fold selective hCA VII activation profiles with respect to all other assayed CAs. In this second youth period for CAAs, innovative pharmacological studies made these lately neglected agents to draw attention in the memory therapy and cognitive neurodegenerative disorders, as well as in therapeutic areas, such as phobias, obsessive-compulsive disorder, generalised anxiety, and post-traumatic stress disordersCitation1,Citation20. As hCA VII is a key isoform involved in brain metabolism, the here identified potent and selective hCA VII activators may be considered of interest for investigations of possible therapeutic applications or as lead compounds in search of more potent and selective CAAs. This work might bring new lights on the intricate relationship between CA activation and brain physiology.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Supuran CT. Carbonic anhydrase activators. Future Med Chem 2018;10:561–73.
- Main RE, Locke A. Activation of carbonic anhydrase by histamine. J Biol Chem 1941;140:LXXXI.
- Clark AM, Perrin DD. A re-investigation of the question of activators of carbonic anhydrase. Biochem J 1951;48:495–503.
- Tu CK, Silverman DN, Forsman C, et al. Role of histidine 64 in the catalytic mechanism of human carbonic anhydrase II studied with a site-specific mutant. Biochemistry 1989;28:7913–18.
- Supuran CT. Carbonic anhydrase activators. Part 4. A general mechanism of action for activators of isozymes I, II and III. Rev Roum Chim 1992;37:411–21.
- Briganti F, Mangani S, Orioli P, et al. Carbonic anhydrase activators: x-ray crystallographic and spectroscopic investigations for the interaction of isozymes I and II with histamine. Biochemistry 1997;36:10384–92.
- Supuran CT, Balaban AT. Carbonic anhydrase activators. Part 8. pKa–activation relationship in a series of amino acid derivatives activators of isozyme II. Rev Roum Chim 1994;39:107–13.
- (a) Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81. (b) Supuran CT. Structure and function of carbonic anhydrases. Biochem J 2016;473:2023–32. (c) Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77.
- Duda D, Tu C, Qian M, et al. Structural and kinetic analysis of the chemical rescue of the proton transfer function of carbonic anhydrase II. Biochemistry 2001;40:1741–8.
- Duda D, Govindasamy L, Agbandje-McKenna M, et al. The refined atomic structure of carbonic anhydrase II at 1.05 A resolution: implications of chemical rescue of proton transfer. Acta Crystallogr D Biol Crystallogr 2003;59:93–104.
- Elder I, Tu C, Ming LJ, et al. Proton transfer from exogenous donors in catalysis by human carbonic anhydrase II. Arch Biochem Biophys 2005;437:106–14.
- Shah GN, Bonapace G, Hu PY, et al. Carbonic anhydrase II deficiency syndrome (osteopetrosis with renal tubular acidosis and brain calcification): novel mutations in CA2 identified by direct sequencing expand the opportunity for genotype-phenotype correlation. Hum Mutat 2004;24:272.
- Ogilvie JM, Ohlemiller KK, Shah GN, et al. Carbonic anhydrase XIV deficiency produces a functional defect in the retinal light response. Proc Natl Acad Sci USA 2007;104:8514–19.
- Datta R, Shah GN, Rubbelke TS, et al. Progressive renal injury from transgenic expression of human carbonic anhydrase IV folding mutants is enhanced by deficiency of p58IPK. Proc Natl Acad Sci USA 2010;107:6448–52.
- van Karnebeek CD, Sly WS, Ross CJ, et al. Mitochondrial carbonic anhydrase VA deficiency resulting from CA5A alterations presents with hyperammonemia in early childhood. Am J Hum Genet 2014;94:453–61.
- Sun MK, Alkon DL. Carbonic anhydrase gating of attention: memory therapy and enhancement. Trends Pharmacol Sci 2002;23:83–9.
- Sun MK, Alkon DL. Pharmacological enhancement of synaptic efficacy, spatial learning, and memory through carbonic anhydrase activation in rats. J Pharmacol Exp Ther 2001;297:961–7.
- Canto de Souza L, Provensi G, Vullo D, et al. Carbonic anhydrase activation enhances object recognition memory in mice through phosphorylation of the extracellular signal-regulated kinase in the cortex and the hippocampus. Neuropharmacology 2017;118:148–56.
- Provensi G, Carta F, Nocentini A, et al. A new kid on the block? Carbonic anhydrases as possible new targets in Alzheimer’s disease. Int J Mol Sci 2019;20:4724.
- Blandina P, Provensi G, Passsani MB, et al. Carbonic anhydrase modulation of emotional memory. Implications for the treatment of cognitive disorders. J Enzyme Inhib Med Chem 2020;35:1206–14.
- (a) Wang J, Ke T, Zhang X, et al. Effects of acetazolamide on cognitive performance during high-altitude exposure. Neurotoxicol Teratol 2013;35:28–33. (b) Supuran CT. An update on drug interaction considerations in the therapeutic use of carbonic anhydrase inhibitors. Expert Opin Drug Metab Toxicol 2020;16:297–307. (c) Supuran CT, Nicolae A, Popescu A. Carbonic anhydrase inhibitors. Part 35. Synthesis of Schiff bases derived from sulfanilamide and aromatic aldehydes: the first inhibitors with equally high affinity towards cytosolic and membrane-bound isozymes. Eur J Med Chem 1996;31:431–8.
- Nestler EJ, Hyman SE. Animal models of neuropsychiatric disorders. Nat Neurosci 2010;13:1161–9.
- Schmidt SD, Costa A, Rani B, et al. The role of carbonic anhydrases in extinction of contextual fear memory. Proc Natl Acad Sci USA 2020;117:16000–8.
- (a) Supuran CT. Carbonic anhydrases: from biomedical applications of the inhibitors and activators to biotechnological use for CO2 capture. J Enzyme Inhib Med Chem 2013;28:229–30. (b) De Simone G, Supuran CT. (In)organic anions as carbonic anhydrase inhibitors. J Inorg Biochem 2012;111:117–29. (c) Supuran CT. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin Drug Discov 2017;12:61–88.
- Gentili D, Lapucci A, Macchia B, et al. Synthesis and beta-adrenergic activity of a series of 3-(substituted-benzylideneaminoxy)propanolamine derivatives. Farmaco 1995;50:519–26.
- Macchia B, Balsamo A, Lapucci A, et al. An interdisciplinary approach to the design of new structures active at the beta-adrenergic receptor. Aliphatic oxime ether derivatives. J Med Chem 1985;28:153–60.
- Schenone S, Bruno O, Ranise A, et al. O-[2-hydroxy-3-(dialkylamino)propyl]ethers of (+)-1,7,7-trimethyl bicyclo[2.2.1]heptan-2-one oxime (camphor oxime) with analgesic and antiarrhythmic activities. Farmaco 2000;55:495–8.
- Krajewska M. Amino alkanol derivatives of oximes. Acta Polonia Pharmaceutica 1977;34:383–9.
- Martani A. New aromatic oxime ether derivatives with β-blocking activity. Farmaco 1975;30:370–9.
- Leclerc G, Mann A, Wermuth CG, et al. Synthesis and beta-adrenergic blocking activity of a novel class of aromatic oxime ethers. J Med Chem 1977;20:1657–62.
- Leclerc G, Bieth N, Schwartz J. Synthesis and beta-adrenergic blocking activity of new aliphatic oxime ethers. J Med Chem 1980;23:620–4.
- Breschi C, Macchia M, Manera C, et al. Conformationally restrained β-blocking oxime ethers. 3. Synthesis and β-adrenergic antagonistic activity of diastereomeric anti and syn 2-(5′-(3′-methyl)isoxazolidinyl)-N-alkylethanolamines. Eur J Med Chem 1996;31:159–63.
- Xu W, Wang J, Liu C, et al. Experimental and computational studies of the isomerization between Z and E isomers of benzaldoximes. Jnl Chinese Chemical Soc 2004;51:1259–66.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73.
- Vullo D, Innocenti A, Nishimori I, et al. Carbonic anhydrase activators: activation of the human isoforms VII (cytosolic) and XIV (transmembrane) with amino acids and amines. Bioorg Med Chem Lett 2007;17:4107–12.
- Pastorekova S, Vullo D, Nishimori I, et al. Carbonic anhydrase activators: activation of the human tumor-associated isozymes IX and XII with amino acids and amines. Bioorg Med Chem 2008;16:3530–6.
- Vullo D, Nishimori I, Innocenti A, et al. Carbonic anhydrase activators: an activation study of the human mitochondrial isoforms VA and VB with amino acids and amines. Bioorg Med Chem Lett 2007;17:1336–40.
- Temperini C, Scozzafava A, Vullo D, et al. Carbonic anhydrase activators. Activation of isozymes I, II, IV, VA, VII, and XIV with l- and d-histidine and crystallographic analysis of their adducts with isoform II: engineering proton-transfer processes within the active site of an enzyme. Chemistry 2006;12:7057–66.
- Temperini C, Scozzafava A, Vullo D, et al. Carbonic anhydrase activators. Activation of isoforms I, II, IV, VA, VII, and XIV with L- and D-phenylalanine and crystallographic analysis of their adducts with isozyme II: stereospecific recognition within the active site of an enzyme and its consequences for the drug design. J Med Chem 2006;49:3019–27.
- Temperini C, Innocenti A, Scozzafava A, et al. Carbonic anhydrase activators: kinetic and x-ray crystallographic study for the interaction of D- and L-tryptophan with the mammalian isoforms I-XIV. Bioorg Med Chem 2008;16:8373–837.
- Saada MC, Vullo D, Montero JL, et al. Carbonic anhydrase I and II activation with mono- and dihalogenated histamine derivatives. Bioorg Med Chem Lett 2011;21:4884–7.
- Saada MC, Vullo D, Montero JL, et al. Mono- and di-halogenated histamine, histidine and carnosine derivatives are potent carbonic anhydrase I, II, VII, XII and XIV activators. Bioorg Med Chem 2014;22:4752–8.
- (a) Supuran CT, Dinculescu A, Balaban AT. Carbonic anhydrase activators. Part 5. CA II activation by 2,4,6-trisubstituted pyridinium cations with 1-(ω-aminoalkyl) side chains. Rev Roum Chim 1993;38:343–9. (b) Pastorekova S, Casini A, Scozzafava A, et al. Carbonic anhydrase inhibitors: the first selective, membrane-impermeant inhibitors targeting the tumor-associated isozyme IX. Bioorg Med Chem Lett 2004;14:869–73.
- Supuran CT, Barboiu M, Luca C, et al. Carbonic anhydrase activators. Part 14. Synthesis of mono- and bis- pyridinium salt derivatives of 2-amino-5-(2-aminoethyl)- and 2-amino-5-(3-aminopropyl)-1,3,4-thiadiazole, and their interaction with isozyme II. Eur J Med Chem 1996;31:597–606.
- Scozzafava A, Supuran CT. Carbonic anhydrase activators – part 21. Novel activators of isozymes I, II and IV incorporating carboxamido and ureido histamine moieties. Eur J Med Chem 2000;35:31–9.
- Briganti F, Scozzafava A, Supuran CT. Novel carbonic anhydrase isozymes I, II and IV activators incorporating sulfonyl-histamino moieties. Bioorg Med Chem Lett 1999;9:2043–8.
- Scozzafava A, Supuran CT. Carbonic anhydrase activators. Part 24. High affinity isozymes I, II and IV activators, derivatives of 4-(4-chlorophenylsulfonylureido-amino acyl)ethyl-1H-imidazole. Eur J Pharm Sci 2000;10:29–41.
- (a) Draghici B, Vullo D, Akocak S, et al. Ethylene bis-imidazoles are highly potent and selective activators for isozymes VA and VII of carbonic anhydrase, with a potential nootropic effect. Chem Commun (Camb) 2014;50:5980–3. (b) Saada MC, Vullo D, Montero JL, et al. Mono- and di-halogenated histamine, histidine and carnosine derivatives are potent carbonic anhydrase I, II, VII, XII and XIV activators. Bioorg Med Chem 2014;22:4752–8.
- (a) Mollica A, Macedonio G, Stefanucci A, et al. Five- and six-membered nitrogen-containing compounds as selective carbonic anhydrase activators. Molecules 2017;22:2178. (b) Maccallini C, Di Matteo M, Vullo D, et al. Indazole, pyrazole, and oxazole derivatives targeting nitric oxide synthases and carbonic anhydrases. ChemMedChem 2016;11:1695–9. (c) Supuran CT, Scozzafava A. Carbonic anhydrase activators: amino acyl/dipeptidyl histamine derivatives bind with high affinity to isozymes I, II and IV and act as efficient activators. Bioorg Med Chem 1999;7:2915–23.
- Vistoli G, Aldini G, Fumagalli L, et al. Activation effects of carnosine- and histidine-containing dipeptides on human carbonic anhydrases: a comprehensive study. Int J Mol Sci 2020;21:1761.
- Akocak S, Lolak N, Bua S, et al. Activation of human α-carbonic anhydrase isoforms I, II, IV and VII with bis-histamine schiff bases and bis-spinaceamine substituted derivatives. J Enzyme Inhib Med Chem 2019;34:1193–8.
- Akocak S, Lolak N, Bua S, et al. α-Carbonic anhydrases are strongly activated by spinaceamine derivatives. Bioorg Med Chem 2019;27:800–4.
- Chiaramonte N, Maach S, Biliotti C, et al. Synthesis and carbonic anhydrase activating properties of a series of 2-amino-imidazolines structurally related to clonidine1. J Enzyme Inhib Med Chem 2020;35:1003–10.
- Angeli A, Vaiano F, Mari F, et al. Psychoactive substances belonging to the amphetamine class potently activate brain carbonic anhydrase isoforms VA, VB, VII, and XII. J Enzyme Inhib Med Chem 2017;32:1253–9.
- Casini A, Caccia S, Scozzafava A, et al. Carbonic anhydrase activators. The selective serotonin reuptake inhibitors fluoxetine, sertraline and citalopram are strong activators of isozymes I and II. Bioorg Med Chem Lett 2003;13:2765–8.
- (a) Abdülkadir Coban T, Beydemir Ş, Gücin İ, et al. Sildenafil is a strong activator of mammalian carbonic anhydrase isoforms I-XIV. Bioorg Med Chem 2009;17:5791–5. (b) Supuran CT. Carbon-versus sulphur-based zinc binding groups for carbonic anhydrase inhibitors? J Enzyme Inhib Med Chem 2018;33:485–95.
- Sugimoto A, Ikeda H, Tsukamoto H, et al. Timolol activates the enzyme activities of human carbonic anhydrase I and II. Biol Pharm Bull 2010;33:301–6.
- (a) Nocentini A, Donald WA, Supuran CT, Human carbonic anhydrases: tissue distribution, physiologic role, and druggability. In: Nocentini A, Supuran CT, eds. Carbonic anhydrases. Amsterdam: Elsevier; 2019. p. 149–185. (b) Supuran CT. Exploring the multiple binding modes of inhibitors to carbonic anhydrases for novel drug discovery. Expert Opin Drug Discov 2020;15:671–86. (c) Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31:345–60.
- Liu X, Lu D, Bowser R, et al. Expression of carbonic anhydrase I in motor neurons and alterations in ALS. Int J Mol Sci 2016;17:1820.
- Halmi P, Parkkila S, Honkaniemi J. Expression of carbonic anhydrases II, IV, VII, VIII and XII in rat brain after kainic acid induced status epilepticus. Neurochem Int 2006;48:24–30.
- Svichar N, Chesler M, Waheed A, et al. Carbonic anhydrases CA4 and CA14 both enhance AE3-mediated Cl-HCO3- exchange in hippocampal neurons. J Neurosci 2009;29:3252–8.
- Ruusuvuori E, Huebner AK, Kirilkin I, et al. Neuronal carbonic anhydrase VII provides GABAergic excitatory drive to exacerbate febrile seizures. Embo J 2013;32:2275–86.
- Karjalainen SL, Haapasalo HK, Aspatwar A, et al. Carbonic anhydrase related protein expression in astrocytomas and oligodendroglial tumors. BMC Cancer 2018;18:584.
- (a) Nocentini A, Alterio V, Bua S, et al. Phenyl(thio)phosphon(amid)ate benzenesulfonamides as potent and selective inhibitors of human carbonic anhydrases II and VII counteract allodynia in a mouse model of oxaliplatin-induced neuropathy. J Med Chem 2020;63:5185–200. (b) Supuran CT. Carbonic anhydrases and metabolism. Metabolites 2018;8:25. (c) Supuran CT. Carbonic anhydrase inhibitors and their potential in a range of therapeutic areas. Expert Opin Ther Pat 2018;28:709–12. (d) Supuran CT. Applications of carbonic anhydrases inhibitors in renal and central nervous system diseases. Expert Opin Ther Pat 2018;28:713–21. (e) Supuran CT, Altamimi ASA, Carta F. Carbonic anhydrase inhibition and the management of glaucoma: a literature and patent review 2013–2019. Expert Opin Ther Pat 2019;29:781–92.