
Abstract
Sulphonamides and their isosteres are classical inhibitors of the carbonic anhydrase (CAs, EC 4.2.1.1) metalloenzymes. The protozoan pathogen Trichomonas vaginalis encodes two such enzymes belonging to the β-class, TvaCA1 and TvaCA2. Here we report the first sulphonamide inhibition study of TvaCA1, with a series of simple aromatic/heterocyclic primary sulphonamides as well as with clinically approved/investigational drugs for a range of pathologies (diuretics, antiglaucoma, antiepileptic, antiobesity, and antitumor drugs). TvaCA1 was effectively inhibited by acetazolamide and ethoxzolamide, with KIs of 391 and 283 nM, respectively, whereas many other simple or clinically used sulphonamides were micromolar inhibitors or did not efficiently inhibit the enzyme. Finding more effective TvaCA1 inhibitors may constitute an innovative approach for fighting trichomoniasis, a sexually transmitted infection, caused by T. vaginalis.
1. Introduction
Trichomonas vaginalis is a protozoan parasite responsible for trichomoniasis, one of the most frequent non-viral sexually transmitted diseases in humansCitation1,Citation2. Treatment of this disease remains almost exclusively based on just one class of drugs, 5-nitroimidazoles (with two available agents, metronidazole and tinidazole), and resistance to these agents is on the rise worldwideCitation3,Citation4. Trichomoniasis may cause a variety of symptoms, from mild to severe, but a large fraction (10–50%) of infected women show no symptoms, and 5–15% of cases may remain undetectable upon examinationCitation1,Citation2. Furthermore, the majority of infected men are totally asymptomatic, making the diagnosis of this disease particularly challengingCitation1,Citation2. T. vaginalis infection may facilitate or worsen other critical pathologies, such as HIV-infectionCitation5 or even prostate cancerCitation6. As a consequence, research on novel drug targets for fighting trichomoniasis has seen an increased interestCitation7–11.
Carbonic anhydrases (CA, EC 4.2.1.1) have recently been identified as new potential targets for finding alternative drugs that may interfere with or abolish crucial steps in the life cycle of protozoan parasitesCitation7–11. Recently, we have characterised a β-class CA, TvaCA1, which is one of the two such enzymes found in the proteome of T. vaginalisCitation10. The other one, TvaCA2, will be described shortly elsewhere. Like the other members of this metalloenzyme superfamilyCitation12–15, TvaCA1 catalyses the interconversion between CO2 and bicarbonate, also generating protons, and is probably a key element of the molecular machinery involved in the pH regulation and metabolism of the parasite. Mammalian hosts, including humans, have several genes encoding α-class CAs in their genomes, whereas they have no β-CA genesCitation12. For this reason, β-CAs may represent an interesting target for finding anti-infectives with a novel mechanism of actionCitation16. Accordingly, Flaherty’s groupCitation17 recently reported that sulphonamide CA inhibitors (CAIs), structurally related to acetazolamide (AAZ), have potent anti-bacterial effects against pathogenic drug resistant bacteria (which normally contain α-, β-, γ- and/or ι-CAs)Citation18,Citation19. They demonstrated this effect in vancomycin-resistant enterococci, validating thus in vivo the bacterial CAs as drug targetsCitation16–17.
Based on these considerations and with the aim to identify new targets for the development of innovative drugs against trichomoniasis, we undertook a detailed study on TvaCA1. We previously determined the X-ray crystal structure of this enzymeCitation10 and described its inhibition with a wide range of inorganic anionsCitation11. Here we continue this study reporting the inhibition of TvaCA1 with a series of simple aromatic/heterocyclic primary sulphonamides as well as with clinically approved/investigational such drugs for a range of pathologies.
2. Materials and methods
2.1. Chemistry
Sulphonamides 1–24 were either commercially available or prepared as reported earlier by our groupCitation20. Clinically used agents AAZ–HCF were the reagents from Sigma-Aldrich (Milan, Italy) with the highest purity available.
2.2. Enzymology
TvaCA1 was a recombinant enzyme obtained in-house as described earlierCitation10. The enzyme was recombinantly produced in E. coli (OneShot® BL21 Star™ (DE3) Chemically Competent Cells, #C601003, Thermo Fisher Scientific, Finland). The enzyme was purified using Ni2+-NTA agarose affinity chromatography resin (Macherey-Nagel GmbH Co., Germany). More detailed information concerning the recombinant protein production and its kinetic and structural characterisation can be found in our previous articleCitation10. The presence of the correct protein in the isolated protein fraction was confirmed by tandem mass spectrometry (MS/MS).
2.3. CA catalytic activity and inhibition assay
An Applied Photophysics stopped-flow instrument was used for assaying the CA catalysed CO2 hydration activityCitation21. Phenol red (at a concentration of 0.2 mM) was used as an indicator, working at the absorbance maximum of 557 nm, with 10 − 20 mM HEPES (pH 7.5, for α-CAs) or TRIS (pH 8.3 for β-CAs) as buffers, and 20 mM NaClO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalysed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor, at least six traces of the initial 5–10% of the reaction were used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitors (10 mM) and dilutions up to 0.01 nM were prepared in distilled-deionised water. Inhibitor and enzyme solutions of concentrations ranging between 5 and 12 nM were preincubated together for 15 min at room temperature prior to assay, in order to allow for the formation of the enzyme–inhibitor complex. The inhibition constants were obtained by non-linear least-squares methods using PRISM 3 as reported earlierCitation22, and represent the mean from at least three different determinations.
3. Results and discussion
TvaCA1 is a β-CA that has an open active siteCitation23, meaning that the water molecule/zinc hydroxide ion, acting as nucleophile in the catalytic cycle, is coordinated to the metal ion, as seen in .
Figure 1. (A) TvaCA1 dimeric structure, with the two monomers shown in green and red, respectivelyCitation10. (B) Active site of the enzyme, with the zinc ion (gray sphere) coordinated by two Cys, one His and one water molecule/hydroxide ion (shown in red). Residues numbering as described by Urbański et al.Citation10.
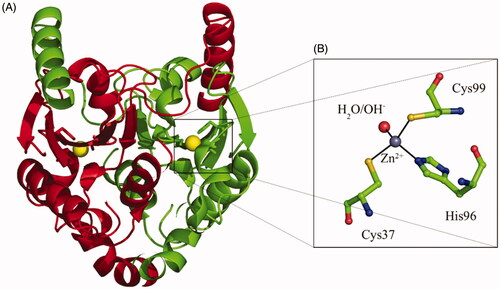
As all β-CAs, TvaCA1 is a dimer with two narrow, channel-like active sites present in the molecule, as seen in . Thus, this enzyme is rather different from the α-CAs present in the human host, which are generally monomeric enzymes with the zinc ion coordinated by three His residues and a water molecule, and possessing a rather ample active site where inhibitors and activators may bindCitation12–14,Citation24. Hence, we decided to investigate the inhibition of TvaCA1 with the main class of organic CA inhibitors, the primary sulphonamides, some of which are clinically used agents as diuretics, antiglaucoma, antiepileptic, antiobesity and antitumor agentsCitation25–30. In particular, a series of such compounds of types 1–24 and AAZ–HCT () were analysed.
Figure 2. Sulphonamides 1–24 and clinically used agents AAZ–HCT investigated as TvaCA1 inhibitors.
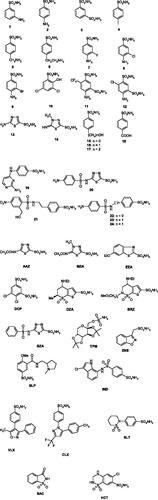
Derivatives 1–24 and AAZ–HCT are simple aromatic/heterocyclic sulphonamides widely used as building blocks for obtaining new families of such pharmacological agentsCitation20,Citation22, or they are clinically used drugs, such as acetazolamide AAZ, methazolamide MZA, ethoxzolamide EZA and dichlorophenamide DCP, which are the classical and systemically acting antiglaucoma CAIs. Dorzolamide DZA and brinzolamide BRZ are topically acting antiglaucoma agents, benzolamide BZA is an orphan drug belonging to this class of pharmacological agents, whereas topiramate TPM, zonisamide ZNS and sulthiame SLT are widely used antiepileptic drugs. Sulpiride SLP and indisulam IND were also shown by our group to belong to this class of pharmacological agents, together with the COX2 “selective” inhibitors celecoxib CLX and valdecoxib VLX. Saccharin and the diuretic hydrochlorothiazide HCT are also known to act as CAIsCitation25–30.
The following structure–activity relationship (SAR) can be drawn from the inhibition data presented in , where hCA II inhibition is also given for comparison, considering that this is the most abundant human isoformCitation12,Citation13 and presumably the major off target enzyme when the use of CAI class anti-infectives are considered:
Table 1. Inhibition of human isoform hCA II, for comparison, and of the protozoan enzyme TvaCA1 with sulphonamides 1–24 and the clinically used drugs AAZ–HCT, measured by a CO2 hydrase, stopped-flow assay.Citation21
(i) Only 14 of the 40 tested derivatives 1–24 and AAZ–HCT inhibited TvaCA1, while the remaining 26 were ineffective at concentrations up to 50 µM in the assay system. The ineffective group of compounds included the clinically used agents DCP, DZA, BRZ, BZA, TPM, ZNS, SLP, IND, VLX, CLX, SLT, SAC and HCT. One should note that apart from SAC, which is a secondary sulphonamide with a rather compact scaffold, the remaining ineffective derivatives possess rather bulky scaffolds and various substituents on which the sulphonamide or sulfamate moieties are appended. This may explain why these compounds do not access easily the narrow channel-like active site of this β-class enzyme, although they generally act as effective hCA II inhibitors. As mentioned above, the active site of the α-CAs is much wider compared to that of the β-CAs. Furthermore, some of the simple derivatives 1–24 were ineffective as TvaCA1 inhibitors including 5, 6, 8, 9, 11, 12, 16–18, 20, and 22–24 (). These sulphonamides belong to heterogeneous classes, such as simple amino/hydroxyl-alkyl-substituted-benzenesulfonamides (5, 6, 16, 17), halogeno-sulfanilamides (8 and 9), 1,3-benzenedisulfonamides (11 and 12), and sulfonylated-sulphonamides with an elongated molecule (20, 22–24). As mentioned for the clinically used agents, some of these derivatives also possess rather bulky scaffolds that probably interfere with their efficient binding within the enzyme active site.
(ii) Sulphonamides 1–4, 7, 10, 13–15, 19, 21 and MZA were micromolar TvaCA1 inhibitors, with inhibition constants ranging between 1.889 and 4.742 µM (). From a structural point of view it may be observed that most of them incorporate compact, simple benzenesulfonamide/thiadiazole sulphonamide scaffolds with few or just one compact substituent (such as 1–4, 7, 13–15 and MZA). However, at least two of these derivatives, 19 and 21, possess more complex scaffolds with elongated molecules which presumably are able to accommodate within the enzyme active site. Thus, it should be in principle possible to find or design even more effective inhibitors considering these two compounds as lead molecules.
(iii) The most effective TvaCA1 inhibitors were acetazolamide AAZ and ethoxzolamide EZA, with KIs of 391 and 283 nM, respectively. However, no inhibitor with an inhibition constant < 100 nM was detected for the Trichomonas enzyme.
4. Conclusions
We report the first inhibition study with sulphonamides of one of the β-CAs, TvaCA1, found in the protozoan parasite T. vaginalis. Only anion inhibitors and other small molecules were reported earlier to act as millimolar TvaCA1 inhibitors. Here we investigated 40 sulphonamides, some of which are clinically used drugs, for their inhibitory interaction with this enzyme. Only 16 of these agents showed inhibitory effects, most of them in the low micromolar range, whereas the most effective TvaCA1 inhibitors were acetazolamide AAZ and ethoxzolamide EZA, with KIs of 391 and 283 nM, respectively. Although no in vivo/ex vivo studies have been performed so far, inhibition of this protozoan enzyme may show anti-infective effects, as was reported for other protozoan species such as Leishmania donovani chagasiCitation31 or Trypanosoma cruziCitation32, for which CAIs belonging to various classes showed potent anti-protozoan activity in vivo. Thus, effective TvaCA1 inhibitors may lead to the development of novel anti-infectives with a diverse mechanism of action.
Acknowledgements
The authors thank Ms. Marianne Kuuslahti for skillful technical assistance. This paper is dedicated to the memory of Dr. Richard Tashian (1922–2020) who contributed with significant discoveries in the carbonic anhydrase field for many decades.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Dessì D, Margarita V, Cocco AR, et al. Trichomonas vaginalis and Mycoplasma hominis: new tales of two old friends. Parasitology 2019;146:1150–5.
- Mercer F, Johnson PJ. Trichomonas vaginalis: pathogenesis, symbiont interactions, and host cell immune responses. Trends Parasitol 2018;34:683–93.
- Muzny CA, Van Gerwen OT, Kissinger P. Updates in trichomonas treatment including persistent infection and 5-nitroimidazole hypersensitivity. Curr Opin Infect Dis 2020;33:73–7.
- (a) Küng E, Fürnkranz U, Walochnik J. Chemotherapeutic options for the treatment of human trichomoniasis. Int J Antimicrob Agents 2019;53:116–27. (b) Alessio C, Nyirjesy P. Management of resistant trichomoniasis. Curr Infect Dis Rep 2019;21:31.
- McClelland RS, Sangare L, Hassan WM, et al. Infection with Trichomonas vaginalis increases the risk of HIV-1 acquisition. J Infect Dis 2007;195:698–702.
- Twu O, Dessi D, Vu A, et al. Trichomonas vaginalis homolog of macrophage migration inhibitory factor induces prostate cell growth, invasiveness, and inflammatory responses. Proc Natl Acad Sci U S A 2014;111:8179–84.
- D'Ambrosio K, Supuran CT, De Simone G. Are carbonic anhydrases suitable targets to fight protozoan parasitic diseases? Curr Med Chem 2018;25:5266–78.
- Vermelho AB, Rodrigues GC, Supuran CT. Why hasn’t there been more progress in new Chagas disease drug discovery? Expert Opin Drug Discov 2020;15:145–58.
- Garcia AR, Oliveira DMP, Claudia F, et al. Leishmania infantum arginase: biochemical characterization and inhibition by naturally occurring phenolic substances. J Enzyme Inhib Med Chem 2019;34:1100–9.
- Urbański LJ, Di Fiore A, Azizi L, et al. S. Biochemical and structural characterisation of a protozoan beta-carbonic anhydrase from Trichomonas vaginalis. J Enzyme Inhib Med Chem 2020; 35:1292–9.
- Urbański LJ, Angeli A, Hytönen VP, et al. Inhibition of the newly discovered β-carbonic anhydrase from the protozoan pathogen Trichomonas vaginalis with inorganic anions and small molecules. J Inorg Biochem 2020;213:111274.
- (a) Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81. (b) Supuran CT. Structure and function of carbonic anhydrases. Biochem J 2016; 473:2023–32. (c) Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77.
- (a) Supuran CT. Carbonic anhydrases and metabolism. Metabolites 2018; 8:25. (b) Supuran CT. Exploring the multiple binding modes of inhibitors to carbonic anhydrases for novel drug discovery. Expert Opin Drug Discov 2020;15:671–86. (c) Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016; 31:345–60.
- (a) Alterio V, Di Fiore A, D’Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012; 112:4421–68. (b) Supuran CT. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin Drug Discov 2017; 12:61–88. (c) Nocentini A, Supuran CT. Advances in the structural annotation of human carbonic anhydrases and impact on future drug discovery. Expert Opin Drug Discov 2019; 14:1175–97. (d) Supuran CT. Structure-based drug discovery of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2012;27:759–72.
- (a) Bua S, Haapanen S, Kuuslahti M, et al. Activation studies of the β-carbonic anhydrase from the pathogenic protozoan Entamoeba histolytica with amino acids and amines. Metabolites 2019;9:26. (b) Angeli A, Donald WA, Parkkila S, Supuran CT. Activation studies with amines and amino acids of the β-carbonic anhydrase from the pathogenic protozoan Leishmania donovani chagasi. Bioorg Chem 2018;78:406–10. (c) Patrikainen MS, Tolvanen MEE, Aspatwar A, et al. Identification and characterization of a novel zebrafish (Danio rerio) pentraxin-carbonic anhydrase. PeerJ 2017;5:e4128.
- Supuran CT, Capasso C. Antibacterial carbonic anhydrase inhibitors: an update on the recent literature. Expert Opin Ther Pat 2020;30:963–982.
- Kaur J, Cao X, Abutaleb NS, et al. Optimization of acetazolamide-based scaffold as potent inhibitors of vancomycin-resistant Enterococcus. J Med Chem 2020;63:9540–62.
- (a) Del Prete S, Bua S, Supuran CT, Capasso C. Escherichia coliγ-carbonic anhydrase: characterisation and effects of simple aromatic/heterocyclic sulphonamide inhibitors. J Enzyme Inhib Med Chem 2020;35:1545–54. (b) Del Prete S, De Luca V, Bua S, et al. The effect of substituted benzene-sulfonamides and clinically licensed drugs on the catalytic activity of CynT2, a carbonic anhydrase crucial for Escherichia coli life cycle. Int J Mol Sci 2020;21:4175. (c) Angeli A, Ferraroni M, Pinteala M, et al. Crystal structure of a tetrameric type II β-carbonic anhydrase from the pathogenic bacterium Burkholderia pseudomallei. Molecules 2020;25:2269.
- (a) Del Prete S, Nocentini A, Supuran CT, Capasso C. Bacterial ι-carbonic anhydrase: a new active class of carbonic anhydrase identified in the genome of the Gram-negative bacterium Burkholderia territorii. J Enzyme Inhib Med Chem 2020;35:1060–8. (b) Gitto R, De Luca L, Mancuso F, et al. Seeking new approach for therapeutic treatment of cholera disease via inhibition of bacterial carbonic anhydrases: experimental and theoretical studies for sixteen benzenesulfonamide derivatives. J Enzyme Inhib Med Chem 2019;34:1186–92. (c) Angeli A, Pinteala M, Maier SS, et al. Inhibition of α-, β-, γ-, δ-, ζ- and η-class carbonic anhydrases from bacteria, fungi, algae, diatoms and protozoans with famotidine. J Enzyme Inhib Med Chem 2019;34:644–50.
- (a) Supuran CT, Nicolae A, Popescu A. Carbonic anhydrase inhibitors. Part 35. Synthesis of Schiff bases derived from sulfanilamide and aromatic aldehydes: the first inhibitors with equally high affinity towards cytosolic and membrane-bound isozymes. Eur J Med Chem 1996;31:431–8. (b) Mishra CB, Tiwari M, Supuran CT. Progress in the development of human carbonic anhydrase inhibitors and their pharmacological applications: where are we today? Med Res Rev 2020;40:2485–565.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73.
- (a) Supuran CT. Carbon- versus sulphur-based zinc binding groups for carbonic anhydrase inhibitors? J Enzyme Inhib Med Chem 2018; 33:485–95. (b) Maresca A, Scozzafava A, Supuran CT. 7,8-Disubstituted- but not 6,7-disubstituted coumarins selectively inhibit the transmembrane, tumor-associated carbonic anhydrase isoforms IX and XII over the cytosolic ones I and II in the low nanomolar/subnanomolar range. Bioorg Med Chem Lett 2010;20:7255–8. (c) Innocenti A, Gülçin I, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. Antioxidant polyphenols effectively inhibit mammalian isoforms I-XV. Bioorg Med Chem Lett 2010;20:5050–3. (d) De Simone G, Supuran CT. (In)organic anions as carbonic anhydrase inhibitors. J Inorg Biochem 2012;111:117–29.
- Covarrubias AS, Bergfors T, Jones TA, Högbom M. Structural mechanics of the pH-dependent activity of beta-carbonic anhydrase from Mycobacterium tuberculosis. J Biol Chem 2006; 281:4993–9.
- (a) Supuran CT. Carbonic anhydrase activators. Future Med Chem 2018;10:561–73. (b) Blandina P, Provensi G, Passsani MB, et al. Carbonic anhydrase modulation of emotional memory. Implications for the treatment of cognitive disorders. J Enzyme Inhib Med Chem 2020;35:1206–14.
- (a) Carta F, Supuran CT. Diuretics with carbonic anhydrase inhibitory action: a patent and literature review (2005–2013). Expert Opin Ther Pat 2013; 23:681–91. (b) Supuran CT. Applications of carbonic anhydrases inhibitors in renal and central nervous system diseases. Expert Opin Ther Pat 2018;28:713–21.
- (a) Supuran CT. Carbonic anhydrase inhibitors and their potential in a range of therapeutic areas. Expert Opin Ther Pat 2018;28:709–12. (b) Scozzafava A, Supuran CT, Carta F. Antiobesity carbonic anhydrase inhibitors: a literature and patent review. Expert Opin Ther Pat 2013;23:725–35. (c) Di Cesare Mannelli L, Micheli L, Carta F, et al. Carbonic anhydrase inhibition for the management of cerebral ischemia: in vivo evaluation of sulfonamide and coumarin inhibitors. J Enzyme Inhib Med Chem 2016;31:894–9.
- (a) Supuran CT, Altamimi ASA, Carta F. Carbonic anhydrase inhibition and the management of glaucoma: a literature and patent review 2013–2019. Expert Opin Ther Pat 2019;29:781–92. (b) Supuran CT. The management of glaucoma and macular degeneration. Expert Opin Ther Pat 2019;29:745–7.
- (a) McDonald PC, Winum JY, Supuran CT, Dedhar S. Recent developments in targeting carbonic anhydrase IX for cancer therapeutics. Oncotarget 2012;3:84–97. (b) Akocak S, Alam MR, Shabana AM, et aal. PEGylated bis-sulfonamide carbonic anhydrase inhibitors can efficiently control the growth of several carbonic anhydrase IX-expressing carcinomas. J Med Chem 2016;59:5077–88. (c) McDonald PC, Chia S, Bedard PL, et al. A phase 1 study of SLC-0111, a novel inhibitor of carbonic anhydrase IX, in patients with advanced solid tumors. Am J Clin Oncol 2020;43:484–90.
- (a) Angeli A, Carta F, Nocentini A, et al. Carbonic anhydrase inhibitors targeting metabolism and tumor microenvironment. Metabolites 2020;10:412. (b) Supuran CT, Alterio V, Di Fiore A, et al. Inhibition of carbonic anhydrase IX targets primary tumors, metastases, and cancer stem cells: three for the price of one. Med Res Rev 2018;38:1799–836.
- (a) Carta F, Scozzafava A, Supuran CT. Sulfonamides: a patent review (2008–2012). Expert Opin Ther Pat 2012;22:747–58. (b) Scozzafava A, Carta F, Supuran CT. Secondary and tertiary sulfonamides: a patent review (2008–2012). Expert Opin Ther Pat 2013;23:203–13.
- (a) Angeli A, Etxebeste-Mitxeltorena M, Sanmartín C, et al. Tellurides bearing sulfonamides as novel inhibitors of leishmanial carbonic anhydrase with potent antileishmanial activity. J Med Chem 2020;63:4306–14. (b) Nocentini A, Osman SM, Almeida IA, et al. Appraisal of anti-protozoan activity of nitroaromatic benzenesulfonamides inhibiting carbonic anhydrases from Trypanosoma cruzi and Leishmania donovani. J Enzyme Inhib Med Chem 2019;34:1164–71. (c) da Silva Cardoso V, Vermelho AB, Ricci Junior E, et al. Antileishmanial activity of sulphonamide nanoemulsions targeting the β-carbonic anhydrase from Leishmania species. J Enzyme Inhib Med Chem 2018;33:850–7.
- (a) Vermelho AB, da Silva Cardoso V, Ricci Junior E, et al. Nanoemulsions of sulfonamide carbonic anhydrase inhibitors strongly inhibit the growth of Trypanosoma cruzi. J Enzyme Inhib Med Chem 2018;33:139–46. (b) Vermelho AB, Capaci GR, Rodrigues IA, et al. Carbonic anhydrases from Trypanosoma and Leishmania as anti-protozoan drug targets. Bioorg Med Chem 2017;25:1543–55. (c) Nocentini A, Cadoni R, Dumy P, et al. Carbonic anhydrases from Trypanosoma cruzi and Leishmania donovani chagasi are inhibited by benzoxaboroles. J Enzyme Inhib Med Chem 2018;33:286–9.