
Abstract
The binding mode of aromatic sulphonamides and clinically licenced drugs to the three carbonic anhydrase (CA, EC 4.2.1.1) isoforms from the human pathogen V. cholerae was here thouroghly characterised by a joint docking and molecular dynamics in silico protocol. In fact, VchCA, VchCAβ, and VchCAγ are crucial in the pathogen life cycle and growth and represent innovative targets to fight V. cholerae proliferation overcoming the spreading chemoresistance to the available drugs. A set of 40 sulphonamides/sulfamates VchCAs inhibitors was studied using the proteins homology built 3 D models unveiling the key and stable interactions responsible for a potent CA inhibition. This study has the aim to offer insights and guidelines for the future rational design of potent and selective inhibitors targeting CA isoforms from V. cholerae or other human pathogens.
1. Introduction
1.1. Cholera disease
Vibrio spp. are bacteria present in freshwater, estuarine and marine environments that prefer the warm and brackish waterCitation1. Among the ∼12 pathogenic species for humans of the >100 described Vibrio spp., Vibrio cholerae is the unique rod shape Gram-negative bacterium that provokes cholera, a disease endemic in low income countriesCitation1,Citation2. Annually, cholera affects more than 2–4 million people worldwide with 21,000–143,000 deaths, half of them being children under 5 years oldCitation3–5. The infection occurs mainly by the faecal-oral route through contaminated food, or poorly sanitised waterCitation6,Citation7, or through the person-to-person close contactCitation8,Citation9. V. cholerae enters in the gastrointestinal tract and reaches the small intestineCitation10. Several intestinal environmental factors such as bicarbonateCitation11, bile, unsaturated fatty acids, and reduced oxygen levels promote the co-transcription of toxin-co-regulated pilus (Tcp), cholera toxin (CT) and other colonization-associated genes (all encoded by regulon toxT), that allow the pathogen proliferationCitation12. Using the filamentous surface appendage Tcp, V. cholerae is able to bind the same adjacent bacterial cells and to tightly adhere to enterocytes without disrupting the mucosal integrityCitation10,Citation13,Citation14. Instead, the pathogen-secreted toxin CT, composed of two subunits ctxA and ctxB, recognises and binds the sialylated glycosphingolipid GM1 on the cytoplasmatic membrane of enterocytes with the pentameric ctxB subunitCitation15. After endocytosis, CT enters in the endoplasmic reticulum (ER) via a retrograde transport, where the subunits are dissociatedCitation15. The enzymatic ctxA subunit released in the cytosol, upon allosteric activation by ADP ribosylation factor 6 (ARF6), is able to trigger the G-protein coupled receptor and consequently the adenylyl cyclase (AC)Citation15. The high levels of produced cAMP stimulate the protein kinase A (PKA)-dependent phosphorylation of the cystic fibrosis transmembrane receptor (CFTR), responsible for the efflux of water and ions into the lumen of the small intestine, leading to diarrheaCitation15. The profuse watery diarrhoea, together with vomiting and gastroenteritis, are the main clinical symptoms of cholera disease that, if untreated, results in death due to dehydration within 1–2 daysCitation16,Citation17. To date, the long-term solutions to prevent cholera are the surveillance, sanitisation of the water, good hygiene practices, social mobilisation monitoring, and oral cholera vaccinesCitation18,Citation19. On the other hand, the infection is treated by prompt administration of oral/intravenous rehydration solution (ORS)Citation20–22, appropriate antibiotics (such as azithromycin and ciprofloxacin)Citation23,Citation24 and zincCitation25. While the administration of ORS is a fundamental but symptomatic therapy, the use of antibiotics is important to eradicate the cause of the illness. However, the spreading drug resistance to antimicrobial agents is threatening the efficacy of current chemotherapy, making the development of new antibiotic drugs with different mechanisms of action essentialCitation26.
1.2. Vibrio cholerae carbonic anhydrases
Bicarbonate is an important virulence factor for V. cholerae as it is a positive effector for toxT activity, promoting the transcription of genes that encode for Tcp, CT and other proteins implicated in proliferationCitation11,Citation27–29. These genes expression is significatively reduced by the addition of carbonic anhydrase inhibitors (CAIs)Citation27–29. Thus, it is probable that V.cholerae uses the carbonic anhydrases (CAs, EC 4.2.1.1) system to accumulate bicarbonate into the cell for activating its virulence, as the bicarbonate levels are very high in the upper small intestine colonised by the pathogen and this bacterium does not encode bicarbonate transporter proteins in its genomeCitation27–29.
Such evidences make CAs interesting targets to prevent V. cholerae proliferation, offering the possibility to develop antibacterial drugs with an innovative mechanism of action to contrast the disease.
In detail, CAs are a superfamily of ubiquitous metalloenzymes, present in all life kingdoms, that catalyse the reversible hydration of carbon dioxide (CO2) into bicarbonate ion (HCO3−) and a protonCitation30. To date, eight genetically unrelated families of CAs called α, β, γ, δ, η, ζ, θ and ιCitation31–38 have been identified, but only α-, β-, γ- and ι-CAs are present in prokaryotesCitation28,Citation39–43. In microorganisms, these CAs are involved in photosynthesis (cyanobacteria), biosynthesis of amino acids, fatty acids, and nucleic acids, but also in proliferation, survival, and differentiationCitation44.
The genome of V. cholerae encodes for three CAs, VchCA, VchCAβ, and VchCAγ respectively belonging to the α-, β- and γ-class. This suggests the important role of these enzymes in the pathogen physiologyCitation28,Citation44,Citation45.
VchCA (α-CA) consists of 239 amino acids, and shows a 30% identity with the two humans (h) α-CAs I and II. Moreover, VchCA maintains basic characteristics common to most α-CAs, that are the three histidine residues coordinating the zinc ion (H104, H106, and H123), a proton shuttle histidine residue (H79), and the gate-keeping glutamate-threonine dyad (E110, T189)Citation46. No X-ray crystal structure of this enzyme is available so far.
The X-ray crystallography of VchCAβ (β-CA; PDB 5CXK)Citation47 showed a tetrameric structure with four active sites, composed of monomers of 222 amino acids eachCitation48–50. Furthermore, VchCAβ preserves the common features of the β-class CAs that are the two cysteines and the histidine residue (C42, C101, and H98 from a same monomer) coordinated to the zinc ion, and the aspartate-arginine dyad (D44, and R46) responsible for the opening/closing of the active siteCitation48–50. In fact, β-CAs can exist in a type I (open active site) or type II (closed active site) enzyme, depending on the pHCitation48–50. When the pH is < 8.0 the aspartate residue of the dyad coordinates the zinc ion as a fourth ligand in place of the water molecule/hydroxide ion, thus disabling the CO2 hydration reaction (type-II form). At pH > 8.0 the aspartate residue forms a salt bridge with the arginine of the dyad, allowing the zinc ion to be coordinated by a water molecule/hydroxide ion (type-I form)Citation48–50. This pH regulation suggests that β-CAs activity is presumably regulated by the substrates and in particular by HCO3- concentrationsCitation48–50. Hence, when Vibrio colerae reaches the upper small intestine, the high levels of bicarbonate can promote the type-II to type-I VchCAβ form conversion, assisting the virulence process.
VchCAγ is a trimeric enzyme with monomers formed by 184 amino acids (chains A, B and C) assembled to form three different active sites. As in the other γ-CAs, the zinc atom is coordinated by three conserved histidine residues (H65, and H94 from a chain and H89 from another chain) and a proton shuttle is present nearby (H68)Citation51. As for the α-class CA from this organism, no X-ray crystal structure of the enzyme is available so far.
Kinetic parameters gathered in show that VchCA (Kcat = 8.2 × 105 s−1) is more active than VchCAγ (Kcat = 7.4 × 105 s−1) and VchCAβ (Kcat = 3.3 × 105 s−1), and all of them are more active than hCA I (Kcat = 2.0 × 105 s−1)Citation45,Citation52–55.
Table 1. Kinetic parameters for the CO2 hydration reaction of α-CAs human cytosolic isozymes hCA I and II and VchCA measured at 20 °C and pH 7.5 in 10 mM HEPES buffer and 20 mM Na2SO4, and VchCAβ and VchCAγ measured at 20 °C, pH 8.3 in 20 mM TRIS buffer and 20 mM NaClO4Citation45,Citation52–55.
Among many investigated chemotypes, primary sulphonamide derivatives stood out as the most potent CAIs to dateCitation31,Citation56. Hence, a set of 38 primary aromatic/aliphatic sulphonamides (1–24, AAZ, MZA, EZA, DCP, DZA, BRZ, BZA, ZNS, SLP, IND, VLX, CLX, SLT, HCT), one secondary sulphonamides (SAC), and one sulphamate (TPM) shown in , were evaluated for the inhibition of VchCA, VchCAβ and VchCAγ to characterise each isozyme response to inhibitors in search of new generation antiinfectives against V. cholerae ()Citation45.
Figure 1. Structure of sulphonamides/sulfamates previously investigated as VchCAs inhibitorsCitation45.
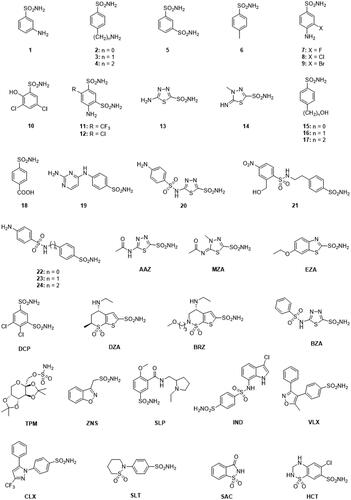
Table 2. Inhibition data of human isoforms hCA I and hCA II, and VchCA, VchCAβ and VchCAγ from V. cholerae with sulphonamides 1–24 and the clinically used drugs AAZ-HCT by a stopped-flow CO2 hydrase assayCitation45.
Many nanomolar inhibitors were identified against the three CAs, with efficacy against VchCA (KIs 0.59->1000 nM) > VchCAγ (KIs 66.2–6223 nM) > VchCAβ (KIs of 68.1->10000 nM)Citation45. Here, a thorough interaction study was carried out in silico with derivatives in and the three VchCA isoforms.
2. Material and methods
2.1. Carbonic anhydrase inhibition
Results, discussion and methods of the CA inhibition assay for compounds 1–24 and used drugs AAZ-HCT were previously reportedCitation45.
2.2. Molecular modelling
The homology models of VchCAα, VchCAβ and VchCAγCitation57 were prepared using the Protein Preparation Wizard tool implemented in the Schrödinger suiteCitation58. The energy minimisation protocol with a root mean square deviation (RMSD) value of 0.30 Å was applied using force field OPLS3e. The ligand structures were prepared by MaestroCitation58b and evaluated for their ionisation states at pH 7.4 ± 0.5 with EpikCitation58c. The conjugate gradient method in MacromodelCitation58e was used for energy minimisation (maximum iteration number: 2500; convergence criterion: 0.05 kcal mol−1 Å−1). The software Glide was used for dockingCitation58f. Grids were centred on the centroids of the zinc-coordinating residues and ligands were docked using standard precision mode (SP). The best pose of a subset of compounds to the three VchCAs, evaluated in terms of anchorage, hydrogen bond interactions and hydrophobic contacts, was submitted to a MD simulation using Desmond and the OPL3e force fieldCitation58g,Citation59. Specifically, the system was solvated in an orthorhombic box using TIP4PEW water molecules, extended 15 Å away from any protein atom. It was neutralised adding chlorine and sodium ions. The simulation protocol included a starting relaxation step followed by a final production phase of 100 ns. In particular, the relaxation step comprised the following: (a) a stage of 100 ps at 10 K retaining the harmonic restraints on the solute heavy atoms (force constant of 50.0 kcal mol−1 Å−2) using the NPT ensemble with Brownian dynamics; (b) a stage of 12 ps at 10 K with harmonic restraints on the solute heavy atoms (force constant of 50.0 kcal mol−1 Å−2), using the NVT ensemble and Berendsen thermostat; (c) a stage of 12 ps at 10 K and 1 atm, retaining the harmonic restraints and using the NPT ensemble and Berendsen thermostat and barostat; (f) a stage of 12 ps at 300 K and 1 atm, retaining the harmonic restraints and using the NPT ensemble and Berendsen thermostat and barostat; (g) a final 24 ps stage at 300 K and 1 atm without harmonic restraints, using the NPT Berendsen thermostat and barostat. The final production phase of MD was run using a canonical NPT Berendsen ensemble at temperature 300 K. During the MD simulation, a time step of 2 fs was used while constraining the bond lengths of hydrogen atoms with the M-SHAKE algorithm. The atomic coordinates of the system were saved every 100 ps along the MD trajectory. Protein and ligand RMSD values, ligand torsions evolution and occupancy of intermolecular hydrogen bonds and hydrophobic contacts were computed along the production phase of the MD simulation with the Simulation Interaction Diagram tools implemented in Maestro.
3. Docking and molecular dynamics
For an exhaustive comprehension of the inhibition profile of compounds 1–24, AAZ, MZA, EZA, DCP, DZA, BRZ, BZA, ZNS, TPM, SLP, IND, VLX, CLX, SLT, SAC, HCT and to understand the key interactions for their recognitions within VchCA, VchCAβ, and VchCAγ, an in silico investigation was carried out applying a computational protocol that includes docking studies, MM-GBSA refinements, and MD calculations.
Sulphonamides and bioisosteres (i.e. sulfamates and sulfamides) act as zinc-binders CAIs against the humanCitation56,Citation61 and bacterialCitation62 α-CAs, binding the zinc ion by the deprotonated nitrogen atom (SO2NH-) according to a tetra-coordinated geometry around the metal ion. Aromatic compounds such as benezenesulfonamides and 1,3,4-thiadiazole-2-sulphonamides show a better inhibitory activity than aliphatic sulphonamide derivatives as their binding mode is stabilised by vdW contacts involving conserved lipophilic residues in the inner active site (namely L198, V121, V143 in hCA II) and the aromatic portion bearing the zinc-binding group.
X-ray studies on β-CAs pointed out that aromatic sulphonamides and sulfamides occupied the fourth coordination site site around the zinc ion that in β-CAs is in a pseudotetrahedral coordination environment. Indeed, the deprotonated nitrogen atom (SO2NH-) is coordinated to the Zn and the aromatic ring is stabilised by a π-π stacking interaction with the side chain of an aromatic residue nearby (i.e. phenylalanine or tyrosine)Citation63,Citation64. Instead no X-ray crystallography data exist which show the binding mode of sulphonamide to γ-CAs. However, it is reasonable to assume that primary sulphonamide derivatives act as fourth ligand of the zinc coordination sphere replacing the water molecule also in γ-CAs. Interestingly, in a recent modelling study performed on sulphonamide inhibitors against the β- and γ-CA isoforms from E. faeciumCitation65, the inhibitors were predicted to act as metal binders against the γ-CA class adopting both a tetrahedral and pentameric coordination.
With the exception for the closed, type II, form of β-CA from Vibrio cholerae, to date in the PDBCitation66 there are no solved structures for VchCAα,VchCAγ and type I/open VchCAβ, both in apo form and or in complex with ligands. In this study, the homology-built models of the three classes of Vibrio cholerae CAs, already obtained for a previous investigation, were used to shed light on the binding mode of sulphonamide derivatives to all CA classes of from this pathogen. First, the ligands were docked within the binding cavity of the three CAs, then the stability of the binding poses was assessed by molecular dynamic simulations. The results are presented according to each enzyme isoform.
3.1. Vchca
All docking solutions for compounds in localise the ligands at the bottom of the conical cavity of the enzyme. Here the SO2NH- moiety coordinated around the zinc ion according to a tetrahedral geometry (Figure S1, Supporting Information). Moreover, the sulphonamide NH- and S=O groups are in H-bond contact with the side chain hydroxyl group of T189 (N−⋯H-O) and with the backbone of the same residue (O…H-N) respectively. The binding orientations of both the benzenesulphonamide and the 1,3,4-thiadiazole-2-sulphonamide inhibitors are stabilised by van der Waals (vdW) interactions that occur between the phenyl ring and the hydrophobic residues L188, V135, and V125 (Figure S1A). In addition 1,3,4-thiadiazole-2-sulphonamides form an H-bond by the N3 atom of the heterocycle with T190 OH group (Figure S1B), which might contribute to the generally increased inhibition profile shown by these derivatives compared to the benzenesulphonamides.
100 ns long MD simulations were carried out to monitor the structural flexibility of the poses. Derivatives 20 (KI = 0.59 nM), EZA (KI = 0.69 nM), and BRZ (KI = 2.5 nM) were considered as representatives among the compounds in having the best inhibition profiles, while 2 (KI = 471 nM) was chosen due to its worst inhibition profile (). Noteworthy, all the bound poses from docking totally maintained the coordination the zinc ion and the H-bond network with T189 for more than 96% of the MD (). In addition, H-bonds involving the N3 atom of the 1,3,4-thiadiazole 20 and 1,3-benzo[d]thiazole EZA ring persist for the 62%, and 58% of the simulation course (). The sulphonamide linker in 20 is involved in a H-bond network with the carboxyamide and the protonated amino groups in the side chains of Q102 and K101 respectively (). Moreover, due to the net negative charge of the linker sulphonamide at the physiological pH, 20 forms a salt bridge interaction with K101, while further contributes to the stabilisation of the pose come from the water bridged H-bonds that occur between the Q82 side chain and the NH2 group and the heterocyclic N4 and the P191 carbonyl group (hetN4⋯HO-H⋯O = C-P191; ).
Figure 2. Most populated binding conformation along the MD trajectory for (A) 20 (blue), (B) EZA (tan), (C) BRZ (green), and (D) 2 (pink) within VchCA active site. H-bonds and salt bridge interactions are depicted as black and magenta dashed lines, respectively. The occupancy over the MD simulation of interactions not involving the zinc-binding group is indicated as percentage.
Water bridged H-bonds also occur for EZA () and BRZ (N-H…O-H T190; H3C-O…H-N Q82; ). The ligands are further stabilised by a wide ensemble of interactions, mainly 15–55% stable vdW interactions involving L188, P192, L133, W23 (). For 59% of the MD simulation, the endocyclic BRZ sulphonamide S=O is engaged in an H-bond interaction with the Q102 side chain NH2.
The MD study of sulphanilamide 2 highlighted 65% stable vdW interactions with L188, and water bridged H-bonds that the outer NH2 can stably form with hydrophilic active site residues, such as Q102 ().
Outcomes from MD computation may also provide insights for understanding the inhibition profile of other benzenesulfonamides 1, 3, 4, 6–9, 19, 21, and 23 (Figures S2-S3, Supporting Information). Similarly to derivative 2, the amine function of 1 (KI = 440 nM), 3 (KI = 447 nM) and 4 (KI = 402 nM) is oriented towards the hydrophilic half of the active site, not able to form direct H-bonds with the protein (Figure S2A-B). Instead, the substitution of the aromatic amine of 2 with a methyl moiety to give compound 6 (KI = 219 nM) increased the inhibition profile, probably because of the increased vdW interactions with P191 and P192 (Figure S2A). The halogenation of sulphanilamide 2 in position 3 with a fluorine, chlorine, or bromine atom to give compound 7–9 (KI = 133–199 nM) enhances the hydrophobic contacts with the lipophilic half of the active site in the order Br > Cl > F (Figure S2C). The elongation of the tail of sulphanilamide 2 by a 2-amino-pyrimidin-4-yl in 19 (KI = 4.7 nM) allows the outer amine group to form direct H-bonds, namely with the C=O of P191, that together with other probable water-mediated interactions could improve the binding to the target (Figure S2D). Likewise, the hydroxymethyl group of derivative 21 (KI = 54.5 nM) engages an H-bond with Q82 side chain NH2 (Figure S3A). The binding mode of compound 23 (KI = 71.5 nM) is shown in Figure S3B as representative of the set 22–24 (KI = 52.1–71.5 nM). The sulphonamide linker S = O group receives an H-bond by the NH2 of Q102 side chain. Residue K101 may be involved in the interaction with the ligand, that is, an H-bond with the S=O group for compounds 22–24, and a salt bridge interaction with the N- moiety of compound 22). Derivatives 13 (KI = 23.5 nM) and 14 (KI = 12.1 nM) are better inhibitors than sulphanilamide 2 probably because of the three H-bonds with T189 and T190 stabilising the scaffold of 1,3,4-thiadiazole-2-sulphonamide derivatives (Figure S3C). The presence of a N4-methyl group in derivative 14 enhances the hydrophobic contacts with the lipophilic half of the active site (P191, P192, and L188), justifying the twofold inhibition profile as compared to that one of 13. Moreover, the clinically used acetylated compounds AAZ (KI = 6.8 nM) and MZA (KI = 3.6 nM) are 4-fold more active than 13 and 14, owing to additional H-bond that the acetyl C = O moiety can receive from the NH2 of Q102 side chain (Figure S3D). Similarly to its precursor 13, BZA (KI = 4.2 nM), showed an H-bond between the ligand S = O group and Q102 (Figure S4A). DZA (KI = 6.3 nM) adopted a similar binding mode as the structurally resembling BRZ (Figure 2C), whereby the NH2+ moiety is involved in H-bond with P191the carbonyl group C = O and the endocyclic sulphonamide engages polar contacts with the hydrophilic half (Figure S4B). IND (KI = 8.1 nM), uniquely having a reversed –SO2NH- linker, showed a binding orientation in which the sulphonamide NH moiety donates an H-bond to the P191 backbone C=O and the indolic ring is stabilised by a π-π stacking with the indole of the W23 (Figure S4C). All poses computed for SLP feature strains occurring in the contact between the ligand pyrrolidine and Q102, applicable to explain a KI value above 1000 nM (Figure S4D).
3.2. Vchcaβ
The binding site of VchCAβ is narrower than those of VchCA and VchCAγ. This made the binding mode prediction more challenging and experimentally led to the generally less favourable inhibition profile of all compounds against this isoform (). According to literature, the SO2NH− was found as coordinated around the zinc ion at the dimeric interface, engaging H-bonds with the OH group of Y83, NH2 of Q33 side chain, and the carboxylic function of D44 (Figure S5, Supporting Information). Moreover, the aromatic ring bearing the zinc-binding group (benzene/1,3,4-thiadiazole-2-sulphonamide) is stabilised by a π-π interaction with Y83 aromatic ring and by vdW contacts with V59.
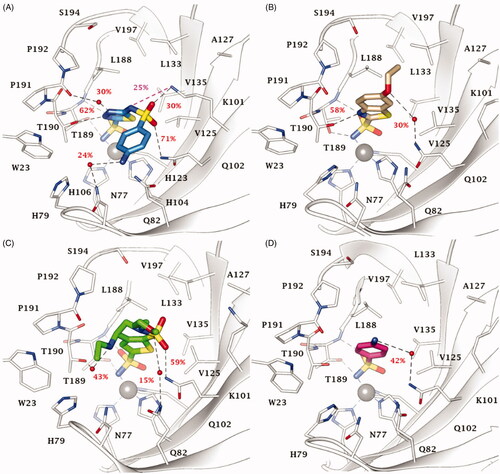
MD simulations performed on 13 (KI = 68.1 nM), the best VchCAβ inhibitor in , and on three compounds with medium inhibition profile 15 (KI = 349 nM), 24 (KI = 361 nM), and 18 (KI = 515 nM) confirmed the total permanency of the metal coordination and the high stability of the interaction network involving the aromatic sulphonamide core and the aminoacidic residues D44, Q33, Y83 and V59, with which the SO2NH- moiety is both in H-bond contacts (D44 and Q33) and forms π-π stacking (Y83) and vdW interactions (V59: stable for 24–93% of the MD; ).
Figure 3. Most populated binding conformation along the MD trajectory for (A) 13 (aquamarine), (B) 15 (pink), (C) 24 (grey), and (D) 18 (orange) within VchCAβ active site. H-bonds and π-π stacking interactions are depicted as black and cyan dashed lines, respectively. The occupancy over the MD simulation of interactions not involving the zinc-binding group is indicated as percentage.
Additionally, the binding orientation of 13 is firmly held within the binding site by water bridged H-bonds involving G102. It is likely that these interactions contribute to make the positioning of thiadiazole derivatives witin the binding site more stable as compared to the benzenesulfonamides (). Among the latter, the phenol derivative 15 also forms a water-mediated H-bond with G102 ().
The short active site of VchCAβ obliges inhibitor 24 to fold at the tail level; as a result the outer amine group holds for 51% of the MD course H-bond distance with D109 carboxylic function, while sulphonamide linker NH forms an H-bond with the C = O group of G102 (). Moreover, hydrophobic contacts persist with P111, L113, I116 and A106 for 16–30% of the MD course.
Similarly to 15, compound 18 forms a water bridged H-bond with G102 C=O by the p-carboxy function (); however, the presence of the charged group COO- nearby the lipophilic area of the active site might be the cause of the weakening of the binding interaction up to a KI value of 515 nM. In the case of 16 (KI = 304 nM), and 17 (KI = 3530 nM), the chain elongation is effective only for n = 1 (16), while in the alkylamine analogues 3 and 4 (with KI > 10000 nM) the presence of some ligand strains, allowed by the docking algorithm, together with the proximity of the charged amine group to the lipophilic part of VchCAβ active site, is responsible for the decreasing in the inhibition profile. Steric hindrance effects prevent the complementarity of the compounds 10 and 11 with the target although the second sulphonamide group in 11 directly binds with the hydroxyl side chain group of Y83 through an H-bond interaction. Modelling the flexible positioning of 13 and 14 (KI = 82.3 nM) within pointed out the detrimental effect of the N4-methylation of the thiadiazole ring, as it disables the water-bridged H-bond stabilisation observed with 13 (Figure S7A). Further, N-acetylation of 13 and 14 to give derivatives AAZ (Figure S7B) and MZA (Figure S7C) produces steric strains within the VchCAβ active site that cause a drop of inhibitory efficacy. Similarly, it was observed for EZA (Figure S7D).
3.3. Vchcaγ
Within the cylindrical active site of VchCAγ all sulphonamide inhibitors bind the zinc ion according to a tetrahedral geometry (Figure S8, Supporting Information). Here, both the NH- and S=O groups of the sulphonamide moiety engage H-bonds with the C=O and NH2 moieties of Q59 side chain, respectively, with the NH- group also acting as an acceptor of an H-bond by the OH of Y159.
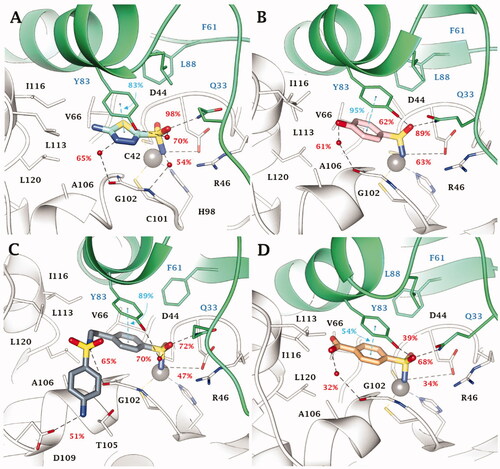
No significant differences are observed in the inhibition profile of 1,3,4-thiadiazole and benzenesulfonamide inhibitors in VchCAγ. MD simulations performed on some of the most effective inhibitors, 4 (KI = 69.0 nM), DZA (KI = 87.3 nM), IND (KI = 91.3 nM), and 5 (KI = 93.6 nM) () pointed out the key role of H68 in the stabilisation of aromatic (benzene/1,3,4-thiadiazole) sulphonamides through H-bond and π-π stacking interactions. The analysis of the trajectories unequivocally demonstrates the stability of these interactions which are maintained for most of the simulation. In addition, both water molecules and salt bridges occurring between charged groups of the ligands and the target also play a role in the stabilisation of the ligand-target conformations. These are the case for compounds 4 (SO…H-OH…HO S162, NH3+…-OOC D112), DZA (-NH2+…-OOC D112, SO…H-OH…O = C I122, SO…H-N M106) and the clinical inhibitor IND (-NH2+…-OOC D112, SO…H-OH…H-N N70, SO…H-N N73).
Figure 4. Most populated binding conformation along the MD trajectory for (A) 4 (spring green), (B) DZA (forest green), (C) IND (yellow), and (D) 5 (orchid) within VchCAγ active site. H-bonds, salt bridge, π-π stacking interactions, and halogen bonds are depicted as black, magenta, cyan, and green dashed lines, respectively. The occupancy over the MD simulation of interactions not involving the zinc-binding group is indicated as percentage.
Moreover, in this latter, the sulphonamide linker and indole NH groups form direct or water bridged H-bonds with the carboxylic group of the D112 (). A 48% stable halogen bond is also established by the ligand chlorine atom with the C=O group of I122 backbone. Also the sulphonamide linker S=O groups of derivative 5 are implicated in the binding to the protein, i.e. forming direct or water bridge H-bonds with the backbone NH of M106 and A124 ().
Interesting flexible ligand docking results were derived also for compounds not investigated by MD. The outcomes from docking allow to shed light on the effects of the substitution position as well as the chain elongation and the linker length. The better inhibition profile of 2 (KI = 95.3 nM) compared to 1 (KI = 672 nM, Figure S9A) arose from the failure for 1 to establish H-bond interaction with the side chain carboxylic group of D112.
Inhibitor 3 showed a similar binding mode with its homolog 4 (), with a charged H-bond forming between the ligand NH3+ and the D112 carboxylic moieties (Figure S9B). The substitution in para position of derivatives 6–9 facilitates vdW contacts with the enzymatic counterpart and, as a result, inhibition profiles with respect to the leads (Figures S9C-D).
The 1,3,4-thiadiazole-2-sulphonamides 13 (KI = 59.2 nM) and 14 (KI = 69.9 nM), potent VchCAγ inhibitors, formed H-bonds between the amine group of the ligands and the COO- of D112 (Figure S10A). On the contrary, this contact is prevented by the steric hindrance of the acetyl pendant in AAZ (KI = 473 nM) and MZA (KI = 494 nM, Figure S10B). Notably, the elongation of the aliphatic chain in derivatives 15–17 (KI = 88.5 − 556-6223 nM) increasingly prevents the stability of the H-bond between the OH in the hydroxyl alkyl chain and the D112 carboxylic moiety (Figure S10C).
Compound 11 showed a similar network of interactions as 5 observed so far but, more than this, further vdW interactions between the CF3 substituent and T67 side chain increase its inhibitory efficacy. The formation of H-bond contacts together with polar interactions of the outer amine group and the neighbour residues, resulted in 24 (KI = 87.1 nM) > 23 (KI = 273 nM) > 22 (KI = 902 nM)
As a result of this in silico analysis, it can be pointed out that heteroaromatic sulphonamide CAIs show greater VchCA and VchCAβ inhibition than benzenesulphonamides, as a result of additional direct or water-mediated H-bonds engaged by the N atoms on the heterocycle with T189 and P191 in VchCA and G102 in VchCAβ. In contrast, no analogue stabilisation can occur in the wider VchCAγ active site providing insights about the comparable VchCAγ inhibitory profiles shown by these two types of aromatic sulphonamides.
The joint docking/MD study also suggests that small, not unwieldy CAIs (e.g. 13 and 14) can more efficiently accommodate and bind in the narrow VchCAβ active site, inducing a greater inhibition than bulky derivatives. Further, it was shown that derivatives able to attain and interact with D109 through H-bonds (e.g. 24) showed an increased VchCAβ inhibition potency. Similarly, residue D122 in VchCAγ was identified as a key residue for H-bond/salt bridge interactions for increasing the binding stability and inhibition of the γ-class isozyme.
4. Conclusions
V. cholerae encodes for three CAs (VchCA, VchCAβ, and VchCAγ) that are crucial in the pathogen life cycle and growth. These enzymes are interesting targets to prevent V. cholerae proliferation. They offer the possibility to develop antibacterial drugs with an innovative mechanism of action for overcoming the spreading chemoresistance to the available drugs. A set of 40 aromatic sulphonamides and clinically licenced drugs (shown in ) were previously evaluated for the inhibition of VchCA, VchCAβ and VchCAγ to characterise each isozyme response to inhibitors in search of a new generation antiinfectives for the treatment of the disease. For the first time, this extended panel of CA inhibition profiles was here characterised at the molecular level by a thorough in silico study to point the structural parameters featuring each isozyme inhibition. Using the homology built 3 D structure of the three VchCAs, a joint docking and MD protocol was adopted to unveil the key and stable interactions responsible for a potent CA inhibition. This study might offer insights and be of crucial relevance in the rational design of new potent and selective inhibitors targeting CA isoforms from V. cholerae or other human pathogens.
Supplemental Material
Download PDF (1.9 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Related Research Data
References
- Baker-Austin C, Oliver JD, Alam M, et. al. Vibrio spp. infections. Nat Rev Dis Primers 2018;4:8.
- Baker-Austin C, Trinanes J, Gonzalez-Escalona N, et al. Non-Cholera vibrios: the microbial barometer of climate change. Trends Microbiol 2017;25:76–84.
- World Health Organization. Weekly epidemiological record. 2016;21:421–28.
- World Health Organization. Weekly epidemiological record. 2018;93:489–500.
- Zuckerman JN, Rombo L, Fisch A. The true burden and risk of cholera: implications for prevention and control. Lancet Infect Dis 2007;7:521–30.
- World Health Organization. Weekly epidemiological record 2010;85:117–28.
- Rabbani GH, Greenough WB. III. Food as a vehicle of transmission of cholera. J Diarrhoeal Dis Res 1999;17:1–9.
- Sugimoto JD, Koepke AA, Kenah EE, et al. Household transmission of Vibrio cholerae in Bangladesh. PLoS Negl Trop Dis 2014;8:e3314e3314.
- Goh KT, Teo SH, Lam S, et al. Person-to-person transmission of cholera in a psychiatric hospital. J Infect 1990;20:193–200.
- Gangarosa EF, Beisel WR, Benyajati C, et al. The nature of the gastrointestinal lesion in asiatic cholera and its relation to pathogenesis: a biopsy study. Am J Trop Med Hyg 1960;9:125–35.
- Abuaita BH, Withey JH. Bicarbonate induces vibrio cholerae virulence gene expression by enhancing ToxT activity. Infect Immun 2009;77:4111–20.
- Weber GG, Klose KEK. The complexity of ToxT-dependent transcription in Vibrio cholerae. Indian J Med Res 2011;133:201–6.
- Kirn TJ, Lafferty MJ, Sandoe CM, et al. Delineation of pilin domains required for bacterial association into microcolonies and intestinal colonization by Vibrio cholerae. Mol Microbiol 2000;35:896–910.
- Herrington DA, Hall RH, Losonsky G, et al. Toxin, toxin-coregulated pili, and the toxR regulon are essential for Vibrio cholerae pathogenesis in humans. J Exp Med 1988;168:1487–92.
- Fishman PH, Atikkan EE. Mechanism of action of cholera toxin: effect of receptor density and multivalent binding on activation of adenylate cyclase. J Membr Biol 1980;54:51–60.
- De SN, Chatterje DN. Enterotoxicity of bacteria-free culture-filtrate of Vibrio cholerae. Nature 1959;183:1533–4.
- Levine MM, Kaper JB, Black RE, et al. New knowledge on pathogenesis of bacterial enteric infections as applied to vaccine development. Microbiol Rev 1983;47:510–50.
- World Health Organization. Prevention and control of cholera. WHO 2020 https://www.who.int/health-topics/cholera#tab=tab_2.
- World Health Organization. Treatment diarrhoea: manual: a manual for physicians other senior health workers, 4th rev. World Health Organization 2005.
- Alam NH, Ashraf H. Treatment of infectious diarrhea in children. Paediatr Drugs 2003;5:151–65.
- Ahmed T, Ali M, Ullah MM, et al. Mortality in severely malnourished children with diarrhoea and use of a standardized management protocol. Lancet 1999;353:1919–22.
- World Health Organization. Clinical management of acute diarrhoea: WHO/UNICEF joint statement. World Health Organization 2004.
- Lindenbaum J, Greenough WB, Islam MR. Antibiotic therapy of Cholera in children. Bull World Health Organ 1967;37: 871–83.
- Kaushik JS, Gupta P, Faridi MM, et al. Single-dose azithromycin versus ciprofloxacin for cholera in children: a randomized controlled trial. Indian Pediatr 2010;47:309–15.
- Roy SK, Hossain MJ, Khatun W, et al. Zinc supplementation in children with cholera in Bangladesh: randomised controlled trial. BMJ 2008;336:266–8.
- Brown ED, Wright GD. Antibacterial drug discovery in the resistance era. Nature 2016;529:336–43.
- Del Prete S, Isik S, Vullo D, et al. DNA cloning, characterization, and inhibition studies of an α-carbonic anhydrase from the pathogenic bacterium vibrio cholerae. J Med Chem 2012;55:10742–8.
- Del Prete S, De Luca V, Scozzafava A, et al. Biochemical properties of a new α-carbonic anhydrase from the human pathogenic bacterium, vibrio cholerae. J Enzyme Inhib Med Chem 2014;29:23–7.
- Vullo D, Isik S, Del Prete S, et al. Anion inhibition studies of the α-carbonic anhydrase from the pathogenic bacterium vibrio cholerae. Bioorg Med Chem Lett 2013;23:1636–8.
- Maren TH. Carbonic anhydrase: chemistry, physiology, and inhibition. Physiol Rev 1967;47:595–781.
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81.
- Ferry JF. The gamma class of carbonic anhydrases. Biochim Biophys Acta 2010;1804:374–81.
- Kikutani S, Nakajima K, Nagasato C, et al. Thylakoid luminal θ-carbonic anhydrase critical for growth and photosynthesis in the marine diatom Phaeodactylum tricornutum. Proc Natl Acad Sci USA 2016;113:9828–33.
- Del Prete S, Vullo D, Fisher GM, et al. Discovery of a new family of carbonic anhydrases in the malaria pathogen Plasmodium falciparum-the η-carbonic anhydrases. Bioorg Med Chem Lett 2014;24:4389–96.
- Alterio V, Langella E, Viparelli F, et al. Structural and inhibition insights into carbonic anhydrase CDCA1 from the marine diatom Thalassiosira weissflogii. Biochimie 2012;94:1232–41.
- Supuran CT, Capasso C. Biomedical applications of prokaryotic carbonic anhydrases. Expert Opin Ther Pat 2018;28:745–54.
- Jensen EL, Clement R, Kosta A, et al. A new widespread subclass of carbonic anhydrase in marine phytoplankton. Isme J 2019;13:2094–106.
- Del Prete S, Nocentini A, Supuran CT, et al. Bacterial ι-carbonic anhydrase: a new active class of carbonic anhydrase identified in the genome of the Gram-negative bacterium Burkholderia territorii. J Enzyme Inhib Med Chem 2020;35:1060–8.
- Smith KS, Ferry JG. Prokaryotic carbonic anhydrases. FEMS Microbiol Rev 2000;24:335–59.
- Del Prete S, Vullo D, De Luca V, et al. Biochemical characterization of recombinant β-carbonic anhydrase (PgiCAb) identified in the genome of the oral pathogenic bacterium Porphyromonas gingivalis. J Enzyme Inhib Med Chem 2015;30:359–70.
- Nishimori I, Vullo D, Minakuchi T, et al. Anion inhibition studies of two new β-carbonic anhydrases from the bacterial pathogen Legionella pneumophila. Bioorg Med Chem Lett 2014;24:1127–32.
- Vullo D, Del Prete S, Osman SM, et al. Sulfonamide inhibition studies of the γ-carbonic anhydrase from the oral pathogen Porphyromonas gingivalis. Bioorg Med Chem Lett 2014;24:240–4.
- Del Prete S, De Luca V, Vullo D, et al. Biochemical characterization of the γ-carbonic anhydrase from the oral pathogen Porphyromonas gingivalis. PgiCA. J Enzyme Inhib Med Chem 2014;29:532–7.
- Cummins EP, Selfridge AC, Sporn PH, et al. Carbon dioxide-sensing in organisms and its implications for human disease. Cell Mol Life Sci 2014;71:831–45.
- Del Prete S, Vullo D, De Luca V, et al. Comparison of the sulfonamide inhibition profiles of the α-, β- and γ-carbonic anhydrases from the pathogenic bacterium Vibrio cholerae. Bioorg Med Chem Lett 2016;26:1941–6.
- Di Fiore A, D'Ambrosio K, Ayoub J, et al. α-Carbonic anhydrases. In: Supuran CT, Nocentini A, editors. Carbonic anhydrases. biochemistry and pharmacology of an evergreen pharmaceutical target. Amsterdam: Elsevier; 2019;19–54.
- Ferraroni M, Del Prete S, Vullo D, et al. Crystal structure and kinetic studies of a tetrameric type II β-carbonic anhydrase from the pathogenic bacterium Vibrio cholerae. Acta Crystallogr D Biol Crystallogr 2015;71:2449–56.
- Murray AB, McKenna R, β-Carbonic anhydrases. In: Supuran CT, Nocentini A, editors. Carbonic Anhydrases. Biochemistry and pharmacology of an evergreen pharmaceutical target. Amsterdam: Elsevier; 2019;55–77.
- Supuran CT, Capasso C. Carbonic anhydrases from pathogens: bacterial carbonic anhydrases and their inhibitors as potential antiinfectives. In: Supuran CT, Nocentini A, editors. Carbonic anhydrases. Biochemistry and pharmacology of an evergreen pharmaceutical target. Amsterdam: Elsevier; 2019;387–417.
- Bonardi A, Gratteri P, Nocentini A. Carbonic anhydrases from pathogens: fungal carbonic anhydrases and their inhibitors as potential antifungal agents. In: Supuran CT, Nocentini A, editors. Carbonic anhydrases. Biochemistry and pharmacology of an evergreen pharmaceutical target. Amsterdam: Elsevier; 2019;419–48.
- Ferraroni M. γ-Carbonic anhydrases. In: Supuran CT, Nocentini A, editors. Carbonic anhydrases. Biochemistry and pharmacology of an evergreen pharmaceutical target. Amsterdam: Elsevier; 2019;79–105.
- Del Prete S, Vullo D, Di Fonzo P, et al. Sulfonamide inhibition profile of the γ-carbonic anhydrase identified in the genome of the pathogenic bacterium Burkholderia pseudomallei the etiological agent responsible of melioidosis. Bioorg Med Chem Lett 2017;27:490–5.
- Del Prete S, Vullo D, Osman SM, et al. Sulfonamide inhibition study of the carbonic anhydrases from the bacterial pathogen Porphyromonas gingivalis: the β-class (PgiCAb) versus the γ-class (PgiCA) enzymes. Bioorg Med Chem 2014;22:4537–43.
- De Luca V, Vullo D, Del Prete S, et al. Cloning, characterization and anion inhibition studies of a γ-carbonic anhydrase from the Antarctic bacterium Colwellia psychrerythraea. Bioorg Med Chem 2016;24:835–40.
- De Luca V, Vullo D, Del Prete S, et al. Cloning, characterization and anion inhibition studies of a new γ-carbonic anhydrase from the Antarctic bacterium Pseudoalteromonas haloplanktis. Bioorg Med Chem 2015;23:4405–9.
- Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31:345–60.
- Bonardi A, Nocentini A, Cadoni R, et al. Benzoxaboroles: new potent inhibitors of the carbonic anhydrases of the pathogenic bacterium Vibrio cholerae. ACS Med Chem. Lett 2020; doi:https://doi.org/10.1021/acsmedchemlett.0c00403.
- Schrödinger Suite Release 2019-1, Schrödinger, LLC, New York, NY, 2019:(a) Prime, v.5.5; (b) Maestro v.11.9; (c) Epik, v.4.7; (d) Impact, v.8.2; (e) Macromodel v.12.3. (f) Glide, v.8.2; (g) Desmond, v.5.7; (h) Jaguar, v. 10.3.
- a. Pustenko A, Nocentini A, Gratteri P, et al. The antibiotic furagin and its derivatives are isoform-selective human carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2020;35:1011–20; b. Said MA, Eldehna WM, Nocentini A, et al. Synthesis, biological and molecular dynamics investigations with a series of triazolopyrimidine/triazole-based benzenesulfonamides as novel carbonic anhydrase inhibitors. Eur J Med Chem 2020;185:111843; c. Nocentini A, Bonardi A, Gratteri P, et al. Steroids interfere with human carbonic anhydrase activity by using alternative binding mechanisms. J Enzyme Inhib Med Chem 2018;33:1453–9.
- a. Abdelrahman MA, Ibrahim HS, Nocentini A, et al. Novel 3-substituted coumarins as selective human carbonic anhydrase IX and XII inhibitors: Synthesis, biological and molecular dynamics analysis. Eur J Med Chem 2020;209:112897; b. Abo-Ashour MF, Eldehna WM, Nocentini A, et al. 3-Hydrazinoisatin-based benzenesulfonamides as novel carbonic anhydrase inhibitors endowed with anticancer activity: Synthesis, in vitro biological evaluation and in silico insights. Eur J Med Chem 2019;184:111768.
- Bonardi A, Nocentini A, Bua S, et al. Sulfonamide inhibitors of human carbonic anhydrases designed through a three-tails approach: improving ligand/isoform matching and selectivity of action. J Med Chem 2020;63:7422–44.
- Modakh YK, Liu YC, Machuca MA, et al. Structural basis for the inhibition of helicobacter pylori α-carbonic anhydrase by sulfonamides. PLoS One 2015;10:e0127149.
- Huang S, Hainzl T, Grundstrom C, et al. Structural studies of β-carbonic anhydrase from the green alga coccomyxa: inhibitor complexes with anions and acetazolamide. PLoS One 2011;6:e28458
- Murray AB, Aggarwal M, Pinard M, et al. Structural mapping of anion inhibitors to β-Carbonic Anhydrase psCA3 from Pseudomonas aeruginosa. ChemMedChem 2018;13:2024–9.
- Kaur J, Cao X, Abutaleb NS, et al. Optimization of acetazolamide-based scaffold as potent inhibitors of vancomycin-resistant enterococcus. J Med Chem 2020;63:9540–62.
- Berman HM, Westbrook J, Feng Z, et al. The Protein Data Bank. Nucleic Acids Res 2000; 28:235–42.