
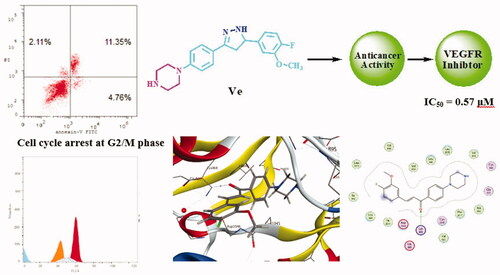
Abstract
New piperazine–chalcone hybrids and related pyrazoline derivatives have been designed and synthesised as potential vascular endothelial growth factor receptor-2 (VEGFR-2) inhibitors. The National Cancer Institute (NCI) has selected six compounds to evaluate their antiproliferative activity in vitro against 60 human cancer cells lines. Preliminary screening of the examined compounds indicated promising anticancer activity against number of cell lines. The enzyme inhibitory activity against VEGFR-2 was evaluated and IC50 of the tested compounds ranged from 0.57 µM to 1.48 µM. The most potent derivatives Vd and Ve were subjected to further investigations. A cell cycle analysis showed that both compounds mainly arrest HCT-116 cell cycle in the G2/M phase. Annexin V-FITC apoptosis assay showed that Vd and Ve induced an approximately 18.7-fold and 21.2-fold total increase in apoptosis compared to the control. Additionally, molecular docking study was performed against VEGFR (PDB ID: 4ASD) using MOE 2015.10 software and Sorafenib as a reference ligand.
Graphical Abstract
1. Introduction
Cancer is a major global health problem characterised by uncontrolled growth of abnormal cells. Although cancer research resulted in a range of innovative and promising approaches, the medications used as therapies have strong drawbacks and cancer is expected to be the leading cause of death in the futureCitation1–3. Targeted cancer treatments have recently been approved to treat specific cancers such as melanoma, renal, colon, lung, ovary, central nervous system, breast, and leukaemiaCitation4. Targeted chemotherapy requires several strategies, including angiogenesis inhibition, which has proved to be a successful technique for tumour growthCitation5. Angiogenesis is an important physiological process in which the pre-existing vessels form new blood vessels. It is a vital physiological process that happens during inflammation and wound healingCitation6,Citation7. New blood vessels penetrate tumour masses and provide them with oxygen and nutrients which promote tumour progression and metastasis in pathological angiogenesisCitation8. Therefore, blocking angiogenesis could be a promising strategy to inhibit growth of tumours with lower adverse effects than other typical chemotherapiesCitation5.
The vascular endothelial growth factor (VEGF) isoforms were particularly attractive targets to inhibit angiogenesisCitation9,Citation10. The expression of VEGF during embryonic stages is high and is thought to play a crucial role in new (vasculogenesis) or pre-existing blood vessels (angiogenesis)Citation11. For certain solid tumours, overexpression of VEGF contributes to increased tumour growth and metastasis, which may be attributed to improved nutrient replenishment availability for the metabolising cellCitation12,Citation13. Vascular endothelial growth factor receptor-2 (VEGFR-2) is a subtype of tyrosine kinase receptor VEGF family (VEGFR-TK)Citation14,Citation15. It is responsible for normal and abnormal changes in vascular endothelial cellsCitation16,Citation17. VEGFR-2 inhibition will affect tumour cell blood supply, inhibiting its development, proliferation, and metastasis. VEGFR signalling pathway inhibition is a key therapeutic target for tumour inhibitionCitation18,Citation19.
N-aryl piperazine derivatives are important organic compounds that have recently attracted considerable interest for their anti-cancer activityCitation20–23. On the other hand, pyrazole a simple aromatic five-membered ring contains two adjacent nitrogen atoms is included in many derivatives that display a variety of pharmacological activities such as anti-Alzheimer diseaseCitation24, anticonvulsantCitation25,Citation26, anti-tubercularCitation27,Citation28, anti-microbialCitation29,Citation30, anti-inflammatory, and analgesicCitation31,Citation32. Numerous pyrazole derivatives have proved their anti-cancer efficacy against different types of cancerCitation33–38. Chalcones, (1,3-diaryl-2-propene-1-ones) derivatives that can conventionally be synthesised by Claisen–Schmidt condensationCitation39 remained a curiosity among researchers due to their diversified biological activitiesCitation40, such as antimalarialCitation41, anti-histaminicCitation42, anti-diabeticCitation43, anti-inflammatoryCitation44, and anti-neoplastic activityCitation45,Citation46.
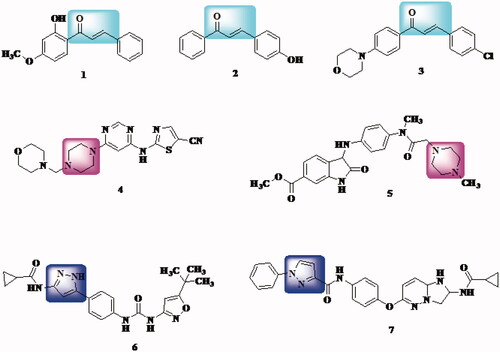
On the other hand, many chalcone derivatives (compound 1Citation47, compound 2Citation48, and compound 3Citation49) along with pyrazole derivatives (compound 4Citation50 and compound 5Citation51) and piperazine derivatives (compound 6Citation52 and compound 7Citation53) display potential inhibitory activity of VEGFR-2 kinase which is an essential factor in angiogenesis (). Thus, chalcone and analogues demonstrated potential inhibitory activity of VEGFR could be considered as important cancer prevention targetsCitation54–56.
Figure 1. Examples of chalcones, pyrazoles, and piperazine derivatives as VEGFR inhibitors.
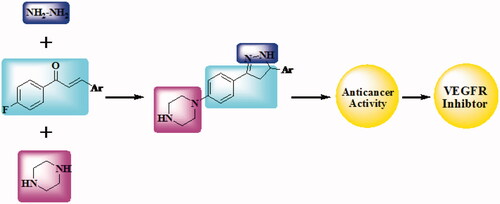
Molecular hybridisation is one of the effective chemotherapeutic agent production techniques that require the synthesis of two distinct bioactive units. In the present research, the design and synthesis of novel hybrid compounds bearing piperazine and chalcone or piperazine and pyrazoline () are our target. Newly synthesised compounds have been submitted for evaluation of their anticancer activity to the National Cancer Institute (NCI). Six compounds were selected by NCI, and 60 lines of human cancer cells were screened in vitro. The inhibitory activity had been also tested against VEGFR-2.
Figure 2. Design of newly synthesised derivatives as VEGFR inhibitor.
2. Results and discussion
2.1. Chemistry
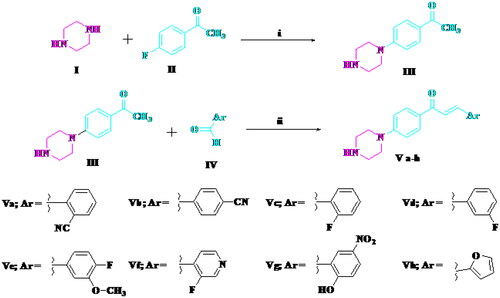
Compounds Va–h were synthesised via the reaction of 4′-piperazino-acetophenone III and the appropriate aldehyde (Scheme 1). The reaction of the chalcone Va, Ve, and Vf and hydrazine hydrate afforded pyrazoline derivatives VIa–c (Scheme 2). The structures of all newly synthesised compounds were confirmed by various methods of spectroscopic analysis, such as 1H NMR, 13C NMR, IR, and mass spectrometry.
Scheme 1. Synthesis of target compounds Va–h. Reagents and conditions: (i) DMSO, heating at 110 °C, 24 h; (ii) alcoholic NaOH (10%), stirring 5 h.
Scheme 2. Synthesis of target compounds VIa–c. Reagents and conditions: (i) hydrazine hydrate, absolute ethanol, reflux 12 h.
2.2. Biological activity
2.2.1. Screening of anticancer activity
Six compounds were selected by the NCI, according to NCI’s DTP selection guidelinesCitation57, for evaluation of their anticancer activity at a single-dose of 10 µM against 60 human tumours cell lines. The compounds’ screening findings are summarised in . Data analysis resulting from the primary assay showed that chalcone derivatives Vd, Ve, and Vf showed moderate to good inhibitory activity mainly against leukaemia and colon cancer cell lines.
Table 1. In vitro growth inhibitory percent for compounds Vd, Ve, Vf, VIa, VIb, and VIc at 10 µM concentration, towards the 60 subpanel cancer cell lines, results were given as a percentage of cell growth promotion.
As illustrated in , compound Vf bearing 3-fluoropyridine moiety showed weak anti-proliferative activity against non-small cell lung cancer HOP-92, leukaemia K-562, colon cancer HT29, leukaemia SR, and breast cancer MCF7 cancer cell lines, with cell growth promotion (82.34%, 85.84%, 86.42%, 92.99%, 99.38%; cell growth inhibition: 17.66%, 14.16%, 13.85%, 7.01%, and 0.62%, respectively). It also showed moderate activity against leukaemia HL-60(TB) (cell growth promotion 79.28%; cell growth inhibition: 20.72%), leukaemia CCRF-CEM (78.16%; cell growth inhibition: 21.84%), and colon cancer HCT-116 (cell growth promotion 73.87%; cell growth inhibition: 26.13%).
Replacing 3-fluoropyridine of compound Vf with 3-fluorophenyl moiety, compound Vd significantly increased growth inhibition against number of cancer cell lines, such as leukaemia HL-60(TB), non-small cell lung cancer HOP-92, leukaemia CCRF-CEM, leukaemia SR, and breast cancer MCF7 with percent growth inhibition of 39.17%, 41.59%, 41.82%, 44.28%, and 48.28%, respectively, compared to that of compound Vf (20.72%, 17.66%, 21.84%, 7.01%, and 0.62%, respectively). In addition, compound Vd, showed good to excellent activity against colon cancer HCT-116, colon cancer HT29, and leukaemia K-562 with percent growth inhibition of 57.92%, 69.56%, and 83.45%, respectively.
Furthermore, replacing 3-fluorophenyl moiety with 4-fluoro-3-methoxyphenyl moiety compound Ve markedly increase growth inhibition towards many cancer cell lines. It showed cell growth promotion for non-small cell lung cancer HOP-92 (55.00%; cell growth inhibition: 45.00%), leukaemia SR (46.22%; cell growth inhibition: 53.78%), breast cancer MCF7 (34.68%; cell growth inhibition: 65.32%), leukaemia HL-60(TB) (28.91%; cell growth inhibition: 71.09%), colon cancer HCT-116 (27.43%; cell growth inhibition: 72.57%), leukaemia CCRF-CEM (22.15%; cell growth inhibition: 77.85%), and leukaemia K-562 (12.37%; cell growth inhibition: 87.63%). From previous results, we can conclude that chalcone derivative bearing 4-fluoro-3-methoxyphenyl moiety (Ve) was the most potent chalcone towards leukaemia CCRF-CEM, leukaemia HL-60(TB), leukaemia K-562, leukaemia SR non-small cell lung cancer HOP-92, colon cancer HCT-116, and breast cancer MCF7 cell lines.
On the other hand, pyrazole analogues VIa, VIb, and VIc showed promising cytotoxicity towards a variety of cancer cell lines. Compound VIa bearing benzonitrile moiety exhibited a weak growth inhibition against several cancer cell lines as it displayed cell growth promotion for leukaemia SR (94.43%; cell growth inhibition: 5.57%), colon cancer HT29 (87.83%; cell growth inhibition: 12.17%), colon cancer HCT-116 (85.83%; cell growth inhibition: 14.17%), and non-small cell lung cancer HOP-92 (83.18%; cell growth inhibition: 16.82%). In addition, it showed good inhibitory activity against many cancer cell lines with cell growth promotion for leukaemia K-562 (78.12%; cell growth inhibition: 21.88%), non-small cell lung cancer A549/ATCC (78.06%; cell growth inhibition: 21.94%), leukaemia CCRF-CEM (71.21%; cell growth inhibition: 28.79%), and non-small cell lung cancer NCI-H522 (69.51%; cell growth inhibition: 30.49%).
Replacing benzonitrile moiety with 3-fluoropyridine moiety, compound VIc increased anti-proliferative activity against few cancer cell lines such as colon cancer HCT-116, non-small cell lung cancer HOP-92 and colon cancer HT29 with percent of cell growth inhibition of 26.18%, 27.58%, and 39.84%, respectively.
Finally, compound VIb with 4-fluoro-3-methoxyphenyl moiety was the most potent pyrazole derivative and exhibited moderate to good inhibitory activity towards many cancer cell lines. It showed percent of cell growth promotion for colon cancer HCT-116, leukaemia K-562, non-small cell lung cancer HOP-92, and colon cancer HT29 of 61.10%, 58.92%, 57.13%, and 20.27%, respectively (cell growth inhibition: 38.9%, 41.08%, 42.87%, and 79.73%, respectively).
2.2.2. Vascular endothelial growth factor receptor-2 inhibition
VEGFR is considered as an important target for the development of potential anti-cancer candidatesCitation54–56. Therefore, compounds Vd, Ve, Vf, VIa, VIb, and VIc were further investigated for their ability to inhibit VEGFR-2 using colorimetric assay of human VEGFR-2 ELISA (enzyme-linked immunosorbent assay) and Sorafenib as a reference drug. Results were presented as half maximal inhibitory concentration (IC50) values (). IC50 of the tested compounds ranged from 0.57 µM to 1.48 µM. Compound Ve was the most potent VEGFR-2 inhibitor among the tested compounds with an IC50 value of 0.57 µM which was comparable to results of Sorafenib that had IC50=0.51 µM. In addition, compound Vd showed significant VEGFR-2 inhibitory activity with IC50=0.80 µM. These results supported that VEGFR-2 could be a possible target for anti-tumour activity of our tested compounds.
Table 2. In vitro VEGFR-2 inhibitory assay.
2.2.3. Cell cycle analysis
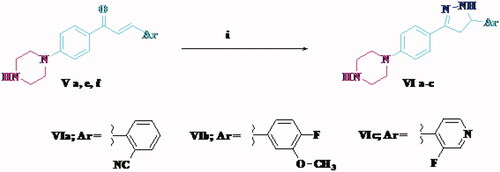
Most of cytotoxic compounds exert their anti-proliferative effect via arresting the cell cycle at certain phase. Flow cytometric analysis is considered a valuable method for determining and analysing the cell cycle parametersCitation58. In this study, compounds Vd and Ve as the most potent derivatives were selected to explore their effect on cell cycle progression and induction of apoptosis in HCT-116 cell line using the standard concentration of 10 µM. The effect on the cell cycle distribution was assessed by a DNA flow cytometry analysis and the cell cycle parameters were compared to untreated control cells in HCT-116 cells which had been incubated with 10 µM of Vd and Ve compounds and the results are shown in and . The results revealed that, the percentage of HCT-116 cells at G2/M phase markedly increased from 15.44% to 50.44% and 46.85% after incubation with compound Vd and Ve, respectively. On the other hand, the percentage of HCT-116 cells at G1 phase decreased from 54.38% in control to 21.78% for compound Vd and 24.57% for compound Ve indicating that compounds Vd and Ve induced cell arrest at G2/M phase. The percentage of cell death at pre-G1 phase for compounds Vd and Ve was 16.54% and 18.22%, respectively.
Figure 3. Effect of compounds Vd and Ve on the phases of cell cycle of HCT-116 cells.
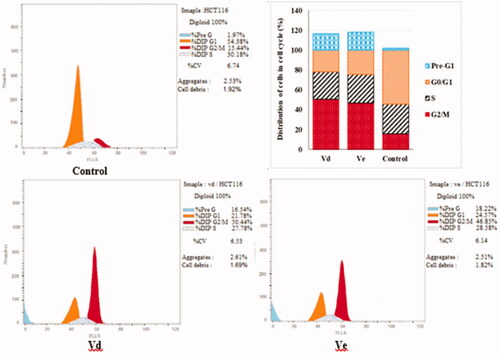
Table 3. Effect of compounds Vd and Ve on the phases of cell cycle of HCT-116 cells.
2.2.4. Annexin V-FITC apoptosis assay
Double staining assay of annexin-V/propidium iodide (PI) was used to investigate the mode of induced HCT-116 cell death when treated with the tested compounds Vd and Ve. HCT-116 cells were treated for 24 h with 10 µM from each tested compound. The results obtained are outlined in and . The percentage of apoptosis caused by the Vd and Ve compounds respectively was (16.54 and 18.22). We can also conclude that in early stage treatment of HCT-116 cells with Vd and Ve compounds results in an increase in the percentage of apoptotic cells from 0.51% for control untreated cells to be 3.92 and 4.76, respectively. In late stage, the percentage of apoptotic cells was 10.32–11.35% compared to control (0.25%). The results indicate that the tested Vd and Ve induced apoptosis in HCT-116 cell line.
Figure 4. Effect of compounds Vd and Ve on the percentage of annexin V-FITC-positive staining in HCT-116 cells. The experiments were done in triplicates. The four quadrants identified as: LL: viable; LR: early apoptotic; UR: late apoptotic; UL: necrotic.
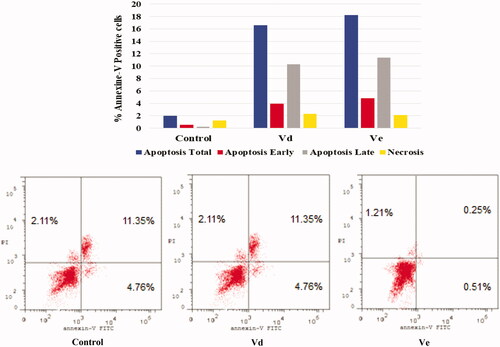
Table 4. Apoptosis and necrosis percent induced by compounds Vd and Ve in HCT-116 cells.
2.3. Molecular docking
Molecular modelling is considered as an important tool to study molecular interactions of certain ligands and binding site of the corresponding protein. The ligand–protein interaction behaviour at the active site was estimated based on the docking score function as implemented in MOE 2015.10Citation59. In this study, six active potential anticancer compounds Vd, Ve, Vf, VIa, VIb, and VIc were subjected to molecular docking studies using MOE program on the 3D structure of VEGFR using Sorafenib as reference compound. Binding free energy data obtained after the docking procedure showed that the tested compounds exhibit favourable docked complexes with the active site of target protein. The tested compounds Vd, Ve, Vf, VIc, VIb, and VIc exhibited interactions with the VEGFR active site to different extents and the docking score free energy of the tested compounds found to be this order: Ve>VIa>Vd>Vf>VIc>VIb, as shown in . Also, the other scoring parameters such as rmsd_refine, E_conf, E_place, and E_refine, indicated that the tested compounds were correctly docked in the biding site as the reference ligand Sorafenib.
Table 5. Docking energy scores (kcal/mol) derived from the MOE for compounds Vd–f, VIa–c, and the reference ligand Sorafenib.
Based on the obtained scoring results, compounds Ve, VIa, and Vd showed the highest binding affinity to the VEGFR-2 active site. Compound Ve was the best among the synthesised compounds with docking score (–7.9154 kcal/mol) compared to the reference ligand Sorafenib docking score (–11.1354 kcal/mol), and it formed a direct interaction in the active site, similar to that of Sorafenib. Also, compounds VIa and Vd showed a good docking score (–7.4371 kcal/mol) and (–7.3974 kcal/mol), respectively, compared to Sorafenib. reveals that the tested compounds VIa and Vd along with Sorafenib reacted with important amino acids in the active binding site. The carbonyl group of the chalcone moiety in both compounds Ve and Vd acts as H-bond acceptor and formed a H-bonding with LYS868 (). On the other hand, the reference compound Sorafenib formed several hydrogen bonding with the active side nearby amino acids GLU885, CYS919, ASP1046, and PHE1047 ().
Figure 5. Docking of compounds Vd, Ve and the reference ligand Sorafenib into VEGFR active sites. (a) Compounds Vd. (b) Compound Ve. (c) Sorafenib.
3. Experimental
3.1. Chemistry
All melting points were uncorrected and measured by Electro thermal IA 9000 series digital melting point apparatus at the Micro-analytical Center, Cairo University (Giza, Egypt). IR spectra had been reported on FT. IR 670-Nicolet spectrophotometer-Nexus (Thermo Scientific, Madison, WT); determination of 1H NMR and 13C NMR spectra on a JEOL AS NMR spectrometer (Tokyo, Japan). Mass spectra were measured on Finnigan Mat SSQ 7000 mode EI 70 ev (Thermo Inst. Sys. Inc., Waltham, MA). Thin-layer chromatography was performed using chloroform/methanol (10:1, v/v) on thin-layer chromatographic plates of silica gel 60 F254 (Merck, Kenilworth, NJ), and the spots were observed for a few seconds by exposure to UV lamps at λ254 nm and used to monitor the reaction time. 1-(4-(Piperazin-1-yl)phenyl)ethan-1-one III was prepared as reported methodCitation60.
3.2. General method for preparation of (Va–h)
A mixture of 4′-piperazinoacetophenone III (0.01 mol) and the corresponding aldehyde derivatives IV, namely, 2-cyanobenzaldehyde, 4-cyanobenzaldehyde, 2-flurobenzaldehyde, 3-flurobenzaldehyde, 4-fluro-3-methoxy benzaldehyde, 3-fluroisonicotinaldehyde, 2-hydroxyl-5-nitrobenzaldehyde, and 2-furaldehyde (0.01 mol) was dissolved in 10% alcoholic sodium hydroxide (25 mL) and stirred for 5 h at room temperature. The precipitate was filtered, washed with water, dried, and crystallised from ethanol to give the target compounds Va–h, respectively.
3.2.1. 2-(3-Oxo-3-(4-(piperazin-1-yl)phenyl)prop-1-enyl)benzonitrile (Va)
Yield 70%, m.p. 150 °C. Analysis calculated for C20H19N3O; Calc.: % C, 75.69; H, 6.03; N, 13.24; found: % C, 75.74; H, 6.12; N, 13.10. IR: υmax./cm−m 3230 (NH), 3020 (C–H aromatic), 2200 (CN), 1700 (C=O), 1580 (C=C). 13C NMR (DMSO-d6): 189, 145, 138, 137,133, 132, 130, 128, 127, 126, 121, 112, 115, 109, 54, 45. 1H NMR: δ 2.1 (s, 1H, NH, D2O exchangeable), 2.4–3.0 (m, 8H, piperazinyl protons), 6.8 (d, 1H, J= 15.0 Hz, CH═), and 7.0–8.0 (m, 9H, Ar-H, CH═). MS: m/z (% relative intensity)=317 (M+, 20%), 215 (100%).
3.2.2. 4-(3-Oxo-3–(4-(piperazin-1-yl)phenyl)prop-1-enyl)benzonitrile (Vb)
Yield 75%, m.p. 170 °C. Analysis calculated for C20H19N3O; calcd.: % C, 75.69; H, 6.03; N, 13.24; found: % C, 75.72; H, 6.10; N, 13.15. IR: υmax./cm−1 3240 (NH), 3010 (C–H aromatic), 2210 (CN), 1720 (C═O), 1590 (C═C). 13C NMR (DMSO-d6): 190, 149, 139, 136,133, 132, 128, 127, 121, 118, 112, 111, 55, 46. 1H NMR: δ 2.0 (s, 1H, NH, D2O exchangeable), 2.5–3.1 (m, 8H, piperazinyl protons), 6.8 (d, 1H, J= 15.2 Hz, CH═), and 7.3–8.1 (m, 9H, Ar-H, CH═). MS: m/z (% relative intensity)=317 (M+, 7%), 77 (100%).
3.2.3. 3-(2-Fluorophenyl)-1-(4-(piperazin-1-yl)phenyl)prop-2-en-1- one (Vc)
Yield 70%, m.p. 176 °C. Analysis calculated C19H19FN2O; calcd.: % C, 73.53; H, 6.17; N, 9.03; found: % C, 73.49; H, 6.34; N, 9.12. IR: υmax./cm−1 3310 (NH), 3000 (C–H aromatic), 1720 (C═O), 1590 (C═C). 13C NMR (DMSO-d6): 192, 161, 145, 138, 132, 130, 129, 127, 125, 123, 121, 115, 112, 55, 46. 1H NMR: δ 2.2 (s, 1H, NH, D2O exchangeable), 2.7–3.3 (m, 8H, piperazinyl protons), 6.7 (d, 1H, J= 15.5 Hz, CH═), and 7.1–7.9 (m, 9H, Ar-H, CH═). MS: m/z (% relative intensity)=310 (M+, 50%), 291 (100%).
3.2.4. 3-(3-Fluorophenyl)-1-(4-(piperazin-1-yl)phenyl)prop-2-en-1- one (Vd)
Yield 80%, m.p. 200 °C. Analysis calculated C19H19FN2O; calcd.: % C, 73.53; H, 6.17; N, 9.03; found: % C, 73.59; H, 6.25; N, 9.10. IR: υmax./cm−1 3315 (NH), 3010 (C–H aromatic), 1720 (C═O), 1600 (C═C). 13C NMR (DMSO-d6): 189, 162, 145, 137, 136, 133, 131, 127, 124, 122, 115, 114, 113, 55, 47. 1H NMR: δ 2.3 (s, 1H, NH, D2O exchangeable), 2.6–3.1 (m, 8H, piperazinyl protons), 6.8 (d, 1H, J= 15.0 Hz, CH═), and 7.2–7.7 (m, 9H, Ar-H, CH═). MS: m/z (% relative intensity)=310 (M+, 100%).
3.2.5. 3-(4-Fluoro-3-methoxyphenyl)-1-(4-(piperazin-1-yl)phenyl) prop-2-en-1-one (Ve)
Yield 70%, m.p. 180 °C. Analysis calculated C20H21FN2O2; calcd.: % C, 70.57; H, 6.22; N, 8.23; found: % C, 70.50; H, 6.19; N, 8.27. IR: υmax./cm−1 3400 (NH), 3050 (C–H aromatic), 1730 (C═O), 1600 (C═C). 13C NMR (DMSO-d6): 190, 151, 149, 145, 137, 132, 131, 127, 123, 121, 117, 113, 112, 57, 50, 45. 1H NMR: δ 2.4 (s, 1H, NH, D2O exchangeable), 2.8–3.3 (m, 8H, piperazinyl protons), 3.80 (s, 3H, OCH3), 7.0 (d, 1H, J= 15.1 Hz, CH═), and 7.1–7.8 (m, 8H, Ar-H, CH═). MS: m/z (% relative intensity)=340 (M+, 24%), 85 (100%).
3.2.6. 3-(3-Fluoropyridin-4-yl)-1-(4-(piperazin-1-yl)phenyl)prop-2- en-1-one (Vf)
Yield 75%, m.p. 225 °C. Analysis calculated C18H18FN3O; calcd.: % C 69.44; H, 5.83; N, 13.50; found: % C, 69.49; H, 5.95; N, 13.61. IR: υmax./cm−1 3450 (NH), 3040 (C–H aromatic), 1720 (C═O), 1590 (C═C). 13C NMR (DMSO-d6): 187, 156, 146, 144, 139, 136, 133, 131, 128, 126, 118, 113, 53, 45. 1H NMR: δ 2.3 (s, 1H, NH, D2O exchangeable), 2.7–3.4 (m, 8H, piperazinyl protons), 6.7 (d, 1H, J= 15.0 Hz, CH═CH), and 6.9–7.8 (m, 8H, Ar-H, CH═). MS: m/z (% relative intensity)=311 (M+, 100%).
3.2.7. 3-(2-Hydroxy-5-nitrophenyl)-1-(4-(piperazin-1-yl)phenyl)prop-2-en-1-one (Vg)
Yield 65%, m.p. 185 °C. Analysis calculated C19H19N3O4; calcd.: % C 64.58; H, 5.42; N, 11.89; found: % C, 64.50; H, 5.37; N, 11.83. IR: υmax./cm−1 3400 (NH), 3320 (OH), 3010 (C–H aromatic), 1700 (C═O), 1590 (C═C). 13C NMR (DMSO-d6): 195, 163, 141, 140, 139, 131, 129, 126, 125, 120, 119, 116, 112, 54, 45. 1H NMR: δ 2.0 (s, 1H, NH, D2O exchangeable), 2.4–3.1 (m, 8H, piperazinyl protons), 6.6 (d, 1H, J= 15.2 Hz, CH═), 7.3–7.9 (m, 8H, Ar-H, CH═), and 9.1 (s, 1H, O-H). MS: m/z (% relative intensity)=353 (M+, 10%), 290 (100%).
3.2.8. 3-(Furan-2-yl)-1-(4-(piperazin-1-yl)phenyl)prop-2-en-1-one (Vh)
Yield 70%, m.p. 190 °C. Analysis calculated C17H18N2O2; calcd.: % C 72.32; H, 6.43; N, 9.92; found: % C 72.37; H, 6.52; N, 9.90. IR: υmax./cm−1 3390 (NH), 3030 (C–H aromatic), 1700 (C═O), 1600 (C═C). 13C NMR (DMSO-d6): 188, 151, 140, 137, 130, 127, 126, 120, 114, 112, 110, 50, 45. 1H NMR: δ 2.1 (s, 1H, NH, D2O exchangeable), 2.9–3.5 (m, 8H, piperazinyl protons), 6.7 (d, 1H, J= 15.5 Hz, CH═), and 7.0–7.8 (m, 8H, Ar-H, CH═). MS: m/z (% relative intensity)=282 (M+, 30%), 197 (100%).
3.3. General method for the preparation of VIa–c
A mixture of the chalcone Va, Ve, and Vf (0.006 mol) and hydrazine hydrate (0.006 mol, 98%) in absolute ethanol (30 mL) was heated for 12 h under reflux. The reaction was cooled, the formed precipitate was filtered off and crystallised from ethanol to give compounds VIa–c, respectively.
3.3.1. 2-(3-(4-(Piperazin-1-yl)phenyl)-4,5-dihydro-1H-pyrazol-5-yl)benzonitrile (VIa)
Yield 64%, m.p. 165 °C. Analysis calculated C20H21N5; calcd.: % C 72.48; H, 6.39; N, 21.13; found: % C 72.54; H, 6.42; N, 21.10. IR: υmax./cm−1 3320 (NH), 3030 (C–H aromatic), 2220 (CN), 1600 (C═C). 13C NMR (DMSO-d6): 151, 146, 134, 133, 131, 129, 128, 127, 125, 115, 112, 111, 54, 47, 45, 42. 1H NMR: δ 2.1 (s, 1H, NH, D2O exchangeable), 2.4–2.9 (m, 8H, piperazinyl protons), 3.3 (dd, 1H, J= 11.4, 5.1 Hz, pyrazoline), 3.4 (dd, 1H, J= 11.3, 6.1 Hz, pyrazoline), 5.0 (dd, 1H, J = 11.0, 5.5 Hz, pyrazoline), 7.0–7.7 (m, 8H, Ar-H), and 9.0 (s, NH, D2O exchangeable). MS: m/z (% relative intensity)=331 (M+, 14%), 77 (100%).
3.3.2. 1-(4-(5-(4-Fluoro-3-methoxyphenyl)-4,5-dihydro-1H-pyrazol- 3-yl)phenyl) piperazine (VIb)
Yield 70%, m.p. 195 °C. Analysis calculated C20H23FN4O; calcd.: % C 67.78; H, 6.54; N, 15.81; found: % C 67.70; H, 6.59; N, 15.74. IR: υmax./cm−1 3400 (NH), 3040 (C–H aromatic), 1590 (C═C). 13C NMR (DMSO-d6): 152, 150, 148, 140, 133, 130, 125, 120, 116, 112, 111, 56, 52, 50, 46, 43. 1H NMR: δ 2.3 (s, 1H, NH, D2O exchangeable), 2.6–3.0 (m, 8H, piperazinyl protons), 3.2 (dd, 1H, J= 11.3, 6.1 Hz, pyrazoline), 3.3 (dd, 1H, J= 11.7, 5.9 Hz, pyrazoline), 3.8 (s, 3H, OCH3), 5.1 (dd, 1H, J= 11.9, 6.1 Hz, pyrazoline), 6.9–7.5 (m, 7H, Ar-H), and 8.5 (s, NH, D2O exchangeable). MS: m/z (% relative intensity)=354 (M+, 100%).
3.3.3. 1-(4-(5-(3-Fluoropyridin-4-yl)-4,5-dihydro-1H-pyrazol-3-yl) phenyl) piperazine (VIc)
Yield 70%, m.p. 135 °C. Analysis calculated C18H20FN5; calcd.: % C 66.44; H, 6.20; N, 21.52; found: % C 66.49; H, 6.25; N, 21.548 IR: υmax./cm−1 3390 (NH), 3020 (C–H aromatic), 1595 (C═C). 13C NMR (DMSO-d6): 152, 150, 147, 139, 138, 133, 130, 125, 124, 111, 54, 47, 45, 44. 1H NMR: δ 2.3 (s, 1H, NH, D2O exchangeable), 2.7–3.2 (m, 8H, piperazinyl protons), 3.4 (dd, 1H, J = 11.2, 5.0 Hz, pyrazoline), 3.6 (dd, 1H, J= 11.6, 5.9 Hz, pyrazoline), 5.2 (dd, 1H, J= 12.0, 5.7 Hz, pyrazoline), 7.0–7.9 (m, 7H, Ar-H), and 8.8 (s, NH, D2O exchangeable). MS: m/z (% relative intensity)=325 (M+, 19%), 229 (100%).
3.4. In vitro cytotoxicity
In vitro cytotoxicity was performed in NCI according to reported methodCitation61.
3.5. VEGFR-2 inhibition assay
IC50s of Vd, Ve, Vf, VIa, VIb, and VIc compounds were evaluated in vitro using colorimetric assay of human VEGFR-2 ELISA (enzyme-linked immunosorbent assay) kits (HTScan® VEGF Receptor 2 Kinase Assay Kit). It includes active VEGFR-2 kinase (a biotinylated peptide substrate and a phospho-tyrosine antibody) for detection of the phosphorylated form of the substrate peptide. On a 96-well plate, a particular VEGFR-2 antibody was seeded and 100 µL of the normal solution or compound tested was applied, incubated at room temperature for 2.5 h and washed.
Then, 100 µL of the prepared biotin antibody was added, incubated for an additional 1 h at room temperature and washed. Following, 100 µL of streptavidin solution was added at room temperature, incubated for 45 min and then, 100 µL of TMB substrate solution was applied and incubated at room temperature for 30 min. Finally, 50 µL stop solution was added and the absorption was measured at 450 nm instantly. The standard curve, the X-axis concentrations, and the Y-axis absorbance were drawn.
3.6. Cell cycle analysis
HCT-116 cells were seeded at concentrations of 1 × 105 cells per well in a six-well plate, then incubated for 24 h. The cells were treated for 24 h with vehicles (0.1% DMSO) or 10 µM of Vd or Ve compounds. Using ice-cold, 70% ethanol at 4 °C, cells were harvested and fixed for 12 h after that. Ethanol removal and cold PBS washing of the cells were done. Then incubated in 0.5 mL of PBS containing 1 mg/mL Ranse for 30 min at 37 °C. In the dark, the cells were stained with PI for 30 min. Flow cytometer was then used to detect contents of DNACitation62.
3.7. Annexin V-FITC apoptosis assay
For this study, annexin V-FITC/PI apoptosis detection kit was used; HCT-116 cells were stained with annexin V fluorescein isothiocyanate (FITC) and PI counter-stained. 1 × 105 HCT-116 cells were 48 h incubated with compound Vd or Ve, trypsinised, washed with phosphate-buffered saline (PBS), stained in the dark at 37 °C for 15 min. Then, analysed with a cytometer of FACS calibre flowCitation63.
3.8. Molecular docking
Molecular docking simulation studies were performed using molecular operating environment (MOE®) version 2015.10. The vascular endothelial growth factor receptor (VEGFR) (PDB 4ASD) was used as a receptor for the docking study and Sorafenib as a reference drug.
3.8.1. Target compounds optimisation
Using the MOE program builder interface, the tested compounds Vd, Ve, Vf, VIa, VIb, and VIc were created into a 3D model. The target structures were checked by 2D depiction and formal charges on atoms, and then a conformational search was conducted for the target compounds. All conformers were subjected to energy minimisation done with MOE until an RMSD gradient of 0.01 kcal/mol and an RMS distance of 0.1 Å with MMFF94X were automatically measured as a force-field and the partial charges. The database of target compounds was then saved as MDB file for use in the calculations for molecular docking.
3.8.2. Optimisation of VEGFR active site
The VEGFR has been prepared for docking experiments by adding hydrogen atoms and their standard geometry. The atom's connections and types were checked with automatic correction for any errors that existed. Selection of the receptor and its potential atoms has been fixed. MOE Alpha Site Finder used all default items to search for the active site in the receptor structure, and then dummy atoms were created from the alpha spheres obtained.
3.8.3. Docking of the target compounds to the VEGFR active sites
Docking of the tested compounds' conformational database was performed using MOE-Dock software. To ensure a reasonable docking accuracy and to determine the effect of the water molecules, the co-crystallised ligand in the VEGFR (PDB 4ASD) was docked to its corresponding protein (in the absence and in the presence of water) and the RMSD values were determined between the co-crystallised ligand and docked pose. The success rates obtained were highly excellent where the active site of the VEGFR was calculated from the binding of co-crystallised ligand and saved as MOE file. The active site file of the VEGFR was then loaded, and the docking tool was used. The program specifications have been adjusted to the dummy atoms as docking site, triangle matcher as placement methodology, London dG as scoring methodology that have been adjusted to its default values. The MDB file of the ligands to be docked (Sorafenib and target compounds) was loaded, and calculations for docking were run automatically. The poses obtained were studied and the poses which had the best ligand–receptor interactions were selected and stored for calculating energy.
4. Conclusions
In summary, novel piperazine–chalcone hybrids and related pyrazoline analogues were synthesised and six of them were selected at a single dose concentration (10−1 M) by NCI (Bethesda, MD) to test their in vitro anticancer activity against full 60 lines of human cancer cells. VEGFR-2 enzyme inhibitory assay was performed to investigate the mechanism of anticancer activity of the tested compounds. While, all tested compounds demonstrate good inhibitory activity against VEGFR-2, the most active compounds were Vd and Ve that have been shown to be able to cause cell cycle arrest during the G2/M process and inducing apoptosis in HCT-116 cells. The present research has led to the discovery of new cytotoxic compounds that target the VEGFR-2. Furthermore, a molecular docking study of selected compounds was carried out and confirmed that compounds Vd and Ve exhibited a direct interaction with the VEGFR.
Supplemental Material
Download PDF (1.3 MB)Acknowledgements
The authors are thankful to National Cancer Institute (NCI) of the United States for performing anticancer evaluation over the 60-cancer cell line.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Varmus H. The new era in cancer research. Science 2006;312:1162–5.
- Gibbs JB. Mechanism-based target identification and drug discovery in cancer research. Science 2000;287:1969–73.
- Kerru N, Singh P, Koorbanally N, et al. Recent advances (2015–2016) in anticancer hybrids. Eur J Med Chem 2017;142:179–212.
- Gerber DE. Targeted therapies: a new generation of cancer treatments. Am Fam Phys 2008;77:311–9.
- Kerbel RS. Tumor angiogenesis: past, present and the near future. Carcinogenesis 2000;21:505–15.
- Polverini PJ. The pathophysiology of angiogenesis. Crit Rev Oral Biol Med 1995;6:230–47.
- Baeriswyl V, Christofori G. The angiogenic switch in carcinogenesis. Semin Cancer Biol 2009;19:329–37.
- Fidler IJ. Angiogenesis and cancer metastasis. Cancer J 2000;6:S134–S41.
- Shawver LK, Lipson KE, Fong TAT, et al. Receptor tyrosine kinases as targets for inhibition of angiogenesis. Drug Discov Today 1997;2:50–63.
- Traxler P. Tyrosine kinases as targets in cancer therapy – successes and failures. Expert Opin Ther Targets 2003;7:215–34.
- Abhinand CS, Raju R, Soumya SJ, et al. VEGF-A/VEGFR2 signaling network in endothelial cells relevant to angiogenesis. J Cell Commun Signal 2016;10:347–54.
- Hoeben A, Landuyt B, Highley MS, et al. Vascular endothelial growth factor and angiogenesis. Pharmacol Rev 2004;56:549–80.
- Ferrara N. Vascular endothelial growth factor: basic science and clinical progress. Endocr Rev 2004;25:581–611.
- Lohela M, Bry M, Tammela T, et al. VEGFs and receptors involved in angiogenesis versus lymphangiogenesis. Curr Opin Cell Biol 2009;21:154–65.
- Shibuya M. Vascular endothelial growth factor (VEGF) and its receptor (VEGFR) signaling in angiogenesis: a crucial target for anti- and pro-angiogenic therapies. Genes Cancer 2011;2:1097–105.
- Daniel JE, La S, Julie B, et al. Novel 2,3-dihydro-1,4-benzoxazines as potent and orally bioavailable inhibitors of tumor-driven angiogenesis. J Med Chem 2008;51:1695–705.
- Machado VA, Peixoto D, Costa R, et al. Synthesis, antiangiogenesis evaluation and molecular docking studies of 1-aryl-3-[(thieno[3,2-b]pyridin-7-ylthio)phenyl]ureas: discovery of a new substitution pattern for type II VEGFR-2 Tyr kinase inhibitors. Bioorg Med Chem 2015;23:6497–509.
- Rajagopalan M, Balasubramanian S, Ramaswamy A, et al. Pharmacophore based 3D-QSAR modeling and free energy analysis of VEGFR-2 inhibitors. J Enzyme Inhib Med Chem 2013;28:1236–46.
- Elsayed NMY, Serya RAT, Tolba MF, et al. Design, synthesis, biological evaluation and dynamics simulation of indazole derivatives with antiangiogenic and antiproliferative anticancer activity. Bioorg Chem 2019;82:340–59.
- Muresan-Pop M, Chereches G, Borodi G, et al. Structural characterization of 5-fluorouracil & piperazine new solid forms and evaluation of their antitumor activity. J Mol Struct 2020;1207:127842.
- Mao ZW, Zheng X, Lin YP, et al. Design, synthesis and anticancer activity of novel hybrid compounds between benzofuran and N-aryl piperazine. Bioorg Med Chem Lett 2016;26:3421–4.
- Patel RV, Mistry B, Syed R, et al. Chrysin–piperazine conjugates as antioxidant and anticancer agents. Eur J Pharm Sci 2016;88:166–77.
- Mehtap T, Halise IG, Kenjiro B, et al. Synthesis and biological evaluation of some new mono Mannich bases with piperazines as possible anticancer agents and carbonic anhydrase inhibitors. Bioorg Chem 2019;90:103095.
- Gokhan-Kelekci N, Yabanoglu S, Kupeli E, et al. A new therapeutic approach in Alzheimer disease: some novel pyrazole derivatives as dual MAO-B inhibitors and anti-inflammatory analgesics. Bioorg Med Chem 2007;15:5775–86.
- Abdel-Aziz M, Abuo-Rahma GA, Hassan AA. Synthesis of novel pyrazole derivatives and evaluation of their antidepressant and anticonvulsant activities. Eur J Med Chem 2009;44:3480–7.
- Kaushik D, Khan SA, Chawla G, et al. N′-[(5-chloro-3-methyl-1-phenyl-1H-pyrazol-4-yl)methylene] 2/4-substituted hydrazides: synthesis and anticonvulsant activity. Eur J Med Chem 2010;45:3943–9.
- Khunt RC, Khedkar VM, Chawda RS, et al. Synthesis, antitubercular evaluation and 3D-QSAR study of N-phenyl-3-(4-fluorophenyl)-4-substituted pyrazole derivatives. Bioorg Med Chem Lett 2012;22:666–78.
- Pathak RB, Chovatia PT, Parekh HH. Synthesis, antitubercular and antimicrobial evaluation of 3-(4-chlorophenyl)-4-substituted pyrazole derivatives. Bioorg Med Chem Lett 2012;22:5129–33.
- Malladi S, Isloor AM, Peethambar SK, et al. Synthesis and antimicrobial activity of some new pyrazole containing cyanopyridone derivatives. Der Pharm Chem 2012;4:43–52.
- Puthran D, Poojary B, Purushotham N, et al. Synthesis of novel Schiff bases using 2-amino-5-(3-fluoro-4-methoxyphenyl)thiophene-3-carbonitrile and 1,3-disubstituted pyrazole-4-carboxaldehydes derivatives and their antimicrobial activity. Heliyon 2019;5:e02233.
- Vijesh AM, Isloor AM, Shetty P, et al. New pyrazole derivatives containing 1,2,4-triazoles and benzoxazoles as potent antimicrobial and analgesic agents. Eur J Med Chem 2013;62:410–5.
- Zabiulla GA, Mohammed YHE, et al. Design, synthesis and molecular docking of benzophenone conjugated with oxadiazole sulphur bridge pyrazole pharmacophores as anti-inflammatory and analgesic agents. Bioorg Chem 2019;92:103220.
- Koca İ, Özgür A, Coşkun KA, et al. Synthesis and anticancer activity of acyl thioureas bearing pyrazole moiety. Bioorg Med Chem 2013;21:3859–65.
- Abdellatif KR, Abdelall EK, Abdelgawad MA, et al. Synthesis and anticancer activity of some new pyrazolo[3,4-d]pyrimidin-4-one derivatives. Molecules 2014;19:3297–309.
- Dawood KM, Eldebss TMA, El-Zahabi HSA, et al. Synthesis of some new pyrazole-based 1,3-thiazoles and 1,3,4-thiadiazoles as anticancer agents. Eur J Med Chem 2013;70:740–9.
- Tu CH, Lin WH, Peng YH, et al. Pyrazolylamine derivatives reveal the conformational switching between type I and type II binding modes of anaplastic lymphoma kinase (ALK). J Med Chem 2016;59:3906–19.
- Thomas R, Mary YS, Resmi KS, et al. Two neoteric pyrazole compounds as potential anti-cancer agents: synthesis, electronic structure, physico-chemical properties and docking analysis. J Mol Struct 2019;1181:455–66.
- Bennani FE, Doudach L, Cherrah Y, et al. Overview of recent developments of pyrazole derivatives as an anticancer agent in different cell line. Bioorg Chem 2020;97:103470–62.
- Farooqui AA. Phytochemicals, signal transduction, and neurological disorders. New York, USA: Springer Science & Business Media; 2012.
- Mahapatra DK, Asati V, Bharti SK. Chalcones and their therapeutic targets for the management of diabetes: structural and pharmacological perspectives. Eur J Med Chem 2015;92:839–65.
- Israf DA, Khaizurin TA, Syahida A, et al. Cardamonin inhibits COX and iNOS expression via inhibition of p65NF-kB nuclear translocation and Ik-B phosphorylation in RAW 264.7 macrophage cells. Mol Immunol 2007;44:673–9.
- Kantevari S, Addla D, Bagul PK, et al. Synthesis and evaluation of novel 2-butyl-4-chloro-1-methylimidazole embedded chalcones and pyrazoles as angiotensin converting enzyme (ACE) inhibitors. Bioorg Med Chem 2011;19:4772–81.
- Romagnolo DF, Selmin OI. Flavonoids and cancer prevention: a review of the evidence. J Nutr Gerontol Geriatr 2012;31:206–38.
- Chen M, Brøgger CS, Zhai L, et al. The novel oxygenated chalcone, 2,4-dimethoxy-4′-butoxychalcone, exhibits potent activity against human malaria parasite Plasmodium falciparum in vitro and rodent parasites Plasmodium berghei and Plasmodium yoelii in vivo. J Infect Dis 1997;176:1327–33.
- Winter E, Gozzi GJ, Chiaradia-Delatorre LD, et al. Quinoxaline-substituted chalcones as new inhibitors of breast cancer resistance protein ABCG2: polyspecificity at B-ring position. Drug Des Dev Ther 2014;8:609–19.
- Debarshi KM, Sanjay KB, Vivek A. Anti-cancer chalcones: structural and molecular target perspectives. Eur J Med Chem 2015;98:69–114.
- Lee YS, Lim SS, Shin KM, et al. Anti-angiogenic and anti-tumor activities of 2′-hydroxy-4′-methoxychalcone. Biol Pharm Bull 2006;29:1028–31.
- Varinska L, Wijhe M, Belleri M, et al. Anti-angiogenic activity of the flavonoid precursor 4-hydroxychalcone. Eur J Pharmacol 2012;691:125–33.
- Guo F, Feng L, Huang C, et al. Prenylflavone derivatives from Broussonetia papyrifera, inhibit the growth of breast cancer cells in vitro and in vivo. Phytochem Lett 2013;6:331–6.
- Sisko JT, Tucker TJ, Bilodeau MT, et al. Potent 2-[(pyrimidin-4-yl)amine]-1,3-thiazole-5-carbonitrile-based inhibitors of VEGFR-2 (KDR) kinase. Bioorg Med Chem Lett 2006;16:1146–50.
- Peng F-W, Liu D-K, Zhang Q-W, et al. VEGFR-2 inhibitors and the therapeutic applications thereof: a patent review (2012–2016). Expert Opin Ther Pat 2017;27:987–1004.
- Mainolfi N, Karki R, Liu F, Anderson K. Evolution of a new class of VEGFR-2 inhibitors from scaffold morphing and redesign. ACS Med Chem Lett 2016;7:363–7.
- Matsumoto S, Miyamoto N, Hirayama T, et al. Structure-based design, synthesis, and evaluation of imidazo[1,2-b]pyridazine and imidazo[1,2-a]pyridine derivatives as novel dual c-Met and VEGFR2 kinase inhibitors. Bioorg Med Chem 2013;21:7686–98.
- Shibuya M. Vascular endothelial growth factor receptor-2: its unique signaling and specific ligand, VEGF-E. Cancer Sci 2003;94:751–6.
- Sun YS. Cancer Center, South China Institute of Botany, Chinese Academy of Sciences Assignee. Use of 20,40 dihydroxy-60-methoxy-30,50 dimethyl-chalcone for preparing anticancer medicine, Patent CN1454895 A; 2003.
- Rizvi SUF, Siddiqui HL, Nisar M, et al. Discovery and molecular docking of quinolyl-thienyl chalcones as anti-angiogenic agents targeting VEGFR-2 tyrosine kinase. Bioorg Med Chem Lett 2012;22:942–4.
- Molecular operating environment (MOE), 10. Montreal: Chemical Computing Group Inc.; 2009.
- Ibrahim MK, Taghour MS, Metwaly AM, et al. Design, synthesis, molecular modeling and anti-proliferative evaluation of novel quinoxaline derivatives as potential DNA intercalators and topoisomerase II inhibitors. Eur J Med Chem 2018;155:117–34.
- DTP selection guidelines; 2020. Available from: https://dtp.cancer.gov/organization/dscb/compoundSubmission/structureSelection.htm.
- Liu F, Dawadi S, Maize KM, et al. Structure-based optimization of pyridoxal 5′-phosphate-dependent transaminase enzyme (BioA) inhibitors that target biotin biosynthesis in Mycobacterium tuberculosis. J Med Chem 2017;60:5507–20.
- Monks A, Scudiero D, Skehan P, et al. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J Natl Cancer Inst 1991;83:757–66.
- Wang J, Lenardo MJ. Roles of caspases in apoptosis, development, and cytokine maturation revealed by homozygous gene deficiencies. J Cell Sci 2000;113:753–7.
- Lo KKW, Lee TKM, Lau JSY, et al. Luminescent biological probes derived from ruthenium(II) estradiol polypyridine complexes. Inorg Chem 2008;47:200–8.