
Abstract
One of the systems responsible for maintaining cellular redox homeostasis is the thioredoxin-dependent system. An equally important function of this system is the regulation of the expression of many proteins by the transcription factor NF-κB or the apoptosis regulating kinase (ASK-1). Since it has been shown that the Trx-dependent system can contribute to both the enhancement of tumour angiogenesis and growth as well as apoptosis of neoplastic cells, the search for compounds that inhibit the level/activity of Trx and/or TrxR and thus modulate the course of the neoplastic process is ongoing. It has been shown that many naturally occurring polyphenolic compounds inactivate elements of the thioredoxin system. In addition, the effectiveness of Trx is inhibited by imidazole derivatives, while the activity of TrxR is reduced by transition metal ions complexes, dinitrohalobenzene derivatives, Michael acceptors, nitrosourea and ebselen. In addition, research is ongoing to identify new selective Trx/TrxR inhibitors.
Introduction
The maintenance of redox balance is important in living organisms. Cellular metabolism, including the metabolism of xenobiotics, generates reactive oxygen/nitrogen species. Their formation and cellular effects under physiological conditions is controlled by antioxidant systems – mainly the glutathione or thioredoxin-dependent systems – which work independently, but can complement each other, particularly in cases involving pathological factors. The thioredoxin-dependent system is of particular relevance in the detoxification of harmful metabolites such as lipid peroxides and in the regulation of gene expression and modulation of cell signalling pathways. The possibility of modulating signalling pathways with exogenous substances/compounds that target the thioredoxin-dependent system creates a real possibility of extending the use of current pharmacotherapies, as well as fuel the search for new cancer pharmacotherapies, to activate apoptotic pathways in neoplastic cells.
Structure and function of the Thioredoxin-Dependent system
Reactive oxygen species (ROS) are generated as a byproduct of metabolism in living organisms. Redox homeostasis is achieved by cellular systems involving macromolecular compounds including proteins that either prevent the production of these reactive species, or regulate their effects. One such system involves three cooperating redox proteins: thioredoxin (Trx), thioredoxin reductase (TrxR), thioredoxin peroxidase (TPx), and NADPH (the latter is a source of protons) ()Citation1. The central element of this system is Trx, which is capable of reducing disulphide bonds in proteins due to the presence of two cysteines in its active centerCitation2.
Figure 1. Scheme of functioning of thioredoxin-dependent system.
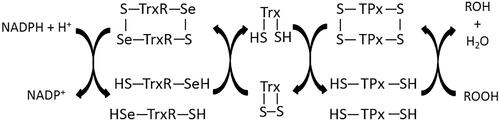
There are two different isoforms of Trx – a cytosolic isoform (Trx1) and a mitochondrial isoform (Trx2), which are found in different tissues of the human bodyCitation3,Citation4.
Trx1 is a 12 kDa proteinCitation5 that is biosynthesized as a 105-amino acid propeptide, which through an activation process, undergoes removal of an N-terminal methionine, leading to the generation of the 104-amino acid active protein that has valine at its N-terminusCitation6. The mitochondrial Trx isoform is biosynthesized as an 18 kDa protein containing 166 amino acids with a 60-amino acid sequence at its N-terminal that is responsible for directing the newly synthesised Trx molecule into the mitochondrion. After reaching the mitochondrion, the protein is hydrolysed with a 60-amino acid peptide cleavage, resulting in the 12.2 kDa active Trx2 proteinCitation7.
Given the metabolic significance of Trx, the most important element of the molecule is its active centre (-Cys32-Gly-Pro-Cys35-) as it is responsible for the antioxidant function of the protein. The cysteines located in the active centre are arranged in such a way that Cys32 is more exposed to the outside while Cys35 is hidden in the coreCitation8. This spatial distribution is important for the reducing action of Cys32, which leads to a disulphide bond between Trx and the protein to be reduced. Cys35 is responsible for the separation of the reduced protein molecule and the creation of a disulphide bond within the active Trx centre ()Citation9. The conservation of this sequence in the active centre is essential for the proper functioning of Trx and any amino acid change in the sequence (e.g. substitution of proline with serine or threonine) leads to a change in protein conformation, resulting in decreased stability of the molecule, which in turn, significantly affects the reduction potential of TrxCitation10. Apart from the cysteine residues located in the active centre of Trx, there are three additional cysteines in the structure of the protein at positions 62, 69, and 73. Therefore, in a situation of Trx deficiency and/or under oxidative conditions, it is possible to use other thiol groups for Trx reduction activities through the formation of a second disulphide bond between Cys62 and Cys69. Further oxidation on the other hand, would lead to the formation of a homodimer with a disulphide bond between the cysteines at position 73 of two Trx moleculesCitation11.
Figure 2. Scheme of protein reduction by thioredoxin-dependent system.
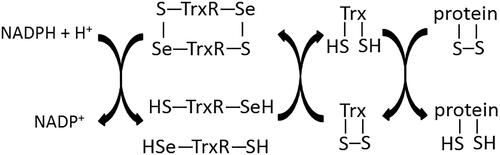
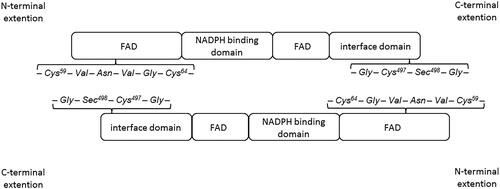
The enzyme responsible for reducing the oxidised form of Trx is thioredoxin reductase (TrxR). The reducing enzyme contains a FAD prosthetic group, NADPH binding domain, interface domain, and an active centre located in the C-terminusCitation4. The TrxR active centre contains one cysteine and one selenocysteine (-Gly-Cys497-Sec498-Gly-) and the FAD domain contains the sequence -Cys59-Val-Asn-Val-Gly-Cys64-, which enables electron transfer during a redox reaction. An important element for TrxR activity is the Sec498 located at the enzyme’s C-terminus. It has been shown that recombinant mutant TrxR containing cysteine instead of selenocysteine in the active centre showed a 100 times lower catalytic constant value in comparison to the wild type Sec-containing enzyme. It was also observed that the substitution of redox inactive serine in place of selenocysteine completely inactivates the enzymeCitation12,Citation13.
The formation of the enzyme as a homodimer is important for TrxR biological activity. TrxR molecules are arranged in space such that the C-terminal active site of one molecule is adjacent to cysteines present in the redox-active sequence at the N-terminal element of another molecule () – this is crucial for the function of the enzyme because such a spatial organisation allows for the free flow of electrons during the redox reactionCitation14,Citation15. After recruitment of NADPH via the NAPDH binding domain of the enzyme, electrons are transferred to the FAD domain at the N-terminal end of the protein. The active redox sequence of one subunit of the enzyme transfers electrons to the active site located at the interface domain of the other subunitCitation4.
Figure 3. Scheme of arrangement of thioredoxin reductase homodimerCitation4.
In humans, TrxR occurs in three isoformsCitation16: the TrxR1 isoform which is present in the cytosol (55 kDa; 499 amino acids), the TrxR2 mitochondrial isoform (56.2 kDa; 521 amino acids), and TrxR3 (65 kDa; 560 amino acids) which localises to the nucleus. TrxR2 contains a domain composed of 33 amino acids situated at the N-terminal portion of the enzyme, which is responsible for mitochondrial translocation of enzyme molecules. TrxR3 can reduce both Trx and glutathione (GSH) and is therefore referred to as thioredoxin glutathione – reductase (TGR)Citation17–20. It contains a TGR structure at the N-terminal motif which contains an additional glutaredoxin domain so that the enzyme can act as a thioredoxin reductase, glutathione reductase, and glutaredoxin, the purpose of which is to reduce mixed disulphides in proteins produced by glutathionylation, among other processesCitation21.
The effective action of the Trx-dependent system is also linked to the presence of thioredoxin peroxidase (TPx), which in cooperation with Trx, can reduce lipid peroxides and hydrogen peroxideCitation22,Citation23. TPx belongs to a family of proteins called peroxiredoxins (Prx), which contain in their active centre, a cysteine (Prx-SH) that is prone to oxidation. The peroxiredoxin molecule is converted to its oxidised form (Prx-SOH) as a result of peroxide reduction, which in reaction with another reduced peroxiredoxin molecule, forms a disulphide bond (Prx-S-S-Prx) with simultaneous release of a water molecule. TPx contains the Cys47 and Cys170 residues, which participate in peroxide reduction. The large distance between the cysteines makes it impossible for the formation of an intramolecular disulphide bond and as a result, the oxidised form of TPx, similar to TrxR, occurs in the form of dimers in which two intermolecular bonds in the Cys47-Cys170 system are formedCitation23. The effective action of TPx requires the presence of reduced Trx. The reduced form of Trx reduces TPx, which can then reduce peroxide molecules in the cell, protecting other molecules from the oxidative effects of ROSCitation22–24.
Although antioxidant activity is the main function of the thioredoxin-dependent system, the system also participates in cofactor activities and regulates the expression of many genesCitation3. Trx supports the transcriptional activity of the nuclear factor NFκB – which can stimulate the growth of cancer cells – by promoting its interaction with DNA through reduction of the disulphide bond in one of the NFκB subunitsCitation25. Trx also binds to Apoptosis signal-regulating kinase (ASK1) – which regulates apoptosis under oxidative stress conditions – to cause its degradationCitation26. However, in the presence of ROS or tumour necrosis factor alpha (TNFα), Trx and ASK1 are released. ASK1 stimulates cellular growth through the activation of kinases such as p38 and JNK, which stimulate the activity of the proapoptotic protein Bax and simultaneously inhibit the anti-apoptotic factor Bcl-2. This promotes the activation of caspase 3, which triggers apoptosisCitation27. Moreover, Trx, through the Reduction-oxidation factor 1 (Ref-1), modulates the activity of the transcription factor activator protein 1 (AP-1) to regulate the expression of genes involved in cell growth in response to exogenous stimuli such as ionising radiationCitation28. It has also been observed that TNFα stimulates the biosynthesis of TrxR, which by reducing Trx, leads to an increase in NFκB activityCitation25,Citation29,Citation30.
In many pathological conditions such as arterial hypertension, insulin resistance or abdominal obesity – which are part of the metabolic syndrome – and in chronic obstructive pulmonary disease (COPD), asthma, cystic fibrosis, as well as in Alzheimer’s and Parkinson’s disease, and in some cancers, increased expression of Trx and TrxR significantly disturbs redox homeostasisCitation30. It has been suggested that oxidative stress leading to increased TrxR biosynthesis may inhibit apoptosis, resulting in initiation of the neoplastic processCitation31. It has also been observed that Trx sensitises tumour cells to growth factors, increasing their proliferation. Therefore, it has been suggested that overexpression of Trx-dependent elements may intensify the process of oncogenesisCitation29. Therefore, some preparations used in cancer pharmacotherapy are characterised by the ability to permanently bind one of the components of the Trx-TrxR system to inhibit Trx or TrxR activityCitation4.
On the other hand, in vivo studies have shown that Trx-dependent neurotoxin-reducing effects are necessary to affect botulinum nerve cells (BoNT) and tetanus neurotoxin (TeNT). It has been observed that blocking of impulse conduction by BoNT and TeNT occurs only when the interchain disulphide bond in the toxin structure is reduced, which is possible in the presence of a reduced form of TrxCitation32. As BoNT- and TeNT-producing Clostridia bacteria are widespread, humans are often exposed to these toxins when wounds are infected or when contaminated food is eaten, which in serious cases, can lead to death. Consequently, the search for inhibitors of the Trx-TrxR system is also necessary in the context of treatment for neurotoxin poisoning, which is a serious toxicological and pharmacological problemCitation32. That is why for many years, research to find effective but selective methods of modifying the function of the Trx-TrxR system has been important. As a consequence, many natural chemical/physical factors and a whole range of synthetic compounds, mainly transition metal ions complexes, have been explored to target this system.
Modification of the Trx-TrxR System
Natural physicochemical factors modulating Trx – TrxR system activity
The biological activity of the Trx-TrxR system depends primarily on endogenous blocking inhibitors that bind at least one of the system’s components. An example of such a Trx-binding molecule is the Thioredoxin Interacting Protein (TXNIP, also called thioredoxin binding protein-2, TBP-2 or vitamin D3 stimulated protein 1, VDUP-1), which binds to TrxCitation6. TXNIP has 391 amino acidsCitation33, a molecular weight of 46 kDa, and contains two cysteines (at positions 63 and 247), which allow it to react directly with Trx. This interaction is key in regulating the Trx-dependent system and in regulating cellular homeostasisCitation34. The formation of a disulphide bond between Cys247 in TXNIP and Cys32 in Trx leads to the formation of a stable complex and blockade of the Trx catalytic centre, resulting in inhibition of the antioxidant action of the redox proteinCitation35. However, a TXNIP mutation that results in substitution of cysteine with serine at position 247 results in the elimination of its inhibitory effect on Trx. It is also important that Trx-TXNIP binding only occurs when Trx is in the reduced formCitation35,Citation36. Under physiological conditions, TXNIP is mainly located in the cell nucleus and cytosol, where it regulates the expression of Trx1. In contrast, oxidative stress causes nuclear TXNIP to be transported to the mitochondria where it can inhibit the action of both Trx isoformsCitation37,Citation38.
It is believed that in some cancers, TXNIP may act as a tumour suppressor as the overexpression of TXNIP leads to significant Trx binding, which disrupts redox homeostasis in tumour cells. Inactivation of Trx leads to oxidative stress, inhibits cell proliferation, and activates cascades of signalling pathways involving kinases that regulate apoptosisCitation39. Interestingly, in many cancers, a decrease in TXNIP expression is observed. For example, it has been shown that TXNIP expression in acute myelogenous leukaemia (AML) cells isolated from patients is decreased by up to 11 times and levels are also reduced in primary breast and colon cancersCitation39,Citation40. Therefore, to prevent and pharmacologically treat tumours, compounds that inhibit Trx-dependent system activity are being sought, and mainly those with natural origins.
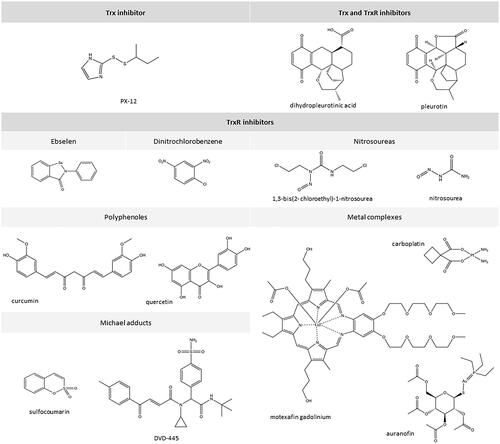
It has been found that the enzymatic activity of TrxR can be inhibited by some natural polyphenolic compounds including curcumin ()Citation41. Curcumin causes covalent modification of the C-terminal TrxR fragment, namely alkylation of both cysteine and selenocysteine. This irreversibly inhibits the activity of the enzyme, causing TrxR to lose its ability to reduce proteins through Trx, which promotes an increase in oxidative stress in cellsCitation41,Citation42. Similar effects have been observed with quercetin and myricetin, which like curcumin, can cause irreversible inhibition of TrxR activityCitation43. For myricetin, it has been observed that a TrxR mutant containing cysteine in position 498 does not prevent the oxidation of this flavanol. This suggests that the selenocysteine in the active centre of TrxR is crucial for the interaction between TrxR and flavanolsCitation44. Curcumin-induced alkylation of TrxR can have effects analogous to NADPH oxidase that involve significant increases in ROS production and increased oxidative stressCitation41.
Figure 4. Chemical structures of selected inhibitors of Trx-dependent system.
The only physical factor that has been found to inactivate TrxR is ultraviolet radiation. It has been shown that both UVA and UVB radiation from sunlight, even at a dose below the minimum erythema dose, causes inactivation of TrxR, which in turn, significantly reduces the antioxidant effect of the TrxR systemCitation45.
Among naturally occurring chemical compounds, some support the Trx-dependent system, leading to increased expression of its components. Among these is cannabidiol (CBD) – a compound found in the extracts of the Cannabis sativa L. plant – which has no psychoactive effect but has antioxidant, anti-inflammatory, and anticancer activityCitation46. In vitro studies have shown that at a concentration of 4 µM, CBD leads to an increase in both Trx and TrxR expression, while reducing the expression of GSH-dependent antioxidant system components including GSH itself, as well as GSH reductase and peroxidase, suggesting that CBD action may promote the activity of the Trx-TrxR systemCitation47.
Synthetic compounds that inhibit the activity of the Trx – TrxR system
In addition to naturally occurring Trx-dependent system inhibitors, many synthetic compounds can also modify the functioning of the Trx-TrxR system by inhibiting the biological activity of Trx or its reductase. This includes compounds from the group of disulphide imidazole derivatives, naphthoquinone derivatives and Michael acceptors as well as several organic and inorganic complex compounds containing transition metal ions as the central atom () ()Citation3.
Table 1. Summary of Trx-TrxR system inhibitors.
Synthetic inhibitors of Trx and TrxR
Spiroketal naphthoquinone derivatives such as pleurotin can be used to inhibit the Trx-TrxR system ()Citation48–50. Some compounds from this group can preferentially act as Trx inhibitors while others more effectively inactivate TrxR. Moreover, some of the compounds in this group are effective inhibitors of the Trx-TrxR system although they do not interact directly with either Trx or TrxR. This suggests that in addition to inhibiting the biological activity of Trx or TrxR, spiroketal derivatives of naphthoquinone may affect other cellular metabolic processes, e.g. by reducing HIF-1 expression levelsCitation51–53.
Synthetic inhibitors of Trx
Compounds from the group of alkyl-2-imidazolyl disulphides, including 1-methylpropyl-2-imidazolyl disulphide (known as PX-12 in formulation), which has been tested in clinical trials as a Trx1 inhibitor, can modulate the functioning of the Trx-TrxR systemCitation54,Citation55. PX-12 has been tested in the treatment of advanced, therapy-resistant cancers (e.g. non-small cell lung cancer, pancreatic, breast, and gastrointestinal cancers, as well as multiple myeloma)Citation56–57. The action of this group of chemicals is based on the thiol alkylation reaction of Cys32 and Cys35 of the catalytic centre and Cys73 outside the catalytic centre of Trx that they induce. It has been observed that the PX-12-mediated thiol alkylation reactions of Trx within Cys32 and Cys35 are reversible and rapid whereas reaction with Cys73 is irreversible and much slowerCitation58. This is important for the action of the Trx-TrxR system because the reaction with the cysteine residue located beyond the Trx catalytic centre causes the protein to be permanently inactivated. This results in a decrease in the expression of factors whose activity is associated with Trx antioxidant activity directly (e.g. factors induced by HIF-1α under hypoxia) as well as indirectly (e.g. vascular endothelial growth factor, VEGF, which is induced by HIF-1α)Citation56,Citation59. Additionally, inhibition of Trx and impairment of its binding to ASK-1 leads to the activation of mitogen-activated protein kinase (MAPK)Citation60. Together, these effects lead to a significant induction of apoptosis. In the case of simultaneous administration of PX-12 and cis-diaminodichloroplatin (a TrxR inhibitor), increased inhibition of tumour growth was observed in comparison to administration of platinum complex onlyCitation56,Citation59,Citation60 ().
Figure 5. Connection of the thioredoxin-dependent system to the cascade of transformations leading to cell apoptosis [according to MatsuzawaCitation61 in own modification].
![Figure 5. Connection of the thioredoxin-dependent system to the cascade of transformations leading to cell apoptosis [according to MatsuzawaCitation61 in own modification].](/cms/asset/06f379bf-5520-41bf-b14e-93216b642d36/ienz_a_1867121_f0005_c.jpg)
Synthetic inhibitors of TrxR
A significant group of compounds that can inhibit the activity of TrxR include complexes containing metal ions, mainly transient ones. Among chemicals that inhibit TrxR activity, some of the most effective are platinum and gold complexesCitation62,Citation63. Platinum complexes (cis-platinum, carboplatin) – which are used in the treatment of many cancers – irreversibly inhibit the activity of TrxR while maintaining the activity of other reductases (e.g. glutaredoxin or GSH reductase). This is because platinum complexes show a greater affinity for the selenol group (present in the TrxR active centre) compared to the thiol group (located in the GSH reductase active centre)Citation64. Gold compounds also inhibit TrxR activity, likely based on a similar mechanism as platinum complexesCitation65. In vitro studies have shown that auranofin – a complex containing gold at +1 oxidation level – is capable of inhibiting TrxR at a low, nanomolar level concentration whereas such an effect on glutathione reductase (GR) was only observed at higher micromolar concentrationsCitation66. Differences in auranofin concentrations needed to inhibit these enzymes were attributed to the presence of selenocysteine in the C-terminal motif of TrxR and its absence in GR. Additionally, it has been observed that during auranofin application, hydrogen peroxide production and cytochrome C secretion increases, which results in a change in the redox environment and consequently, apoptosisCitation67,Citation68. It has been suggested that gold ions that bind to cysteine and selenocysteine in the C-terminal motif of TrxR create a -Se-Au-S- motif, blocking the possibility of the enzyme to form disulphide bondsCitation68.
In addition, auranofin – apart from increasing the generation of hydrogen peroxide – can also lead to an increase in intracellular concentrations of calcium ions. Since an increase in ROS promotes the transport of calcium ions across the cell membraneCitation69, it is suggested that hydrogen peroxide may cause the release of intracellular calcium ions, which are involved in regulating metabolic functions in cellsCitation70. Calcium ions regulate the expression of various cytokines including IL-2 and IL-8, as well as COX2 and prostaglandin E2, influencing inflammatory processes in cells. Calcium gradients also participate in the homeostasis of permeability barriersCitation71,Citation72. Based on circular dichroism spectra, it was found that calcium ions acting on the TrxR1 isoform cause a change in the conformation of the enzyme, which leads to a reduction in its cytosolic activity, whereas such changes were not observed for TrxR2. Moreover, it is suggested that the oxidised form of Trx, acting by way of feedback, may partially protect TrxR against inactivating calcium ionsCitation73.
Lanthanum (III) chloride (LaCl3) also shows an inhibitory effect on TrxR activity, similar to that of calcium ions, which can be explained by the similarities between Ca2+ and La3+ ions. Like calcium ions, lanthanum ions have a stronger affinity for the cytosolic TrxR isoform than the mitochondrial reductase, which manifests itself in lower required concentrations to reduce TrxR activity, and increasing ROS levels only affect relatively low concentrations of lanthanum ionsCitation74,Citation75. Higher concentrations of La3+ ions (similar to calcium ions) inhibit TrxR, whereas no increase in ROS levels are observed, which suggests that lanthanum compounds may also act as scavengers of ROS. It is important to note that LaCl3 does not react directly with cysteine or selenocysteine in TrxR but forms a link with the enzyme outside its active centre, which changes the spatial arrangement of the polypeptide TrxR chain. This is explained by the fact that according to the theory of hard and soft acids and bases (HSAB), the La3+ ion is a hard acid that reacts with groups classified as hard bases (e.g. carboxylate ions or hydroxyl groups)Citation74. Additionally, it has been observed that LaCl3 does not show an inhibitory effect on GR activity, even when significant concentrations of the salt are usedCitation75.
Other compounds that inhibit TrxR activity include organic complexes containing tin (Sn) or ruthenium (Ru) ions at +4 and +3, and +2 oxidation levels, respectivelyCitation76,Citation77. Like other metal ions, ruthenium compounds inhibit the activity of only the cytosolic isoform of the TrxR, while the mitochondrial isoform does not show significant changes in activity even after administration of high concentrations of rutheniumCitation76. However, some Ru (II) compounds show an antiproliferative effect on melanoma cells similar to the effects of cisplatin, whereas other ruthenium compounds have more significant anti-proliferative effects than cis-platin. In the case of multi-drug resistant lung cancer cells insensitive to cisplatin, ruthenium complexes have been shown to significantly inhibit TrxR activity. Also, these compounds were shown to inhibit the activity of TrxR more effectively than auranofinCitation78. Tin compounds also inhibit the activity of TrxR; however, this activity is much weaker compared to gold or platinum compoundsCitation77.
The activity of TrxR is also inhibited by motexafin gadolinium (MGd), which is a drug used in anticancer therapy. Direct binding of MGd to TrxR has been observed, but not in the active centre of the enzymeCitation79. It has also been found that in the presence of suitable proteins (e.g. some metallothioneins), the administration of MGd may lead to the release of zinc as a byproduct of redox reactions, which is also an inhibitor TrxR. Thus, in addition to its direct inhibitory effects, MGd can also inhibit TrxR indirectly through generation of zinc ionsCitation79,Citation80.
In addition to the abovementioned organometallic compounds, TrxR activity is also inhibited by compounds containing rhodium (Rh) and copper (Cu) ions. It has been shown that compound complexes containing Rh(I) in their structure that inhibit the activity of TrxR also have an antiproliferative effect on cancer cells such as breast adenoma and colorectal cancer cellsCitation81,Citation82. However, copper (I) alkyl phosphine complexes have greater cytotoxicity towards cancer cells than analogous compounds containing Au + and Ag + as a central ion, and higher cytotoxicity than the reference compound cisplatinCitation83. The fact that Cu(I) complexes inhibit TrxR activity to a much lesser extent than the reference compound suggests that the toxic action of Cu-containing complexes against cancer cells does not result from apoptosis, but is caused by impairments to systems that regulate redox homeostasis.
Ebselen – an organic compound capable of reducing lipid peroxides and H2O2 – is another type of Trx inhibitor that results in the production of selenolic acid (EbSeOH). In mammalian cells, this compound is a substrate for TrxR, which reduces EbSeOH to an active selenolic form (EbSeH). Ebselen is a selective inhibitor that exclusively inhibits the activity of the bacterial form of TrxR, leading to an increase in ROS production, ultimately resulting in the death of bacterial cells. This is important for the potential use of ebselen for the treatment of bacterial infections, but the compound also has a protective effect on mammalian cellsCitation84.
The ability to interact and modify thiol and selenium TrxR groups is exhibited by electrophilic organic compounds from the groups of nitrosoureas, dinitrohalobenzenes, and polyphenolsCitation3,Citation85. These compounds may also bind to other functional groups, effectively inhibiting not only the activity of TrxR, but also blocking the activity of GR in reactions involving NADHCitation53. TrxR modified in this way cannot reduce molecules, and oxidised Trx cannot bind ASK-1, leading to the initiation of a signalling cascade resulting in apoptosisCitation86,Citation87. However, in vitro studies have shown that these reactions are reversible by the addition of an appropriately reduced protein. One of the most significant features of nitrosourea derivatives is their ability to cross the blood-brain barrier and influence the Trx-dependent system in brain cellsCitation88,Citation89.
Another pharmacologically important group of TrxR inhibitors are compounds that are classified as Michael acceptors. These include compounds containing electron-withdrawing groups (for instance ester or carbonyl group) connected with alkenes which interact directly with the active centre of the TrxRCitation90. The compound signed as DVD-445 deserves special attention because of its preferential effect on easily proliferating cancer cellsCitation90. This effect was noticed during a cell viability assay performed on human keratinocytes (HaCaT)Citation90,Citation91 as well as neuroblastoma (SH-SY5Y)Citation90,Citation91 and glioblastoma (U87 and U87-TxR) cell linesCitation92. Despite the effective selective action of DVD-445 against cancer cells, it has been observed that neuroblastoma cells remain viable at approximately 80%Citation90–92. In addition, some DVD-445 analogues also show inhibitory activity against TrxR activity, but not in cancer cellsCitation90. Therefore, the search for other TrxR inhibitors that will simultaneously inhibit the action of other pro-survival enzymes in relation to cancer cells is ongoing. Such a group of compounds includes the sulfocoumarinsCitation93 and other Michael acceptors which inhibit both thioredoxin reductase and carbonic anhydraseCitation94. The presence of electron withdrawing groups in their structures makes them act as Michael acceptors and thus have the ability to inhibit the activity of TrxRCitation93. A similar effect was observed for a synthetic compound that contains both the Michael acceptor group (responsible for the inhibition of TrxR) and the motif responsible for inhibiting carbonic anhydrase, which is very important in the process of carcinogenesis, because carbonic anhydrase protects cancer cells against acidification and hypoxia, preventing them death. This "hybrid" exhibits significant cytotoxicity to the pancreatic cancer cell line (PANC-1) compared to co-administered combinations of TrxR inhibitors and carbonic anhydrase inhibitorsCitation94. Consequently, due to the efficacy and preference for cancer cells, the synthesis and further research on Michael acceptors indicate the possible emergence of a compound effective in the treatment of highly multi-drug resistant cancers ().
Table 2. Summary of compounds inhibiting the efficiency of thioredoxin reductase.
Conclusion
The Trx-TrxR system is essential to maintaining cellular redox homeostasis. Cellular metabolism in normal physiological, as well as pathophysiological, conditions including carcinogenesis, depends on the efficiency of this system. Since the Trx-dependent system can both contribute to the intensification of angiogenesis, tumour growth, resistance to therapy, as well as drive changes that result in the death of cancer cells by apoptosis, the search for compounds that are inhibitors of Trx and/or TrxRs could modulate the tumoral activity of the Trx-dependent system. The functions of the Trx- and GSH-dependent systems complement each other, so a decrease in the performance of a Trx-dependent system usually increases the performance of a GSH-dependent system. Therefore, ideal inhibitors would simultaneously limit the function of both antioxidant systems.
Disclosure statement
No potential competing interest was reported by the author(s).
References
- Miller CG, Holmgren A, Arnér ES, Schmidt EE. NADPH-dependent and -independent disulfide reductase systems. Free Radic Biol Med 2018;127:248–61.
- Hashemy SI, Holmgren A. Regulation of the catalytic activity and structure of human thioredoxin 1 via oxidation and S-nitrosylation of cysteine residues. J Biol Chem 2008;283:21890–8.
- Gromer S, Urig S, Becker K. The thioredoxin system-from science to clinic. Med Res Rev 2004;24:40–89.
- Holmgren A, Lu J. Thioredoxin and thioredoxin reductase: current research with special reference to human disease. Biochem Biophys Res Commun 2010;396:120–4.
- Chen X, Tang W, Liu S, et al. Thioredoxin-1 phosphorylated at T100 is needed for its anti-apoptotic activity in HepG2 cancer cells. Life Sci 2010;87:254–60.
- Ahsan MK, Lekli I, Ray D, et al. Redox regulation of cell survival by the thioredoxin superfamily: an implication of redox gene therapy in the heart. Antioxid Redox Signal 2009;11:2741–58.
- Powis G, Mustacich D, Coon A. The role of the redox protein thioredoxin in cell growth and cancer. Free Radic Biol Med 2000;29:312–22.
- Fujiwara N, Fujii T, Fujii J, Taniguchi N. Roles of N-terminal active cysteines and C-terminal cysteine-selenocysteine in the catalytic mechanism of mammalian thioredoxin reductase. J Biochem 2001;129:803–12.
- Roos G, Foloppe N, Van Laer K, et al. How thioredoxin dissociates its mixed disulfide. PLoS Comput Biol 2009;5:e1000461.
- Collet JF, Messens J. Structure, function, and mechanism of thioredoxin proteins. Antioxid Redox Signal 2010;13:1205–16.
- Watanabe R, Nakamura H, Masutani H, Yodoi J. Anti-oxidative, anti-cancer and anti-inflammatory actions by thioredoxin 1 and thioredoxin-binding protein-2. Pharmacol Ther 2010;127:261–70.
- Chen W, Jiang Z, Lin N, et al. Evaluation of N-acetyl-S-(p-chlorophenylcarbamoyl) cysteine as an irreversible inhibitor of mammalian thioredoxin reductase1. J Enzyme Inhib Med 2016;31:229–35.
- Zhong L, Arnér ES, Holmgren A. Structure and mechanism of mammalian thioredoxin reductase: the active site is a redox-active selenolthiol/selenenylsulfide formed from the conserved cysteine-selenocysteine sequence. Proc Natl Acad Sci USA 2000;97:5854–9.
- Cortes-Bratti X, Bassères E, Herrera-Rodriguez F, et al. Thioredoxin 80-activated-monocytes (TAMs) inhibit the replication of intracellular pathogens. PLoS One 2011;6:e16960
- Lee S, Kim SM, Lee RT. Thioredoxin and thioredoxin target proteins: from molecular mechanisms to functional significance. Antioxid Redox Signal 2013;18:1165–207.
- Zhang B, Zhang J, Peng S, et al. Thioredoxin reductase inhibitors: a patent review. Expert Opin Ther Pat 2017;27:547–56.
- Krasavin M, Žalubovskis R, Grandāne A, et al. Sulfocoumarins as dual inhibitors of human carbonic anhydrase isoforms IX/XII and of human thioredoxin reductase. J. Enzyme Inhib. Med 2020;35:506–10.
- Brandstaedter C, Fritz-Wolf K, Weder S, et al. Kinetic characterization of wild-type and mutant human thioredoxin glutathione reductase defines its reaction and regulatory mechanisms. Febs J 2018;285:542–58.
- Rendón JL, Miranda-Leyva M, Guevara-Flores A, et al. Insight into the mechanistic basis of the hysteretic-like kinetic behavior of Thioredoxin-Glutathione Reductase (TGR). Enzyme Res 2018;2018:3215462– 17.
- Miranda-Vizuete A, Damdimopoulos AE, Pedrajas JR, et al. Human mitochondrial thioredoxin reductase: cDNA cloning, expression and genomic organization. Eur J Biochem 1999;261:405–12.
- Dobrovolska O, Rychkov G, Shumilina E, et al. Structural insights into interaction between mammalian methionine sulfoxide reductase B1, and thioredoxin. BioMed Res Int 2012;2012:1–9.
- Rhee SG, Chae HZ, Kim K. Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic Biol Med 2005;38:1543–52.
- Rhee SG. Overview on peroxiredoxin. Mol Cells 2016;39:1–5.
- Liao J, Wang K, Yao W, et al. Cloning, expression and antioxidant activity of a thioredoxin peroxidase from Branchiostoma belcheri tsingtaunese. PLoS One 2017;12:e0175162
- Yoshioka J, Schreiter ER, Lee RT. Role of thioredoxin in cell growth through interactions with signaling molecules. Antioxid Redox Signal 2006;8:2143–51.
- Liu Y, Min W. Thioredoxin promotes ASK1 ubiquitination, and degradation to inhibit ASK1-mediated apoptosis in a redox activity-independent manner. Circ Res 2002;90:1259–66.
- Wu Y, Qian Z, Fu S, et al. IcarisideII improves left ventricular remodeling in spontaneously hypertensive rats by inhibiting the ASK1-JNK/p38 signaling pathway. Eur J Pharmacol 2018;819:68–79.
- Karimpour S, Lou J, Lin LL, et al. Thioredoxin reductase regulates AP-1 activity as well as thioredoxin nuclear localization via active cysteines in response to ionizing radiation. Oncogene 2002;21:6317–27.
- Lincoln DT, Al-Yatama F, Mohammed FM, et al. Thioredoxin and thioredoxin reductase expression in thyroid cancer depends on tumour aggressiveness. Anticancer Res 2010;30:767–75.
- Mahmood DFD, Abderrazak A, El Hadri K, et al. The thioredoxin system as a therapeutic target in human health and disease. Antioxid Redox Signal 2013;19:1266–303.
- Arner ES, Holmgren A. The thioredoxin system in cancer. Semin Cancer Biol 2006;16:420–6.
- Pirazzini M, Zanetti G, Lista F, et al. The thioredoxin reductase-Thioredoxin redox system cleaves the interchain disulphide bond of botulinum neurotoxins on the cytosolic surface of synaptic vesicles. Toxicon 2015;107:32–6.
- Hu C, Zhang H, Qiao Z, et al. Loss of thioredoxin 2 alters mitochondrial respiratory function and induces cardiomyocyte hypertrophy. Exp Cell Res 2018;372:61–72.
- Yoshihara E, Masaki S, Matsuo Y, et al. Thioredoxin/Txnip: redoxisome, as a redox switch for the pathogenesis of diseases. Front Immunol 2014;4:514.
- Zhou J, Chng WJ. Roles of thioredoxin binding protein (TXNIP) in oxidative stress, apoptosis and cancer. Mitochondrion 2013;13:163–9.
- Yodoi J, Masutani H, Nakamura H. Redox regulation by the human thioredoxin system. Biofactors 2001;15:107–11.
- Lane T, Flam B, Lockey R, Kolliputi N. TXNIP shuttling: missing link between oxidative stress, and inflammasome activation. Front. Physiol 2013;4:50.
- Saxena G, Chen J, Shalev A. Intracellular shuttling and mitochondrial function of thioredoxin-interacting protein. J Biol Chem 2010;285:3997–4005.
- Zhou J, Bi C, Cheong LL, et al. The histone methyltransferase inhibitor, DZNep, up-regulates TXNIP, increases ROS production, and targets leukemia cells in AML. Blood 2011;118:2830–9.
- Morrison JA, Pike LA, Sams SB, et al. Thioredoxin interacting protein (TXNIP) is a novel tumor suppressor in thyroid cancer. Mol Cancer 2014;13:62
- Fang J, Lu J, Holmgren A. Thioredoxin reductase is irreversibly modified by curcumin: a novel molecular mechanism for its anticancer activity. J Biol Chem 2005;280:25284–90.
- Cai W, Zhang B, Duan D, et al. Curcumin targeting the thioredoxin system elevates oxidative stress in HeLa cells. Toxicol Appl Pharmacol 2012;262:341–8.
- Rodriguez-Garcia A, Hevia D, Mayo JC, et al. Thioredoxin 1 modulates apoptosis induced by bioactive compounds in prostate cancer cells. Redox Biol 2017;12:634–47.
- Lu J, Papp LV, Fang J, et al. Inhibition of mammalian thioredoxin reductase by some flavonoids: implications for myricetin and quercetin anticancer activity. Cancer Res 2006;66:4410–8.
- Jones DA. Rosacea, reactive oxygen species, and azelaic acid. J Clin Aesthet Dermatol 2009;2:26–30.
- Atalay S, Dobrzyńska I, Gęgotek A, Skrzydlewska E. Cannabidiol protects keratinocyte cell membranes following exposure to UVB and hydrogen peroxide. Redox Biol 2020;36:101613
- Jastrząb A, Gęgotek A, Skrzydlewska E. Cannabidiol regulates the expression of keratinocyte proteins involved in the inflammation process through transcriptional regulation. Cells 2019;8:827.
- Berdicevsky I, Kaufman G, Newman DJ, Horwitz BA. Preliminary study of activity of the thioredoxin inhibitor pleurotin against Trichophyton mentagrophytes: a novel anti-dermatophyte possibility. Mycoses 2009;52:313–7.
- Sweeney M, Coyle R, Kavanagh P, et al. Discovery of anti-cancer activity for benzo [1, 2, 4] triazin-7-ones: very strong correlation to pleurotin, and thioredoxin reductase inhibition. Bioorg Med Chem 2016;24:3565–70.
- Sandargo B, Thongbai B, Praditya D, et al. Antiviral 4-hydroxypleurogrisein, and antimicrobial pleurotin derivatives from cultures of the nematophagous basidiomycete Hohenbuehelia grisea. Molecules 2018;23:2697.
- Wipf P, Hopkins TD, Jung JK, et al. New inhibitors of the thioredoxin-thioredoxin reductase system based on a naphthoquinone spiroketal natural product lead. Bioorg Med Chem Lett 2001;11:2637–41.
- Powis G, Wipf P, Lynch SM, et al. Molecular pharmacology and antitumor activity of palmarumycin-based inhibitors of thioredoxin reductase. Mol Cancer Ther 2006;5:630–6.
- Mukherjee A, Martin SG. The thioredoxin system: a key target in tumour, and endothelial cells. Br J Radiol 2008;81:S57–S68.
- Baker AF, Dragovich T, Tate WR, et al. The antitumor thioredoxin-1 inhibitor PX-12 (1-methylpropyl 2-imidazolyl disulfide) decreases thioredoxin-1 and VEGF levels in cancer patient plasma. J Lab Clin Med 2006;147:83–90.
- Baker AF, Adab KN, Raghunand N, et al. A phase IB trial of 24-hour intravenous PX-12, a thioredoxin-1 inhibitor, in patients with advanced gastrointestinal cancers. Invest New Drugs 2013;31:631–41.
- Ramanathan RK, Stephenson JJ, Weiss GJ, et al. A phase I trial of PX-12, a small-molecule inhibitor of thioredoxin-1, administered as a 72-hour infusion every 21 days in patients with advanced cancers refractory to standard therapy. Invest New Drugs 2012;30:1591–6.
- Raninga PV, Di Trapani G, Vuckovic S, et al. Inhibition of thioredoxin 1 leads to apoptosis in drug-resistant multiple myeloma. Oncotarget 2015;6:15410–24.
- Li GZ, Liang HF, Liao B, et al. PX-12 inhibits the growth of hepatocelluar carcinoma by inducing S-phase arrest, ROS-dependent apoptosis, and enhances 5-FU cytotoxicity. Am J Transl Res 2015;7:1528–40.
- Naranjo-Suarez S, Carlson BA, Tobe R, et al. Regulation of HIF-1α activity by overexpression of thioredoxin is independent of thioredoxin reductase status. Mol Cells 2013;36:151–7.
- Yu T, Tang B, Sun X. Development of inhibitors targeting hypoxia-inducible factor 1 and 2 for Cancer Therapy. Yonsei Med J 2017;58:489–96.
- Matsuzawa A, Saegusa K, Noguchi T, et al. ROS-dependent activation of the TRAF6-ASK1-p38 pathway is selectively required for TLR4-mediated innate immunity. Nat Immunol 2005;6:587–92.
- Ortego L, Cardoso F, Martins S, et al. Strong inhibition of thioredoxin reductase by highly cytotoxic gold(I) complexes. DNA binding studies. J Inorg Biochem 2014;130:32–7.
- Cutillas N, Yellol GS, de Haro C, et al. Anticancer cyclometalated complexes of platinum group metals, and gold. Coord Chem Rev 2013;257:2784–97.
- Witte AB, Anestål K, Jerremalm E, et al. Inhibition of thioredoxin reductase but not of glutathione reductase by the major classes of alkylating and platinum-containing anticancer compounds. Free Radic Biol Med 2005;39:696–703.
- Hickey JL, Ruhayel RA, Barnard PJ, et al. Mitochondria-targeted chemotherapeutics: the rational design of gold(I) N-heterocyclic carbene complexes that are selectively toxic to cancer cells and target protein selenols in preference to thiols. J Am Chem Soc 2008;130:12570–1.
- Gandin V, Fernandes AP, Rigobello MP, et al. Cancer cell death induced by phosphine gold(I) compounds targeting thioredoxin reductase. Biochem Pharmacol 2010;79:90–101.
- Hwang-Bo H, Jeong JW, Han MH, et al. Auranofin, an inhibitor of thioredoxin reductase, induces apoptosis in hepatocellular carcinoma Hep3B cells by generation of reactive oxygen species. Gen Physiol Biophys 2017;36:117–28.
- Scalcon V, Bindoli A, Rigobello MP. Significance of the mitochondrial thioredoxin reductase in cancer cells: an update on role, targets and inhibitors. Free Radic Biol Med 2018;127:62–79.
- Harper MT. Auranofin, a thioredoxin reductase inhibitor, causes platelet death through calcium overload. Platelets 2019;30:98–104.
- Kamsler A, Segal M. Hydrogen peroxide as a diffusible signal molecule in synaptic plasticity. Mol Neurobiol 2004;29:167–78.
- Minke B. (2006). TRP channels and Ca2+ signaling. Cell Calcium 2006;40:261–75.
- Lee SE, Lee SH. Skin barrier and calcium. Ann Dermatol 2018;30:265–75.
- Rigobello MP, Vianello F, Folda A, et al. Differential effect of calcium ions on the cytosolic and mitochondrial thioredoxin reductase. Biochem Biophys Res Commun 2006;343:873–8.
- Zhang J, Li X, Han X, et al. Targeting the thioredoxin system for cancer therapy. Trends Pharmacol Sci 2017;38:794–808.
- Citta A, Folda A, Scutari G, et al. Inhibition of thioredoxin reductase by lanthanum chloride. J Inorg Biochem 2012;117:18–24.
- Mura P, Camalli M, Bindoli A, et al. Activity of rat cytosolic thioredoxin reductase is strongly decreased by trans-[bis (2-amino-5-methylthiazole) tetrachlororuthenate (III)]: first report of relevant thioredoxin reductase inhibition for a ruthenium compound. J Med Chem 2007;50:5871–4.
- de Oliveira KN, Andermark V, Onambele LA, et al. Organotin complexes containing carboxylate ligands with maleimide and naphthalimide derived partial structures: TrxR inhibition, cytotoxicity and activity in resistant cancer cells. Eur J Med Chem 2014;87:794–800.
- Zeng L, Chen Y, Liu J, et al. Ruthenium(II) complexes with 2-phenylimidazo[4,5-f][1,10]phenanthroline derivatives that strongly combat cisplatin-resistant tumor cells. Sci. Rep 2016;6:19449.
- Hashemy SI, Ungerstedt JS, Avval FZ, Holmgren A. Motexafin gadolinium, a tumor-selective drug targeting thioredoxin reductase and ribonucleotide reductase. J Biol Chem 2006;281:10691–7.
- Magda D, Miller RA. Motexafin gadolinium: a novel redox active drug for cancer therapy. Semin Cancer Biol 2006;16:466–76.
- Oehninger L, Küster LN, Schmidt C, et al. A chemical–biological evaluation of rhodium (I) N-heterocyclic carbene complexes as prospective anticancer drugs. Chem Eur J 2013;19:17871–80.
- Zhang JJ, Muenzner JK, el Maaty MAA, et al. A multi-target caffeine derived rhodium(i) N-heterocyclic carbene complex: evaluation of the mechanism of action. Dalton Trans 2016;45:13161–8.
- Santini C, Pellei M, Papini G, et al. In vitro antitumour activity of water soluble Cu(I), Ag(I) and Au(I) complexes supported by hydrophilic alkyl phosphine ligands. J Inorg Biochem 2011;105:232–40.
- Ren X, Zou L, Lu J, Holmgren A. Selenocysteine in mammalian thioredoxin reductase and application of ebselen as a therapeutic. Free Radic Biol Med 2018;127:238–47.
- Ju Y, Wu L, Yang G. Thioredoxin 1 regulation of protein S-desulfhydration. Biochem Biophys Rep 2016;5:27–34.
- Arnér ES, Zhong L, Holmgren A. Preparation and assay of mammalian thioredoxin and thioredoxin reductase. Methods in enzymology 1999;300:226–39.
- Gitler C, Danon A. (Eds.). Cellular implications of redox signaling. London: Imperial College Press; 2003.
- Purich D, The inhibitor index: a desk reference on enzyme inhibitors, receptor antagonists, drugs, toxins, poisons, biologics, and therapeutic leads. Boca Raton: CRC Press; 2017.
- Schallreuter KU, Gleason FK, Wood JM. The mechanism of action of the nitrosourea anti-tumor drugs on thioredoxin reductase, glutathione reductase, and ribonucleotide reductase. Biochim Biophys Acta Mol Cell Res 1990;1054:14–20.
- Jovanović M, Zhukovsky D, Podolski-Renić A, et al. Novel electrophilic amides amenable by the Ugi reaction perturb thioredoxin system via thioredoxin reductase 1 (TrxR1) inhibition: identification of DVD-445 as a new lead compound for anticancer therapy. Eur J Med Chem 2019;181:111580.
- Jovanović M, Zhukovsky D, Podolski-Renić A, et al. Further exploration of DVD-445 as a lead thioredoxin reductase (TrxR) inhibitor for cancer therapy: optimization of potency and evaluation of anticancer potential. Eur J Med Chem 2020;191:112119.
- Jovanović M, Dragoj M, Zhukovsky D, et al. Novel TrxR1 inhibitors show potential for glioma treatment by suppressing the invasion and sensitizing glioma cells to chemotherapy. Front Mol Biosci 2020;7:586146.
- Krasavin M, Žalubovskis R, Grandāne A, et al. Sulfocoumarins as dual inhibitors of human carbonic anhydrase isoforms IX/XII and of human thioredoxin reductase. J Enzyme Inhib Med Chem 2020;35:506–10.
- Krasavin M, Sharonova T, Sharoyko V, et al. Combining carbonic anhydrase and thioredoxin reductase inhibitory motifs within a single molecule dramatically increases its cytotoxicity. J Enzyme Inhib Med Chem 2020;35:665–71.
- Marzano C, Gandin V, Folda A, et al. Inhibition of thioredoxin reductase by auranofin induces apoptosis in cisplatin-resistant human ovarian cancer cells. Free Radic Biol Med 2007;42:872–81.
- Luo Z, Yu L, Yang F, et al. Ruthenium polypyridyl complexes as inducer of ROS-mediated apoptosis in cancer cells by targeting thioredoxin reductase. Metallomics 2014;6:1480–90.